

Abstract
Epithelial-to-mesenchymal transition (EMT) and its reversed process mesenchymal-to-epithelial transition (MET) play a critical role in epithelial plasticity during development and cancer progression. Among important regulators of these cellular processes are non-coding RNAs (ncRNAs). The imprinted DLK1-DIO3 locus, containing numerous maternally expressed ncRNAs including the lncRNA maternally expressed gene 3 (MEG3) and a cluster of over 50 miRNAs, has been shown to be a modulator of stemness in embryonic stem cells and in cancer progression, potentially through the tumor suppressor role of MEG3. In this study we analyzed the expression pattern and functional role of ncRNAs from the DLK1-DIO3 locus in epithelial plasticity of the breast. We studied their expression in various cell types of breast tissue and revisit the role of the locus in EMT/MET using a breast epithelial progenitor cell line (D492) and its isogenic mesenchymal derivative (D492M). Marked upregulation of ncRNAs from the DLK1-DIO3 locus was seen after EMT induction in two cell line models of EMT. In addition, the expression of MEG3 and the maternally expressed ncRNAs was higher in stromal cells compared to epithelial cell types in primary breast tissue. We also show that expression of MEG3 is concomitant with the expression of the ncRNAs from the DLK1-DIO3 locus and its expression is therefore likely indicative of activation of all ncRNAs at the locus. MEG3 expression is correlated with stromal markers in normal tissue and breast cancer tissue and negatively correlated with the survival of breast cancer patients in two different cohorts. Overexpression of MEG3 using CRISPR activation in a breast epithelial cell line induced partial EMT and enriched for a basal-like phenotype. Conversely, knock down of MEG3 using CRISPR inhibition in a mesenchymal cell line reduced the mesenchymal and basal-like phenotype of the cell line. In summary our study shows that maternally expressed ncRNAs are markers of EMT and suggests that MEG3 is a novel regulator of EMT/MET in breast tissue. Nevertheless, further studies are needed to fully dissect the molecular pathways influenced by non-coding RNAs at the DLK1-DIO3 locus in breast tissue.
Keywords: DLK1-DIO3 locus, MEG3, ncRNAs, epithelial plasticity, breast progenitor cells
Introduction
Breast cancer is the most common cancer in women and the second most common cancer overall (Ghoncheh et al., 2016). Despite major advances in diagnosis and treatment of cancer in recent years, metastasis and development of resistance to cancer therapies continues to be a challenge, causing over 90% of all cancer-related deaths (Ben-Jacob et al., 2012). A major contributing factor to metastasis and drug resistance is the heterogeneity and plasticity of the cells within tumors (Dagogo-Jack and Shaw, 2018). Epithelial-to-mesenchymal transition (EMT), is a developmental process that can be hijacked by cancer cells (Zeisberg and Kalluri, 2004; Moustakas and Heldin, 2007; Radisky et al., 2007). Generally, cells undergoing EMT, acquire increased migration and invasive properties and show increased resistance to apoptosis (Robson et al., 2006; Cao et al., 2016). Through these processes, EMT is considered a major mediator of phenotypic plasticity in cancer cells, metastatic formation and drug resistance (Mani et al., 2008; Scheel and Weinberg, 2012; Ansieau, 2013; Nieto et al., 2016; Lu and Kang, 2019). Recently, hybrid E/M (or partial EMT) cells have been shown to have even more metastatic and stem cell potential compared to the full epithelial or mesenchymal phenotype (Pastushenko et al., 2018). A reversed program, mesenchymal-to–epithelial transition (MET) is considered to facilitate colonization in secondary sites and reverse the plastic mesenchymal phenotype back to an epithelial state (Lu and Kang, 2019). This, however, is debated and further studies will increase our knowledge of the role of EMT/MET in cancer progression and metastasis.
EMT can be initiated through intrinsic factors such as expression of EMT related transcription factors (SNAI1, SNAI2, TWIST1, ZEB1, or ZEB2), cadherin switch from E-cadherin (CDH1) to N-cadherin (CDH2) or through epigenetic mechanisms. It can also be brought on by extrinsic factors derived from the microenvironment, such as secreted soluble factors: transforming growth factor-β (TGF-β), epidermal growth factor (EGF), fibroblast growth factors (FGFs), hepatocyte growth factor (HGF) or Wnt signaling factors (Moustakas and Heldin, 2007; Peinado et al., 2007; De Craene and Berx, 2013; Wang and Zhou, 2013; Williams et al., 2019).
Non-coding RNAs (ncRNAs) are among intrinsic regulators of EMT (Zaravinos, 2015). It is increasingly apparent that the ncRNAs are crucial in normal development and disease, but its mechanistic mode of action is largely unknown (Liz and Esteller, 2016). The two major classes of non-coding RNAs are long non-coding RNA (lncRNAs) and microRNA (miRNAs). Accumulating evidence suggests that lncRNAs function in a broad range of cellular processes such as cell growth, survival, migration, invasion and differentiation (Mercer et al., 2009; Sun et al., 2013; Di Gesualdo et al., 2014; Fatica and Bozzoni, 2014). LncRNAs are defined by the size of their transcripts and are longer than 200 nucleotides (nt), with no protein-coding function (Eades et al., 2014). Unlike microRNAs, lncRNAs are poorly conserved, but function in a regulatory network at the transcriptional, post-transcriptional, and translational level. miRNAs are 22 nt long RNA molecules that regulate expression post-transcriptionally primarily by binding to three prime untranslated region (3′UTR) of target genes (Bartel, 2009).
The imprinted DLK1-DIO3 locus located on chromosome 14 contains three paternally expressed protein-coding genes (DLK1, RTL1, DIO3) and numerous maternally expressed non-coding genes, including the lncRNA maternally expressed gene 3 (MEG3), and a cluster of over 50 miRNAs (Zhang et al., 2010; Dill and Naya, 2018; Baulina et al., 2019; Li et al., 2019).
The DLK1-DIO3 locus has been described as an important contributor to pluripotency and stemness in embryonic stem cells (ESCs) (Kaneko et al., 2014). It discriminates between mouse induced pluripotent stem cells (iPCS) and mouse ESCs, where genes from the locus were strongly repressed in iPSC clones compared to ES clones (Liu et al., 2010; Stadtfeld et al., 2010a). Furthermore, activation of maternally expressed genes from the locus is a strong indicator of the developmental potential of iPSC (Kang et al., 2009). miRNAs from the DLK1-DIO3 locus have been shown to promote pluripotency by inhibition of differentiation and stimulation of self-renewal in mouse ES cells (Moradi et al., 2017) and were found to be increased in tumor-originating cancer cells from lung adenocarcinoma (Valdmanis et al., 2015).
MEG3 is a potential tumor suppressor gene in several cancer types, mainly through the observation that MEG3 expression is lower in various tumor tissues compared with non-tumor tissues of the same origin (Sheng et al., 2014; Sun et al., 2014, 2016; Yin et al., 2015; Chak et al., 2017; Molina-Pinelo et al., 2018). The tumor suppressor role of MEG3 is ascribed to stabilization of p53 with inhibition of proliferation and promotion of apoptosis (Zhang et al., 2003, 2010; Zhou et al., 2007; Wang et al., 2012; Sun et al., 2016).
MEG3 was reported to positively regulate EMT in lung (Terashima et al., 2017) and ovarian (Mitra et al., 2017) cancer. Furthermore, MEG3 has been shown to contribute to the development of osteosarcoma through increased migration, invasion and decreased apoptosis (Wang and Kong, 2018). Higher levels of MEG3 were detected in plasma from colorectal cancer patients compared with non-cancerous controls (Liu et al., 2019).
D492 is a primary breast epithelial cell line, immortalized with the E6 and E7 oncogenes from the human papilloma virus 16 (Gudjonsson et al., 2002). Therefore, the p53 protein, which mediates the previously described tumor suppressor role of MEG3, is repressed in this cell line. D492 can generate both luminal and basal/myoepithelial cells in monolayer and 3D culture, expressing luminal or myoepithelial keratins such as keratin 19 and keratin 14, respectively. Furthermore, when D492 cells are co-cultured with endothelial cells, they, can generate spindle-shaped colonies with EMT phenotype. D492M (mesenchymal) was established from one such spindle-shaped colony (Sigurdsson et al., 2011). D492M is a phenotypically stable EMT cell line. It has lost epithelial markers such as keratins, E-cadherin and TP63, and gained expression of mesenchymal markers such as N-cadherin (Sigurdsson et al., 2011; Hilmarsdottir et al., 2015). D492M has acquired classical properties of cancer stem cells, such as increased CD44/CD24 ratio, anchorage independent growth, resistance to apoptosis and increased migration/invasion (Sigurdsson et al., 2011). D492 serves as a model for branching morphogenesis and together D492 and D492M represent a unique EMT model of isogenic cell lines with an epithelial and mesenchymal phenotype, respectively (Briem et al., 2019b). The ability of D492 to undergo mesenchymal transition upon endothelial stimulation makes it a valuable cell model to study EMT induced by extrinsic factors, although it is important to note that neither D492 nor D492M are tumorigenic in mice.
In this study, we describe a new role for the DLK1-DIO3 locus in EMT and phenotypic plasticity of breast cells. Following EMT in breast epithelial cell lines, expression of the ncRNAs at the DLK1-DIO3 locus was increased. In addition, MEG3 was highly expressed in stromal cells in breast tissue and its expression correlated with decreased survival in breast cancer. Moreover, increased expression of the ncRNAs at the DLK1-DIO3 locus in a breast epithelial progenitor cell line promoted cellular plasticity and induced partial EMT. Collectively, our study provides a further understanding of the role of the DLK1-DIO3 locus in cellular phenotype of breast cells and might provide important insight into novel therapeutic targets aimed at overcoming heterogeneity and therapy resistance in breast cancer.
