

Abstract
Genetic variants are often not predictive of the phenotypic outcome. Individuals carrying the same pathogenic variant, associated with Mendelian or complex disease, can manifest to different extents, from severe to mild to no disease. Improving the accuracy of predicted clinical manifestations of genetic variants has emerged as one of the biggest challenges in precision medicine, which can only be addressed by understanding the mechanisms underlying genotype-phenotype relationships. Efforts to understand the molecular basis of these relationships have identified complex systems of interacting biomolecules that underlie cellular function. Here, we review recent advances in how modelling cellular systems as networks of interacting proteins has fueled identification of disease-associated processes, delineation of underlying molecular mechanisms, and prediction of the pathogenicity of variants. This review is intended to be inspiring for clinicians, geneticists and network biologists alike who aim to jointly advance our understanding of human disease and accelerate progress towards precision medicine.
Graphical Abstract
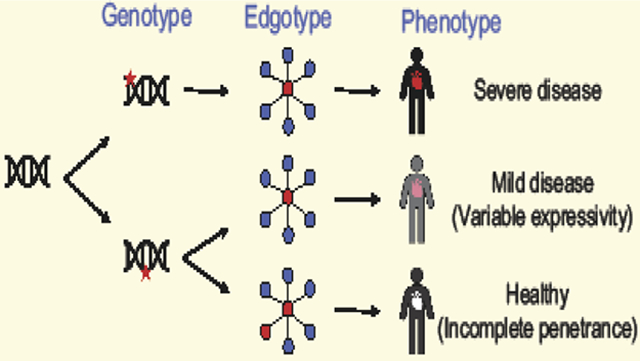
A network-based approach towards precision medicine
The cause of many diseases has been pinpointed to mutations in single genes, leading to apparent linear “one gene-one disease” relationships. However, the presence of a disease-associated mutation does not always result in manifestation of the disease. This phenomenon, where only a subset of individuals carrying a pathogenic mutation develop disease, is called incomplete penetrance and is increasingly being observed even in arguably simplistic Mendelian disorders. One such example is cystic fibrosis, an autosomal recessive pulmonary disorder associated with mutations in CFTR. While almost all patients suffering from cystic fibrosis have mutations in both CFTR alleles, not all individuals with disease-associated mutations in CFTR alleles manifest cystic fibrosis. The one gene-one disease paradigm is unable to explain why only ~ one-third males and one-twentieth females with mutations in both CFTR alleles develop the disease [1,2]. This example of incomplete penetrance is not the exception, but rather it appears to be the rule, even among well-studied “single gene” diseases [3]. Even in cases of very high penetrance, such as Huntington’s Disease, where a 40+ CAG triplet nucleotide repeat (encoding glutamine) in HTT leads to the disease in most probands [4], the age-of-onset and severity of the disease in an individual depend on variants in other modifier genes [5]. Surprisingly, even apparently healthy individuals carry 40 to 110 genetic mutations classified by the Human Gene Mutation Database (HGMD) as disease-associated, among which ~ 10–20% are found as homozygous [6], underscoring the ubiquity of incomplete penetrance. The observed widespread prevalence of incomplete penetrance strongly challenges a linear model of genotype-phenotype relationships (Figure 1).
Figure 1.
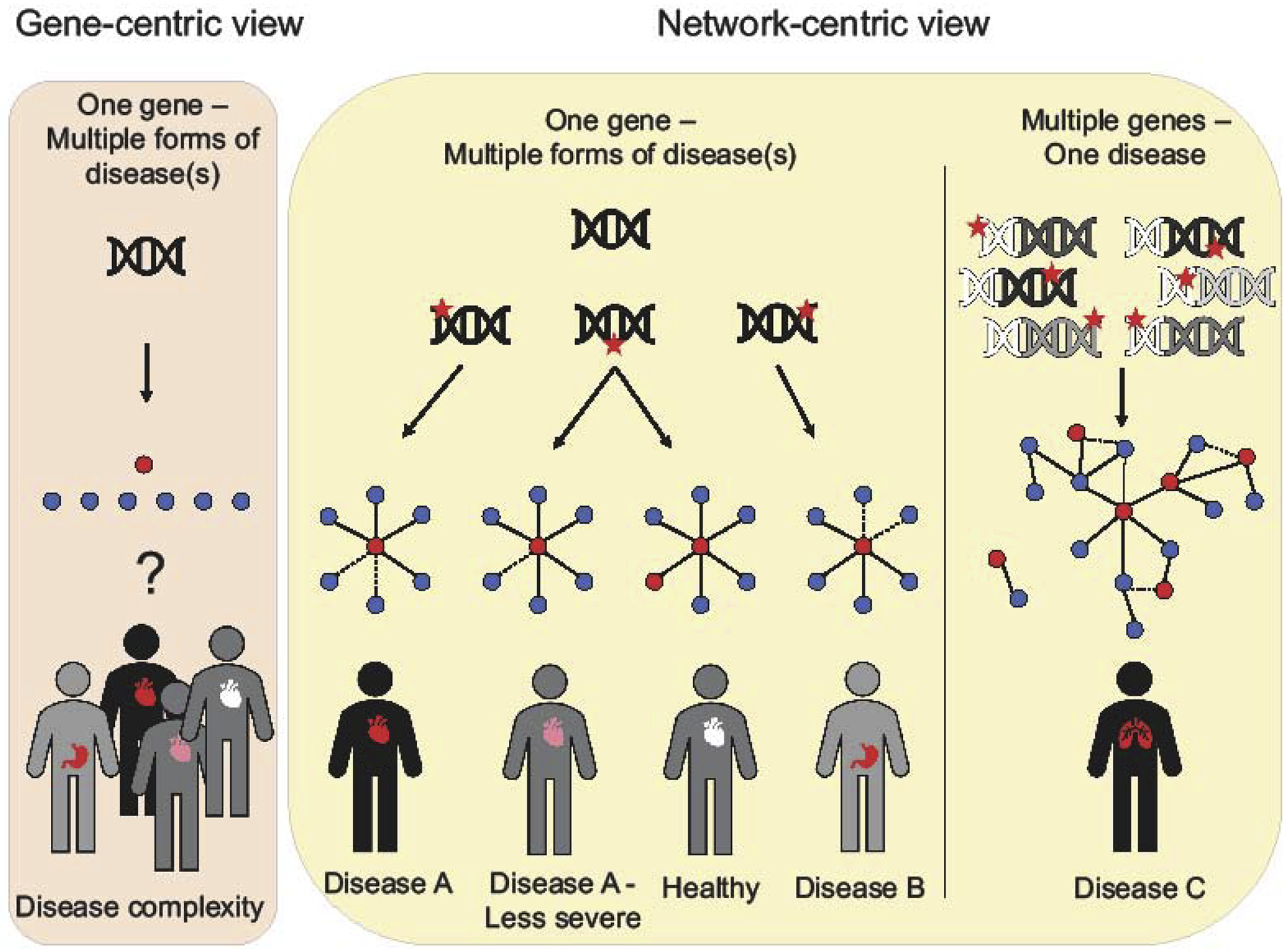
Comparison of a gene-centric versus network-centric approach to precision medicine. Red stars indicate mutations, either in non-coding (white DNA) or coding regions (colored DNA). Proteins with coding mutations or non-coding mutations, that affect their expression, in networks are shown as red nodes and their interaction partners as blue nodes. Solid lines indicate protein-protein interactions, dotted lines perturbed ones.
A growing body of scientific work suggests that at the root of these “non-linear” relationships is a complex interplay between genetic and epigenetic factors that is mediated by interactions between regulatory DNA sites, non-coding RNA, proteins and other molecules in the cell [7]. Together, these interactions constitute the cellular “interactome” which can be modelled as networks [7]. In order to accurately predict the phenotype from the genotype, it is important to be able to capture, model and manipulate these complex molecular relationships mediated by interactome networks.
