

Abstract
In biomedical applications of machine learning, relevant information often has a rich structure that is not easily encoded as real-valued predictors. Examples of such data include DNA or RNA sequences, gene sets or pathways, gene interaction or coexpression networks, ontologies, and phylogenetic trees. We highlight recent examples of machine learning models that use structure to constrain model architecture or incorporate structured data into model training. For machine learning in biomedicine, where sample size is limited and model interpretability is crucial, incorporating prior knowledge in the form of structured data can be particularly useful. The area of research would benefit from performant open source implementations and independent benchmarking efforts.
Introduction
It can be challenging to distinguish signal from noise in biomedical datasets, and machine learning methods are particularly hampered when the amount of available training data is small. Incorporating biomedical knowledge into machine learning models can reveal patterns in noisy data [1] and aid model interpretation [2]. Biological knowledge can take many forms, including genomic sequences, pathway databases, gene interaction networks, and knowledge hierarchies such as the Gene Ontology [3]. However, there is often no canonical way to encode these structures as real-valued predictors. Modelers must creatively decide how to encode biological knowledge that they expect will be relevant to the task.
Biomedical datasets often contain more input predictors than data samples [4,5]. A genetic study may genotype millions of single nucleotide polymorphisms (SNPs) in thousands of individuals, or a gene expression study may profile the expression of thousands of genes in tens of samples. Thus, it can be useful to include prior information describing relationships between predictors to inform the representation learned by the model. This contrasts with non-biological applications of machine learning, where one might fit a model on millions of images [6] or tens of thousands of documents [7], making inclusion of prior information unnecessary.
We review approaches that incorporate external information about the structure of desirable solutions to learn from biomedical data. One class of commonly used approaches learns a representation that considers the context of each base pair from raw sequence data. For models that operate on gene expression data or genetic variants, it can be useful to incorporate networks or pathways describing relationships between genes. We also consider other examples, such as neural network architectures that are constrained based on biological knowledge.
There are many complementary ways to incorporate heterogeneous sources of biomedical data into the learning process, which have been covered elsewhere [8,9]. These include feature extraction or representation learning before modeling and/or other data integration methods that do not necessarily involve customizing the model itself.
Sequence models
Early neural network models primarily used hand-engineered sequence features as input to a fully connected neural network [10,11] (Figure 1). As convolutional neural network (CNN) approaches matured for image processing and computer vision, researchers leveraged biological sequence proximity similarly. CNNs are a neural network variant that groups input data by spatial context to extract features for prediction.
Figure 1.

Contrasting approaches to extracting features from DNA or RNA sequence data. Early models defined features of interest by hand based on prior knowledge about the prediction or clustering problem of interest, such as GC content or sequence melting point, as depicted in the left branch in the figure. Convolutional models, depicted in the right branch, use sequence convolutions to derive features directly from sequence proximity, without requiring quantities of interest to be identified before the model is trained. Red or blue emphasis denotes inputs to the predictive model (either the hand-defined numeric features on the left or the outputs of convolutional filters on the right).
The definition of ‘spatial context’ is specific to the input: one might group image pixels that are nearby in 2D space, or genomic base pairs that are nearby in the linear genome. In this way, CNNs consider context without making strong assumptions about exactly how much context is needed or how it should be encoded; the data inform the encoding. A detailed description of how CNNs are applied to sequences can be found in Angermueller et al. [12].
Applications in regulatory biology
Many early applications of deep learning to biological sequences were in regulatory biology. Early CNNs for sequence data predicted binding protein sequence specificity from DNA or RNA sequence [13], variant effects from noncoding DNA sequence [14], and chromatin accessibility from DNA sequence [15].
Recent sequence models take advantage of hardware advances and methodological innovation to incorporate more sequence context and rely on fewer modeling assumptions. BPNet, a CNN that predicts transcription factor binding profiles from DNA sequences, accurately mapped known locations of binding motifs in mouse embryonic stem cells [16••]. BPNet considers 1000 base pairs of context around each position when predicting binding probabilities with a technique called dilated convolutions [17], which is particularly important because motif spacing and periodicity can influence binding. cDeepbind [18•] combines RNA sequences with information about secondary structure to predict RNA binding protein affinities. Its convolutional model acts on a feature vector combining sequence and structural information, using context for both to inform predictions. APARENT [19] is a CNN that predicts alternative polyadenylation (APA) from a training set of over three million synthetic APA reporter sequences. These diverse applications underscore the power of modern deep learning models to synthesize large sequence datasets.
