

Abstract
Objective.
Iaccarino et al [1] exposed one hour of light flickering at 40 Hz to awake 5XFAD Alzheimer’s Disease (AD) mouse models, generating action potentials at 40 Hz, activating ~54% of microglia to colocalize with Aβ plaque, acutely, and clearing ~ 50% of Aβ plaque after seven days, but only in the visual cortex.
Hypothesis.
Transcranially delivered, focused ultrasound (tFUS) can replicate the results of Iaccarino et al [1] but throughout its area of application.
Methods.
We exposed sedated 5XFAD mice to tFUS (2.0 MHz carrier frequency, 40 Hz pulse repetition frequency, 400 microsecond-long pulses, spatial peak pulse average value of 190 W/cm2). Acute studies targeted tFUS into one hemisphere of brain centered on its hippocampus for one hour. Chronic studies targeted comparable brain in each hemisphere for one hour/day for five days.
Results.
Acute application of tFUS activated more microglia that colocalized with Aβ plaque relative to sham ultrasound (36.0 +/− 4.6% versus 14.2 +/− 2.6% [mean +/− standard error], z = 2.45, p < 0.014) and relative to the contralateral hemisphere of treated brain (36.0 +/− 4.6% versus 14.3 +/− 4.0%, z = 2.61, p < 0.009). Chronic application over five days reduced their Aβ plaque burden by nearly half relative to paired sham animals (47.4 +/− 5.8%, z = 2.79, p < 0.005).
Conclusion.
Our results compare to those of Iaccarino et al [1] but throughout the area of ultrasound-exposed brain. Our results also compare to those achieved by medications that target Aβ, but over a substantially shorter period of time. The proximity of our ultrasound protocol to those shown safe for non-human primates and humans may motivate its rapid translation to human studies.
Graphical Abstract
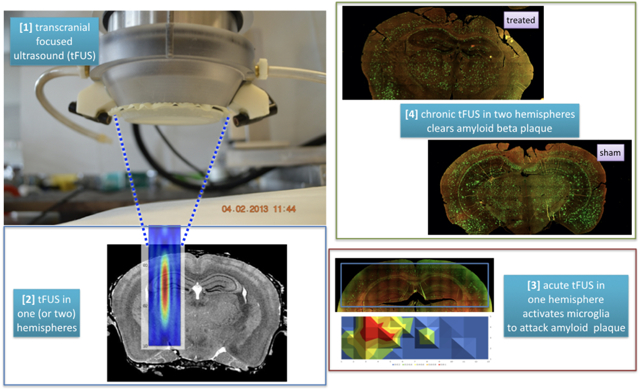
Introduction.
Dementia incurs devastating individual human cost with an increasingly substantial societal burden, with Alzheimer’s Disease (AD) representing the clinically most diagnosed form of dementia, seconded by vascular dementia [2]. AD’s histopathological definition and associated hypothesis guides the majority of existing treatment paradigms to date: excessive distribution of the protein amyloidβ (Aβ) external to neurons and subsequently evolving neurofibrillary tangles within neurons lead to eventual neuronal damage and associated decline in clinical signs and symptoms [2,3]. Unfortunately, medications that target reduction of excessive Aβ have failed thus far when tested primarily on late-stage AD patients [4–6]. Here we contribute to device-based means of reducing Aβ plaque burden through activation of microglia, a recently active field that seeks to treat dementia based on a range of hypotheses (excessive Aβ; cerebral hypotension; inadequate autophagy; among others). Of likely interest, because of the complementary biophysical mechanisms through which ultrasound can have influence, future studies may one day create an optimal protocol that can address dementia through multiple disease mechanisms.
Light and sound.
Iaccarino et al [1] showed that intermittent stimulation of the visual cortex of 5XFAD AD mouse models with 40 Hz flickering light caused 53.9% of microglia there to both activate and co-localize with Aβ plaque, acutely. Application of this same protocol for one hour per day for seven days reduced insoluble Aβ1–40 and Aβ1–42 by 43.7% and 57.9% respectively, with comparable reduction of soluble Aβ as compared to sham exposure. This occurred only in the visual cortex, the portion of brain activated by the light. This same group [7] has shown that chronic auditory and visual stimulation at 40 Hz reduced Aβ plaque in auditory and visual cortex, the CA1, and medial prefrontal cortex by ~56, 72, 70 and 58%, respectively, relative to no stimulation.
Generalization of these 40 Hz protocols to the entire brain may open up new avenues for clearing Aβ, possibly neurofibrillary tangles, and may therefore lead to improved patient outcomes. Ultrasound has the potential to do so.
Disruption of the blood brain barrier (BBBD) with ultrasound plus acoustic contrast agents.
BBBD with ultrasound plus acoustic contrast agents can improve histopathology and behavior in mouse models of AD. Burgess et al [8] applied their technique to TgCRND8 mice once a week for three consecutive weeks followed by a week of behavioral testing. They observed a reduction in Aβ burden by 19% and improved behavior by 50% relative to controls. Leinenga and Gotz [9] applied their specific ultrasound protocol, necessarily with contrast agents [10], to two-year-old APP23 mice for several minutes/day, four days/week, two weeks/month for a total of two months. The BBBD they induced reduced Aβ burden by 58% relative to controls. Of note, their recent extension of this work shows promising reduction of tau and associated improvement in motor function through activation of intraneuronal autophagy [11].
Unfortunately, BBB disruption is not without risk. For example, use of osmotic agents that disrupt the BBB within an entire hemisphere of brain can produce well-documented problems in the initial hours or days such as epileptic seizure among other serious issues [12]. BBBD via ultrasound plus contrast agents also has the potential to cause unwanted effects including sterile inflammation [13]. There exists an on-going debate in the literature on this subject, with attention to possible transient versus permanent biological effects, appropriate doses and types of acoustic contrast agents, among other issues [13–16]. Also of interest, 0.5% or fewer of patients who receive acoustic contrast agents experience excruciating lower back pain due to an adverse reaction between the agents and the kidney (see the packaging on the following acoustic contrast agents: Definity, Optison and Sonovue). Nonetheless, initial pre-clinical studies of individual episodes of BBBD in small volumes of human brain have not produced any meaningful clinical nor radiological adverse events [17]. Moreover, Aryal et al [18], Horodyckid et al [19] and Pandit et al [11] have demonstrated that repeated BBBD with ultrasound plus contrast agents within a range of volumes of rat, nonhuman primate and AD mouse brain, respectively, have produced neither histopathological or behavioral deficits.
Enhancement of cerebrovascular flow with ultrasound.
Eguchi et al [20] delivered transcranial and unfocused ultrasound with a pulse repetition frequency (PRF) of 1 kHz and pulse length of 16 microseconds into 5XFAD mice for twenty minutes per day at each of three targets in brain, for three days per week, one week per month, for three months. This enhanced cerebral blood flow and net co-localization of activated and non-activated microglia with Aβ plaque by 15% and reduced Aβ plaque burden by nearly 40% relative to control mice. They also observed an increase by 15% in spontaneous alternations in the Y maze, a metric for improved short-term memory. Importantly, their studies connected these observations causally to production of endothelial nitric oxide synthase (eNOS) – a potent endogenous vasodilator – consistent with peripheral application of ultrasound [21]. These are promising results, especially with the possibility of greater Aβ plaque clearance if amendments to their ultrasound protocol could activate more microglia while retaining its ability to dilate the cerebrovasculature.
Can ultrasound without contrast agents improve AD histopathology in vivo through activation of central neurons a la Iaccarino et al [1] and Martorell et al [7]?
There exists rapidly evolving literature demonstrating that transcranially delivered ultrasound can temporarily and safely activate central neural circuits, shown in rodents, sheep, primates, and humans [22,23]. Here we demonstrate that transcranially delivered, focused ultrasound (tFUS), pulsed at 40 Hz and delivered without acoustic contrast agents, can activate neurons in the brains of AD mice, can activate a substantial percentage of microglia acutely, and, after five days of daily application clear nearly 50% of Aβ plaque across a large area of the brains of 5XFAD AD mice.
Methods.
Overview of experimental procedures.
Animal model.