Materials and Methods
Cell Lines
Both D492 and D492M were cultured in H14 medium, as described previously (Gudjonsson et al., 2002; Sigurdsson et al., 2011) in flasks coated with collagen I (Advanced BioMFatrix, 5005-B). HEK-293T cell were cultured in Dulbecco’s Modified Eagle Medium (DMEM), high glucose, GlutaMAX (TM), pyruvate (Gibco, 31966), supplemented with 10% Fetal bovine serum (FBS), penicillin and streptomycin (Gibco, 15140-122). Primary Human umbilical vein endothelial cells (HUVECs) were obtained from Landspitali, University Hospital in Reykjavik, Iceland, (with informed consent, approved by Landspitali Ethical Committee No. 35/2013), cultured in Endothelial Growth Medium 2 (EGM2) media (Lonza, CC-3162) supplemented with growth factors and 5% FBS, further referred to as EGM5 medium as previously described (Sigurdsson et al., 2011). HMLE (Elenbaas et al., 2001) is epithelial progenitor cell line, from which was derived mesenchymal cell line HMLEmes after stable induction of EMT-TF (Mani et al., 2008). HMLE and HMLEmes were cultured in chemically defined HMLE media, containing DMEM/F12 with penicillin and streptomycin and growth factors Insulin (Sigma, I1882) 10 μg/ml, EGF (Peprotech, AF-100-15) 10 ng/ml, Hydrocortisone (Sigma, H0888) 500 ng/ml.
Primary human luminal-epithelial cells (LEP), myoepithelial cells (MEP), breast endothelial cells (BRENCs) and fibroblast were isolated from breast reduction mammoplasties (with informed consent, approved by the Icelandic National Bioethics Committee VSN-13-057) as previously described (Sigurdsson et al., 2011) and maintained in chemically defined medium 3 (CDM3) and chemically defined medium 4 (CDM4) as previously described (Pechoux et al., 1999; Ingthorsson et al., 2010). All cells were maintained in an incubator with 5% CO2 at 37°C.
3D Cultures/Mammosphere Assays
3D cultures were carried out in a 48-well plate format (Corning, 353078) in growth factor reduced reconstituted basement membrane rBM (further referred to as Matrigel, Corning, 354230). 5–10 × 103 cells were seeded in 150 μl of Matrigel per well. Plate was incubated in 5% CO2 at 37°C for 15 min to solidify the Matrigel and then 300 μl of H14 media was added on top. The cells were grown for 3 weeks and pictures were taken on day 1, 7, 14, and 21. Cell culture media was changed three times per week. The colonies were quantified at day 14. The total number of cells was converted into percentage.
For co-culture experiments, 0.5 × 103 of the epithelial cells were co-cultured with 1 × 105 of endothelial cells (HUVECs) and were resuspended in 150 μl of Matrigel. Plate was incubated in 5% CO2 at 37°C for 15 min to solidify the Matrigel and then 300 μl EGM5 media was added on top. HUVECs cultured in Matrigel are viable, however, quiescent, having supporting role in the epithelial cells’ proliferation. The effect of MEG3 was quantified by counting all colonies bigger than 100 μm.
Total RNAseq and Analysis of the Data
The gene microarray expression analysis from D492 and D492M was published previously from our group by Sigurdsson and colleagues (Sigurdsson et al., 2011) and the total RNA-sequencing comparing D492 and D492M was published by Halldorsson and colleagues (Halldorsson et al., 2017).
The RNA was extracted using Tri-Reagent (Thermo Fisher Scientific, AM9738) from 5 replicates for each cell line. Whole Transcriptome Sequencing of D492MKD–CTRL and D492MKD–MEG3 was performed in deCODE genetics (Reykjavik, Iceland). RNA sequencing reads were mapped to the reference genome (Ensembl primary assembly, version GRCh38) using STAR version 2.6.1 (Dobin et al., 2013). The program htseq-count (Anders et al., 2015) was used to quantify how many reads match each gene in an annotation file (Ensemble version GRCh38.96). The data from htseq-count was imported into R (R Development Core Team, 2015) and differential expression (DE) analysis on D492MKD–CTRL vs D492KD–MEG3 was performed using DESeq2 (Love et al., 2014). Prior to DE analysis, genes with expression less than two reads were discarded. P-values were corrected for multiple testing using the false discovery rate (FDR) method. To compare gene expression from D492MKD–CTRL vs D492MKD–MEG3 a volcano plot was generated. P value cut-off of 0.05 was applied. Volcano plot over all data (p < 0.05) was made in R using the EnhancedVolcano package from BioConductor. The top ten most upregulated and downregulated genes according log2 fold change were labeled. Gene Set Enrichment Analysis (GSEA) was applied to identify enrichment of gene signatures. Comparative analysis was investigated using the “Hallmark” database. The list of significantly expressed pathways is presented as a bar plot.
Quantitative RT-PCR Analysis
Total RNA was extracted with Tri-Reagent (Thermo Fisher Scientific, AM9738). 1 μg of RNA of each sample was reverse transcribed into complementary DNA (cDNA), using Random Hexamers (Thermo Fisher Scientific, N8080127) and SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, 18090-200) kit and subjected to quantitative real time PCR (qRT-PCR) using Sybr Green dye Luna® Universal qPCR Master Mix (NEB, M3003L) or TaqMan probes Luna® Universal Probe qPCR Master Mix (NEB, M3004L) according to manufacturer’s protocol. GAPDH was used as control for gene expression. For assaying the relative expression of each gene, the 2–ΔΔCt was determined using an ABI 7500 instrument (Applied Biosystems).
List of Primers
TaqMAN: ZEB1 (Thermo Fisher Scientific, Hs00232783_m1), ZEB2 (Thermo Fisher Scientific, Hs00207691_m1), SNAI1 (Thermo Fisher Scientific, Hs00195591_m1), SNAI2 (Thermo Fisher Scientific, Hs00950344_m1), TWIST1 (Thermo Fisher Scientific, Hs01675818_s1), GAPDH (Thermo Fisher Scientific, 4326317E).
SYBR Green: KRT14 (IDT, Hs.PT.58.4592110), KRT19 (IDT,Hs.PT.58.4188708), MEG3 ex 10-11 (IDT, Hs.PT.58.25190740), GAPDH (IDT, Hs.PT.39a.22214836), KRT5 (IDT, Hs.PT.58.14446018), TP63 (IDT, Hs.PT.58.2966111), CDH3 (IDT, Hs.PT.58.39234242).
Small RNAseq
The Microarray of small RNA data was published previously by our group by Hilmarsdottir and colleagues (Hilmarsdottir et al., 2015) and the small RNAseq data was published previously by Briem and colleagues (Briem et al., 2019a).
miRNA qRT PCR
Total RNA was extracted with Tri-Reagent (Thermo Fisher Scientific, AM9738). The RNA was reverse transcribed using miRCURY LNA RT Kit (Qiagen, 339340) for cDNA synthesis reactions, according to manufacturer’s protocol. Quantitative RT-PCR analysis of miRNAs was performed using miRCURY LNA SYBR Green PCR Kit (Qiagen, 339346), according to manufacturer’s protocol. Gene expression levels were quantified using primers for: hsa-miR-127-3p (Qiagen, YP00204048), hsa-miR-409-3p (Qiagen, YP00204358), hsa-miR-411-5p (Qiagen, YP00204531), hsa-miR-493-3p (Qiagen, YP00204557). Normalization was done with U6 snRNA (Qiagen, YP00203907). The 2–ΔΔCt was used determined using ABI 7500 instrument (Applied Biosystems) to calculate the relative expression of each gene.
Allele Specific Expression Analysis
Total RNA was extracted with Tri-Reagent (Thermo Fisher Scientific, AM9738) and reverse transcription done using 1 μg of DNase I-treated total RNA using random hexamers (Thermo Fisher Scientific, N8080127) and SuperScript II Reverse Transcriptase (Thermo Fisher Scientific, 18064022) according to the manufacturer’s instructions. PCR primers were designed using Primer3 and Pyrosequencing primers were designed using PyroMark Assay Design 2.0 (Qiagen). The reverse PCR primer had a 5′-biotin modification and was HPLC-purified. Primers were synthesized by IDT 5′-TGGCCTTTTCTTCTCCTGAA, 5′-/5Biosg/TGACACATGGAAAGCACCAT and sequencing primer 5′-TCCGGGGTTACTGCCCT-3′. Polymerase chain reactions were performed in 50 μl using 10 ng of diluted cDNA or 10 ng of DNA, 1 U DreamTaq DNA polymerase (Fermentas, EP0701), 1X PCR buffer, 200 μM of dNTPs and 0,5 μM of each PCR primer. The following PCR protocol was used: 94°C for 2 min, followed by 50 cycles of 94°C for 1 min, 60°C for 1 min, 72°C for 1 min and 72°C for 9 min. To check the quality of the amplification, PCR products were analyzed by gel electrophoresis. Pyrosequencing were sequenced using the PyroMark Q24 system (Qiagen), following the manufacturer’s instructions. For the ASE SNP, DNA and RNA (cDNA) were pyrosequenced simultaneously. The proportions of individual alleles for the SNP were obtained using the PyroMark Q24 software version 1.0.10 (Qiagen). Genomic DNA from D492M was examined to confirm the heterozygosity.
Clinical Cohort
RNA from breast cancer patients (diagnosed in the years 1987–2003) and relevant patient data was obtained from the Department of Pathology Landspitali – The National University Hospital of Iceland. Informed consent was obtained from patients involved in this study according to the national guidelines. The study was approved by The Icelandic Data Protection Commission (2001/523 and 2002/463) as well as the National Bioethics Committee of Iceland (VSN-11-105-V2). 119 samples were used in the study assigned to the following subgroup: 33 luminal A, 24 luminal B, 22 Basal, 12 ErbB2, 10 Normal and 18 not classified. cDNA was synthesized from 2 μg of total RNA using Random Hexamers primers (Thermo Fisher Scientific, N8080127) and RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher). MEG3 mRNA expression level was measured with the previously described qRT-PCR primers and TBP (Applied Biosystems, 4326322E) was used as a reference gene.
Western Blot Assay
Cells were washed with cold Phosphate Buffered Saline (PBS) and lysed in radio immunoprecipitation assay (RIPA) buffer with phosphatase and protease inhibitors (Halt Protease Inhibitor Cocktail, Thermo Fisher Scientific, 78430) for 10 min on ice and scraped with cell scraper. Protein concentration was measured using Bradford reagent (BioRad, 5000002). Equal amounts of protein (5–15 μg) were separated on 10% NuPage Bis-Tris gels (Invitrogen, NP0301PK2) with NuPage MES running buffer (Thermo Fisher Scientific, NP0002) and transferred with NuPage Transfer buffer (Thermo Fisher Scientific, NP0006-1) to polyvinylidene fluoride (PVDF) membranes Millipore Imobilion-FL transfer membrane, pore size 0,45 μM (Millipore, IPFL00010). The membranes were blocked with Odyssey Blocking buffer (TBS) (LiCor, 927-500) and incubated with primary antibodies overnight at 4°C. List of antibodies: keratin 14 (KRT14; Abcam, Ab15461), keratin 19 (KRT19; Abcam, Ab7754), P-cadherin (CDH3; Cell signaling, CS2130), tumor protein p63 (TP63, Abcam, Ab124762), keratin 5/6 (KRT5/6; Invitrogen, 180267), Actin (Licor, 926-42212). Actin was used as loading control. Secondary antibodies were mouse or rabbit IRDey (Li-Cor 926-32213, 926-32212, respectively) used at 1:10.000 for 1 h at room temperature (RT) and detected and quantified using the Odyssey Infrared Imaging System (Li-Cor Fluorescent signal was detected by Odyssey image system (Li-Cor) and converted to gray scale.