Models that consider sequence context have also been applied to epigenetic data. DeepSignal [20] is a CNN that uses contextual electrical signals from Oxford Nanopore single-molecule sequencing data to predict 5mC or 6 mA DNA methylation status. MRCNN [21] uses sequences of length 400, centered at CpG sites, to predict 5mC methylation status. Deep learning models have also been used to predict gene expression from histone modifications [22,23•]. Here, a neural network model consisting of long short-term memory (LSTM) units was used to encode the long-distance interactions of histone marks in both the 3′ and 5′ genomic directions. In each of these cases, proximity in the linear genome helped model the complex interactions between DNA sequence and epigenome.
Applications in variant calling and mutation detection
Identification of genetic variants also benefits from models that include sequence context. DeepVariant [24••] applies a CNN to images of sequence read pileups, using read data around each candidate variant to accurately distinguish true variants from sequencing errors. CNNs have also been applied to single molecule (PacBio and Oxford Nanopore) sequencing data [25], using a different sequence encoding that results in better performance than DeepVariant on single molecule data. However, many variant calling models still use hand-engineered sequence features as input to a classifier, including current state-of-the-art approaches to insertion/deletion calling [26,27]. Detection of somatic mutations is a distinct but related challenge to detection of germline variants, and has also recently benefitted from use of CNNs [28].
Network-based and pathway-based models
Rather than operating on sequences, many machine learning models in biomedicine operate on inputs that lack intrinsic order. Models may make use of gene expression data matrices from RNA sequencing or micro-array experiments in which rows represent samples and columns represent genes. To account for relationships between genes, one might incorporate known interactions or correlations when making predictions or generating a low-dimensional representation of the data (Figure 2). This is comparable to the manner in which sequence context pushes models to consider nearby base pairs similarly.
Figure 2.

The relationships between genes provide structure that can be incorporated into machine learning models. One common approach is to use a network or collection of gene sets to embed the data in a lower-dimensional space, in which genes that are in the same gene sets or that are well-connected in the network have a similar representation in the lower-dimensional space. The embedded data can then be used for classification or clustering tasks. The ‘x’ values in the data table represent gene expression measurements.
Applications in transcriptomics
Models built from gene expression data can benefit from incorporating gene-level relationships. One form that this knowledge commonly takes is a database of gene sets, which may represent biological pathways or gene signatures for a biological state of interest. PLIER [29••] uses gene set information from MSigDB [30] and cell type markers to extract a representation of gene expression data that corresponds to biological processes and reduces technical noise. The resulting gene set-aligned representation accurately decomposed cell type mixtures. MultiPLIER [31] applied PLIER to the recount2 gene expression compendium [32] to develop a model that shares information across multiple tissues and diseases, including rare diseases with limited sample sizes. PASNet [33] uses MSigDB to inform the structure of a neural network for predicting patient outcomes in glioblastoma multiforme (GBM) from gene expression data. This approach aids interpretation, as pathway nodes in the network with high weights can be inferred to correspond to certain pathways in GBM outcome prediction.
Gene-level relationships can also be represented with networks. Network nodes typically represent genes and real-valued edges may represent interactions or correlations between genes, often in a tissue or cell type context of interest. Network-based stratification [34] is an early example of a method for utilizing gene interaction network data in this manner, applied to improve subtyping across several cancer types. More recently, netNMF-sc [35••] incorporates coexpression networks [36] as a smoothing term for dimension reduction and dropout imputation in single-cell gene expression data. The coexpression network improves performance for identifying cell types and cell cycle marker genes, as compared to using raw gene expression or other single-cell dimension reduction methods. Combining gene expression data with a network-derived smoothing term also improved prediction of patient drug response in acute myeloid leukemia [37•] and detection of mutated cancer genes [38]. PIMKL [39•] combines network and pathway data to predict disease-free survival from breast cancer cohorts. This method takes as input both RNA-seq gene expression data and copy number alteration data, but can also be applied to gene expression data alone.
Gene regulatory networks can also augment models for gene expression data. These networks describe how the expression of genes is modulated by biological regulators such as transcription factors, microRNAs, or small molecules. creNET [40••] integrates a gene regulatory network, derived from STRING [41], with a sparse logistic regression model to predict phenotypic response in clinical trials for ulcerative colitis and acute kidney rejection. The gene regulatory information allows the model to identify the biological regulators associated with the response, potentially giving mechanistic insight into differential clinical trial response. GRRANN [42], which was applied to the same data as creNET, uses a gene regulatory network to inform the structure of a neural network. Several other methods [43,44] have also used gene regulatory network structure to constrain the structure of a neural network, reducing the number of parameters to be fit and facilitating interpretation.