The genetic complexity behind most phenotypes also illustrates that the disease associated mechanisms in individuals with the same or similar phenotype can be very different, and thus require genetically informed diagnosis and adapted treatment for optimal therapeutic outcomes. This idea forms the basic motivation for Precision Medicine, i.e. prevention, diagnosis, and treatment strategies that take individual variability into account [8]. In this review, we will discuss recent advances in how network-based approaches have contributed towards addressing the two main challenges in precision medicine: 1) accurate diagnosis of the genetic basis of a disease, and 2) understanding of the molecular mechanisms that result in disease pathology. While networks that model different kinds of functional relationships between biomolecules, i.e. coexpression, genetic interaction profile, or co-localization networks, have been successfully used for this purpose [9], here, we focus on concepts and recent applications of protein-protein interaction (PPI) networks for the characterization of both genetic variation and disease mechanisms. While improved understanding of the biophysical and topological properties of PPIs furthered their prediction [10,11], the most significant advances in using PPIs to understand human disease have been achieved using experimentally derived PPIs, which will be the focus of this review.
In the light of a growing understanding of non-linearity of genotype-phenotype relationships, we propose that the term ‘disease-causing variant’ is potentially unfitting and inapt. Current methodologies to identify a ‘causal variant’ are probabilistic, rather than deterministic, relying upon pedigree and epidemiology data. Moreover, as described above, even truly causal variants can be incompletely penetrant and not always result in disease. Erroneous identification of the causal variant can mislead patients about the disease, or worse, start them on an inappropriate treatment path. Therefore, we have made a conscious choice to use the term ‘disease-associated variants’ to refer to pathogenic variants from here on.
Advances in mapping human protein interaction networks
An accurate network analysis depends on the availability of comprehensive, unbiased and high-quality protein interaction data. Notable efforts to generate human PPI resources can be distinguished into those that systematically map PPIs at proteome-scale and those that curate protein interactions from the scientific literature. Approaches to systematically map human protein interactions have been developed since the mid-1990s with two distinct and complementary methodologies that clearly stand out. The first method consists of systematically testing every possible pair of proteins for interaction by expressing both partners exogenously in a cell-heterologous system [12–14]. While determining PPIs out of their endogenous cellular context, this approach is not limited by the set of proteins expressed in a given human cell line. Using this technology, the largest such screening effort interrogated 17,500-by-17,500 human protein pairs, generating the first human reference interactome map (HuRI) with about 53,000 binary and mostly direct protein interactions (interactome-atlas.org) [15]. The second technique systematically and exogenously expresses each human protein in a cell line and then affinity-purifies the protein and its associated interaction partners to identify protein complex data. The largest such effort, known as BioPlex, has generated data from about 6,000 affinity purifications corresponding to 56,000 protein associations (bioplex.hms.harvard.edu) [16,17]. These and other human protein interactome mapping efforts have been reviewed in more detail elsewhere [7,18].
Significant effort has gone into the development and application of curation standards [19] as well as workflows to capture PPI information from a rapidly growing body of scientific literature. Despite these achievements, applicability of literature-curated resources is impacted by the heterogeneity of experimental methods and protocols used to detect protein interactions, as well as variation in the reporting of such methodological information, including processing and quality control of raw data. Differences in quality might underlie significantly lower retest rates of PPIs with just one literature-curated piece of experimental evidence compared to PPIs with multiple pieces of evidence [14]. Furthermore, the protein interactome has been interrogated by the scientific community in a very uneven way, mostly focusing on a few genes of particularly high interest, which introduced an immense study bias [14].
When compared to literature-curated data, systematically generated maps produce high quality data at proteome-scale while reducing data heterogeneity and ascertainment bias (reviewed in [18]). For example, HuRI and BioPlex detect protein interactions just as well for highly versus poorly studied proteins, and thus connect disease-associated proteins much more uniformly, regardless of how well they or their interaction partners have been studied (Figure 2) [14–16]. This provides new opportunities to prioritize disease candidate proteins and to reveal molecular disease mechanisms. Of course, the screening platforms are not without technical biases of their own, such as increased detection of PPIs for more highly expressed proteins or depletion of transmembrane proteins [14–16]. Technical biases in systematic PPI maps and especially the aforementioned study bias in literature-curated datasets can significantly confound correlations between gene properties such as essentiality, intolerance to loss-of-function mutations, or age, and the number of protein interactions (degree) [15,20,21]. For example, do essential proteins appear to have a higher number of interactions in a PPI network because it reflects their importance for cellular function, or, because essential proteins are much better studied and more highly expressed, thus resulting in more curated and detected interactions [15,18,21,22]? Similar to other disciplines such as the social sciences, the network biology community must carefully control correlative analyses for possible confounders originating from biases in the underlying data.
Figure 2.
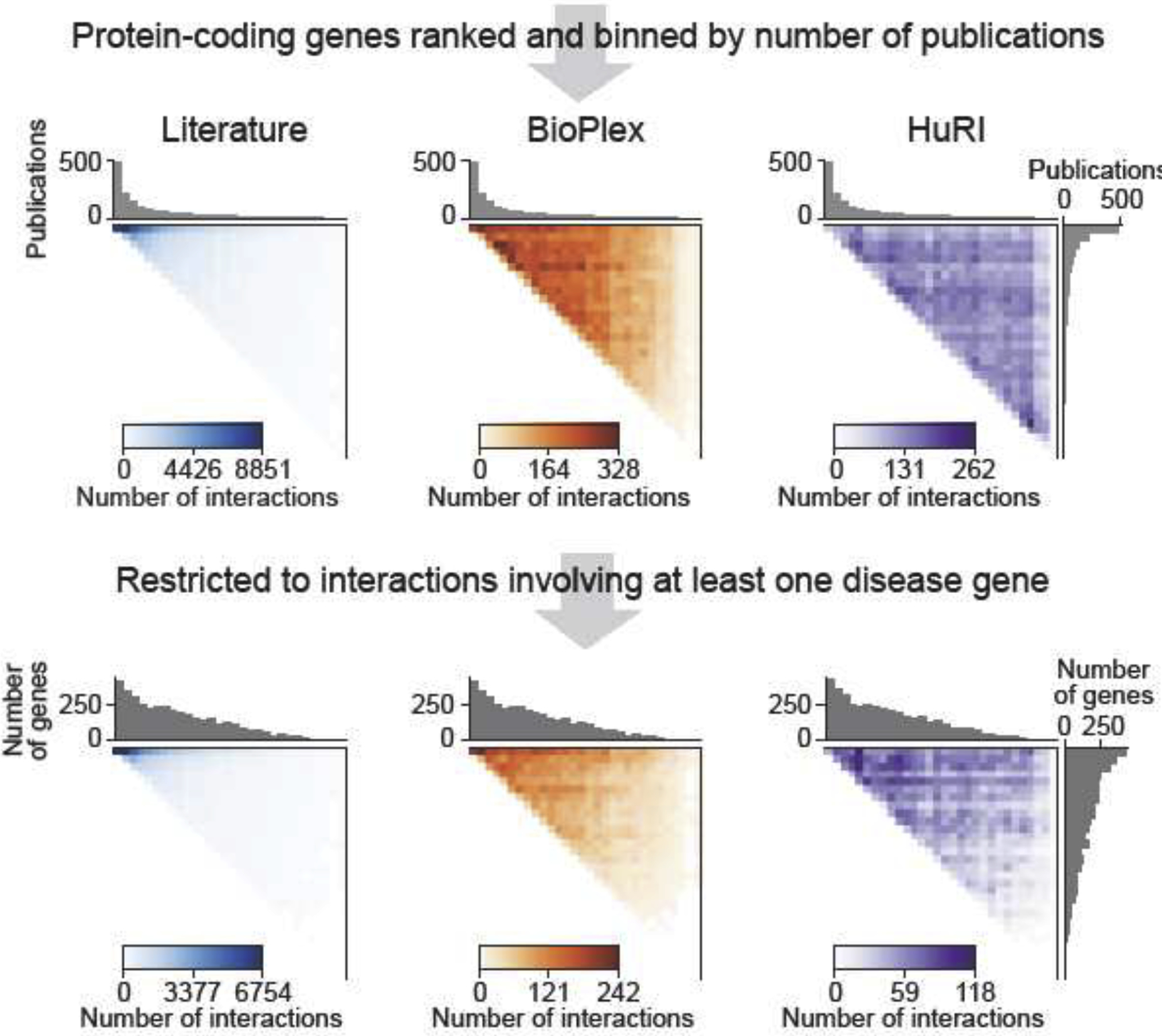
Coverage of the human protein-coding genes in different network maps. Coverage of all human protein-coding genes and disease genes (source OMIM) with protein-protein interaction data from different resources, as a function of how well genes have been studied. Literature-curated PPIs have been obtained from Mentha [89], BioPlex2.0 and HuRI from their respective websites (see text). Color ranges were set from 0 to maximum number of PPIs observed in a pair of gene bins for each respective heatmap.