All animal procedures were approved by the University of Washington Institutional Animal Care and Use Committee under protocol #4084–08, and conformed to applicable national guidelines. We used, along with Eguchi et al [20] and Iaccarino et al [1] six-month-old male 5XFAD (C57BL/6) transgenic mice ([24], MMRRC stock #34840; Jackson Laboratory, Maine, USA). This model overexpresses mutant human APP(695) with the Swedish (K670N/M671L), Florida (I716V), and London (V717I) Familial AD (FAD) mutations and human PS1 with two FAD mutations, M146L and L286V [25]. 5XFAD (C57BL/6) mice present with Aβ plaque emerging significantly between 2 and 4 months of age [24,26].
Time line of acute study.
We treated five sedated mice acutely with tFUS on the left side of their brains with the ultrasound focus placed at 3.5 mm posterior of Bregma, −2.2 mm from midline, 1.9 mm below the skin surface, into the center of the hippocampus. We applied tFUS for one hour followed by one hour of further sedation until sacrifice. We exposed four sedated age-matched animals to sham ultrasound under the same pre/post ultrasound procedures.
Time line of chronic study.
We treated five sedated mice chronically with tFUS on the left and right side of the brain with the ultrasound focus placed at 3.5 mm posterior of Bregma, +/− 2.2 mm from midline, 1.9 mm below the skin surface, into the center of each hippocampus. We applied ultrasound for one hour to each hippocampus followed by one hour of further sedation and then returned the mice to their cages. We did so every day for four days. On the fifth day we repeated that procedure then allowed the animal to stay sedated for another hour before euthanasia. We exposed five sedated age-matched animals to sham ultrasound under the same pre/post ultrasound procedures as the treated animals.
Detail of experimental procedures.
Anesthesia.
After initial exposure to isoflurane at 1.5–3% and placement within the experimental setup (see below), mice then received a subcutaneous injection of medetomidine [27] at 0.1 ug/gram at which point we weaned them off of isoflurane to approximate the awake state used by Iaccarino et al [1]. Mice received booster shots of medetomidine as necessary to maintain sedation.
Experimental setup – acute and chronic studies without EEG.
After induction of an anesthetic plane using isoflurane, we placed the treatment animal on a heating pad and into a mouse stereotaxic headpiece (Stoelting Digital Lab Standard). We then applied ophthalmic ointment (Puralube Vet Ointment, Dechra) to their eyes followed by a depilatory cream (Nair) to the top of the mouse head in order to remove its fur. Then, using a 3D-printed pointer attached to the distal face of the ultrasound transducer (see below), we used the digital stereotax guidance system to guide placement of the ultrasound focus within the hippocampus as described above. We then raised the transducer and changed the anesthesia to medetomidine. After achieving a stable sedated state, we placed ultrasound gel (Aquasonic Clear, Clinton Township, MI, USA) on the mouse head, lowered the distal tip of the transducer’s water filled cone into the gel, then started the ultrasound.
Experimental setup – acute study with EEG.
After removing hair with Nair we injected lidocaine subcutaneously into the dorsal aspect of the mouse skull, surgically exposed the skull, then drilled holes for placement of electrodes in each of auditory, visual and somatosensory cortex in each hemisphere, thereby avoiding the hippocampus to preserve its structure for histological analysis. We created custom made EEG electrode arrays by soldering five silver (0.0130 inch coated/0.010 inch bare diameter) electrode wires as well as one silver (0.0190 inch coated/0.015 inch bare diameter) ground wire. We connected that array into a preamplifier chip itself connected to a biosignal analog converter (Pinnacle Technology, Lawrence, Kansas, USA). EEG signals propagated from the chip into PowerLab, hence into LabChart (AD Instruments, Colorado Springs, CO, USA) for real-time monitoring of the data and to facilitate post-hoc analysis with MATLAB (MathWorks, Natik, MA, USA). We used copper mesh to wrap the majority of our equipment, as well as the mouse, thereby removing ambient 60 Hz signals and the 40 Hz electromagnetic pulse sent from the transducer.
Ultrasound equipment.
We used a two-annular 2 MHz transducer (Sonic Concepts, Woodinville WA, USA, model # H148 array S/N 003) with a cone placed on its distal surface that contained degassed water and capped with a thin membrane (Saran Wrap, S.C. Johnson & Son, Racine, WI, USA) actuated with supporting equipment as described in Mehic et al [28]. We initiated and controlled ultrasound delivery through use of LabChart and PowerLab (ADInstruments, Inc, Colorado Springs, CO).
Quantification of the ambient sound field.
We measured the ambient noise at the position of the treated and of the sham-treated animals, under conditions of ultrasound on and ultrasound off, using SpectrumView 2.3 (Oxford Wave Research, Ltd, Oxford, England) mounted on an iPhone 6s (Apple, Inc. Cupertino, CA). For each condition we captured five, 15-second recordings. We quantified the pressure level (in Decibels, defined relative to a fixed internal standard, designated as ‘dBFS’) at 40 Hz at every second within that time span using an on-board Fast Fourier Transform.
Ultrasound parameters.
Within each target location(s) we applied tFUS at 40 Hz with a carrier frequency of 2.0 MHz and pulse lengths of 400 microseconds, for five seconds followed by five seconds of no ultrasound, for one hour. We used ultrasound with a spatial peak pulse average intensity (I_sppa) value of 190 W/cm2 as define over the five seconds the ultrasound was turned on. This intensity value translates to the derated FDA limit for diagnostic ultrasound (190 W/cm2). When integrated over the time the ultrasound was turned on, this translates into a spatial peak temporal average intensity (I_spta) value of 3.0 W/cm2, above the derated FDA limit for diagnostic ultrasound (0.72 W/cm2) and consistent with a range of ultrasound studies dedicated to brain activation. We calibrated the device using an needle hydrophone (Onda, Sunnyvale, CA, USA, model # HNR-500 S/N 1890) in degassed water. Figure 1 shows our ultrasound equipment and a mathematical model of the maximum pressure field at the focus. Worth noting, neither the copper mesh nor ultrasound propagation through ex vivo skull appreciably attenuated the ultrasound field, well less than 5% of the maximum pressure value achieved in water (data not shown).
Figure 1. Montage of our experimental tFUS device.