Applications in genetics
Approaches that incorporate gene set or network structure into genetic studies have a long history [45,46]. Recent applications include expression quantitative trait loci (eQTL) mapping studies, which aim to identify associations between genetic variants and gene expression. netReg [47] implements a graph-regularized dual LASSO algorithm for eQTL mapping [48] in a publicly available R package. This model smoothens regression coefficients simultaneously based on networks describing associations between genes (target variables in the eQTL regression model) and between variants (predictors in the eQTL regression model). eQTL information is also used in conjunction with genetic variant information to predict phenotypes, in an approach known as Mendelian randomization (MR). In [49], a smoothing term derived from a gene regulatory network is used in an MR model. The model with the network smoothing term, applied to a human liver dataset, more robustly identifies genes that influence enzyme activity than a network-agnostic model. As genetic datasets grow, we expect that researchers will continue to develop models that leverage gene set and network databases.
Other models incorporating biological structure
Knowledge about biological entities is often organized in an ontology, which is a directed graph that encodes relationships between entities (see Figure 3 for a visual example). The Gene Ontology (GO) [3] describes the relationships between cellular subsystems and other attributes describing proteins or genes. DCell [50••] uses GO to inform the connectivity of a neural network predicting the effects of gene deletions on yeast growth. DCell performs comparably to an unconstrained neural network for this task. Additionally, it is easier to interpret: a cellular subsystem with high neuron outputs under a particular gene deletion can be inferred to be strongly affected by the gene deletion, providing a putative genotype-phenotype association. DeepGO [51•] uses a similar approach to predict protein function from amino acid sequence with a neural network constrained by the dependencies of GO. However, a follow-up paper by the same authors [52] showed that this hierarchy-aware approach can be outperformed by a hierarchy-naive CNN, which uses only amino acid sequence and similarity to labeled training set proteins. This suggests a trade-off between interpretability and predictive accuracy for protein function prediction.
Figure 3.
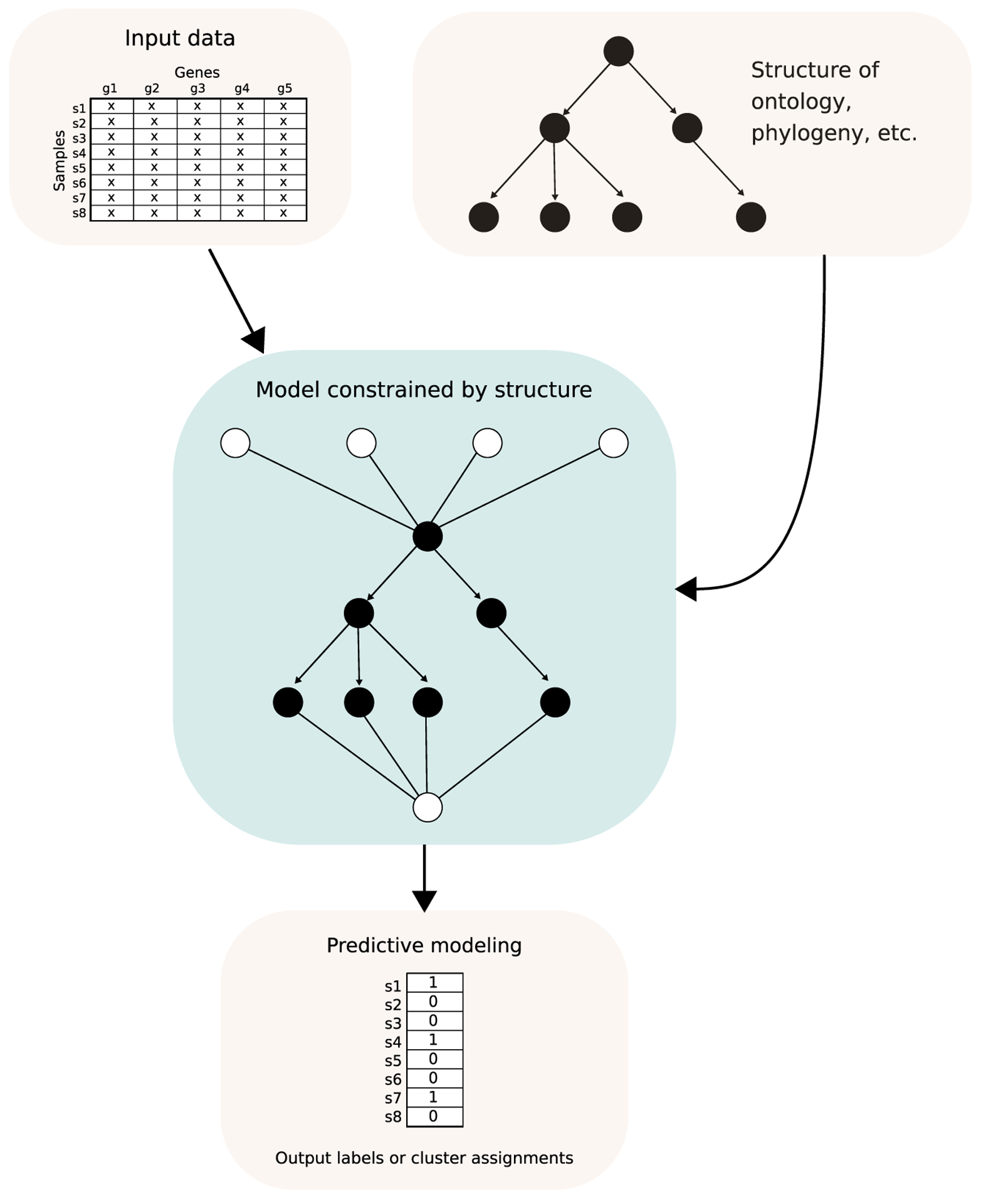
Directed graph-structured data, such as an ontology or phylogenetic tree, can be incorporated into machine learning models. Here, the connections in the neural network used to predict a set of labels parallel those in the tree graph. This type of constraint can also be useful in model interpretation: for example, if the nodes in the right tree branch have high neuron outputs for a given sample, then the subsystem encoded in the right branch of the tree graph is most likely important in making predictions for that sample. The ‘x’ values in the data table represent gene expression measurements.