The products of genes that are associated with a given disease often work together in the same or related biological processes that underlie the disease and these proteins tend to exhibit significantly higher connectivity among each other compared to randomized networks [23,24]. This observation paired with the fact that currently available protein interaction data are still highly incomplete [15,18], motivated more targeted protein interaction mapping efforts to increase the detection of PPIs between candidate disease genes. For example, protein interaction networks have been generated for ciliopathies [25], Parkinson’s Disease [26], and Autism Spectrum Disorder [27], expanding the molecular subnetworks for these diseases and enabling greater connectivity between implicated genomic elements. Various consortia have been proposed to more deeply map the protein interaction networks involving genes associated with cancer and psychiatric diseases [28,29].
Networks to identify genes and biological processes involved in disease
Despite identification of hundreds, and in some cases thousands, of genetic loci, most of the genes and biological processes that underlie complex diseases remain unknown. A genetic locus identified by genome-wide association (GWA) studies can contain hundreds of genes. Similarly, the burden of potentially damaging, missense rare variants (with an allele frequency less than 1%) is distributed across hundreds of protein-coding genes. Identification of disease-associated genes from this pool of candidate genes is crucial to begin to understand disease mechanisms. Disease-associated genes from i.e. different loci jointly contribute to the dysfunction of specific biological processes in disease. A pathway enrichment analysis between candidate genes has the potential to identify such disease-associated genes and the biological processes affected during disease. However, annotation of genes with their respective cellular functions is far from complete and biased towards well-characterized genes [30].
An alternative method to identify disease-associated genes from GWA loci, which does not depend on pre-existing functional annotations, consists of assessing the closeness of candidate genes in, ideally systematically generated, protein interaction networks. The potential of using network data to discover novel functional connections between disease-associated genes is highlighted by a recent study of the Commander complex. This complex was hitherto unknown but was identified in systematically generated protein complex data [31,32]. Subunits of this complex are highly conserved across metazoa and have been implicated in several developmental disorders [33].
The concept of disease modules has been put forward to describe the observation that genes that are associated with the same disease tend to be closer to each other in the network [24,34]. Approaches to identifying disease modules using biophysical, coexpression, and gene regulatory networks have been reviewed in detail elsewhere [9,35,36]. The concept of disease modules can be used to identify new disease genes based on their closeness to known ‘core’ disease genes. For example, it has previously been shown that proteins encoded by core cancer genes interact with each other more frequently than expected by chance in a systematically generated human protein interactome map [14]. Similarly, candidate cancer genes from GWA studies were found to interact with the core cancer genes in the network more often than expected by chance, and this information was used to prioritize candidate cancer genes from GWA loci. A similar methodology was used to identify novel Type 1 Diabetes genes [37]. Search of proteins within GWA loci, which interacted with proteins encoded by known diabetes genes, led to the identification of Huntingtin Interacting protein, HIP14. Downstream experiments established HIP14 as an anti-apoptotic protein required for beta-cell survival and insulin secretion [37].
Interactions between proteins of genes in GWA loci or of genes containing variants detected in whole genome and exome sequencing studies can be used to identify disease-relevant functional relationships between genes and affected biological processes even without relying on interactions with known disease genes. For example, a study integrated type 1 diabetes GWA data with protein interaction data to construct networks that might be relevant for the disease [38]. Some of the proteins in the networks harbored variants already known to be associated with diabetes in other studies. Selected candidate genes from this network analysis were enriched among differentially expressed genes in response to cytokine stimulation in insulin-secreting INS-1 beta-cells [38].
Complex diseases can also be associated with rare coding variants [39–41], however, identification of the disease-associated rare variant(s) and thus, the corresponding gene(s) is highly challenging. One possible way to detect such genes relies on the assumption that many of the proteins harboring disease-associated rare variants would likely work together in the same biological process(es), and thus protein interactions between identified candidates can be explored to further prioritize genes and gain insights into the disease biology. Indeed, using this approach various studies have identified novel disease-associated mechanisms [42,43]. For example, network data was employed to study the genes and biological processes that are likely affected by inherited and de novo variants from individuals diagnosed with Autism Spectrum Disorder (ASD) [44]. Genes carrying inherited coding variants, expected to have smaller effect sizes and reduced penetrance compared to de novo variants, were found to be enriched for cellular processes related to ion transport, the cell cycle, and the microtubule cytoskeleton, whereas genes carrying de novo variants showed enrichment for transcriptional and chromatin regulation. Interestingly, despite different functional enrichments, genes from both types of genetic variation were connected with each other in a protein interaction network suggesting that the disease mechanisms associated with either inherited or de novo variants ultimately converge to affect similar cellular function. This further demonstrates the ability of network data to uncover the biological processes underlying the disease.
Despite these and other successes, we would also like to point out three areas that require further development and increased caution.
The first area is related to the implications of study bias. Literature-curated protein interaction networks have frequently been used to perform network-based analyses in the biomedical field. However, inclusion of data curated from numerous studies, which specifically tested proteins for interaction that are associated with the same biological process or disease, have generally resulted in very uneven coverage of the interactome in which certain groups of genes appear more densely and closely connected than they might actually be within the context of the full interactome. Thus, detection of significant connectivity between disease genes when based on curated protein interaction data might at least in some cases be driven by study bias in the underlying network data. Whereas great attention has been paid to batch effects in the genomics and transcriptomics field, we argue that more attention has to be paid to similar confounders in network data [15].
A second area, which warrants more attention, is related to reporting of network biology findings. Often, the description of the experimental and computational procedures in published network analyses lacks information to evaluate the soundness of the conclusions being made. For example, it is important to describe whether and how randomizations were performed to calculate significances of reported observations. Was the number of interaction partners of every protein held constant during the network randomization process, and were proteins sampled while respecting the degree distribution in the original set of selected proteins?
Finally, a third area which has the most potential to advance the identification of disease mechanisms is to better utilize the information encoded in PPI datasets. Protein interaction network-based approaches inform on the actual biophysical relationships between the proteins and thus provide mechanistic insights. However, this potential is masked by nondiscriminatory aggregation of curated and predicted protein interaction data into second generation network databases [45] and analysis software without full transfer of information about the original experiment (e.g. co-elution profiling, affinity-purification, binary PPI assay) or other source (e.g. predicted based on orthologs or structural modelling) that reported the interaction. As a consequence, valuable data that can be used to generate testable hypotheses on the function of a reported interaction is lost. We suggest that aggregated PPI resources would benefit in their application if interactions were annotated with information on their likely biophysical and functional properties, i.e. whether an interaction is indirect or direct, conserved across species, involved in signaling, metabolism or other processes [46,47].
We are confident that all three points can relatively easily be addressed by the scientific community, thereby further increasing the impact of network-based discoveries in the area of precision medicine.