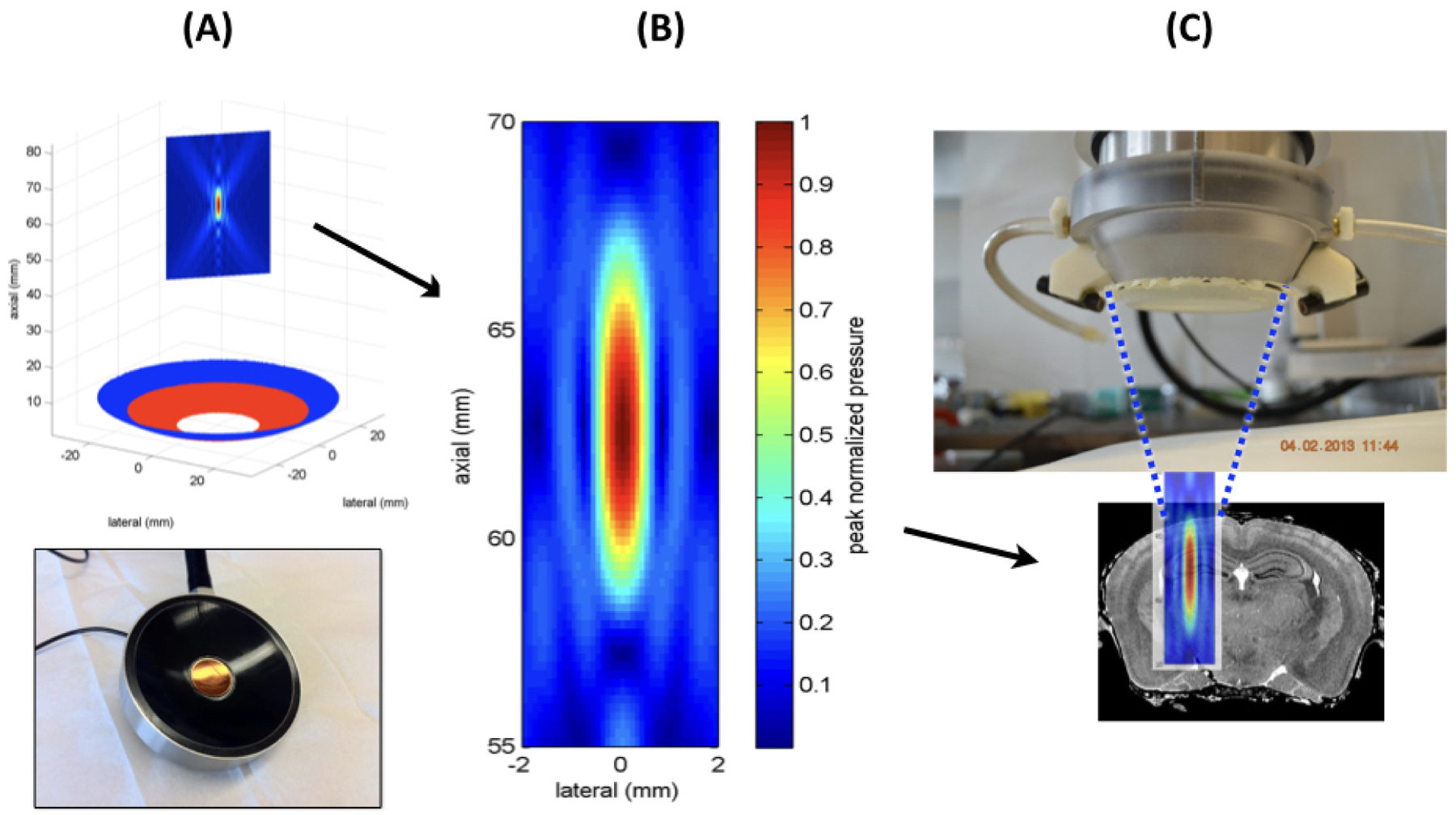
(A) This figure shows the transducer portion of the entire tFUS device below a mathematical representation of the two-annular transducer (red and blue circles) beneath a planar representation of the focal volume of ultrasound. (B) This figure shows a closeup of a mathematical representation of the maximum pressure field generated within the focal region of our ultrasound source as if it were measured in water. (C) This figure shows the working end of the tFUS device (top) with water-filled cone and closing membrane over ultrasound field of (B) itself superimposed upon a coronal slice of mouse brain, to scale.
Detail of analysis methods.
EEG.
We used continuous wavelet transforms (CWT) via MATLAB (Mathworks, Natick, MA) to analyze the difference in EEG signal between two electrodes, thereby allowing identification of brain-activation structure in both time and frequency.
Histopathology.
Staining for Aβ and microglia followed the histological procedures of Iaccarino et al [1]; staining for eNOS followed published protocols [29]. From each mouse we took a coronal slice of brain centered on 3.5 mm posterior of Bregma and −2.2 mm from midline – the planar location of the ultrasound focus. We report all our histological results with respect to one or both hemispheres of these coronal slices. Note that we placed brain tissue slices from paired treated and sham mouse brains on the same slides to insure they experienced the same exposure to staining media.
Imaging.
We used a Zeiss AxioZoom V16 microscope with a fully automated xyz stage controller and z-stack and tiling capabilities with multichannel imaging, capable of capturing confocal-like images with an attached AxioCam 506 monochrome camera. We used the ZEN core software for the microscope to scan the slides, following its intrinsic Tiling method for acquisition and Tiling & Stitching method for processing. We scanned histological images at 160X magnification thereby producing a minimum of 200 tiles per histological sample, each measuring 700 microns to a side. We used ZEN 3.0 blue edition to capture the fluorescent intensity of Aβ plaque and of eNOS then used the Fiji version of ImageJ to analyze the scanned images.
Analysis of activated microglia – choice of metric.
We used canonical discriminant analysis (CDA) to identify the morphological characteristics of the microglia most efficacious for our final analysis of activated microglia. CDA analysis offers a conservative means of identifying statistically significant variables within a given data set [30]. Specifically, we applied CDA to the several metrics of microglia activation (area, roundness, aspect ratio, solidity and circularity) defined as indicative of activated microglia in the literature [31,32]. We used histological data from two acutely treated mice and one sham mouse, data separate from those we used in our final, more substantial analysis. We quantified six total regions per brain, one where we applied ultrasound, two adjacent regions ipsilateral to ultrasound application, and three on the contralateral side of the brain. This analysis showed that there exists a subset of the dependent variables that varied significantly over the sham plus ultrasound treated histological data set (F=238 p≤ 0.0001). Further analysis revealed roundness and aspect ratio, together, as the major contributors to this global statistical significance in a way unique to sections of brain that received ultrasound acutely as different from the others (F= 6.18 p < 0.0001, see Figure 2). The average aspect ratio at the point of application measured 1.963 +/− 0.029 as compared to the contralateral side of the brain (2.306 +/− 0.049) and as compared to the value for sham-exposed mouse brain (2.320 +/− 0.017), with statistically significance (z = −7.35 p < 0.0001). The average roundness at the point of ultrasound application measured 0.5616 +/− 0.0074 as compared to the contralateral side of the brain (0.49961 +/− 0.0084) and as compared sham-exposed mouse brain (0.5011 +/− 0.0029). These differences were also statistically significant (z = 7.35 p < 0.0001). We therefore used roundness and aspect ratio to discriminate between active and nonactive microglia.
Figure 2. Sample images of active (A-D) and inactive (E-H) microglia.

These representative images show morphology typical of what we found in our analysis. For each, we measured their aspect ratio and roundness, the two metrics for glia activation that arose out of our canonical discrimination analysis. Active Microglia. (A) Aspect ratio = 1.643, Roundness = 0.609. (B) Aspect ratio = 1.72, Roundness = 0.582. (C) Aspect ratio = 1.54, Roundness = 0.649. (D) Aspect ratio =1.64, Roundness = 0.608. Inactive Microglia. (E) Aspect ratio = 2.653, Roundness = 0.377. (F) Aspect ratio = 2.653, Roundness = 0.377. (G) Aspect ratio = 4.61, Roundness = 0.217. (H) Aspect ratio = 2.64, Roundness = 0.379.
Analysis of activated microglia – identification.
The CDA analysis discussed above gave us a metric to use to identify activated microglia in our data. With those results in hand, we used ImageJ to identify by eye activated microglia in each image tile, typically 3–10 per tile. Our analysis covered an area measuring [1.0 cm] × [2.4–3.4 mm], depending upon the size of the brain slice, thereby encompassing ~40–75 sub-images. This procedure allowed us to take into account imaging artifacts such as mis-combined adjacent microglia or fragmented large microglia. We encountered this issue in less than ~5% of our analysis.
Identification of Aβ plaque.
Plaque size varied from under 10 microns (twenty times an image pixel) to over 50 microns, with a significant change in size distribution at around ten microns (Figure 3). Pilot analysis of colocalization of active microglia with plaque (method described below) for a subset of our histological samples using ten versus twenty micron cutoff values produced comparable results. Specifically, using a 10 micron cutoff we observed a 1.50 increase in colocalization in the treated versus contralateral side [t = 3.82, p < 0.0002] while with a 20 micron cutoff we observed a 1.51 increase [t = 3.62, p < 0.0004)]. With this analysis in hand, we used a cut-off value of ten microns for our colocalization analysis, also consistent other published analysis e.g., [7,9].
Figure 3.