Phylogenetic trees, or hierarchies describing the evolutionary relationships between species, can be useful for a similar purpose. glmmTree [53] uses a phylogenetic tree describing the relationship between microorganisms to improve predictions of age based on gut microbiome data. The same authors combine a similar phylogeny smoothing strategy with sparse regression to model caffeine intake and smoking status based on microbiome data [54]. Phylogenetic trees can also describe the relationships between subclones of a tumor, which are fundamental to understanding cancer evolution and development. Using a tumor phylogeny inferred from copy number aberration (CNA) sequencing data as a smoothing term for deconvolving tumor subclones provided more robust predictions than a phylogeny-free model [55]. The tree structure of the phylogeny and the subclone mixture model are fit jointly to the CNA data.
Depending on the application, other forms of structure or prior knowledge can inform predictions and interpretation of the model’s output. CYCLOPS [56] uses a circular node autoencoder [57] to order periodic gene expression data and estimate circadian rhythms. The authors validated the method by correctly ordering samples without temporal labels and identifying genes with known circadian expression. They then applied it to compare gene expression in normal and cancerous liver biopsies, identifying drug targets with circadian expression as candidates for chronotherapy. NetBiTE [58••] uses drug-gene interaction information from GDSC [59], in addition to protein interaction data, to build a tree ensemble model with splits that are biased toward high-confidence drug-gene interactions. The model predicts sensitivity to drugs that inhibit crucial signaling pathways in cancer, showing improved predictive performance compared to random forests, another commonly used tree ensemble model.
Conclusions and future directions
As the quantity and richness of biomedical data have increased, sequence repositories and interaction databases have expanded and become more robust. This raises opportunities to integrate these resources into the structure of machine learning models. There have been several past attempts to benchmark and compare approaches to integrating structured data into predictive models in biomedicine, including the evaluation in Ref. [60] and more recent studies in Refs. [61] and [62]. Extending and broadening benchmarking efforts such as these will be vital, improving our understanding of the suitability of problem domains and datasets for the classes of methods described in this review.
Many methods described in this review have open-source implementations available; however, increased availability of performant and extensible implementations of these models and algorithms would facilitate further use and development. In the future, we foresee that incorporating structured biomedical data will become commonplace for improving model interpretability and boosting performance when sample size is limited.
Acknowledgement
The authors would like to thank Daniel Himmelstein for a critical reading of the manuscript and helpful discussion.