Networks to decipher disease mechanisms
Identification of disease-associated variants that confer higher risks for particular traits or phenotypes is a necessary, but far from sufficient step towards the dissection of disease mechanisms. Understanding the mechanism(s) of pathogenesis of such variants requires that we link them with perturbations of the functions of the corresponding gene products or non-coding RNA function, the surrounding network of molecular interactions, cellular processes, and finally the organismal phenotype. Over 50,000 pathogenic coding mutations have been identified in approximately 4,000 Mendelian disease genes [48]. Yet, for most of these mutations, our understanding of their impact on gene product functions and the resulting disease associated mechanisms falls short at the very first step. In this section, we discuss examples of how experimental and computational network-based methodologies are starting to uncover the functional effects of disease-associated variants.
A network-based approach can help identify the interactions, and corresponding functions perturbed by mutations, and in doing so provide insights into the mechanisms underlying the disease. Contrary to common belief, missense and even some nonsense variants often result in incomplete loss-of-function even when they are associated with rare Mendelian diseases. One way to globally assess compromised or complete loss-of-function is to assay whether mutated proteins are bound by chaperones [49]. In a recent survey it was found that about 70% of 2,200 mostly pathogenic variants remain free of any chaperone association, indicating that they encode fully folded and stable protein products [50]. Therefore, instead of complete loss-of-function, disease-associated variants likely impair a specific subset of the encoded protein’s functions and it is this impairment that underlies the disease. Such variants, that affect only specific interactions, or edges, while leaving most other interactions unperturbed, are called “edgetic” [51]. Testing a few hundred disease-associated variants showed that while 25% of them perturbed all the tested interactions, another 30% were edgetic, i.e. they specifically perturbed just a subset of the interactions mediated by the wild type protein [50]. In comparison, naturally segregating variants hardly perturbed any protein interactions [50]. Accordingly, disease-associated missense mutations have been found to be enriched both in the core of proteins and on their interaction interfaces [51–54]. These observations provide strong evidence that both destabilization of proteins and edgetic perturbation of specific protein interactions can mediate disease physiology. Conceptually, all known interactions e.g. protein-protein or DNA-protein interactions, and/or those mediating biochemical activities such as kinase, deacetylase activities or others can be systematically profiled, or edgotyped, for both wild-type and mutated products to begin to dissect mechanisms underlying disease [51].
One of the key strengths of the concept of network-biology to understand genotype-phenotype relationships is its ability to provide insights into mechanisms underlying complex, non-linear phenomena such as pleiotropy. Pleiotropy describes the ability of a gene to mediate multiple functions [55]. A gene can exhibit pleiotropy by interacting with biomolecules involved in different biological processes [51]. Different mutations in a pleiotropic gene can result in physiologically distinct diseases by edgetically perturbing different subsets of interactions mediated by the encoded protein [51,56]. In one such case mutations in a pleiotropic gene encoding slow muscle alpha-tropomyosin, TPM3, can lead to two distinct diseases, fiber-type disproportion myopathy and nemaline myopathy, through hitherto unknown mechanisms. Mutations that are associated with fiber-type disproportion myopathy perturb five of ten interaction partners of the wild type protein [50]. One of the perturbed interactions is between TPM3 and troponin, which has been shown to be vital for transduction of calcium-induced signals required for muscle contraction. Other perturbed interaction partners i.e. heat shock transcription factor 2, HSF2, and coiled-coil alpha-helical rod protein1, CCHCR1, are important for myotube generation and cytoskeleton organization, respectively. In comparison, a mutation in TPM3 that results in nemaline myopathy does not perturb any of these edges, proposing a potential association between edgetic perturbation and physiological outcome.
Studying the effects of mutations on protein interactions has not only helped to understand the molecular mechanisms underlying monogenic diseases, it also provided mechanistic insights into diseases associated with multiple genes, mutations in which together contribute to the manifestation or severity of a disease in an individual [57]. For example, retinitis pigmentosa, a common ciliopathy in which dysfunction of cilia in retina results in severe inflammation, can be associated with mutations in USH2A, encoding a membrane protein, usherin. However, the severity of the disease depends on mutations in other genes. A study showed that two sisters, both carrying the same pathogenic mutation in USH2A, manifested retinitis to different degrees [58]. The sister with the more severe and early onset form of retinitis also carried a mutation in a protein interactor of USH2A called PDZD7. Unlike a single mutation in USH2A, mutations in both USH2A and PDZD7 perturbed the edge between the two proteins suggesting a critical role of the interaction in the severity of the disease [58]. Mutations in PDZD7 have been found to co-occur with mutations in USH2A in patients suffering from Usher’s syndrome, another ciliopathy affecting the inner ear [58] suggesting a similar role of the protein interaction in the digenic mode of inheritance of the disease. In contrast, restoration of perturbed protein interactions by compensatory mutations in the same protein [59] or its interaction partner [60] has the potential to restore the disease phenotype or reduce its severity. This requirement for co-occurrence of mutations on genes encoding interacting proteins represents a molecular mechanism that might underlie incomplete penetrance of disease variants.
Although at lower frequency, pathogenic variants can also lead to a gain-of-interaction resulting in disease. GLUT1 deficiency syndrome, a disorder characterized by seizures and intellectual disability with onset in early infancy, is associated with mutations in a major glucose transporter in brain, GLUT1. A disease-associated mutation, P485L, creates a dileucine motif that leads to a gain-of-interaction of GLUT1 with clathrin adaptor protein AP-2, culminating in GLUT1 mislocalization and loss of glucose transport [61]. Another such example is in the gene encoding p110 alpha, the catalytic subunit of phosphatidylinositol 3-kinase alpha, PIK3CA, which is frequently mutated in human cancers. One of the hot-spot mutations in PIK3CA, encoding E545K, leads to its gain-of-interaction with insulin receptor substrate 1, IRS1 [62]. The gained interaction stabilizes the mutant PIK3CA leading to its constitutive activation and tumor growth. Abrogation of the interaction in both GLUT1 and PIK3CA reverted the disease phenotype in model systems. We want to point out that such gain-of-interactions involving disease proteins are an attractive target for therapeutic intervention.
These examples also highlight the diversity in which pathogenic variants can alter protein function. Capturing this diversity more systematically and at higher throughput requires assessment of that variant not only for its potential to perturb or gain PPIs but also for its impact on protein stability and expression levels, protein localization, binding to other molecules, such as DNA, RNA, and metabolites, etc. Availability of wild type Open Reading Frames (ORFs) and generation of resources of mutant ORFs in multipurpose vectors is essential for efficient profiling of variants across these many molecular and functional assays [50,63,64].
In parallel to advances on the experimental side, there have been computational efforts that use protein interaction data to predict disease mechanisms. Predicting the perturbation of specific protein interactions by a variant requires structural information on the interaction interfaces that mediate the protein interactions under consideration. Several resources have been created in which protein interaction data have been annotated with structural information on interaction interfaces [65–67]. Genetic variants have been mapped onto these structurally resolved protein interactions to predict edgetic perturbation of interactions. For example, colorectal cancer is associated with mutations in several members of the mismatch repair machinery including MLH1 and PMS2. The mismatch repair activity of MLH1 depends on its interaction with PMS2. Three cancer associated mutations in MLH1 mapped onto a predicted interaction interface and instead of affecting the stability of MLH1, edgetically perturbed the interaction between MLH1 and PMS2 [66]. Another example is Rapp Hodgkin Syndrome, an ectodermal dysplasia associated with mutations in a member of the p53 family of transcription factors, TP63. Most pathogenic mutations in TP63 tend to cluster on an interface that has been predicted to mediate interaction with another p53 like protein, TP73 [66], indicating a role of this interaction in pathogenesis of the disease. In another case, known mutations associated with neurodevelopmental disorders tend to cluster on the interaction interface between guanine nucleotide exchange factor TRIO and GTPase RAC1 strongly suggesting a role of the interaction in the disease [68].
Similar to experimental interaction profiling described above, predicting edgetic perturbation by mutations can also sometimes provide insights into mechanisms underlying disease pleiotropy. For example, mutations in a protein involved in transducing signals from cell surface receptors to the actin cytoskeleton, WASP, can lead to three different disorders, Wiskott-Aldrich syndrome (WAS), X-linked thrombocytopenia (XLT), or clinically more distinct X-linked neutropenia (XLN) [69]. While mutations associated with WAS and XLT are enriched on one interface of WASP and were predicted to perturb an interaction between WASP and vasodilator-stimulated phosphoprotein, VASP, mutations that are associated with XLN are enriched on another interface and were shown to perturb an interaction between WASP and a cell cycle GTPase, CDC42 [66].