Cell Migration Assay
Cell migration was examined by using trans-well Boyden chambers with 8 μm pore size (Corning, 353097). Briefly, 3 × 103 cells were resuspended in 250 μl H14 medium and seeded on the trans-well inserts in 24-well plate (Corning, 353047). H14 media with 10% FBS was added to the lower chamber, below filter. Cells were incubated for 48 h in 5% CO2 at 37°C. Non-migratory cells from the upper part of the filter were removed with cotton swab and washed 3 times with 1× PBS. The filters were then fixed with methanol and stained with DAPI (Sigma, D9542-1MG). Cells were photographed in three random fields EVOS FL Auto 2 imaging system (ThermoFisher). Pictures were analyzed with ImageJ Software.
Low Attachment Assay
Anchorage independent growth was examined using 24-well ultra-low attachment plates (Corning, 3473). Briefly, D492 and D492M cells were single cell filtered and 500 cells/well were seeded into EGM5 media and cultured for 9 days. The growth of colonies was quantified under the microscope, counting all the colonies bigger than 40 μm.
Apoptosis Assay
Resistance to chemically induced apoptosis was examined by inducing the cells with 10 μM camptothecin [CPT, Sigma-Aldrich, C9911)] in 96-well plate format (Corning, 353072). and quantified using IncuCyte Caspase-3/7 Reagents (Essen Bioscience, 4440) on IncuCyte Zoom (Essen Bioscience) according to the manufacturer’s instructions.
Lentivirus Packaging and Transfection
The packaging of lentiviral expression constructs into pseudoviral particles, was performed with the psPAX2 (Addgene, 12260) and PMDG.2 (Addgene, 12259) plasmids using Turbofect (Thermo Fisher Scientific, R05319) in HEK-293T cells. The supernatant was harvested after 48 and 72 h and filtered through 0,45 μm pore filter. For infection, cells were plated on T25 flasks, so they were 70–80% confluent following day and were infected with 1 ml of viral particles and 1 ml of fresh media in the presence of 8 μg/ml polybrene. Lentivirus-transduced cells were selected with antibiotics or sorted by FACS (Sony SH800), based on fluorescent dye to obtain stable pool of clones. The altered expression of MEG3 was determined by qRT-PCR.
The list of lentiviral expression constructs (plasmids) used in the study and their selection marker (with final concentration in case of antibiotics): pLenti_sgRNA(MS2)_zeo (Zeocin Invitrogen 4 μl/ml), pLenti_dCas9-VP64_Blast (Blasticidin, 2 μg/ml), pLenti_dCas9-KRAB_mCherry (mCherry fluorescence), SAM MS2-P65-HSF1 Plasmids (Hygromycin 1 μl/ml).
CRISPRi/CRISPRa
To perform CRISPRi and CRISPRa, two vectors were used. First, vector with dCAS9 with effector domain KRAB (pLenti_dCas9-KRAB_mCherry, Genscript) and VP64 (pLenti_dCas9-VP64_Blast, Genscript) effector domain for CRISPRi and CRISPRa, respectively, was incorporated, using lentiviral transfection. Subsequently, vector with designed gRNA targeting specific site of our gene of interest MEG3 was incorporated, in second round of lentiviral transfection. In case of gain of function studies with CRISPRa, one additional helper plasmid SAM (SAM MS2-P65-HSF1 Plasmids, Genscript) was used to further increase activation.
The sequence of gRNA for overexpression of MEG3: Guide 1: GCTCTCCGCCGTCTGCGCTA, the sequence of gRNA for downregulation of MEG3: Guide 2: GCGGGTGAGGGATCCTCTCGT, the sequence of gRNA for negative control: GCTTAGTTACGCGTGGACGA were cloned into pLenti_sgRNA(MS2)_zeo (Genscript).
Statistical Analysis
Statistical differences of qRT-PCRs (Figures 1E,F, Figures 5A,B, and Figures 7A–C) and functional assay (Figures 8B–E) between samples were assessed with unpaired Student t-test. Statistical differences in Figure 8A was calculated using multiple unpaired Student t-test per row. Statistical differences of quantifications of western blots (Figures 7B,C) among samples were assessed using one-way ordinary ANOVA, followed by Tukey’s multiple comparison test. Statistical differences in Figure 4A (left) was calculated using Kruskal Wallis Test (one-way ANOVA on ranks). Statistical analysis of qRT-PCRs in Figure 2 were assessed with One-way ANOVA with Dunnett’s multiple comparisons test. All statistical analyses were performed in GraphPad Prism. P-values below 0,05 were considered significant (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ****p ≤ 0.0001).
FIGURE 1.

The DLK1-DIO3 locus is upregulated in breast epithelial stem cells undergoing EMT. (A) D492 and D492M generate branching and mesenchymal structures in 3D culture, respectively. In 2D culture, D492 is cuboidal in shape and D492M is more spindle shaped. Scale bar = 100 μm. (B) Majority of the top upregulated miRNAs in D492M are from the DLK1-DIO3 locus. Microarray heat map showing top 25 upregulated miRNAs in D492M compared to D492. 15 of them are from the DLK1-DIO3 locus (highlighted in orange). (C) LncRNAs from the DLK1-DIO3 locus are among the most upregulated in D492M. RNAseq data showing top ten differentially expressed lncRNA, with lncRNAs from DLK1-DIO3 locus (MEG3 and MEG8) highlighted in orange. (D) Schematic figure of the DLK1-DIO3 locus. The DLK1-DIO3 locus is located on chromosome 14 and is imprinted. It contains three paternally expressed protein coding genes (DLK1, RTL1, and DIO3) and many maternally expressed non-coding genes, among them lncRNAs (MEG3 and MEG8) and over 50 miRNAs, among them MIR127 and MIR493 located in cluster A and MIR409 and MIR411 located in cluster B and numerous C/D-box-containing small nucleolar RNAs (snoRNAs). DMR – differentially methylated region, filled circles represent methylated DMRs, and unfilled represent unmethylated DMRs. (E) Upregulation of selected ncRNA from the DLK1-DIO3 locus verified with qRT-PCR. Graphs showing higher expression of MEG3 in D492M compared to D492 (left) and higher expression of four representative miRNAs (MIR127, MIR493, MIR409, and MIR411) at the DLK1-DIO3 locus in D492M compared to D492 (right). Results shown as mean ± SD. Unpaired t-test was used to test significance: **p ≤ 0.01; ***p ≤ 0.001; n = 3. (F) ncRNAs from the DLK1-DIO3 locus are upregulated in HMLEmes. qRT-PCR showing higher expression of MEG3 and representative miRNAs (MIR127, MIR493, MIR409, and MIR411) from DLK1-DIO3 locus in HMLEmes compared to HMLE. Results shown as mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; n = 3.
FIGURE 5.

Concomitant expression of non-coding RNAs from DLK-DIO3 locus with MEG3. (A) Overexpression and knock-down of MEG3 in D492 and D492M, respectively. qRT-PCR confirming upregulation of MEG3 in D492 (D492MEG3) compared to D492 with scrambled control (D492CTRL; left). Phase contrast pictures of D492CTRL and D492MEG3 (below). qRT-PCR confirming knock-down of MEG3 in D492M (D492MKD–MEG3) compared to D492M with scrambled control (D492MKD–CTRL; right). Results shown as mean ± SD. Unpaired t-test was used to test significance: ***p ≤ 0.001; ****p ≤ 0.0001; n = 3. Phase contrast pictures of D492MKD–CTRL and D492MKD–MEG3 (below). Scale bar = 100 μm. (B) miRNAs form the DLK1-DIO3 locus are upregulated with overexpression of MEG3 and downregulated with knock-down of MEG3. qRT-PCR shows increased expression of four representative miRNAs from the DLK1-DIO3 locus in D492MEG3 compared to D492CTRL (left) and their decreased expression in D492MKD–MEG3 compared to D492MKD–CTRL (right). Results shown as mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05 **p ≤ 0.01; ****p ≤ 0.0001; n = 3.
FIGURE 7.

MEG3 induce partial EMT. (A) MEG3 increases expression of EMT transcription factors in D492 and the knock down of MEG3 decrease expression of TF-EMT in D492M. qRT-PCR showing D492MEG3 increased expression of transcription factors (TF) SNAI2 compared to D492CTRL (left) and decreased expression of EMT related TF SNAI2, ZEB1, ZEB2 and TWIST1 in D492MKD–MEG3 compared to D492MKD–CTRL (right). Results shown as mean ± SD. Unpaired t-test was used to test significance: **p ≤ 0.01; ***p ≤ 0.001; n = 3. (B) MEG3 increases expression of myoepithelial marker KRT14 and decrease expression of luminal epithelial marker KRT19 on mRNA level. qRT-PCR showing D492MEG3 has increased expression of KRT14 and decrease expression of KRT19 compared to D492CTRL (left). D492MKD–MEG3 has decreased expression of KRT14 compared to D492MKD–CTRL (right). qRT-PCR results shown as mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05; ***p ≤ 0.001; n = 3. (C) qRT-PCR results confirmed on protein level. Representative pictures of western blot (WB) with its quantification (below). D492MEG3 has increased protein level of KRT14 and decreased protein level of KRT19 compared to D492CTRL. WB results shown as mean ± SD. One-way ordinary ANOVA, followed by Tukey’s multiple comparison test was used to test significance: **p ≤ 0.01; ***p ≤ 0.001; n = 3. (D) MEG3 increase expression of myoepithelial markers TP63 and CDH3 and knock-down of MEG3 decrease expression of myoepithelial marker KRT5, on mRNA level. qRT-PCR showing D492MEG3 has increased expression of TP63 and CDH3 compared to D492CTRL (left). D492MKD–MEG3 has decreased expression of KRT5 compared to D492MKD–CTRL (right). qRT-PCR results shown as mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05; **p ≤ 0.01; n = 3. (E) qRT-PCR results confirmed on protein level. Representative pictures of western blot (WB) with its quantification (below). D492MEG3 has increased protein level of CDH3 (P-cad), TP63 (p63) and KRT5 compared to D492CTRL. D492MKD–MEG3 has decreased protein level of TP63 compared to D492MKD–CTRL. WB results shown as mean ± SD. One-way ordinary ANOVA, followed by Tukey’s multiple comparison test was used to test significance: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; n = 3.