Funding
This work was funded by grants from the National Human Genome Research Institute (R01 HG010067), the Gordon and Betty Moore Foundation (GBMF 4552), the National Cancer Institute (R01 CA237170) and Alex’s Lemonade Stand Foundation (CCDL) to CSG.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Lenore Cowen, Trey Ideker, Raphael Benjamin J, Sharan Roded: Network propagation: a universal amplifier of genetic associations. Nat Rev Genet 2017. 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 2.Yu Michael K, Ma Jianzhu, Fisher Jasmin, Kreisberg Jason F, Raphael Benjamin J, Ideker Trey: Visible Machine Learning for Biomedicine. Cell 2018. 10.1016/j.cell.2018.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res 2018. 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michael KK, Leung Andrew, Delong Babak, Alipanahi, Frey Brendan: Machine learning in genomic medicine: a review of computational problems and data sets. Proc IEEE 2016. 10.1109/jproc.2015.2494198. [DOI] [Google Scholar]
- 5.Romero Adriana, Luc Carrier Pierre, Erraqabi Akram, Sylvain Tristan, Auvolat Alex, Dejoie Etienne, Legault Marc-André, Dubé Marie-Pierre, Hussin Julie G, Bengio Yoshua: Diet networks: thin parameters for fat genomics. arXiv 2016. [Google Scholar]
- 6.Jia Deng, Wei Dong, Richard Socher, Li-Jia Li, Kai Li, Li Fei-Fei: ImageNet: a large-scale hierarchical image database, 2009. IEEE Conference on Computer Vision and Pattern Recognition 2009. 10.1109/cvpr.2009.5206848. [DOI] [Google Scholar]
- 7.Maas Andrew L, Daly Raymond E, Pham Peter T, Huang Dan, Ng Andrew Y: Christopher Potts learning word vectors for sentiment analysis. Proceedings of the 49th Annual Meeting of the Association for Computational Linguistics: Human Language Technologies 2011. [Google Scholar]
- 8.Walter Nelson, Marinka Zitnik, Bo Wang, Jure Leskovec, Anna Goldenberg, Roded Sharan: To embed or not: network embedding as a paradigm in computational biology. Front Genet 2019. 10.3389/fgene.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zitnik Marinka, Nguyen Francis, Wang Bo, Leskovec Jure, Goldenberg Anna, Hoffman Michael M: Machine learning for integrating data in biology and medicine: principles, practice, and opportunities. Inf Fusion 2019. 10.1016/j.inffus.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleftogiannis Dimitrios, Kalnis Panos, Bajic Vladimir B: DEEP: a general computational framework for predicting enhancers. Nucleic Acids Res 2014. 10.1093/nar/gku1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR et al. : The human splicing code reveals new insights into the genetic determinants of disease. Science 2014. 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christof Angermueller, Tanel Pärnamaa, Leopold Parts, Oliver Stegle: Deep learning for computational biology. Mol Syst Biol 2016. 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babak Alipanahi Andrew, Delong, Matthew T, Weirauch Frey Brendan J: Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 2015. 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 14.Jian Zhou, Troyanskaya Olga G: Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods 2015. 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley David R, Snoek Jasper, Rinn John L: Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 2016. 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.••.Žiga Avsec, Melanie Weilert, Avanti Shrikumar, Amr Alexandari, Sabrina Krueger, Khyati Dalal, Robin Fropf, Charles McAnany, Julien Gagneur, Anshul Kundaje, Julia Zeitlinger: Deep learning at base-resolution reveals motif syntax of the cis-regulatory code. Cold Spring Harbor Lab 2019. 10.1101/737981. [DOI] [Google Scholar]; This paper describes BPNet, a neural network for predicting transcription factor (TF) binding profiles from raw DNA sequence. The model is able to accurately infer the spacing and periodicity of pluripotency-related TFs in mouse embryonic stem cells, leading to an improved understanding of the motif syntax of combinatorial TF binding in cell development.
- 17.Fisher Yu, Vladlen Koltun: Multi-scale context aggregation by dilated convolutions. arXiv 2015. [Google Scholar]
- 18.•.Gandhi Shreshth, Lee Leo J, Delong Andrew, Duvenaud David, Frey Brendan J: cDeepbind: a context sensitive deep learning model of RNA-protein binding. Cold Spring Harbor Lab 2018. 10.1101/345140. [DOI] [Google Scholar]; cDeepbind is a neural network model for predicting RNA binding protein (RBP) specificity from RNA sequence and secondary structure information. The authors show that this combined approach provides an improvement over previous models that use only sequence information.