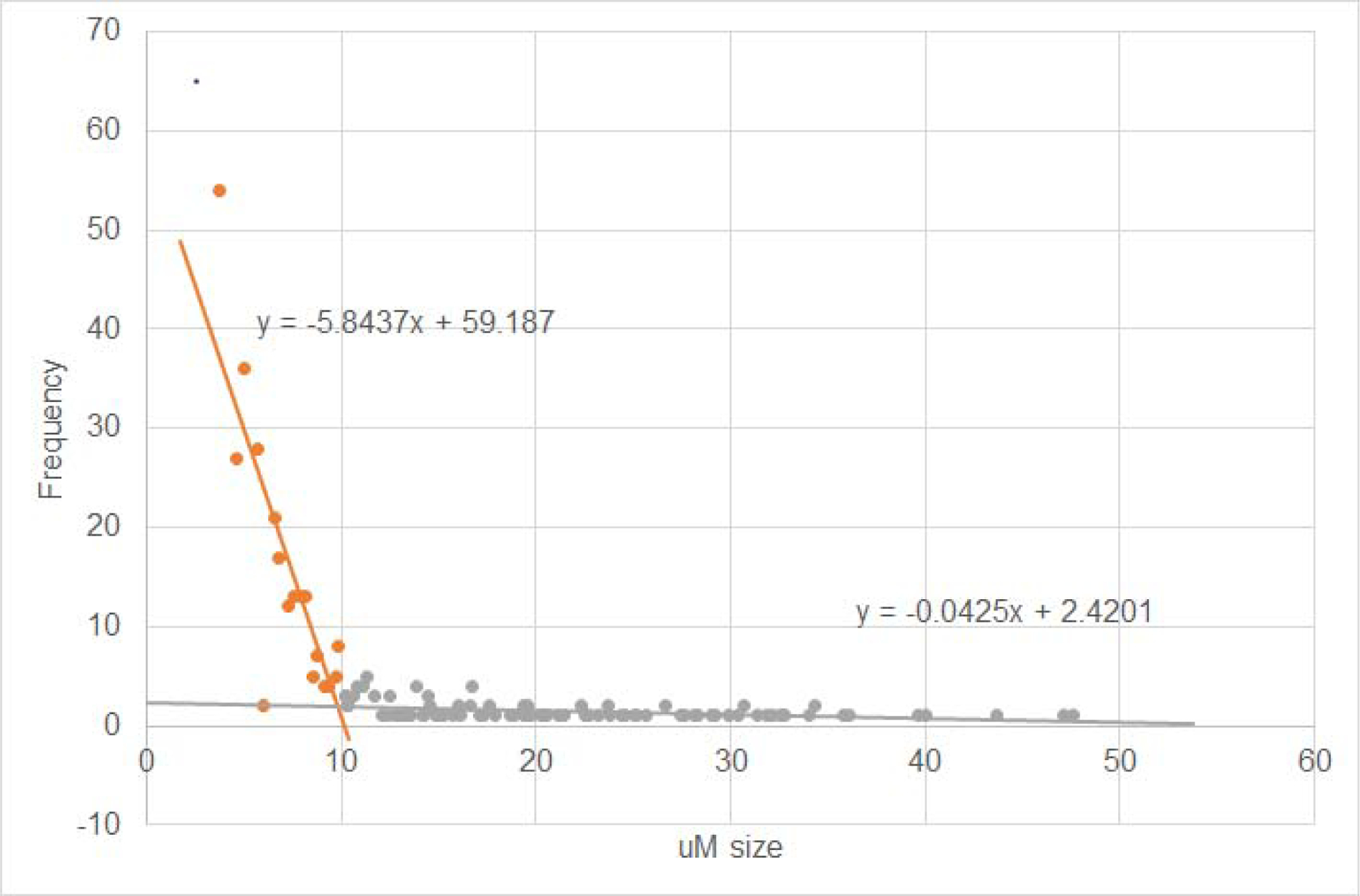
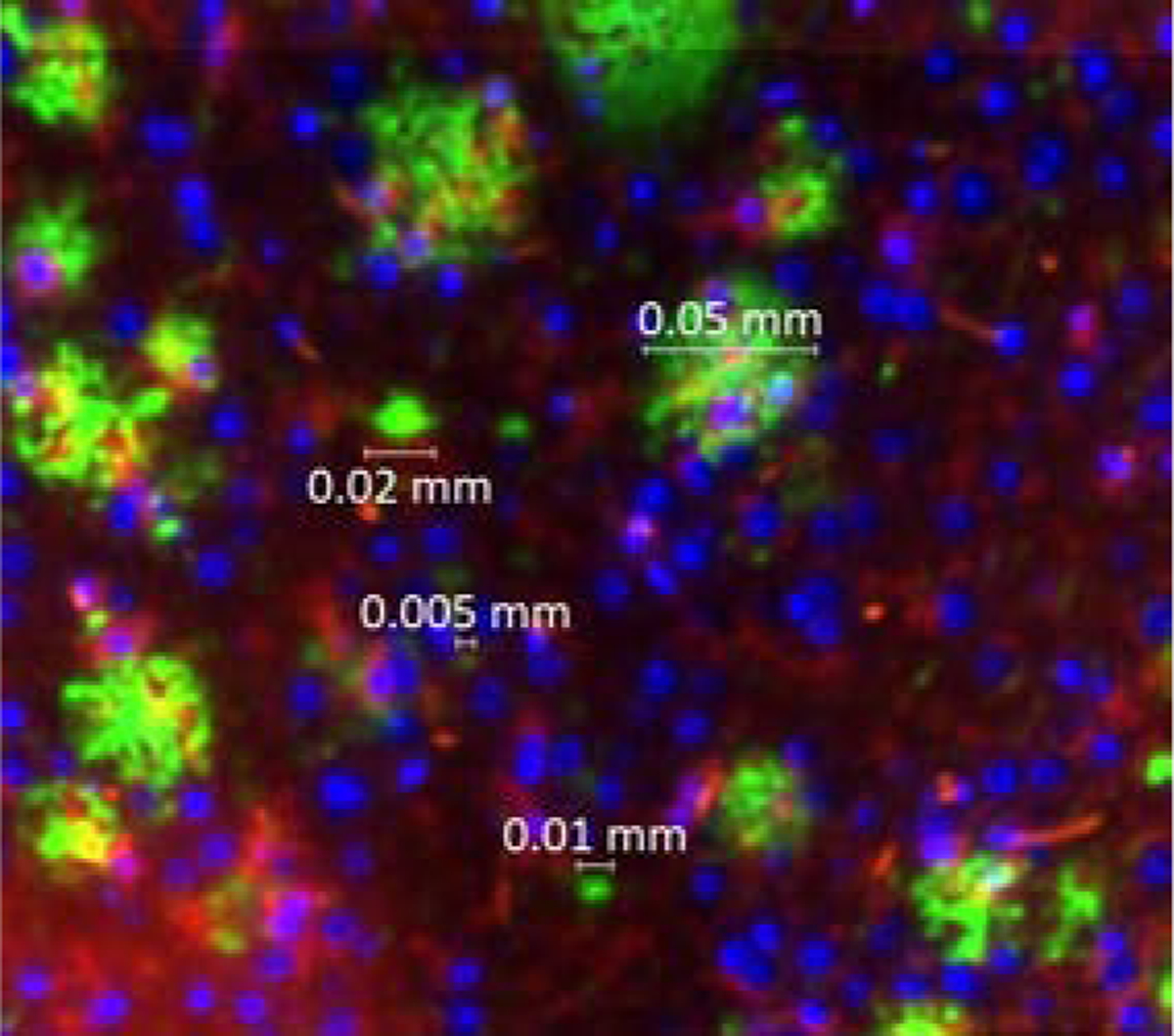
Representative plaque size distribution (A) and examples of plaque (B, in green) across a range of sizes
Analysis of Aβ plaque burden.
To compare the net Aβ plaque burden between matched pairs of treated versus sham-treated mice, we first measured the fluorescent intensity across sub-images of brain as above for activated microglia, thereby producing a mean plus standard error (SE) for that slice. We then divided the mean intensity +/− SE of the treated slice by the intensity of the matched sham and multiplied by 100 to get percentage change in plaque burden for that pair. We then used all paired values of Aβ plaque burden to calculate the net change across all mice. In addition, we measured the net Aβ plaque burden in the CA1 region of the hippocampus by measuring the fluorescent intensity within a region of interest that surrounded it, otherwise following the procedure just outlined for an entire brain slice.
Analysis of activated microglia co-located with Aβ plaque.
We defined colocalization of activated microglia and Aβ plaque as their physical overlap within our histological samples as presented by ImageJ and identified by eye. We checked the analysis of an individual researcher through use of intermittent independent review.
Analysis of eNOS expression.
To compare the net eNOS expression between treated versus sham-treated mice after chronic ultrasound exposure, we followed the procedure for Aβ plaque burden described above. Note that we did not analyze eNOS expression for the acute case because changes in eNOS expression takes at least 18–24 hours after stimulation [21].
Statistics.