FIGURE 8.

MEG3 increases stem cell properties. (A) MEG3 increase resistance to chemically induced apoptosis. Apoptosis assay: D492MEG3 is more resistant to chemically induced apoptosis compared to D492CTRL (left). Data is analyzed on Incucyte Zoom and displayed as Caspase 3/7 object count/mm2. Results are shown as mean ± SD. Multiple unpaired Student t-test per row was used to test significance at 72-h time-point: *p ≤ 0.05; **p ≤ 0.01; n = 6. (B) MEG3 increases migration and knock down of MEG3 decreases migration through trans-well filters. Migration assay: D492MEG3 has increased migration rate compared to D492CTRL (left), with representative pictures on side. D492MKD–MEG3 migrates less compared to D492MKD–CTRL (right), with representative pictures on side. Quantification of number of migratory cells, analyzed using ImageJ software, results shown as mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05; ****p ≤ 0.0001; n = 6. (C) MEG3 increases clonogenic capacity in 3D culture in rBM (reconstitute basement membrane). Mammosphere assay: D492MEG3 has higher clonogenic capacity compared to D492CTRL (left). D492MKD–MEG3 has lower clonogenic capacity compared to D492MKD–CTRL (right). Data shown as % mean ± SD, *p ≤ 0.05; **p ≤ 0.01; n = 3. Scale bar = 500 μm. (D) MEG3 increases clonogenic capacity in low attachment assay. D492MEG3 increases the formation of colonies compared to D492CTRL (left), with representative pictures on side. D492MKD–MEG3 decreases the formation of colonies compared to D492MKD–CTRL (right), with representative pictures on side. Results shown as % of mean ± SD. Unpaired t-test was used to test significance: *p ≤ 0.05; **p ≤ 0.01; n = 3. Scale bars = 200 μm. (D) Co-culture of D492MEG3 with endothelial cells (HUVECs) increases number and size of colonies and forms less branching compared to D492CTRL. Results shown as % of mean ± SD. Unpaired t-test was used to test significance: **p ≤ 0.01; n = 3. Representative pictures scale bars = 200 μm.
FIGURE 4.

MEG3 expression negatively correlates with breast cancer prognosis. (A) High MEG3 expression decreases overall survival in breast tumors. qRT-PCR shows that MEG3 expression (Numbers of tumors per group: ErBB2 = 12, Luminal A = 33, Luminal B = 24, Normal = 10, Basal = 22) is significantly higher in normal-like (NL) breast cancer (left; p = 0.0003). High MEG3 expression is correlated with low overall patient survival (NL tumors omitted). Kruskal Wallis Test (or one-way ANOVA on ranks) was used to test the significance (right; p = 0.01). (B) High MEG3 expression decrease distant metastasis free survival in grade three and Luminal B type tumors. Kaplan-Meier plot showing data from the online GOBO database: high expression of MEG3 decrease DMSF (distant metastasis free survival), of poorly differentiated (grade 3) tumors (left; p = 0.00373) and luminal B type tumor (right; p = 0.04607).
FIGURE 2.

The ncRNA from the DLK1-DIO3 locus are highly expressed in stromal cells and whole tissue compared to epithelial cells. qRT-PCR showing MEG3 expression is higher in breast stromal cells (fibroblasts) and whole tissue than in D492 (left). (LEP – luminal epithelial cells, MEP – myoepithelial cells, BRENCs – breast endothelial cells). Results shown as mean ± SD. One-way ANOVA with Dunnett’s multiple comparisons test was used to test significance: **p ≤ 0.01; ****p ≤ 0.0001; n = 3. Expression of representative miRNAs MIR127 and MIR411 at the DLK1-DIO3 locus is higher in breast fibroblast than in D492 (right). LEP – luminal epithelial cells, MEP - myoepithelial cells. Results shown as mean ± SD. One-way ANOVA with Dunnett’s multiple comparisons test was used to test significance: *p ≤ 0.5; n = 3.
Results
MEG3 Is Highly Expressed in Cell Lines With a Mesenchymal Phenotype and in the Stromal Compartment of Breast Tissue
D492 and D492M are isogenic cell lines with stem cell and mesenchymal properties, respectively. D492 cells acquire cuboidal shape in 2D culture, and form branching structures in 3D culture, akin to terminal duct lobular units (TDLU) in the breast. In contrast, D492M is elongated and spindle-shaped in 2D culture and in 3D culture it forms irregular mesenchymal-like colonies (Figure 1A). We have previously shown that MIR203a and the MIR200 family are downregulated in D492M and their expression is essential for the epithelial phenotype (Hilmarsdottir et al., 2015; Briem et al., 2019a). Of miRNAs upregulated in D492M, the miRNAs at the DLK1-DIO3 locus are prominent. A microarray analysis of miRNA expression demonstrated that 15 of the 25 most highly expressed miRNAs in D492M compared to D492 belong to the DLK1-DIO3 locus (Figure 1B). Furthermore, small RNA sequencing revealed that 33 of the miRNAs belonging to the DLK1-DIO3 miRNA cluster have more than 1,5-fold increased expression in D492M compared to D492 (Supplementary Figure 1). Moreover, total RNA sequencing of D492 and D492M, revealed that MEG3 and MEG8 are amongst the most upregulated lncRNAs in D492M (Figure 1C). The non-coding part of the DLK1-DIO3 locus consists of maternally expressed lncRNAs MEG3 and MEG8 and miRNAs grouped into two clusters (Figure 1D). To confirm the sequencing results, we selected four representative miRNAs from the DLK1-DIO3 locus, two from each cluster (MIR127 and MIR493 from cluster A, MIR409 and MIR411 from cluster B). These miRNAs as well as the lncRNA MEG3 had higher expression, as revealed by qRT-PCR, in D492M compared to D492 (Figure 1E). In another isogenic EMT cell model, HMLE (epithelial) and HMLEmes (mesenchymal variant) both MEG3 and the representative miRNAs were more highly expressed in HMLEmes compared to HMLE (Figure 1F). Thus, our data suggests that increased MEG3 expression is not a stochastic event but consistently associates with EMT induction in breast epithelial cell lines.
Next, we analyzed the expression of MEG3 and miRNAs from the DLK1-DIO3 locus in primary cells from three healthy donors. We found that the expression of MEG3 is higher in purified stromal cells (fibroblasts) than in epithelial cells (D492, luminal epithelial cells, myoepithelial cells and organoids; Figure 2, left). Interestingly, expression of MEG3 in whole breast tissue lysates is closer to fibroblast expression levels than epithelial cells (Figure 2, left). This finding is most likely explained by the richness of stroma in normal breast tissue, whereas organoids contain only the epithelial cells. A similar pattern is seen with the four representative miRNAs, where MIR127 and MIR411 have higher expression in fibroblasts compared to their expression in D492 (Figure 2, right).
We next acquired a list of genes correlated the expression of MEG3 using the GOBO (Gene expression-based Outcome for Breast Cancer Online) dataset and submitted the list to DAVID (the database for annotation, visualization and integrated discovery, version 6.7) (Huang et al., 2009a, b) to identify pathways associated with MEG3. Herein, the expression of MEG3 correlates with expression of extracellular matrix genes, which are in line with the observations of a high expression of MEG3 in cells found in the stromal compartment (Supplementary Figure 2A). Using analysis of publicly available NGS data using MiPanda (Niknafs et al., 2018) we found positive correlation of MEG3 with common EMT markers in normal breast and breast cancer (Supplementary Figure 2B). Many of these have a correlation coefficient > 0.3 (Spearman correlation) which is considered a fair positive correlation (Chan, 2003). Interestingly, even more genes are positively correlated to MEG3 expression in breast cancer as compared to normal breast tissue (Supplementary Figure 2B).
Collectively, the lncRNA MEG3 and miRNAs from DLK1-DIO3 locus are highly expressed in the mesenchymal compartment compared to epithelial breast tissue and their expression positively correlate with numerous mesenchymal genes and EMT markers.
MEG3 Is Imprinted in Both D492 and D492M
The DLK1-DIO3 locus is imprinted and regulated by DNA methylation (Cui et al., 2018). Using pyrosequencing (Harrington et al., 2013) covering a heterozygous SNP (C/T) in MEG3 (rs4906024) we confirmed monoallelic expression of MEG3 in both D492 and D492M, with expression in both cell lines being from the T allele (Figure 3). As both cell lines are diploid at the MEG3 locus on a DNA level a C/T ratio of 50% is expected which is consistent with the 48% C-allele prominence observed. On the mRNA deviation from expected monoallelic expression was not detected as results showed zero C allele expression in D492 and 2% in D492M. Hence, increased expression of MEG3 in D492M is not caused by loss of imprinting. The expression remains monoallelic confirming that the increased expression originates from the non-imprinted allele.
FIGURE 3.

Increased expression of MEG3 in D492M is not caused by loss of imprinting. Allele specific expression analysis using pyro-sequencing shows that the increased expression of MEG3 in D492M is not caused by loss of imprinting, but increased expression of the already expressed allele, the expression remains mono-allelic. Analyzed sequence: GAGCAC/TGTCCCA.