- 19.Bogard Nicholas, Linder Johannes, Rosenberg Alexander B, Seelig Georg: A deep neural network for predicting and engineering alternative polyadenylation. Cell 2019. 10.1016/j.cell.2019.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Ni, Neng Huang, Zhi Zhang, De-Peng Wang, Fan Liang, Yu Miao, Chuan-Le Xiao, Feng Luo, Jianxin Wang: DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 2019. 10.1093/bioinformatics/btz276. [DOI] [PubMed] [Google Scholar]
- 21.Qi Tian, Jianxiao Zou, Jianxiong Tang, Yuan Fang, Zhongli Yu, Shicai Fan: MRCNN: a deep learning model for regression of genome-wide DNA methylation. BMC Genomics 2019. 10.1186/s12864-019-5488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritambhara Singh, Jack Lanchantin, Arshdeep Sekhon, Yanjun Qi: Attend and predict: understanding gene regulation by selective attention on chromatin. Cold Spring Harbor Lab 2018. 10.1101/329334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.•.Arshdeep Sekhon, Ritambhara Singh, Yanjun Qi : DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications. Bioinformatics 2018. 10.1093/bioinformatics/bty612. [DOI] [PubMed] [Google Scholar]; DeepDiff uses a long short-term memory neural network to predict differential gene expression from the spatial structure of histone modification measurements. The network has a multi-task objective, enabling gene expression predictions to be made simultaneously in multiple cell types.
- 24.••.Ryan Poplin, Pi-Chuan Chang, David Alexander, Scott Schwartz, Thomas Colthurst, Ku Alexander, Dan Newburger, Jojo Dijamco, Nam Nguyen, Pegah T Afshar et al. : A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 2018. 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]; This paper describes DeepVariant, a neural network model for distinguishing true genetic variants from errors in next-generation DNA sequencing data. The model adapts techniques from the image processing community to fit a model on images of read pileups around candidate variants, using information about the sequence around the candidate variant site to make predictions about the true genotype at the site.
- 25.Luo Ruibang, Sedlazeck Fritz J, Lam Tak-Wah, Schatz Michael C: A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat Commun 2019. 10.1038/s41467-019-09025-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shariful Islam Bhuyan Md, Pe’er Itsik, Sohel Rahman M: SICaRiO: Short Indel Call filteRing with bOosting. Cold Spring Harbor Lab 2019. 10.1101/601450. [DOI] [Google Scholar]
- 27.Curnin Charles, Goldfeder Rachel L, Marwaha Shruti, Bonner Devon, Waggott Daryl, Wheeler Matthew T, Ashley Euan A: Machine learning-based detection of insertions and deletions in the human genome. Cold Spring Harbor Lab 2019. 10.1101/628222. [DOI] [Google Scholar]
- 28.Ebrahim Sahraeian Sayed Mohammad, Ruolin Liu, Bayo Lau, Karl Podesta, Marghoob Mohiyuddin, Hugo YK Lam: Deep convolutional neural networks for accurate somatic mutation detection. Nat Commun 2019. 10.1038/s41467-019-09027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.••.Mao Weiguang, Zaslavsky Elena, Hartmann Boris M, Sealfon Stuart C, Chikina Maria: Pathway-level information extractor (PLIER) for gene expression data. Nat Methods 2019. 10.1038/s41592-019-0456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a ‘pathway-level information extractor’ (PLIER), a method for reducing the dimension of gene expression data in a manner that aligns with known biological pathways or informative gene sets. The method can also reduce the effects of technical noise. The authors show that PLIER can be used to improve cell type inference and as a component in eQTL studies.
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taroni Jaclyn N, Grayson Peter C, Hu Qiwen, Eddy Sean, Kretzler Matthias, Merkel Peter A, Greene Casey S: MultiPLIER: a transfer learning framework for transcriptomics reveals systemic features of rare disease. Cell Syst 2019. 10.1016/j.cels.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collado-Torres Leonardo, Nellore Abhinav, Kammers Kai, Ellis Shannon E, Taub Margaret A, Hansen Kasper D, Jaffe Andrew E, Langmead Ben, Leek Jeffrey T: Reproducible RNA-seq analysis using recount2. Nat Biotechnol 2017. 10.1038/nbt.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jie Hao, Youngsoon Kim, Tae-Kyung Kim, Mingon Kang: PASNet: pathway-associated sparse deep neural network for prognosis prediction from high-throughput data. BMC Bioinf 2018. 10.1186/s12859-018-2500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matan Hofree, Shen John P, Carter Hannah, Andrew Gross, Trey Ideker: Network-based stratification of tumor mutations. Nat Methods 2013. 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.••.Elyanow Rebecca, Dumitrascu Bianca, Engelhardt Barbara E, Raphael Benjamin J: netNMF-sc: leveraging gene-gene interactions for imputation and dimensionality reduction in single-cell expression analysis. Cold Spring Harbor Lab 2019. 10.1101/544346. [DOI] [PMC free article] [PubMed] [Google Scholar]; netNMF-sc is a dimension reduction method that uses network information to ‘smooth’ a matrix factorization of single-cell gene expression data, such that genes that are connected in the network have a similar low-dimensional representation. Inclusion of network information is particularly useful when analyzing single-cell expression data, due to its ability to mitigate ‘dropouts’ and other sources of variability that are present at the single cell level.