We used Microsoft Excel (Microsoft Excel, version 3.04) for regression analysis (Figure 3). We used STATA/SE15 (Stata Statistical Software, Release #15. StataCorp, LLC, College Station, TX, USA) to apply multivariate and nonparametric statistics as follows. We determined the statistical significance of between-group differences for three or more groups using the Kruskal-Wallis equality-of-populations rank test where appropriate (ambient noise) before determining the statistical significance between two paired groups using the two-sample, non-parametric Wilcoxon rank-sum (Mann-Whitney) test (Figures 5 and 6). We used non-parametric canonical discriminant analysis to identify from among multiple variables those with the highest statistically significant explanatory power for a given data set (Figure 2). We report all results in terms of mean +/− standard error (SE).
Figure 5. Histology and colocalization map for acutely treated or sham treated AD mice.

Representative treated (left column) and corresponding sham treated (right column) histology (top of each figure) and map of colocalized activated microglia and plaque (bottom of each figure). The color scheme refers to the percentage of activated microglia colocalized with plaque. Note the following correspondence between image pairs here and the quantitative analysis results in Table 1. (A) This image corresponds to treated mouse #1 while (B) corresponds to sham treated mouse #1. (C) This image corresponds to treated mouse #5 while (D) corresponds to sham treated mouse #5. Overall, we observed a statistically significant difference between colocalization maps of actual and sham treated mice, e.g., between treated side and the entire sham brain (36.0 +/− 4.6% versus 14.2 +/− 2.6%, z = 0.245, p < 0.014).
Figure 6. Sample histology of Aβ plaque burden for chronically treated versus sham treated AD mice.
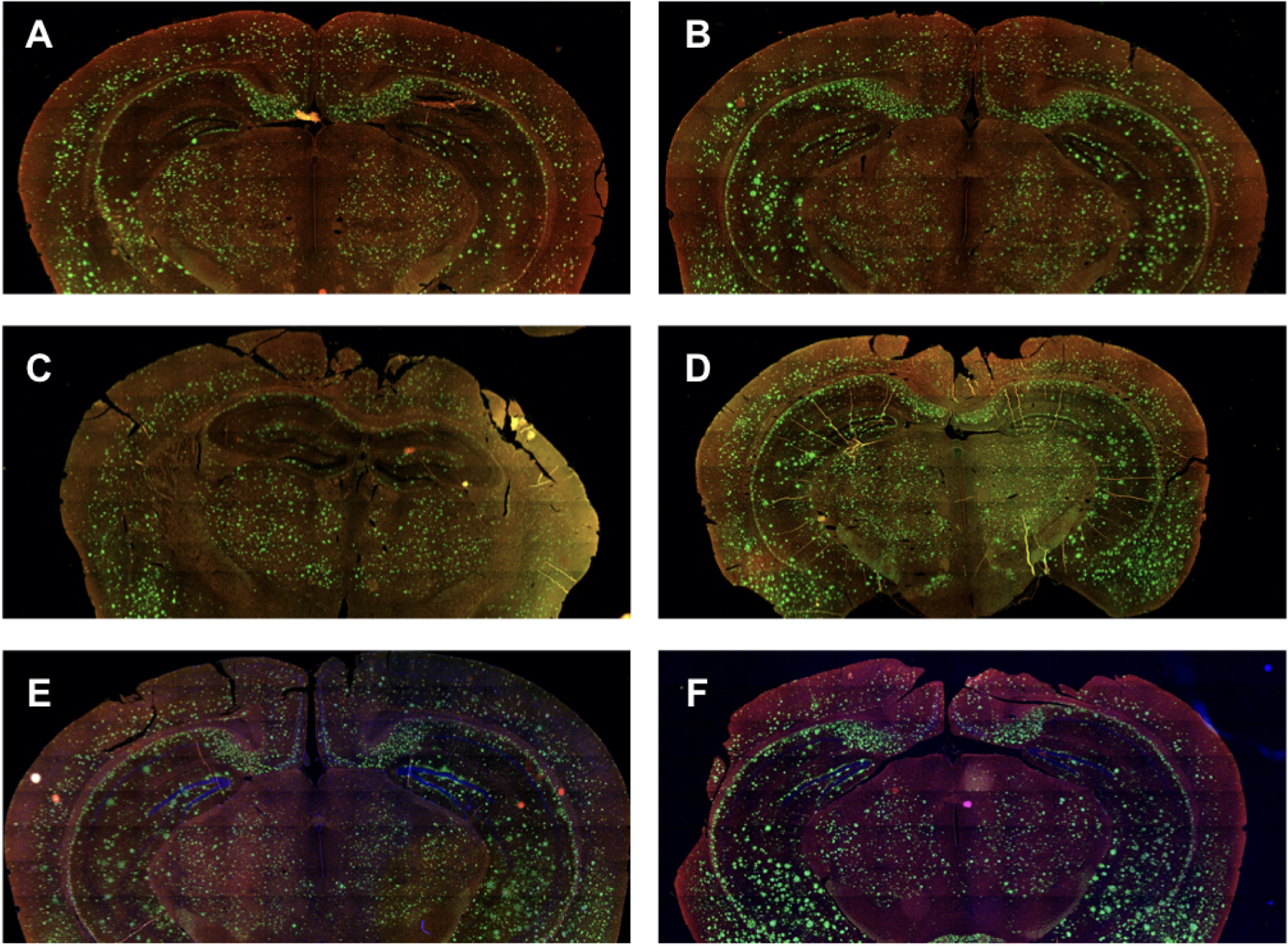
Representative Aβ plaque burden (green) and microglia (red) in treated (left column) and corresponding sham treated (right column) mouse brains. Note the following correspondence between image pairs here and the quantitative analysis results in Table 3. (A) This image corresponds to treated mouse #2 while (B) corresponds to sham treated mouse #2. Together, this pair has the smallest difference in integrated Aβ plaque burden (14.5 +/− 2.6%). (C) This image corresponds to treated mouse #4 while (D) corresponds to sham treated mouse #4. Together, this pair has the largest difference in integrated Aβ plaque burden (66.8 +/− 3.0%). (E) This image corresponds to treated mouse #5 while (F) corresponds to sham treated mouse #5, another image pair with a large difference in integrated Aβ plaque burden (62.9 +/− 0.9%). Overall, we observed a statistically significant reduction in Aβ plaque burden for the treated versus sham mice (47.4 ± 5.8%, z 1/4 − 2.79, p < 0.0005)
Results.
Quantification of the ambient sound field.
Our observations showed a local maximum in ambient pressure in the 30–70Hz range, due to our laboratory fume hood. Kruskal-Wallis analysis of all our data revealed that there exists a statistically significant difference within at least one of our paired sub-group of data. Paired sub-group analysis determined that there did not exist a statistically significant difference in local pressure field amplitude at 40 Hz with the transducer on or off at either the treatment site (−29.85 +/− 2.7 dBFS versus −29.83 +/− 2.0 dBFS, z = −4.26, p < 0.99) or at the sham-treatment site (−26.78 +/− 1.9 versus −27.38 +/− 3.4, z = − 1.12, p < 0.26). Interestingly, the pressure field at 40 Hz was louder at the sham site compared to the treatment site, here shown with ultrasound on (−26.78 +/− 1.9 dBFS versus −29.85 +/− 2.7 dBFS, z = −4.26, p < 0.00001), due to the proximity of the fume hood to the sham treatment site relative to the actual treatment site.
EEG.
CWT analysis of the difference between acute EEG measurements in the left versus right hemisphere of AD mouse brain, with tFUS delivered into the left hemisphere, demonstrated a strong and temporally nonuniform signal at 40 Hz – Figure 4.
Figure 4. Plot of frequency and temporal distribution of brain activity generated by tFUS.

This figure shows our measurement of brain activity between two electrodes generated by transcranial, focused ultrasound whose delivery starts at the 1.0 second mark and ends by 6.0 seconds. Here we have averaged the difference in data streams between each visual cortex of one sedated mouse over a 17 minutes window within an hour-long delivery of tFUS. The frequency with the largest signal is centered on 40 Hz, the pulse repetition frequency of the ultrasound.
eNOS production.