Increased Expression of MEG3 Is Negatively Correlated With Survival of Breast Cancer Patients
EMT has been suggested to promote metastatic behavior of epithelia-originating cancer (Felipe Lima et al., 2016) and, in addition, our data shows association of MEG3 expression with the mesenchymal phenotype. We therefore investigated MEG3 expression levels in different subtypes of breast cancer. We have evaluated the expression of MEG3 in clinically well-defined breast tumors. Herein, normal like (NL) breast tumors had significantly higher expression of MEG3 with a p-value of 0.0003 (Figure 4A, left). Survival analysis of all tumor samples showed reduced, but not significant overall survival in patients with high MEG3 expression. However, as the normal-like tumors have in recent years been subjected to scrutiny as a possible misclassification due to low tumor cellularity and thus, high proportion of normal tissue. In light of our results showing high expression of MEG3 in breast stromal tissue, and uncertainty that measured MEG3 expression in the normal-like subgroup is representative of the primary tumor, we omitted NL breast tumors from the survival analysis (Elloumi et al., 2011; Prat and Perou, 2011; Yersal and Barutca, 2014). The results show significant worse overall survival of patients with high MEG3 expression (Figure 4A, right). Corroborating our findings, using the GOBO database (Ringnér et al., 2011)1, we found that high MEG3 expression reduces distant metastasis free survival (DMSF) of patients with poorly differentiated (grade 3) tumors (Figure 4A, left) and patients with luminal B tumors (Figure 4B, right).
Increased Expression at the DLK1-DIO3 Locus Contributes to the Basal and Mesenchymal Phenotype
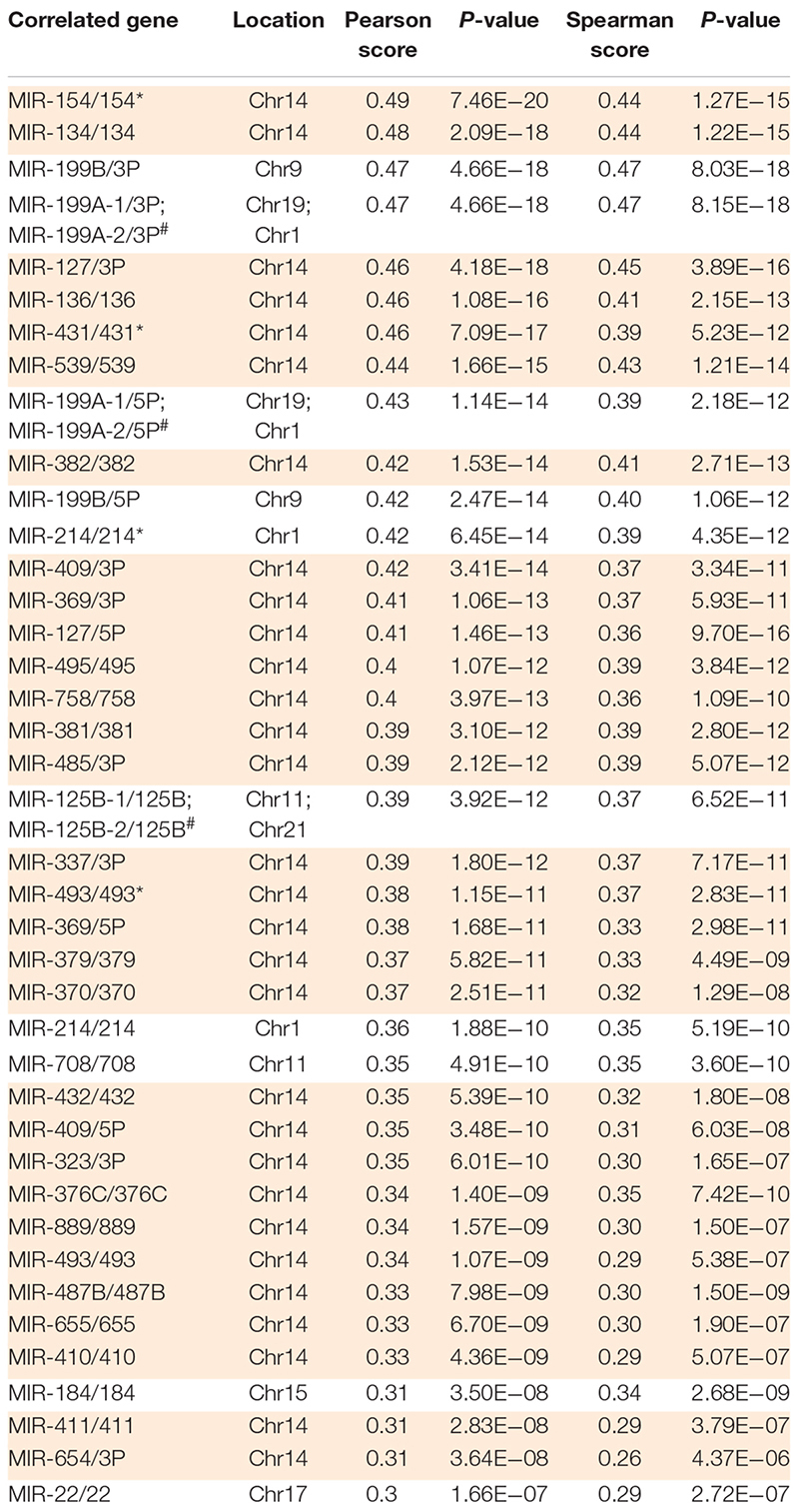
To explore the functional role of MEG3 in D492 and D492M, we established sublines with altered expression of MEG3. Using the CRISPRa approach (Cheng et al., 2013), we generated a D492 cell line with stable overexpression of MEG3 (D492MEG3). A control cell line was generated using a scrambled sgRNA (D492CTRL). Furthermore, we used the CRISPRi approach (Gilbert et al., 2013; Qi et al., 2013), to generate knockdown of MEG3 in D492M (D492MKD–MEG3) and a control cell line was generated using scrambled sgRNA (D492MKD–CTRL). The increase of MEG3 expression was about seven-fold in D492MEG3 compared to D492CTRL as determined by qRT-PCR (Figure 5A, left). Downregulation of MEG3 in D492MKD–MEG3 was more prominent, with about 20-fold reduced expression compared to D492MKD–CTRL (Figure 5A, right). Having established stable overexpression and downregulation of MEG3 in D492 and D492M, we re-evaluated the epithelial/mesenchymal phenotypes of D492 and D492M, respectively. Based on phase contrast images, no obvious difference in phenotype could be seen between D492MEG3 and D492CTRL or D492MKD–MEG3 and D492MKD–CTRL (Figure 5A, below). Interestingly, expression of the representative miRNAs located on the DLK1-DIO3 locus is increased in D492MEG3 compared to D492CTRL, to similar levels as seen in D492M (Figure 5B, left). Conversely, the expression of representative miRNAs is downregulated in D492MKD–MEG3 compared to D492MKD–CTRL (Figure 5B, right). Thus, it appears, that the expression of miRNAs from the DLK1-DIO3 locus is concomitant with MEG3 expression. To test, if that holds true, we used the cBioPortal and explored correlation of MEG3 with miRNAs using data on invasive breast cancer from the Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Network, 2012) we found that of 40 miRNAs that had positive correlation over 0,3 (Person score) with MEG3, 30 were located at the DLK1-DIO3 locus (with other miRNAs from the locus not being in the dataset; Table 1). This suggests that MEG3 may be used as a marker for the expression of ncRNAs from the DLK1-DIO3 locus.
TABLE 1.
MiRNAs from the DLK1-DIO3 locus positively correlate with MEG3 expression.
|
Out of 40 miRNAs that positively correlate with MEG3 in breast cancer (with correlation over 0.3), 30 are from the DLK1-DIO3 locus, highlighted in orange (TCGA, Nature 2012 data set). #Due to sequence similarities, these two miRNAs are indistinguishable in the sequencing data used.
Next, we conducted RNA sequencing of our cell lines with stably altered expression levels of MEG3 focusing on the analysis of D492MKD–CTRL vs D492MKD–MEG3. There were 1235 significantly differentially expressed genes, with symmetric distribution over genes downregulated and upregulated in D492MKD–MEG3, shown in the volcano plot (p < 0.05; Supplementary Figure 3A), with the list of top 30 up and down-regulated genes in D492MKD–MEG3 (Supplementary Figure 3B). To identify unifying biological them from RNA-sequencing data, we performed Gene Set enrichment analysis (GSEA). These gene sets consist of the defined gene lists, based on biological knowledge about biochemical pathways and co-expression data. Using the Hallmark dataset, one of the significantly, downregulated set of genes in D492MKD–MEG3 was the epithelial-mesenchymal transition gene set, with normalized enrichment score (NES) of −2.03 and False discovery rate (FDR) q = 0.023 (Figure 6A). These genes define epithelial-mesenchymal transition, as in wound healing, fibrosis and metastasis. The genes belonging to this gene set are overrepresented toward the top of the ranked list, based on fold change of D492MKD–CTRL vs D492MKD–MEG3 (Figure 6B, right). A manually curated list of mesenchymal genes from the Hallmark EMT dataset that are downregulated in D492MKD–MEG3 is shown in Figure 6B, left. Further analysis of the RNA sequencing data of D492MKD–MEG3 vs D492MKD–CTRL, using common literature-based markers of breast tissue has showed that luminal epithelial markers GATA3 and MUC1 are upregulated, while myoepithelial KRT14, mesenchymal VIM, ZEB2, SNAI2, LAMA1, CDH2, and stem cell MME, CTNNB1 are downregulated with knock down of MEG3 (Figure 6C).
FIGURE 6.

Knock down of MEG3 in D492M decrease mesenchymal markers. (A) Epithelial-mesenchymal transition gene set is enriched pathway in D492MKD–MEG3 (highlighted in orange). Bar plot of Gene Set Enrichment Analysis (GSEA) with Hallmark dataset showing all significantly (False discovery rate-FDR q ≤ 0.05) enriched pathways in D492MKD–MEG3. Gene set Epithelial-mesenchymal transition has normalized enrichment score (NES) of –2.06 and FDR q = 0.014. (B) Knock-down of MEG3 correlates with downregulation of mesenchymal genes relevant for breast cells. Enrichment plot showing the Enrichment Score (ES) of the genes in the Hallmark gene set Epithelial-mesenchymal transition in D492MKD–MEG3. The genes are overrepresented toward the top the ranked list of D492MKD–MEG3 (right). Table with relevant genes for breast cells, significantly (p ≤ 0.05) deregulated, from the Hallmark gene list epithelial-mesenchymal transition showing Log2 fold change (FC; left). (C) Luminal epithelial markers (GATA3 and MUC1) are upregulated, while myoepithelial (KRT14), mesenchymal (CDH2, LAMA1, SNAI2, VIM, and ZEB2) and stem cell (CTNNB1 and MME) are downregulated with knock down of MEG3. Genes from literature-based list of markers significantly (p ≤ 0.05) differentially expressed in D492MKD–MEG3 vs D492MKD–CTRL.