Despite progress in computationally predicting the effect of protein variants based on protein structure information, the subset of human protein interactions with structurally resolved or predicted interaction interfaces remains extremely small (far less than 10,000) and very likely highly biased both towards the set of well characterized disease genes and the protein interactions that are mediated by domain-domain interaction interfaces [65]. More computational and experimental efforts are needed that incorporate protein interactions mediated by disordered regions in proteins [70,71] and that characterize novel types of protein interaction interfaces.
Networks to characterize genetic variation
Concepts
One of the core ideas of precision medicine is that identification of the pathogenic variant within a patient would allow for selection of the most effective clinical intervention. For example, if pathogenic mutations are detected in breast cancer-associated genes such as BRCA1/2 or in cardiomyopathy-associated genes such as LMNA or TNNT2, then highly effective disease-preventing or disease-mitigating interventions can be implemented [72]. However, identification of the pathogenic mutation in an individual, even in cases where we know most of the genes that underlie a disease, remains a highly challenging task because even known disease-associated genes can harbor variants that are entirely benign. Discriminating pathogenic from benign variants is especially difficult as over 95% of the variants detected in human populations are rare and over 50% of all variants have so far been found only once in the human population [73]. The rarity of these variants renders a standard association or enrichment analysis with a control group of healthy individuals virtually infeasible. An average human being, independently of whether they are healthy or not, carries 300–500 coding variants predicted to be highly damaging, with 40–85 in homozygous state distributed throughout their protein-coding genome [6,74]. Moreover, some common coding variants might not be completely benign, as they can increase disease risk or exhibit incomplete penetrance. Identifying truly pathogenic variants among the many rare and common variants in a diseased individual, then, is akin to identifying a needle in a haystack. For most of the variants, sufficient statistical power does not exist to conclusively classify them as pathogenic or benign and ironically, attempts to increase this statistical power by sequencing more individuals has resulted in the identification of even more rare variants [75]. Variants that are rare, appear damaging, but lack sufficient epidemiological or functional evidence, are termed Variants of Uncertain Significance (VUSs) [76]. ClinVar currently reports around 200,000 coding VUSs, four times the number of catalogued pathogenic variants [48].
Computational methods based on protein conservation and structure can be used to predict the functional effects of a large number of variants [77]. However, these programs were found to be inaccurate 30% of the time [78]. On the other hand, functional assays that experimentally assess the effects of variants on the function of a gene products [79] have only been developed for a few disease genes. As reviewed earlier, a growing body of scientific work demonstrates the physiological relevance of protein interactions and how pathogenic but not benign variants perturb interactions that are critical for normal cellular function [50,68]. This observation can be used in a scalable and systematic way to assess the pathogenic potential of the growing number of VUSs. By first mapping the effect of known pathogenic variants on protein interactions or any other types of interactions, we can identify interactions that are perturbed by the majority of the pathogenic variants and hence have a higher likelihood of being relevant for the disease. A VUS, if indeed disease associated, would then tend to perturb the same “likely pathogenic” interactions. On the other hand, a VUS that is actually benign should not perturb any of these “likely pathogenic” interactions (Figure 3). For example, a mutation in the retinoic acid receptor alpha, RARA, found in an Autism proband, was predicted and experimentally confirmed to disrupt interaction with retinoic acid receptor beta, RARB, whereas another mutation found in an unaffected sibling did not [68]. Of note, PolyPhen 2 predicted both mutations to be probably damaging [68]. In another case, edgetic profile similarity was used to correctly predict the effect of a rare variant in the GTPase SEPT12 on sperm motility [80]. Such edgotyping does not rely on pre-existing knowledge of the variant, as required by some computational predictors, or prior understanding of disease mechanisms as required by some functional assays, and is applicable to other types of molecular interactions. Yet, its broad feasibility remains to be demonstrated and, as outlined here, represents only one possible scenario of disease-associated versus benign PPI perturbation.
Figure 3.
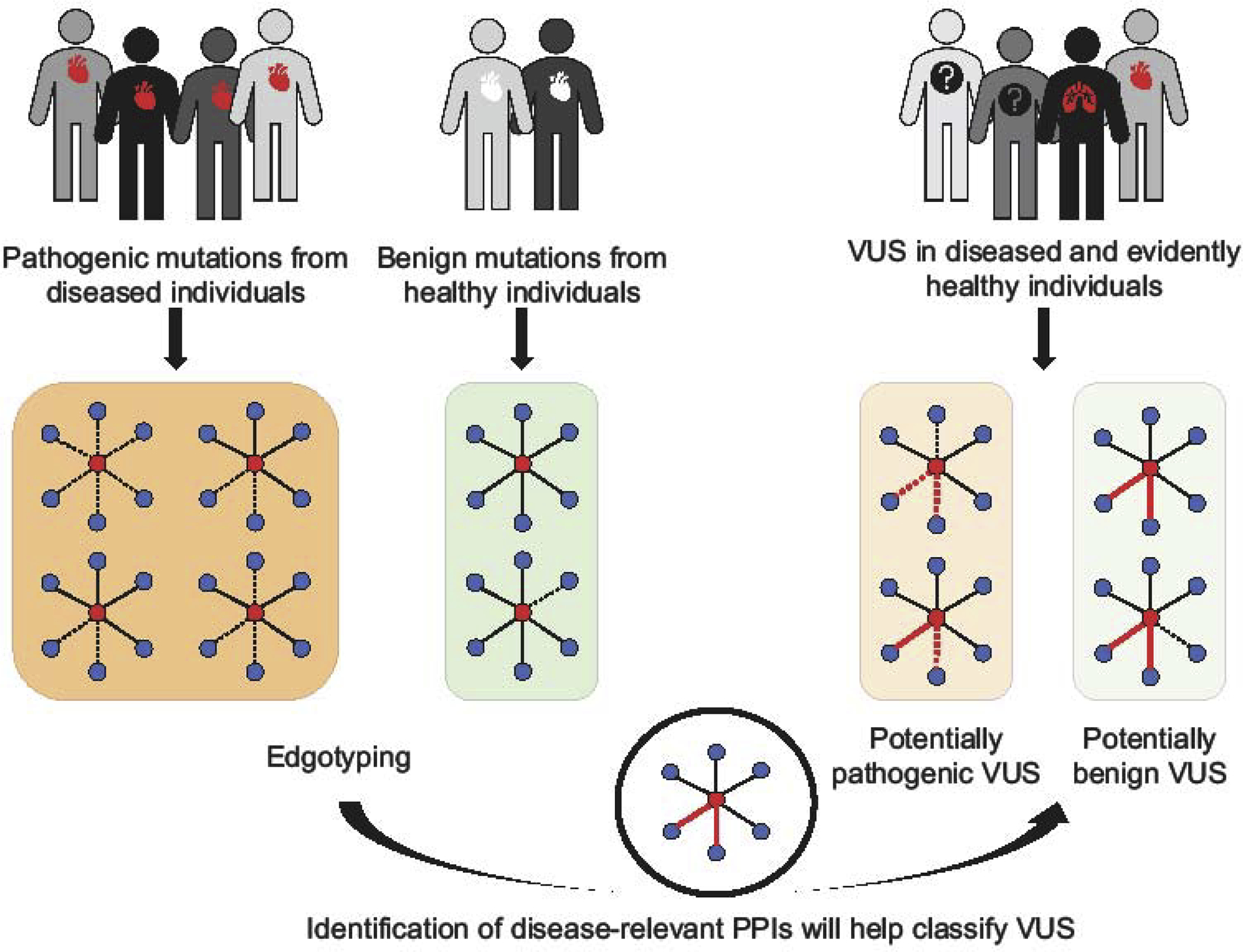
Illustration of an edgotyping approach to predict the pathogenicity of VUSs. Networks are drawn as in Figure 1. Red edges indicate predicted disease-relevant protein interactions.