We did not observe a meaningful production of eNOS in chronically treated brain relative to sham-treated brain. Specifically, we observed that eNOS expression for actual-treated mice was insignificantly less than expression for sham-treated mice: 1.5 +/− 3.8%, z = − 0.028, p < 0.78.
Colocalization of activated microglia and Aβ plaque after acute treatment.
We treated five mice acutely with ultrasound while we sham treated four other mice on the same day as the treated mice. Mouse #2 did not have a corresponding sham mouse treated on the same day; instead we treated it one day after mouse #1 and its sham. Given this close proximity in time we use the same sham data for mouse #1 and mouse #2 for our analysis. In all cases we observed statistically significant activation of microglia that co-localized with Aβ plaque relative to sham (Figure 5 and Table 1). For example, we observed a statistically significant difference between the ultrasound treated side versus the sum of both hemispheres of the age-matched sham mouse for each pair of mice. In addition, aggregating these results across all paired mice, we observed statistically significant differences between the treated side and untreated side (36.0 +/− 4.6% versus 14.3 +/− 4.0%, z = 2.611, p < 0.009) as well as between the treated side and the entire sham brain (36.0 +/− 4.6% versus 14.2 +/− 2.6%, z = 0.245, p < 0.014).
Table 1.
Statistical analysis of colocalization of activated microglia and Aβ plaque for acutely treated 5XFAD mice versus age-matched, sham-treated 5XFAD mice. Note that we used the same sham data for mouse #1 and mouse #2, given that treatment of this sham mouse occurred within 24 hours of each of treated mouse #s 1 and 2.
| Mouse # | Percent colocalized | SE | left vs right | left vs sham | right vs sham | ||||
|---|---|---|---|---|---|---|---|---|---|
| z | p< | z | p< | z | p< | ||||
| 1 | US left | 40.2 | 6.2 | 40.2 | 0.0001 | 4.935 | 0.0001 | −2.253 | 0.0243 |
| US Right | 5.3 | 2.3 | |||||||
| Sham | 8.0 | 1.1 | |||||||
| 2 | US left | 25.4 | 3.8 | 25.4 | 0.002 | 3.779 | 0.0002 | 0.248 | 0.8 |
| US Right | 8.6 | 1.5 | |||||||
| Sham | 8.0 | 1.1 | |||||||
| 3 | US left | 25.4 | 4.9 | 25.4 | 0.004 | 2.673 | 0.008 | −0.812 | 0.417 |
| US Right | 10.1 | 2.1 | |||||||
| Sham | 11.7 | 1.4 | |||||||
| 4 | US left | 39.7 | 2.2 | 39.7 | 0.0001 | 6.367 | 0.0001 | 2.802 | 0.005 |
| US Right | 24.9 | 1.6 | |||||||
| Sham | 19.3 | 1.7 | |||||||
| 5 | US left | 49.2 | 3.7 | 5.283 | 0.0001 | 5.896 | 0.0001 | 1.743 | 0.08 |
| US Right | 22.8 | 1.2 | |||||||
| Sham | 17.8 | 1.6 | |||||||
| all | US left | 36.0 | 4.6 | 2.611 | 0.009 | 2.449 | 0.014 | 0.001 | 1 |
| US Right | 14.3 | 4.0 | |||||||
| Sham | 14.2 | 2.6 | |||||||
Plaque burden after acute treatment.
We did not observe a meaningful change in Aβ plaque burden in acutely treated brain relative to sham-treated brain. Specifically, we observed that the Aβ plaque burden for ipsilateral, treated brain versus contralateral, untreated brain did not differ significantly from one another (3.8 +/− 3.6%, z = 1.00, p < 0.32) nor did the Aβ plaque burden within ipsilateral, treated brain differ from the entirety of sham-treated brain (2.0 +/− 3.0%, z = − 0.39, p < 0.70).
Colocalization of activated microglia and Aβ plaque after chronic treatment.
We treated five mice chronically with ultrasound while sham treating five age-matched mice on the same days. Unlike for the acutely treated mice, only three of five paired cases produced a statistically significant increase in activated microglia that co-localized with Aβ plaque across the entire treated brain for treated mice relative to sham. Aggregated across all mice (Table 2), we observed no statistically significant difference between treated brains and sham treated brains (33.0 +/− 7.4% versus 24.2 +/− 1.4%, z = 0.32, p < 0.75).
Table 2.
Statistical analysis of colocalization of activated microglia and Aβ plaque for chronically treated versus age-matched, sham-treated 5XFAD mice.
| Mouse # | percent colocalized | SE | z | p< | |
|---|---|---|---|---|---|
| 1 | 40 Hz | ||||
| daily | 50.9 | 1.3 | 7.835 | 0.0001 | |
| sham | 26.8 | 1.9 | |||
| 2 | 40 Hz | ||||
| daily | 51.2 | 1.8 | 7.138 | 0.0001 | |
| sham | 27.9 | 1.6 | |||
| 3 | 40 Hz | ||||
| daily | 19.7 | 1.1 | 0.625 | 0.5321 | |
| sham | 20.6 | 1.2 | |||
| 4 | 40 Hz | ||||
| daily | 23.3 | 1.1 | 1.302 | 0.1931 | |
| sham | 21.7 | 1.5 | |||
| 5 | 40 Hz | ||||
| daily | 20.0 | 1 | 2.308 | 0.021 | |
| sham | 23.9 | 1.2 | |||
| all combined | 40 Hz | ||||
| daily | 33.0 | 7.4 | 0.32 | 0.75 | |
| sham | 24.2 | 1.4 |
Plaque burden after chronic treatment.
Each of the five pairs of ultrasound treated AD mice displayed a significant reduction in Aβ plaque burden integrated across the histological slice relative to the sham treated mice – Figure 6 and Table 3. The decrease in Aβ load ranged from 15% to 67% across paired mice while the combined decrease in Aβ plaque load equaled 47.4 +/− 5.8% (z = − 2.79, p < 0.005). When restricted to the CA1 portion of the hippocampus (Table 4), the decrease in Aβ plaque burden within treated mouse brain ranged from 12.6% to 41.3%, equaling across all paired mice to a difference of 28.1 +/− 1.8% (z = − 6.67, p < 0.0001).
Table 3.
Statistical analysis of Aβ plaque for chronically treated versus age-matched, sham-treated 5XFAD mice.
| Paired sample, By mouse number | % reduction in amyloid beta load | SE | z | p< |
|---|---|---|---|---|
| 1 | 59.1 | 1.7 | −5.24 | 0.0001 |
| 2 | 14.5 | 2.6 | −2.662 | 0.0078 |
| 3 | 33.8 | 3.8 | −3.554 | 0.0004 |
| 4 | 66.8 | 3.0 | −3.464 | 0.0005 |
| 5 | 62.9 | 0.9 | −3.317 | 0.0009 |
| all | 47.4 | 5.8 | −2.785 | 0.005 |
Table 4.
Statistical analysis of Aβ plaque within the CA1 portion of the hippocampus for chronically treated versus age-matched, sham-treated 5XFAD mice.
| Paired sample | % reduction in amyloid beta load in CA1 region of hippocampus | SE | z | p< |
|---|---|---|---|---|
| 1 | 41.3 | 1.1 | −3.361 | 0.0008 |
| 2 | 37.6 | 1.6 | −3.361 | 0.0.008 |
| 3 | 21.0 | 2.1 | −3.136 | 0.0017 |
| 4 | 30.1 | 1.2 | −3.576 | 0.0003 |
| 5 | 12.6 | 2.3 | −1.722 | 0.0851 |
| all | 28.1 | 1.8 | −6.674 | 0.0001 |
Discussion
Using the 5XFAD mouse model of Alzheimer’s disease (AD), we observed a net reduction of average Aβ burden across an entire brain slice measuring ~47% after five days of transcranial, focused ultrasound (tFUS), as compared to sham exposure. When restricted to CA1, we observed a reduction in Aβ burden by ~28%. We hypothesize that this reduction in Aβ burden occurred due to activation of microglia (which we observed acutely, but not after 5 days of treatment) and not with enhancement of the vasodilator endothelial nitric oxide synthase (which we measured immediately after chronic treatment but did not observe). This reduction in Aβ burden approaches that achieved by Iaccarino et al [1] as measured in only the visual cortex and of Martorell et al [7], as measured in only discrete segments of brain. This reduction in Aβ burden compares to that achieved by Leinenga and Gotz [9], who used ultrasound plus acoustic contrast agents to disrupt the blood-brain barrier in an intermittent fashion over a period of two months. Finally, our results compare favorably to that of Eguchi et al [20] with their ultrasound delivered intermittently over several months.