Expression of mesenchymal and basal markers was additionally confirmed on RNA level by qRT-PCR and on protein level western blot. Most of the core EMT-related transcription factors (EMT-TF) were affected by MEG3. D492MEG3 has increased expression of SNAI2 compared to D492CTRL (Figure 7A, left). On the other hand, D492MKD–MEG3 has decreased expression of SNAI2, ZEB1, ZEB2 and TWIST1 compared to D492MKD–CTRL (Figure 7A, right). Luminal cytokeratin 19 (KRT19) and basal/myoepithelial cytokeratin 14 (KRT14) are also affected by manipulation of MEG3 expression levels. Thus, D492MEG3 shows increased KRT14 and decreased KRT19 expression compared to D492CTRL on both mRNA (Figure 7B, left) and protein level (Figure 7C, left). D492MKD–MEG3 shows decreased KRT14 expression compared to D492MKD–CTRL (Figure 7B, right). Furthermore, D492MEG3 shows increased expression of other myoepithelial markers such as CDH3 (P-cad), TP63 or KRT5 compared to D492CTRL as determined both at mRNA (Figure 7D, left) and protein level (Figure 7E). Also, D492MKD–MEG3 shows decreased expression of myoepithelial markers KRT5 on mRNA level (Figure 7D, right) and of TP63 on protein level (Figure 7E, middle) compared to D492MKD–CTRL. This suggests that MEG3 expression induces a shift toward a basal/myoepithelial phenotype. However, our cell lines with stably altered expression of MEG3 do not show a significant switch in E-cadherin (CDH1) to N-cadherin (CDH2) expression (Supplementary Figure 4), which may explain why there are no clear changes in morphology.
MEG3 Induces Mesenchymal Properties and Stemness
As MEG3 has previously been ascribed to have a role in pluripotency and stemness (Stadtfeld et al., 2010b; Kaneko et al., 2014), we asked how MEG3 manipulation affects mesenchymal and stem cell properties of D492 and D492M. The expression of both aldehyde dehydrogenase (ALDH1A3) and integrin alpha 6 (ITGA6; Supplementary Figure 6), markers of stemness, is increased in D492MEG3 compared to D492CTRL. Next, we employed several functional assays to assess the effect of MEG3 levels in D492 and D492M on mesenchymal and stem cell properties. D492MEG3 is more resistant to chemically induced apoptosis than D492CTRL (Figure 8A). Migration can be assessed in vitro using the wound healing assay or by trans-well migration where the cells migrate toward a chemo-attractant. In the wound healing assay, D492MEG3 has slightly increased migration rate compared to D492CTRL, while D492MKD–MEG3 has decreased migration rate compared to D492MKD–CTRL (Supplementary Figure 5A). In the trans-well migration assay, D492MEG3 has about two-fold increased migration rate compared to D492CTRL and D492MKD–MEG3 has reduced migration rate compared to D492MKD–CTRL (Figure 8B). MEG3 manipulation, however, did not affect invasion in a transwell invasion assay (Supplementary Figure 5B). We performed mammosphere assays in rBM (reconstituted basement membrane, Matrigel) (Figure 8C) and in low attachment plates (Figure 8D), with comparable results. D492MEG3 increases the formation of colonies compared to D492CTRL while D492MKD–MEG3 decreases the formation of colonies compared to D492MKD–CTRL. In addition, we co-cultured D492MEG3 with endothelial cells (HUVECs) and observed increased size of colonies and less branching compared to D492CTRL (Figure 8E). Finally, manipulation of MEG3 levels slightly affected proliferation rate of D492MKD–MEG3 compared to D492MKD–CTRL (Supplementary Figure 5C).
Discussion
In this study, we show that ncRNAs from the DLK1-DIO3 locus are highly expressed in stromal/mesenchymal cells in the breast and positively correlate with the expression of EMT genes in breast tissue. MEG3 expression was monoallelic in both D492 and D492M and gain and loss of function studies have shown concomitant expression of MEG3 with miRNAs from the DLK1-DIO3 locus, indicating that MEG3 could be used as a marker for the expression of the non-coding RNAs from the locus. MEG3 expression was shown to be negatively correlated with survival of breast cancer patients, particularly with the luminal B subtype. Furthermore, we demonstrate that enhanced MEG3 expression accompanied by increased expression of the ncRNAs at the DLK1-DIO3 locus, contributes to partial EMT more correctly referred to as epithelial plasticity, seen by increased expression of EMT related TFs, increase of basal/mesenchymal markers and enhanced properties such as migration, resistance to apoptosis and clonogenic capacity.
We used an isogenic breast cell line model to study the expression pattern and functional role of ncRNAs, both miRNAs and lncRNAs, in EMT. Of interest was the largest miRNA locus in the human genome and the lncRNA MEG3, both within the DLK1-DIO3 imprinted region on chromosome 14. The non-coding part of the DLK1-DIO3 locus has higher expression in cells with mesenchymal phenotype (D492M) compared to cells with epithelial phenotype (D492). These results were validated in primary breast tissue and in another cellular model of EMT. Furthermore, we have shown that MEG3 expression correlates with expression of extracellular matrix proteins, which are secreted by cells with a mesenchymal phenotype, and with mesenchymal genes in breast tissue. Data from pyrosequencing demonstrate that the expression of MEG3 is monoallelic in both D492 and D492M indicating that the increased expression of MEG3 in D492M is not due to loss of imprinting. We have shown that MEG3 negatively correlates with survival of luminal B breast cancer patients and patients with grade 3 breast cancer. This is in line with a recent study where high expression of MEG3 was identified to be a negative prognostic marker for breast cancer (Yao et al., 2019).
Many studies suggest MEG3 as a tumor suppressor, largely due to the observation that MEG3 expression is lower in tumor tissue compared to normal tissue (Sheng et al., 2014; Sun et al., 2014, 2016; Yin et al., 2015; Chak et al., 2017; Molina-Pinelo et al., 2018). Our data demonstrates that MEG3 expression levels are comparable in whole normal breast tissue and in stroma (fibroblasts), however, the expression of MEG3 in epithelial cells is much lower. There was considerable variation of the MEG3 expression in breast tissue samples that could be partially due to different proportions of subset of fibroblasts associated with ducts vs TDLUs. There are studies confirming existence of, for instance, two distinct functionally specialized lineages of lobular vs ductal fibroblast (Morsing et al., 2016) or myoepithelial cells (Fridriksdottir et al., 2017), which could be identified by specific marker expression. Importantly, relative proportions of stromal and epithelial compartment are different in normal and cancerous human breast tissue. Breast cancers arise in vast majority from epithelial cells, with TDLUs being the predominant site of breast tumor occurrence (Tabar et al., 2014). Therefore, it would be expected that expression of MEG3 is higher in normal breast tissue, as it comprises relatively more stromal cells compared to breast cancer tissue. In line with this, expression of MEG3 from whole breast tissue is distorted as proportions of stroma vs epithelia in normal/cancer tissue are different, resulting in misleading interpretations. Using RNA only from unsorted normal tissue will mainly represents expression of stromal cells. Therefore, it is crucial to use a proper control when comparing expression of genes in normal vs tumor tissue. Single-cell RNA-sequencing or sorted stromal and epithelial cells would give more informative results as it would enable distinctions between epithelial and stromal tissue compartments. In this paper we show that MEG3 expression negatively correlates with survival in breast cancer, particularly in grade three tumors and the luminal B subtype. However, our study does not determine if the high MEG3 expression represents increased stromal infiltration in the tumors or elevated expression in cancer cells.
Another reason for classifying MEG3 as tumor suppressor is its action on stabilization of p53 (Ghafouri-Fard and Taheri, 2019). However, inactivation of p53 is a frequent event in cancer, estimated to have about 50 % occurrence (Gasco et al., 2002; Marine et al., 2006; Haupt and Haupt, 2017). The percentage is even higher, when the inactivation in p53’s regulatory pathways is considered (Joerger and Fersht, 2016). Therefore, the use of cell lines which lack active p53, such as D492 and D492M, offers a different approach, more relevant for studying breast cancer signaling pathways, to study the role of DLK1-DIO3. The role of p53 in the cell is that of a tumor suppressor, impacting acts in proliferation, cell cycle and genomic stability (Mercer, 1992). In D492 cell lines, as could be expected, we did not observe effect on cell proliferation. Recently, Uroda and colleagues’ stated, that cell cycle arrest by MEG3 is exclusively p53-dependent, (Uroda et al., 2019), in line with our suggestions that MEG3 can have a different role in cells lacking p53. Collectively, these observations could explain the conflicting results about role of MEG3 in tumors.
Many imprinted genes are located in clusters regulated by a differentially methylated regions (DMRs) (Bartolomei and Ferguson-Smith, 2011). In our study targeting the MEG3 promoter, we have observed concomitant expression of MEG3 with other miRNAs from the DLK1-DIO3 locus. Our data may support previous studies showing that the MEG3 promoter controls expression of all maternally expressed genes from the DLK1-DIO3 locus (Tierling et al., 2006; Ioannides et al., 2014; Sanli et al., 2018). Zhu et al. (2019) have shown that the MEG3-DMR overlaps with the MEG3 gene promoter and any deletion in this region inactivates both MEG3-DMR and the MEG3 gene. Their data shows, that it is the MEG3-DMR, not the MEG3 gene, which regulates imprinting (and expression). Therefore, by targeting the MEG3 promoter at the MEG3-DMR all the non-coding RNAs at the DLK1-DIO3 locus are inactivated. MEG3 expression can be considered as a marker for the expression of other ncRNAs at the locus.
Cellular plasticity, an important contributor to heterogeneity and drug resistance in breast cancer can be conveyed through EMT/MET (Liu et al., 2014). Partial EMT (p-EMT) may reflect cellular plasticity better than full-EMT and consequently, cells possessing this state adapt more easily to a new environment, which is necessary for cancer cell invasion and metastasis (Thiery, 2002; Tam and Weinberg, 2013; Lambert et al., 2017). Notably, a recent report highlights the importance of the intermediate stages of EMT for the intravasation of tumor cells and for metastasis formation in experimental breast or skin tumors (Pastushenko et al., 2018). Similarly, another study showed that cancer cells might only reach an intermediate EMT stage allowing for increased motility, while keeping its cellular plasticity (Brabletz et al., 2018). It has also been observed that full mesenchymal phenotype (EMT), has a low capacity to form metastasis compared to p-EMT (Schmidt et al., 2015). The essential criteria for aggressive behavior does not need to be a particular phenotype, but rather enhanced cellular plasticity, as is also observed for hybrid E/M cells (Grosse-Wilde et al., 2015). Thus, EMT may be viewed as a trans-differentiation process where epithelial and mesenchymal cells interconvert by passing through an intermediate “stem-like” state (Grosse-Wilde et al., 2018).