Software developed to predict the potential of genetic variants to perturb interactions, can likewise be used to predict the pathogenicity of VUSs [67,81–83]. For example, primary hyperoxaluria, a rare condition characterized by recurrent kidney and bladder stones, is associated with mutations in a glyoxylate and hydroxypyruvate reductase, GRHPR. The enzymatic activity of GRHPR requires dimerization [84]. Two known pathogenic mutations in GRHPR, R302H and E113K, were predicted to affect the binding stability of the homodimer [83]. It is therefore plausible that a VUS, R171H, found in hyperoxaluria patients and predicted to impact the binding affinity of the homodimer, could be potentially pathogenic. However, the biological and clinical validity of the prediction remains to be demonstrated. Literature curated datasets of experimentally derived effects of mutation on protein interaction perturbation [85] can serve as benchmarking tools for such variant effect prediction tools.
Challenges
Using edgotyping to predict the pathogenicity of variants is a promising concept, yet multiple challenges aimed towards increasing the accuracy of these predictions remain to be solved.
Firstly, although being an incredibly rich source of mechanistic information, protein interaction networks are still far from complete, i.e. probably less than 20% of all direct PPIs are known to-date [15]. Therefore, it is likely that a considerable number of disease-relevant interactions remain to be discovered.
Secondly, the vast majority of PPIs have been detected out of their natural context, i.e. either by expressing and testing both interacting proteins in an in vitro or cell-heterologous system or by exogenously expressing and affinity-purifying a bait protein with its partners from a cell line. Consequently, for most protein interactions we lack information on the relevant cellular context, i.e. tissue, cell type, and developmental stage, at which a given PPI is functional. Some of the identified protein interactions, even though biophysically real, might not exist under any physiological context. These ‘pseudointeractions’ [86] might correspond in some cases to ancient interactions that have lost physiological relevance, or, like hidden genetic variation that serves as a reservoir for evolution, may act as a pool of interactions that might become relevant in certain disease or stress conditions where gene expression and protein localization are altered. Rapidly growing but still highly incomplete understanding of physiological states of cell-types and tissues make it challenging to conclusively differentiate between biologically real PPIs and pseudointeractions. Given these limitations, comparing edgetic profiles of benign with pathogenic variants might not always be sufficient to identify disease-associated PPI perturbations. Integration of PPI perturbation data with contextual information, such as gene and protein expression data as well as protein localization data, can be used to further identify disease-associated PPI perturbations as has been attempted in the context of tissue-specific diseases [15]. In fact, for 80% of the 1,000 known tissue-specific diseases, the disease-associated genes seem uniformly expressed across tissues [15]. Yet, mutations in these uniformly expressed genes manifest in only specific tissues. Perturbation of PPIs between a uniformly expressed disease-associated protein and tissue-specific interaction partners can mediate the tissue-specific manifestation of the disease [87]. We recently tested the effects of mutations in ten uniformly expressed genes that are associated with tissue-specific diseases and found that in seven cases the mutations perturbed PPIs with proteins that were significantly preferentially expressed in the disease-associated tissue [15]. Even though these results do not establish a definite disease-associated relationship, the method likely represents an efficient way to identify potentially disease-associated PPIs for further experimental follow-up.
Lastly, perturbation of PPIs is often assumed to result in severe phenotypic outcomes, possibly because of the following observations that have been made: (a) protein interactions appear to be highly conserved, (b) comparatively few coding mutations have been identified in complex diseases, (c) Mendelian disease mutations were found to be enriched both in the core and on interaction interfaces of proteins, and, (d) only a small number of common variants seemed to be able to perturb PPIs. It has therefore been suggested that mutations that perturb PPIs are likely damaging [80], and alternatively, that PPIs perturbed by benign or common variants, are irrelevant and dispensable [88]. However, recent studies show that coding variants display a range of phenotypic effects [39–41] from truly benign to having a small effect on increasing disease risk to being the primary cause of disease. It is thus highly possible that molecular interactions, and their perturbation, will similarly exhibit a continuum of how strongly they impact cellular function. While perturbation of certain PPIs will be the primary cause of a disease, others might “only” increase the disease risk or contribute to inter-individual differences in non-pathogenic phenotypic traits (Figure 4). Variant classification using edgotyping would, therefore, need to account for a continuum of both genetic effects and the functional relevance of PPIs. Annotation of biophysical networks, i.e. via integration with other functional genomic and proteomic data, will help determine the physiological relevance of PPIs and thus, potentially allow for a more accurate prediction of the phenotypic effects of genetic variation.
Figure 4.
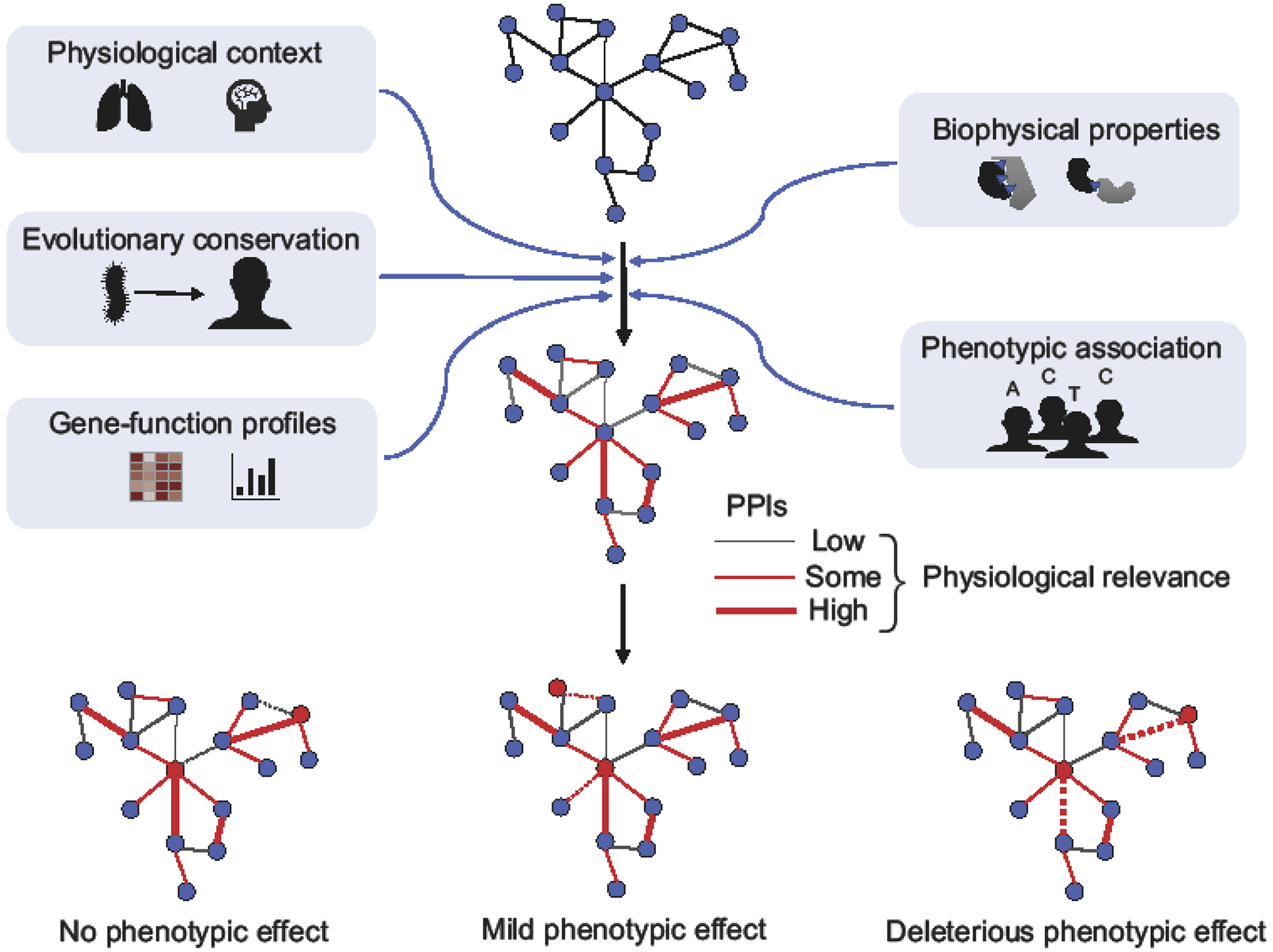
Illustration of an integrative approach to predict the physiological relevance of protein interactions.