- 36.Sunmo Yang, Kim Chan Yeong, Sohyun Hwang, Eiru Kim, Hyojin Kim, Hongseok Shim, Insuk Lee: COEXPEDIA: exploring biomedical hypotheses via co-expressions associated with medical subject headings (MeSH). Nucleic Acids Res 2016. 10.1093/nar/gkw868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.•.Erion Gabriel, Janizek Joseph D, Sturmfels Pascal, Lundberg Scott, Lee Su-In: Learning explainable models using attribution priors. arXiv 2019. [Google Scholar]; This paper describes ‘model attribution priors’, or a method for constraining a machine learning model’s behavior during training with prior beliefs or expectations about the data or problem structure. As an example of this concept, the authors show that incorporation of network data improves the performance of a model for drug response prediction in acute myeloid leukemia.
- 38.Jianing Xi, Ao Li, Minghui Wang: A novel network regularized matrix decomposition method to detect mutated cancer genes in tumour samples with inter-patient heterogeneity. Sci Rep 2017. 10.1038/s41598-017-03141-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.•.Matteo Manica, Joris Cadow, Roland Mathis, Rodríguez Martínez María: PIMKL: pathway-induced multiple kernel learning. NPJ Syst Biol Appl 2019. 10.1038/s41540-019-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the authors present an algorithm for combining gene expression and copy number data with prior information, such as gene networks and pathways or gene set annotations, to predict survival in breast cancer. The weights learned by the model are also interpretable, providing a putative set of explanatory features for the prediction task.
- 40.••.Kourosh Zarringhalam, David Degras, Christoph Brockel, Daniel Ziemek: Robust phenotype prediction from gene expression data using differential shrinkage of co-regulated genes. Sci Rep 2018. 10.1038/s41598-018-19635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes creNET, a regression model for gene expression data that uses information about gene regulation to differentially weight or penalize gene sets that are co-regulated. The authors show that the model can be used to predict phenotype from gene expression data in clinical trials. The model also provides interpretable weights for each gene regulator.
- 41.Szklarczyk Damian, Franceschini Andrea, Wyder Stefan, Forslund Kristoffer, Heller Davide, Huerta-Cepas Jaime, Simonovic Milan, Roth Alexander, Santos Alberto, Tsafou Kalliopi P et al. : STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2014. 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tianyu Kang, Wei Ding, Luoyan Zhang, Daniel Ziemek, Kourosh Zarringhalam: A biological network-based regularized artificial neural network model for robust phenotype prediction from gene expression data. BMC Bioinf 2017. 10.1186/s12859-017-1984-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chieh Lin, Siddhartha Jain, Hannah Kim, Ziv Bar-Joseph: Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res 2017. 10.1093/nar/gkx681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ameen Eetemadi, Ilias Tagkopoulos: Genetic neural networks: an artificial neural network architecture for capturing gene expression relationships. Bioinformatics 2018. 10.1093/bioinformatics/bty945. [DOI] [PubMed] [Google Scholar]
- 45.Wei Z, Li H: Nonparametric pathway-based regression models for analysis of genomic data. Biostatistics 2006. 10.1093/biostatistics/kxl007. [DOI] [PubMed] [Google Scholar]
- 46.Li C, Li H: Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics 2008. 10.1093/bioinformatics/btn081. [DOI] [PubMed] [Google Scholar]
- 47.Dirmeier Simon, Fuchs Christiane, Mueller Nikola S, Theis Fabian J: netReg: network-regularized linear models for biological association studies. Bioinformatics 2017. 10.1093/bioinformatics/btx677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Cheng, Xiang Zhang, Zhishan Guo, Yu Shi, Wei Wang: Graph-regularized dual Lasso for robust eQTL mapping. Bioinformatics 2014. 10.1093/bioinformatics/btu293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bin Gao, Xu Liu, Hongzhe Li, Yuehua Cui: Integrative analysis of genetical genomics data incorporating network structures. Biometrics 2019. 10.1111/biom.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.••.Jianzhu Ma, Michael Ku Yu, Samson Fong, Keiichiro Ono, Eric Sage, Barry Demchak, Roded Sharan, Trey Ideker: Using deep learning to model the hierarchical structure and function of a cell. Nat Methods 2018. 10.1038/nmeth.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents DCell, a neural network model for prediction of yeast growth phenotype from gene deletions. The structure of the neural network is constrained by the relationships encoded in the Gene Ontology (GO), enabling predictions for a given input to be interpreted based on the subsystems of GO that they activate. Thus, the neural network can be seen as connecting genotype to phenotype.