EMT is a complex process and meta-analysis indicates that there are possibly different types of EMT (Liang et al., 2016). We have shown, that by manipulating MEG3 expression, and thus changing the expression of the non-coding genes at the DLK1-DIO3 locus, the majority of these EMT related TFs are affected, indicating an important role of the ncRNAs the DLK1-DIO3 locus in the EMT process. One of the most typical hallmarks of EMT is downregulation of CDH1 (E-cadherin) and epithelial-specific keratins (Peinado et al., 2007). Altered expression of MEG3 does not lead to change of E-cadherin expression and therefore MEG3 may have induced only a partial EMT phenotype. However, it has been shown, that cells with p-EMT phenotype display concomitant expression of epithelial and mesenchymal markers (Armstrong et al., 2011) and loss of E-cadherin is not a prerequisite for EMT (Hollestelle et al., 2013). Cells undergoing collective migration have hybrid EMT phenotype characterized by E-cadherin expression, which helps to maintain cell–cell contacts (Friedl et al., 2012; Aceto et al., 2015). Furthermore, we have shown that altered expression of MEG3 revealed distinct luminal and myoepithelial marker expression. Increased expression of KRT14 and decreased expression KRT19 indicate increased myoepithelial differentiation, which has been connected to a partial EMT phenotype (Petersen et al., 2001). Study on collective migration revealed KRT14 as a key regulator of metastasis (Cheung et al., 2016) and the same applied for collective invasion, which was facilitated by subpopulation of cells expressing KRT14 (Cheung et al., 2013). The observed increase of myoepithelial/basal differentiation in cells with higher expression of MEG3 was supported with altered expression of other markers such as KRT5, TP63, and CDH3.
A key characteristic defining breast stem cells is the ability to form of mammospheres (Dontu et al., 2003; Grosse-Wilde et al., 2015). Morel and colleagues confirmed that human mammary epithelial cells undergoing EMT exhibited better mammosphere-forming capabilities (Morel et al., 2008) and Shimono et al. have shown that mammosphere-forming activity is abrogated in both normal and malignant mammary stem cells when the EMT program is shut down (Shimono et al., 2009). In this study phenotypic differences upon altered MEG3 expression were more prominent in 3D than in 2D cell culture, where MEG3 increased mammosphere formation ability and slightly decreases branching potential in 3D culture. Furthermore, we have shown increased expression of ALDH1A3 and ITGA6, in cells with overexpression of MEG3, supporting role of MEG3 in stemness.
We propose that increased expression of MEG3, and thus increased expression of the ncRNAs at the DLK1-DIO3 locus, in D492 leads to partial EMT phenotype/enhanced plasticity, seen by molecular changes with increased mesenchymal and myoepithelial/basal genes and increased migration and resistance to apoptosis. In contrast, the repression of MEG3, and the maternally imprinted ncRNAs, in D492M leads to decreased mesenchymal and basal gene expression and decreased migration and resistance to apoptosis. Nguyen-Ngoc et al. also demonstrated, that motility can occur in cells that retain an epithelial molecular signature (Nguyen-Ngoc et al., 2012). This supports our observation, that manipulation of MEG3 expression did not affect the morphological phenotype, but rather affected the functional phenotype. These characteristic properties of cells undergoing EMT were originally proposed to occur in breast cancer by Mani and colleagues (Mani et al., 2008), showing that stem-like and p-EMT properties share many characteristics, such as increased migration, resistance and survival (Creighton et al., 2009; Armstrong et al., 2011; Hanahan and Weinberg, 2011).
Increased understanding of branching morphogenesis in the breast and the regulation of EMT and MET may hold the key for future development of methods and drugs that neutralize the invading properties of cancer cells. Currently, there is need for biomarkers to accurately monitor the EMT/MET process that may improve treatment. Prognostic value of MEG3 in human malignancies remains controversial and requires further investigation. Our results and conflicting data from the literature suggest that MEG3 has a complex role in breast tissue.
Data Availability Statement
The RNAseq data for this article has been submitted to GEO, with the GEO accession number GSE142268, see here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142268
Ethics Statement
The studies involving human participants were reviewed and approved by Icelandic National Bioethics Committee VSN-13-057 and VSN-11-105-V2. The Icelandic Data Protection Commission (2001/523 and 2002/463) Landspitali Ethical Committee No. 35/2013. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MM, TG, JB, ZB, EB, GT, and BH: conceptualization and design of the study. ZB, EB, JB, AS, and BH: data acquisition. ZB, AS, EB, GT, SS, and BH: data analysis. ZB, GT, TG, and BH: drafting the manuscript. All authors participated in data interpretation, revision of the manuscript and approved the final version to be published.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Rosa B. Barkardottir, Adalgeir Arason, Bjarni A. Agnarsson, and Oskar Thor Johannsson for their contribution to this work by providing RNA samples from breast cancer patients and pathological and clinical information. We would also like to thank Gudrun Johannesdottir for excellent technical support.
Funding This work was supported by Grants from Landspitali University Hospital Science Fund, University of Iceland Research Fund, Icelandic Science and Technology Policy Council Research Fund no. 1103010061, Icelandic Science and Technology Policy – Grant of Excellence: 52144051, “Vísindasjóður Krabbameinsfélagsins” (Icelandic Cancer Society Science Fund) 2017 and ‘Göngum saman’, a supporting group for breast cancer research in Iceland (www.gongumsaman.is). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00461/full#supplementary-material
References
- Aceto N., Toner M., Maheswaran S., Haber D. A. (2015). En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 1 44–52. 10.1016/j.trecan.2015.07.006 [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P. T., Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansieau S. (2013). EMT in breast cancer stem cell generation. Cancer Lett. 338 63–68. 10.1016/j.canlet.2012.05.014 [DOI] [PubMed] [Google Scholar]
- Armstrong A. J., Marengo M. S., Oltean S., Kemeny G., Bitting R. L., Turnbull J. D., et al. (2011). Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 9 997–1007. 10.1158/1541-7786.mcr-10-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei M. S., Ferguson-Smith A. C. (2011). Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 3:a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulina N., Osmak G., Kiselev I., Popova E., Boyko A., Kulakova O., et al. (2019). MiRNAs from DLK1-DIO3 imprinted locus at 14q32 are associated with multiple sclerosis: gender-specific expression and regulation of receptor tyrosine kinases signaling. Cells 8:133. 10.3390/cells8020133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Jacob E., Coffey D. S., Levine H. (2012). Bacterial survival strategies suggest rethinking cancer cooperativity. Trends Microbiol. 20 403–410. 10.1016/j.tim.2012.06.001 [DOI] [PubMed] [Google Scholar]
- Brabletz T., Kalluri R., Nieto M. A., Weinberg R. A. (2018). EMT in cancer. Nat. Rev. Cancer. 18 128–134. 10.1038/nrc.2017.118 [DOI] [PubMed] [Google Scholar]
- Briem E., Budkova Z., Sigurdardottir A. K., Hilmarsdottir B., Kricker J., Timp W., et al. (2019a). MiR-203a is differentially expressed during branching morphogenesis and EMT in breast progenitor cells and is a repressor of peroxidasin. Mech. Dev. 155 34–47. 10.1016/j.mod.2018.11.002 [DOI] [PubMed] [Google Scholar]
- Briem E., Ingthorsson S., Traustadottir G. A., Hilmarsdottir B., Gudjonsson T. (2019b). Application of the D492 cell lines to explore breast morphogenesis, EMT and cancer progression in 3D culture. J. Mammary Gland Biol. Neoplasia 24 139–147. 10.1007/s10911-018-09424-w [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490 61–70. 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Livas T., Kyprianou N. (2016). Anoikis and EMT: lethal “Liaisons” during cancer progression. Crit. Rev. Oncog. 21 155–168. 10.1615/critrevoncog.2016016955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chak W. P., Lung R. W., Tong J. H., Chan S. Y., Lun S. W., Tsao S. W., et al. (2017). Downregulation of long non-coding RNA MEG3 in nasopharyngeal carcinoma. Mol. Carcinog. 56 1041–1054. 10.1002/mc.22569 [DOI] [PubMed] [Google Scholar]
- Chan Y. H. (2003). Biostatistics 104: correlational analysis. Singapore Med. J. 44 614–619. [PubMed] [Google Scholar]
- Cheng A. W., Wang H., Yang H., Shi L., Katz Y., Theunissen T. W., et al. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 23 1163–1171. 10.1038/cr.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. J., Gabrielson E., Werb Z., Ewald A. J. (2013). Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155 1639–1651. 10.1016/j.cell.2013.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. J., Padmanaban V., Silvestri V., Schipper K., Cohen J. D., Fairchild A. N., et al. (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. U.S.A. 113 E854–E863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton C. J., Li X., Landis M., Dixon J. M., Neumeister V. M., Sjolund A., et al. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. U.S.A. 106 13820–13825. 10.1073/pnas.0905718106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X., Yi Q., Jing X., Huang Y., Tian J., Long C., et al. (2018). Mining prognostic significance of MEG3 in human breast cancer using bioinformatics analysis. Cell. Physiol. Biochem. 50 41–51. 10.1159/000493956 [DOI] [PubMed] [Google Scholar]
- Dagogo-Jack I., Shaw A. T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15 81–94. 10.1038/nrclinonc.2017.166 [DOI] [PubMed] [Google Scholar]
- De Craene B., Berx G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 13 97–110. 10.1038/nrc3447 [DOI] [PubMed] [Google Scholar]