Reference networks of high quality biophysical protein interactions can be integrated with various sources of information to infer which PPIs likely exist under which physiological context and how impactful their function might be. Networks are drawn as in Figure 1. Red edges indicate PPIs of physiological relevance for the cellular context under study and thickness of the edge indicates the extent of physiological relevance. Red nodes indicate mutated proteins.
Conclusion
A non-linear, network-based understanding of genotype-phenotype relationships, as described in this review, cautions against recently suggested, ill-informed applications of genome-editing. No gene functions in isolation or mediates a single function. An incomplete understanding of the complex cellular system is bound to result in misguided and potentially harmful therapeutic interventions. Interrogating this system from a network perspective is essential to better understand disease mechanisms and address long standing questions in incomplete penetrance and missing heritability. Important concepts have been developed and significant achievements are being made by employing a network-based approach to precision medicine. However, the way towards reaching more accurate diagnosis and treatment is long and will benefit from improved physiological models of molecular interaction networks, better tools to interrogate the function of molecular interactions in vivo, and closer collaboration between the network and translational sciences.
Highlights.
One gene-one disease paradigm rarely explains incomplete penetrance.
Cellular systems of interacting molecules mediate genotype-to-phenotype relationships.
Protein-protein interactions are being systematically mapped at proteome-scale.
Protein-protein interaction networks help identify molecular disease mechanisms.
Edgotyping emerges as a tool to characterize variants of uncertain significance.
Acknowledgements
We thank L. Lambourne for help with generating figures and various members of the Vidal lab for critical feedback on the manuscript. Funding: This work was supported by NHGRI grants U41HG001715, P50HG004233. M.V. is a Chercheur Qualifié Honoraire from the Fonds de la Recherche Scientifique (FRS-FNRS, Wallonia-Brussels Federation, Belgium).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
M.V. is a shareholder and scientific advisor of seqWell, Inc.
References
- 1.Sun W, Anderson B, Redman J, Milunsky A, Buller A, McGinniss MJ, Quan F, Anguiano A, Huang S, Hantash F, et al. : CFTR 5T variant has a low penetrance in females that is partially attributable to its haplotype. Genet Med 2006, 8:339–345. [DOI] [PubMed] [Google Scholar]
- 2.Strom CM, Crossley B, Redman JB, Buller A, Quan F, Peng M, McGinnis M, Sun W: Cystic fibrosis screening: lessons learned from the first 320,000 patients. Genet Med Off J Am Coll Med Genet 2004, 6:136–140. [DOI] [PubMed] [Google Scholar]
- 3.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H: Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 2013, 132:1077–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, International Huntington’s Disease Collaborative Group: A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004, 65:267–277. [DOI] [PubMed] [Google Scholar]
- 5.Lee J-M, Wheeler VC, Chao MJ, Vonsattel JPG, Pinto RM, Lucente D, Abu-Elneel K, Ramos EM, Mysore JS, Gillis T, et al. : Identification of genetic factors that modify clinical onset of huntington’s disease. Cell 2015, 162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, et al. : Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet 2012, 91:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vidal M, Cusick ME, Barabási A-L: Interactome networks and human disease. Cell 2011, 144:986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins FS, Varmus H: A new initiative on precision medicine. N Engl J Med 2015, 372:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonawane AR, Weiss ST, Glass K, Sharma A: Network medicine in the age of biomedical big data. Front Genet 2019, 10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. : Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovács IA, Luck K, Spirohn K, Wang Y, Pollis C, Schlabach S, Bian W, Kim D-K, Kishore N, Hao T, et al. : Network-based prediction of protein interactions. Nat Commun 2019, 10:1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rual J-F, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. : Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437:1173–1178. [DOI] [PubMed] [Google Scholar]
- 13.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. : A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005, 122:957–968. [DOI] [PubMed] [Google Scholar]
- 14.Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. : A proteome-scale map of the human interactome network. Cell 2014, 159:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luck K, Kim D-K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, et al. : A reference map of the human protein interactome. bioRxiv 2019, 605451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. : Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. : The BioPlex network: a systematic exploration of the human interactome. Cell 2015, 162:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luck K, Sheynkman GM, Zhang I, Vidal M: Proteome-scale human interactomics. Trends Biochem Sci 2017, 42:342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FSL, Cesareni G, et al. : Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat Methods 2012, 9:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coulomb S, Bauer M, Bernard D, Marsolier-Kergoat M-C: Gene essentiality and the topology of protein interaction networks. Proc Biol Sci 2005, 272:1721–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanic J, Yu X, Wallqvist A, Reifman J: Influence of protein abundance on high-throughput protein-protein interaction detection. PloS One 2009, 4:e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. : High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barabási A-L, Oltvai ZN: Network biology: understanding the cell’s functional organization. Nat Rev Genet 2004, 5:101–113. [DOI] [PubMed] [Google Scholar]
- 24.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabasi A-L: Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347:1257601–1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen T-MT, Texier Y, van Beersum SEC, Horn N, Willer JR, Mans DA, et al. : An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun 2016, 7:11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khurana V, Peng J, Chung CY, Auluck PK, Fanning S, Tardiff DF, Bartels T, Koeva M, Eichhorn SW, Benyamini H, et al. : Genome-scale networks link neurodegenerative disease genes to α-synuclein through specific molecular pathways. Cell Syst 2017, 4:157–170.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L, Broly M, Rodriguez M, Tam S, Trigg SA, et al. : Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat Commun 2014, 5:3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T: The cancer cell map initiative: defining the hallmark networks of cancer. Mol Cell 2015, 58:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willsey AJ, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, Baum TB, Cagney G, Bender KJ, Desai TA, et al. : The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 2018, 174:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandey AK, Lu L, Wang X, Homayouni R, Williams RW: Functionally enigmatic genes: a case study of the brain ignorome. PloS One 2014, 9:e88889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. : A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163:712–723. [DOI] [PubMed] [Google Scholar]
- 32.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. : Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mallam AL, Marcotte EM: Systems-wide studies uncover commander, a multiprotein complex essential to human development. Cell Syst 2017, 4:483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartwell LH, Hopfield JJ, Leibler S, Murray AW: From molecular to modular cell biology. Nature 1999, 402:C47–52. [DOI] [PubMed] [Google Scholar]
- 35.Capriotti E, Ozturk K, Carter H: Integrating molecular networks with genetic variant interpretation for precision medicine. Wiley Interdiscip Rev Syst Biol Med 2019, 11:e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao V, Wong AK, Troyanskaya OG: Enabling precision medicine through integrative network models. J Mol Biol 2018, 430:2913–2923. [DOI] [PubMed] [Google Scholar]