- 51.•.Maxat Kulmanov, Khan Mohammed Asif, Robert Hoehndorf: DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2017. 10.1093/bioinformatics/btx624. [DOI] [PMC free article] [PubMed] [Google Scholar]; Here, the authors describe a method for predicting protein function from amino acid sequence, incorporating the dependency structure of the Gene Ontology (GO) into their neural network used for prediction. Using the GO information provides a performance improvement over similar models that do not incorporate this information.
- 52.Maxat Kulmanov, Robert Hoehndorf: DeepGOPlus: improved protein function prediction from sequence. Bioinformatics 2019. 10.1093/bioinformatics/btz595. [DOI] [Google Scholar]
- 53.Jian Xiao, Li Chen, Stephen Johnson, Yue Yu, Xianyang Zhang, Jun Chen: Predictive modeling of microbiome data using a phylogeny-regularized generalized linear mixed model. Front Microbiol 2018. 10.3389/fmicb.2018.01391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jian Xiao, Li Chen, Yue Yu, Xianyang Zhang, Jun Chen: A phylogeny-regularized sparse regression model for predictive modeling of microbial community data. Front Microbiol 2018. 10.3389/fmicb.2018.03112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lei Haoyun, Lyu Bochuan, Michael Gertz E, Schäffer Alejandro A, Shi Xulian, Wu Kui, Li Guibo, Xu Liqin, Hou Yong, Dean Michael, Schwartz Russell: Tumor copy number deconvolution integrating bulk and single-cell sequencing data. Lect Notes Comput Sci 2019. 10.1007/978-3-030-17083-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anafi Ron C, Francey Lauren J, Hogenesch John B, Kim Junhyong: CYCLOPS reveals human transcriptional rhythms in health and disease. Proc Natl Acad Sci U S A 2017. 10.1073/pnas.1619320114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirby Michael J, Miranda Rick: Circular nodes in neural networks. Neural Comput 1996. 10.1162/neco.1996.8.2.390. [DOI] [PubMed] [Google Scholar]
- 58.••.Ali Oskooei, Matteo Manica, Roland Mathis, Maria Rodriguez Martinez: Network-based biased tree ensembles (NetBiTE) for drug sensitivity prediction and drug sensitivity biomarker identification in cancer. arXiv 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a method for using prior knowledge about drug targets to inform the structure of a tree ensemble model, used for predicting IC50 drug sensitivity from gene expression data. The model also uses a protein interaction network to ‘smooth’ the gene weights, such that genes that are related in the network will have a similar influence on predictions.
- 59.Yang Wanjuan, Soares Jorge, Greninger Patricia, Edelman Elena J, Lightfoot Howard, Forbes Simon, Bindal Nidhi, Beare Dave, Smith James A, Richard Thompson I et al. : Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2012. 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christine Staiger, Sidney Cadot, Raul Kooter, Marcus Dittrich, Tobias Müller, Klau Gunnar W, Wessels Lodewyk FA: A critical evaluation of network and pathway-based classifiers for outcome prediction in breast cancer. PLoS One 2012. 10.1371/journal.pone.0034796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bertin Paul, Hashir Mohammad, Weiss Martin, Boucher Geneviève, Frappier Vincent, Cohen Joseph Paul: Analysis of gene interaction graphs for biasing machine learning models. arXiv 2019. [Google Scholar]
- 62.Mohammad Hashir, Paul Bertin, Martin Weiss, Vincent Frappier, Perkins Theodore J, Boucher Geneviève, Cohen Joseph Paul: Is graph-based feature selection of genes better than random? arXiv 2019. [Google Scholar]