Our results rival reduction in Aβ burden caused by medications applied for a substantially longer period of time. For example. Bhattacharya et al [33] gave galantamine every day for two months to 7-month-old male 5XFAD mice, which reduced hippocampal Aβ plaque burden by 19% relative to sham treatment. Chandra and Pahan [34] produced a 55% reduction in Aβ plaque burden within the hippocampus of 6-month-old female and male 5XFAD mice after a month of daily application of the lipid-lowering drug gemfibrozil (Lopid®).
While we achieved comparable results over a shorter period of time than for medications, our ultrasound approach would benefit from a less onerous application schedule were it ever translated to human use. Recall that we did not observe a difference between the percent of activated microglia colocalized with Aβ plaque in chronically treated versus chronically sham-treated 5XFAD mice when averaged across all five pairs of mice. This suggests to us that ultrasound may have exhausted its ability to activate microglia before the end of our five-day treatment protocol. Perhaps a treatment protocol that lasted for fewer days would produce the same effect with less of a temporal burden. Similarly, our ultrasound protocol may have also worked through the eNOS pathway but only during the first phase of treatment. Future studies will explore these possibilities, through analysis of histological samples taken from a subset of treated versus sham-treated mice at multiple time points during chronic treatment.
Our method along with those of Iaccarino et al [1], Leinenga and Gotz [9], and Burgess et al [8], seek to treat AD under the Aβ hypothesis. Discouragingly, a large number of candidate medications that act to clear Aβ haven’t improved clinical outcomes, at least as tested on late-stage AD patients [4–6]. Among the explanations for this failure, perhaps these patients had already experienced intra-neuronal damage from neurofibrillary tangles that often arises after Aβ build up, plausible since tau burden more closely tracks with clinical signs and symptoms than Aβ burden [2,3]. One of several complementary mechanisms undergoing increased scrutiny thanks to these failures includes cerebrovascular contributions to dementia, including AD. For example, postmortem analysis of clinically assigned AD patients often shows both a range of aberrant protein (Aβ, tau) burden as well as pathological signs of vascular dementia [3]. Also, epidemiological studies show that a higher risk for cardiovascular disease increases the risk for dementia [2,3], as does traumatic brain injury [35,36]. Interestingly, Nortley et al [37] demonstrated that Aβ exposure constricts intra-cerebral blood flow via their action on pericytes, a candidate explanation for reduced cerebral blood flow early in AD progression [38] that relates a growing Aβ burden to disease progression, along with mitochondrial dysfunction among other effects of cerebral ischemia. Furthermore, very recent research [39] used both animal models and post-mortem tissue to show that norepinephrine acts as an important co-factor in the process by which extracellular Aβ leads to the production of intra-neuronal neurofibrillary tangles, itself closely tied to symptom progression. Importantly, their work suggests that extracellular Aβ burden must reduce to nanomolar concentrations in order to meaningfully reduce subsequent production of neurofibrillary tangles, a reduction not yet met by any published drug or process alone. In future work we will therefore explore drug/ultrasound combinations to further reduce the net extracellular Aβ burden. We will also attempt to broaden our approach from a strictly ‘microglia attacks Aβ’ orientation by moving our ultrasound protocol towards that of Eguchi et al [20], whose effect acts primarily via induction of enhanced cerebrovascular blood flow with only minimal activation of microglia. In this way we hope to identify an ultrasound-based protocol, or drug/ultrasound combination, that optimally activates microglia as well as enhances cerebrovascular blood flow, available in principle to treat AD, vascular dementia, as well as mixed Alzheimer’s/vascular dementia.
Limitations.
We used the 5XFAD mouse model of AD due to its emphasis on rapid plaque formation and its use by Iaccarino et al [1], whose light-based stimulus of male 5XFAD mouse brain we emulated in our study. In future in vivo research we will use not only both sexes of mice but also other mouse models. Those mouse models should have substantial tau as well as Aβ burdens, and should exhibit vascular as well as Aβ pathology. For example, Martorell et al [7] showed that auditory stimulation at 40 Hz reduced phosphorylated tau in the P301S mouse model of AD.
We reported our measurement of a temporally nonuniform average signal at 40 Hz: strong for the first three seconds, followed by a short dip in intensity, then closing with a less intense EEG signal until the end of the five seconds of ultrasound application. Such a temporal pattern is not consistent with a false signal produced by the electromagnetic pulse of our ultrasound transducer at 40 Hz, which would have produced a temporally uniform EEG signal. Worthy of future study is study of the details of the brain’s response to ultrasound delivered at 40 Hz. Also, Iaccarino et al [1] showed that other stimulation frequencies in the gamma band (30, 50, and 80 Hz) did not activate microglia – we don’t yet know if that holds true for ultrasound.
Could 40 Hz sound emitted by our ultrasound device have achieved what we observed? This is an important question because Guo et al [40] showed that ultrasound could activate the auditory cortex, thereby creating a startle response, for example, as an alternative explanation to ultrasound’s observed ability to generate movement in sedated or anesthetized animals. For the present case, the possibility of auditory activation has particular interest, since Martorell et al [7] showed that auditory stimulation at 40 Hz activated microglia and cleared Aβ plaque in auditory cortex and the CA1 portion of the hippocampus. This is not a concern here for a variety of reasons. For example, our observed effects occurred throughout the area of ultrasound delivery not just in auditory cortex and CA1 as in Martorell et al [7]. Also, we exposed sham animals to more sound at 40 Hz yet the ultrasound-exposed mice showed a substantial decrease in Aβ plaque burden compared to sham-exposed ultrasound mice. Finally, we observed anatomical asymmetry in activation of microglia acutely – more on the side of ultrasound delivery, less on the contralateral side, despite the fact that we placed the ultrasound device centered above each mouse head, giving sound emitted by the device the opportunity to stimulate each auditory cortex in a comparable way.
Conclusion.
One hour of transcranially delivered, focused ultrasound (tFUS) applied acutely to the brains of a mouse model of Alzheimer’s Disease activated microglia to colocalize with Aβ plaque. Five days of tFUS cleared almost 50% of Aβ plaque, results comparable to published studies of the effects of medications on 5XFAD mice that seek to reduce Aβ burden, but applied over a substantially longer period of time. Our ultrasound protocol has intensity values at or near those of diagnostic ultrasound, similar to the structure of existing ultrasound protocols shown to safety activate the brains of mouse, rat, sheep, rabbit, non-human primate and humans. Given the existence of relatively inexpensive and highly portable diagnostic ultrasound devices (Butterfly IQ, Lumify, SonoSite, Vscan, among others), results such as ours could one day lead to rapid production of tFUS devices capable of addressing Alzheimer’s Disease.
Acknowledgements.
We received funding from NIH, grant # 5R21EY027557-02. We thank Kahte Culevski, Chikodi Ezeokeke, and Madison Selby, members of the Mourad lab, for their contributions to our experiments. We thank Tom Grabowski, Dirk Keen and Martin Darvas, all at the University of Washington, for their helpful conversations. Finally, we thank the reviewers, whose constructive criticism substantially improved our manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
All authors declare that they have no known financial interests or any other conflict of interest with other people or organizations that could inappropriately influence this work.
Ultrasound alone can activate microglia to attack Aβ plaque, acutely, and clear Aβ plaque, after five days of exposure, in vivo.
This kind of ultrasound has parameter values near that of diagnostic ultrasound, making its translation to human studies a plausibly quick affair.