- 37.Berchtold LA, Størling ZM, Ortis F, Lage K, Bang-Berthelsen C, Bergholdt R, Hald J, Brorsson CA, Eizirik DL, Pociot F, et al. : Huntingtin-interacting protein 14 is a type 1 diabetes candidate protein regulating insulin secretion and beta-cell apoptosis. Proc Natl Acad Sci U S A 2011, 108:E681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergholdt R, Brorsson C, Palleja A, Berchtold LA, Fløyel T, Bang-Berthelsen CH, Frederiksen KS, Jensen LJ, Størling J, Pociot F: Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 2012, 61:954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.CHD Exome+ Consortium, EPIC-CVD Consortium, ExomeBP Consortium, Global Lipids Genetic Consortium, GoT2D Genes Consortium, EPIC InterAct Consortium, INTERVAL Study, ReproGen Consortium, T2D-Genes Consortium, The MAGIC Investigators, et al. : Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet 2018, 50:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.International Inflammatory Bowel Disease Genetics Consortium, Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, Andersen V, Cleynen I, Cortes A, et al. : Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The EPIC-InterAct Consortium, CHD Exome+ Consortium, ExomeBP Consortium, T2D-Genes Consortium, GoT2D Genes Consortium, Global Lipids Genetics Consortium, ReproGen Consortium, MAGIC Investigators, Marouli E, Graff M, et al. : Rare and low-frequency coding variants alter human adult height. Nature 2017, 542:186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. : Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segre AV, Roberts AE, Smoot LB, Pu WT, Pereira AC, et al. : Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc Natl Acad Sci 2012, 109:14035–14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruzzo EK, Pérez-Cano L, Jung J-Y, Wang L-K, Kashef-Haghighi D, Hartl C, Singh C, Xu J, Hoekstra JN, Leventhal O, et al. : Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178:850–866.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Stærfeldt HH, et al. : A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 2017, 14:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. : The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010, 38:W214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotlyar M, Pastrello C, Malik Z, Jurisica I: IID 2018 update: context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res 2019, 47:D581–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. : ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016, 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wegele H, Müller L, Buchner J: Hsp70 and Hsp90--a relay team for protein folding. Rev Physiol Biochem Pharmacol 2004, 151:1–44. [DOI] [PubMed] [Google Scholar]
- 50.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, et al. : Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Q, Simonis N, Li Q-R, Charloteaux B, Heuze F, Klitgord N, Tam S, Yu H, Venkatesan K, Mou D, et al. : Edgetic perturbation models of human inherited disorders. Mol Syst Biol 2009, 5:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao M, Zhou H, Skolnick J: Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure 2015, 23:1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engin HB, Kreisberg JF, Carter H: Structure-based analysis reveals cancer missense mutations target protein interaction interfaces. PLOS ONE 2016, 11:e0152929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang F, Petsalaki E, Rolland T, Hill DE, Vidal M, Roth FP: Protein domain-level landscape of cancer-type-specific somatic mutations. PLoS Comput Biol 2015, 11:e1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paaby AB, Rockman MV: The many faces of pleiotropy. Trends Genet TIG 2013, 29:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang J, Von den Hoff J, Lange J, Ren Y, Bian Z, Carels CEL: MSX1 mutations and associated disease phenotypes: genotype-phenotype relations. Eur J Hum Genet EJHG 2016, 24:1663–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gazzo AM, Daneels D, Cilia E, Bonduelle M, Abramowicz M, Van Dooren S, Smits G, Lenaerts T: DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res 2016, 44:D900–D907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schäfer E, Roux A-F, Dafinger C, Bernd A, et al. : PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 2010, 120:1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karras GI, Yi S, Sahni N, Fischer M, Xie J, Vidal M, D’Andrea AD, Whitesell L, Lindquist S: HSP90 shapes the consequences of human genetic variation. Cell 2017, 168:856–866.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, et al. : Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet 2007, 3:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer K, Kirchner M, Uyar B, Cheng J-Y, Russo G, Hernandez-Miranda LR, Szymborska A, Zauber H, Rudolph I-M, Willnow TE, et al. : Mutations in disordered regions can cause disease by creating dileucine motifs. Cell 2018, 175:239–253.e17. [DOI] [PubMed] [Google Scholar]
- 62.Hao Y, Wang C, Cao B, Hirsch BM, Song J, Markowitz SD, Ewing RM, Sedwick D, Liu L, Zheng W, et al. : Gain of interaction with IRS1 by p110α-helical domain mutants is crucial for their oncogenic functions. Cancer Cell 2013, 23:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ORFeome Collaboration: The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat Methods 2016, 13:191–192. [DOI] [PubMed] [Google Scholar]
- 64.Wei X, Das J, Fragoza R, Liang J, Bastos de Oliveira FM, Lee HR, Wang X, Mort M, Stenson PD, Cooper DN, et al. : A massively parallel pipeline to clone DNA variants and examine molecular phenotypes of human disease mutations. PLoS Genet 2014, 10:e1004819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer MJ, Beltrán JF, Liang S, Fragoza R, Rumack A, Liang J, Wei X, Yu H: Interactome INSIDER: a structural interactome browser for genomic studies. Nat Methods 2018, 15:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H: Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol 2012, 30:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mosca R, Tenorio-Laranga J, Olivella R, Alcalde V, Céol A, Soler-López M, Aloy P: dSysMap: exploring the edgetic role of disease mutations. Nat Methods 2015, 12:167–168. [DOI] [PubMed] [Google Scholar]
- 68.Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, Devlin B, Yu H: An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat Genet 2018, 50:1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thrasher AJ, Burns SO: WASP: a key immunological multitasker. Nat Rev Immunol 2010, 10:182–192. [DOI] [PubMed] [Google Scholar]
- 70.Dinkel H, Van Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A, et al. : ELM 2016--data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 2016, 44:D294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tompa P, Davey NE, Gibson TJ, Babu MM: A million peptide motifs for the molecular biologist. Mol Cell 2014, 55:161–169. [DOI] [PubMed] [Google Scholar]
- 72.Hunter JE, Irving SA, Biesecker LG, Buchanan A, Jensen B, Lee K, Martin CL, Milko L, Muessig K, Niehaus AD, et al. : A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med Off J Am Coll Med Genet 2016, 18:1258–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. : Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. : Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manolio TA, Fowler DM, Starita LM, Haendel MA, MacArthur DG, Biesecker LG, Worthey E, Chisholm RL, Green ED, Jacob HJ, et al. : Bedside back to bench: building bridges between basic and clinical genomic research. Cell 2017, 169:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. : Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet 2015, 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adzhubei I, Jordan DM, Sunyaev SR: Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013, Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun S, Yang F, Tan G, Costanzo M, Oughtred R, Hirschman J, Theesfeld CL, Bansal P, Sahni N, Yi S, et al. : An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res 2016, 26:670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM: Variant interpretation: functional assays to the rescue. Am J Hum Genet 2017, 101:315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fragoza R, Das J, Wierbowski SD, Liang J, Tran TN, Liang S, Beltran JF, Rivera-Erick CA, Ye K, Wang T-Y, et al. : Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat Commun 2019, 10:4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li M, Simonetti FL, Goncearenco A, Panchenko AR: MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res 2016, 44:W494–W501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiong P, Zhang C, Zheng W, Zhang Y: BindProfX: assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts. J Mol Biol 2017, 429:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wagih O, Galardini M, Busby BP, Memon D, Typas A, Beltrao P: A resource of variant effect predictions of single nucleotide variants in model organisms. Mol Syst Biol 2018, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Booth MPS, Conners R, Rumsby G, Brady RL: Structural basis of substrate specificity in human glyoxylate reductase/hydroxypyruvate reductase. J Mol Biol 2006, 360:178–189. [DOI] [PubMed] [Google Scholar]
- 85.The IMEx Consortium Curators, del-Toro N, Duesbury M, Koch M, Perfetto L, Shrivastava A, Ochoa D, Wagih O, Piñero J, Kotlyar M, et al. : Capturing variation impact on molecular interactions in the IMEx Consortium mutations data set. Nat Commun 2019, 10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Venkatesan K, Rual J-F, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh K-I, et al. : An empirical framework for binary interactome mapping. Nat Methods 2009, 6:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barshir R, Shwartz O, Smoly IY, Yeger-Lotem E: Comparative analysis of human tissue interactomes reveals factors leading to tissue-specific manifestation of hereditary diseases. PLoS Comput Biol 2014, 10:e1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghadie M, Xia Y: Estimating dispensable content in the human interactome. Nat Commun 2019, 10:3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Calderone A, Castagnoli L, Cesareni G: Mentha: a resource for browsing integrated protein-interaction networks. Nat Methods 2013, 10:690–691. [DOI] [PubMed] [Google Scholar]