Bibliography
- [1].Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016. 10.1038/nature20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Prim 2015;1:15056 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- [3].Azarpazhooh MR, Avan A, Cipriano LE, Munoz DG, Sposato LA, Hachinski V. Concomitant vascular and neurodegenerative pathologies double the risk of dementia. Alzheimer’s Dement 2018;14:148–56. 10.1016/j.jalz.2017.07.755. [DOI] [PubMed] [Google Scholar]
- [4].Gravitz L Drugs: A tangled web of targets. Nature 2011;475:S9–11. 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- [5].Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs 2017;26:735–9. 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement Transl Res Clin Interv 2019;5:272–93. 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martorell AJ, Paulson AL, Suk H-J, Abdurrob F, Drummond GT, Guan W, et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019;177:256–271.e22. 10.1016/j.cell.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Burgess A, Dubey S, Yeung S, Hough O, Eterman N, Aubert I, et al. Alzheimer disease in a mouse model: MR imaging-guided focused ultrasound targeted to the hippocampus opens the blood-brain barrier and improves pathologic abnormalities and behavior. Radiology 2014;273:736–45. 10.1148/radiol.14140245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Leinenga G, Gotz J. Scanning ultrasound removes amyloid- and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med 2015. 10.1126/scitranslmed.aaa2512. [DOI] [PubMed] [Google Scholar]
- [10].Leinenga G, Koh WK, Götz J. Scanning ultrasound in the absence of blood-brain barrier opening is not sufficient to clear β-amyloid plaques in the APP23 mouse model of Alzheimer’s disease. Brain Res Bull 2019;153:8–14. 10.1016/j.brainresbull.2019.08.002. [DOI] [PubMed] [Google Scholar]
- [11].Pandit R, Leinenga G, Götz J. Repeated ultrasound treatment of tau transgenic mice clears neuronal tau by autophagy and improves behavioral functions. Theranostics 2019;9:3754–67. 10.7150/thno.34388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hendricks BK, Cohen-Gadol AA, Miller JC. Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg Focus 2015;38:E10 10.3171/2015.1.FOCUS14767. [DOI] [PubMed] [Google Scholar]
- [13].Sinharay S, Tu T-W, Kovacs ZI, Schreiber-Stainthorp W, Sundby M, Zhang X, et al. In vivo imaging of sterile microglial activation in rat brain after disrupting the blood-brain barrier with pulsed focused ultrasound: [18F]DPA-714 PET study. J Neuroinflammation 2019;16:155 10.1186/s12974-019-1543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kovacs ZI, Tu T-W, Sundby M, Qureshi F, Lewis BK, Jikaria N, et al. MRI and histological evaluation of pulsed focused ultrasound and microbubbles treatment effects in the brain. Theranostics 2018;8:4837–55. 10.7150/thno.24512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kovacs ZI, Kim S, Jikaria N, Qureshi F, Milo B, Lewis BK, et al. Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc Natl Acad Sci 2017;114:E75–84. 10.1073/pnas.1614777114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McMahon D, Hynynen K. Reply to Kovacs et al. : Concerning acute inflammatory response following focused ultrasound and microbubbles in the brain. Theranostics 2018;8:2249–50. 10.7150/thno.25468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lipsman N, Meng Y, Bethune AJ, Huang Y, Lam B, Masellis M, et al. Blood-brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat Commun 2018;9:2336 10.1038/s41467-018-04529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aryal M, Vykhodtseva N, Zhang Y-Z, McDannold N. Multiple sessions of liposomal doxorubicin delivery via focused ultrasound mediated blood–brain barrier disruption: A safety study. J Control Release 2015;204:60–9. 10.1016/j.jconrel.2015.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Horodyckid C, Canney M, Vignot A, Boisgard R, Drier A, Huberfeld G, et al. Safe long-term repeated disruption of the blood-brain barrier using an implantable ultrasound device: a multiparametric study in a primate model. J Neurosurg 2017;126:1351–61. 10.3171/2016.3.JNS151635. [DOI] [PubMed] [Google Scholar]
- [20].Eguchi K, Shindo T, Ito K, Ogata T, Kurosawa R, Kagaya Y, et al. Whole-brain low-intensity pulsed ultrasound therapy markedly improves cognitive dysfunctions in mouse models of dementia - Crucial roles of endothelial nitric oxide synthase. Brain Stimul 2018;11:959–73. 10.1016/j.brs.2018.05.012. [DOI] [PubMed] [Google Scholar]
- [21].Bonow RH, Silber JR, Enzmann DR, Beauchamp NJ, Ellenbogen RG, Mourad PD. Towards use of MRI-guided ultrasound for treating cerebral vasospasm. J Ther Ultrasound 2016;4:6 10.1186/s40349-016-0050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bobola MS, Chen L, Ezeokeke CK, Kuznetsova K, Lahti AC, Lou W, et al. A Review of Recent Advances in Ultrasound, Placed in the Context of Pain Diagnosis and Treatment. Curr Pain Headache Rep 2018;22:60 10.1007/s11916-018-0711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blackmore J, Shrivastava S, Sallet J, Butler CR, Cleveland RO. Ultrasound Neuromodulation: A Review of Results, Mechanisms and Safety. Ultrasound Med Biol 2019;45 10.1016/j.ultrasmedbio.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging 2012;33:196.e29–40. 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- [25].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006;26:10129–40. 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gu L, Wu D, Tang X, Qi X, Li X, Bai F, et al. Myelin changes at the early stage of 5XFAD mice. Brain Res Bull 2018;137:285–93. 10.1016/j.brainresbull.2017.12.013. [DOI] [PubMed] [Google Scholar]
- [27].Wu T, Grandjean J, Bosshard SC, Rudin M, Reutens D, Jiang T. Altered regional connectivity reflecting effects of different anaesthesia protocols in the mouse brain. Neuroimage 2017;149:190–9. 10.1016/j.neuroimage.2017.01.074. [DOI] [PubMed] [Google Scholar]
- [28].Mehić E, Xu JM, Caler CJ, Coulson NK, Moritz CT, Mourad PD. Increased anatomical specificity of neuromodulation via modulated focused ultrasound. PLoS One 2014;9:e86939 10.1371/journal.pone.0086939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ruan G, Ren H, Zhang C, Zhu X, Xu C, Wang L. Cardioprotective Effects of QiShenYiQi Dripping Pills on Transverse Aortic Constriction-Induced Heart Failure in Mice. Front Physiol 2018;9:324 10.3389/fphys.2018.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].American Society of Photogrammetry Z, American Society for Photogrammetry and Remote Sensing AL. Photogrammetric engineering and remote sensing. vol. 66. American Society of Photogrammetry; 2000. [Google Scholar]
- [31].Zanier ER, Fumagalli S, Perego C, Pischiutta F, De Simoni M-G. Shape descriptors of the “never resting” microglia in three different acute brain injury models in mice. Intensive Care Med Exp 2015;3:7 10.1186/s40635-015-0039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fernández-Arjona M del M, Grondona JM, Granados-Durán P, Fernández-Llebrez P, López-Ávalos MD. Microglia Morphological Categorization in a Rat Model of Neuroinflammation by Hierarchical Cluster and Principal Components Analysis. Front Cell Neurosci 2017;11:235 10.3389/fncel.2017.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bhattacharya S, Haertel C, Maelicke A, Montag D. Galantamine slows down plaque formation and behavioral decline in the 5XFAD mouse model of Alzheimer’s disease. PLoS One 2014;9:e89454 10.1371/journal.pone.0089454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chandra S, Pahan K. Gemfibrozil, a Lipid-Lowering Drug, Lowers Amyloid Plaque Pathology and Enhances Memory in a Mouse Model of Alzheimer’s Disease via Peroxisome Proliferator-Activated Receptor α. J Alzheimer’s Dis Reports 2019;3:149–68. 10.3233/ADR-190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Johnson VE, Stewart W, Smith DH. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol 2012;22:142–9. 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schaffert J, LoBue C, White CL, Chiang H-S, Didehbani N, Lacritz L, et al. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer’s disease. Neuropsychology 2018;32:410–6. 10.1037/neu0000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019;365:eaav9518 10.1126/science.aav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 2016;7:11934 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang F, Gannon M, Chen Y, Yan S, Zhang S, Feng W, et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci Transl Med 2020;12:eaay6931 10.1126/scitranslmed.aay6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guo H, Hamilton M, Offutt SJ, Gloeckner CD, Li T, Kim Y, et al. Ultrasound Produces Extensive Brain Activation via a Cochlear Pathway. Neuron 2018;98:1020–1030.e4. 10.1016/j.neuron.2018.04.036. [DOI] [PubMed] [Google Scholar]