- Di Gesualdo F., Capaccioli S., Lulli M. (2014). A pathophysiological view of the long non-coding RNA world. Oncotarget 5 10976–10996. 10.18632/oncotarget.2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill T. L., Naya F. J. (2018). A hearty dose of noncoding RNAs: the imprinted DLK1-DIO3 locus in cardiac development and disease. J. Cardiovasc. Dev. Dis. 5:37. 10.3390/jcdd5030037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., et al. (2003). In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17 1253–1270. 10.1101/gad.1061803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eades G., Zhang Y. S., Li Q. L., Xia J. X., Yao Y., Zhou Q. (2014). Long non-coding RNAs in stem cells and cancer. World J. Clin. Oncol. 5 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B., Spirio L., Koerner F., Fleming M. D., Zimonjic D. B., Donaher J. L., et al. (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15 50–65. 10.1101/gad.828901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elloumi F., Hu Z., Li Y., Parker J. S., Gulley M. L., Amos K. D., et al. (2011). Systematic bias in genomic classification due to contaminating non-neoplastic tissue in breast tumor samples. BMC Med. Genomics 4:54. 10.1186/1755-8794-4-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A., Bozzoni I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15 7–21. 10.1038/nrg3606 [DOI] [PubMed] [Google Scholar]
- Felipe Lima J., Nofech-Mozes S., Bayani J., Bartlett J. M. (2016). EMT in breast carcinoma-a review. J. Clin. Med. 5:65. 10.3390/jcm5070065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridriksdottir A. J., Villadsen R., Morsing M., Klitgaard M. C., Kim J., Petersen O. W., et al. (2017). Proof of region-specific multipotent progenitors in human breast epithelia. Proc. Natl. Acad. Sci. U.S.A. 114 E10102–E10111. 10.1073/pnas.1714063114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P., Locker J., Sahai E., Segall J. E. (2012). Classifying collective cancer cell invasion. Nat. Cell Biol. 14 777–783. 10.1038/ncb2548 [DOI] [PubMed] [Google Scholar]
- Gasco M., Shami S., Crook T. (2002). The p53 pathway in breast cancer. Breast Cancer Res. 4 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafouri-Fard S., Taheri M. (2019). Maternally expressed gene 3 (MEG3): a tumor suppressor long non coding RNA. Biomed. Pharmacother. 118:109129. 10.1016/j.biopha.2019.109129 [DOI] [PubMed] [Google Scholar]
- Ghoncheh M., Pournamdar Z., Salehiniya H. (2016). Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac. J. Cancer Prev. 17 43–46. 10.7314/apjcp.2016.17.s3.43 [DOI] [PubMed] [Google Scholar]
- Gilbert L. A., Larson M. H., Morsut L., Liu Z., Brar G. A., Torres S. E., et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154 442–451. 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Wilde A., Fouquier D’herouel A., Mcintosh E., Ertaylan G., Skupin A., Kuestner R. E., et al. (2015). Stemness of the hybrid Epithelial/Mesenchymal state in breast cancer and its association with poor survival. PLoS One 10:e0126522. 10.1371/journal.pone.0126522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Wilde A., Kuestner R. E., Skelton S. M., Macintosh E., D’herouel A. F., Ertaylan G., et al. (2018). Loss of inter-cellular cooperation by complete epithelial-mesenchymal transition supports favorable outcomes in basal breast cancer patients. Oncotarget 9 20018–20033. 10.18632/oncotarget.25034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson T., Villadsen R., Nielsen H. L., Ronnov-Jessen L., Bissell M. J., Petersen O. W. (2002). Isolation, immortalization, and characterization of a human breast epithelial cell line with stem cell properties. Genes Dev. 16 693–706. 10.1101/gad.952602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson S., Rohatgi N., Magnusdottir M., Choudhary K. S., Gudjonsson T., Knutsen E., et al. (2017). Metabolic re-wiring of isogenic breast epithelial cell lines following epithelial to mesenchymal transition. Cancer Lett. 396 117–129. 10.1016/j.canlet.2017.03.019 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Harrington C. T., Lin E. I., Olson M. T., Eshleman J. R. (2013). Fundamentals of pyrosequencing. Arch. Pathol. Lab. Med. 137 1296–1303. 10.5858/arpa.2012-0463-ra [DOI] [PubMed] [Google Scholar]
- Haupt S., Haupt Y. (2017). P53 at the start of the 21st century: lessons from elephants. F1000Res. 6:2041. 10.12688/f1000research.12682.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmarsdottir B., Briem E., Sigurdsson V., Franzdottir S. R., Ringner M., Arason A. J., et al. (2015). MicroRNA-200c-141 and Np63 are required for breast epithelial differentiation and branching morphogenesis. Dev. Biol. 403 150–161. 10.1016/j.ydbio.2015.05.007 [DOI] [PubMed] [Google Scholar]
- Hollestelle A., Peeters J. K., Smid M., Timmermans M., Verhoog L. C., Westenend P. J., et al. (2013). Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 138 47–57. [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ingthorsson S., Sigurdsson V., Fridriksdottir A., Jr., Jonasson J. G., Kjartansson J., Magnusson M. K., et al. (2010). Endothelial cells stimulate growth of normal and cancerous breast epithelial cells in 3D culture. BMC Res. Notes 3:184. 10.1186/1756-0500-3-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides Y., Lokulo-Sodipe K., Mackay D. J., Davies J. H., Temple I. K. (2014). Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J. Med. Genet. 51 495–501. 10.1136/jmedgenet-2014-102396 [DOI] [PubMed] [Google Scholar]
- Joerger A. C., Fersht A. R. (2016). The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85 375–404. 10.1146/annurev-biochem-060815-014710 [DOI] [PubMed] [Google Scholar]
- Kaneko S., Bonasio R., Saldana-Meyer R., Yoshida T., Son J., Nishino K., et al. (2014). Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 53 290–300. 10.1016/j.molcel.2013.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L., Wang J., Zhang Y., Kou Z., Gao S. (2009). iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell 5 135–138. 10.1016/j.stem.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Lambert A. W., Pattabiraman D. R., Weinberg R. A. (2017). Emerging biological principles of metastasis. Cell 168 670–691. 10.1016/j.cell.2016.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Shen H., Xie H., Ying Y., Jin K., Yan H., et al. (2019). Dysregulation of ncRNAs located at the DLK1DIO3 imprinted domain: involvement in urological cancers. Cancer Manag. Res. 11 777–787. 10.2147/cmar.s190764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L., Sun H., Zhang W., Zhang M., Yang X., Kuang R., et al. (2016). Meta-analysis of EMT datasets reveals different types of EMT. PLoS One 11:e0156839. 10.1371/journal.pone.0156839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Ye D., Chen A., Tan D., Zhang W., Jiang W., et al. (2019). A pilot study of new promising non-coding RNA diagnostic biomarkers for early-stage colorectal cancers. Clin. Chem. Lab. Med. 57 1073–1083. 10.1515/cclm-2019-0052 [DOI] [PubMed] [Google Scholar]
- Liu L., Luo G. Z., Yang W., Zhao X., Zheng Q., Lv Z., et al. (2010). Activation of the imprinted Dlk1-Dio3 region correlates with pluripotency levels of mouse stem cells. J. Biol. Chem. 285 19483–19490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Cong Y., Wang D., Sun Y., Deng L., Liu Y., et al. (2014). Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2 78–91. 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liz J., Esteller M. (2016). lncRNAs and microRNAs with a role in cancer development. Biochim. Biophys. Acta 1859 169–176. 10.1016/j.bbagrm.2015.06.015 [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Kang Y. (2019). Epithelial-Mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 49 361–374. 10.1016/j.devcel.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine J. C., Francoz S., Maetens M., Wahl G., Toledo F., Lozano G. (2006). Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 13 927–934. 10.1038/sj.cdd.4401912 [DOI] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E., Mattick J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10 155–159. 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- Mercer W. E. (1992). Cell cycle regulation and the p53 tumor suppressor protein. Crit. Rev. Eukaryot. Gene Expr. 2 251–263. [PubMed] [Google Scholar]
- Mitra R., Chen X., Greenawalt E. J., Maulik U., Jiang W., Zhao Z., et al. (2017). Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat. Commun. 8:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Pinelo S., Salinas A., Moreno-Mata N., Ferrer I., Suarez R., Andres-Leon E., et al. (2018). Impact of DLK1-DIO3 imprinted cluster hypomethylation in smoker patients with lung cancer. Oncotarget 9 4395–4410. 10.18632/oncotarget.10611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi S., Sharifi-Zarchi A., Ahmadi A., Mollamohammadi S., Stubenvoll A., Gunther S., et al. (2017). Small RNA sequencing reveals Dlk1-Dio3 locus-embedded MicroRNAs as major drivers of ground-state pluripotency. Stem Cell Rep. 9 2081–2096. 10.1016/j.stemcr.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A. P., Lievre M., Thomas C., Hinkal G., Ansieau S., Puisieux A. (2008). Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3:e2888. 10.1371/journal.pone.0002888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsing M., Klitgaard M. C., Jafari A., Villadsen R., Kassem M., Petersen O. W., et al. (2016). Evidence of two distinct functionally specialized fibroblast lineages in breast stroma. Breast Cancer Res. 18:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A., Heldin C. H. (2007). Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 98 1512–1520. 10.1111/j.1349-7006.2007.00550.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Ngoc K. V., Cheung K. J., Brenot A., Shamir E. R., Gray R. S., Hines W. C., et al. (2012). ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. U.S.A. 109 E2595–E2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M. A., Huang R. Y., Jackson R. A., Thiery J. P. (2016). EMT: 2016. Cell 166 21–45. [DOI] [PubMed] [Google Scholar]
- Niknafs Y. S., Pandian B., Gajjar T., Gaudette Z., Wheelock K., Maz M. P., et al. (2018). MiPanda: a resource for analyzing and visualizing next-generation sequencing transcriptomics data. Neoplasia 20 1144–1149. 10.1016/j.neo.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I., Brisebarre A., Sifrim A., Fioramonti M., Revenco T., Boumahdi S., et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556 463–468. 10.1038/s41586-018-0040-3 [DOI] [PubMed] [Google Scholar]
- Pechoux C., Gudjonsson T., Ronnov-Jessen L., Bissell M. J., Petersen O. W. (1999). Human mammary luminal epithelial cells contain progenitors to myoepithelial cells. Dev. Biol. 206 88–99. 10.1006/dbio.1998.9133 [DOI] [PubMed] [Google Scholar]
- Peinado H., Olmeda D., Cano A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7 415–428. 10.1038/nrc2131 [DOI] [PubMed] [Google Scholar]
- Petersen O. W., Lind Nielsen H., Gudjonsson T., Villadsen R., Ronnov-Jessen L., Bissell M. J. (2001). The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 3 213–217. 10.1186/bcr298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., Perou C. M. (2011). Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 5 5–23. 10.1016/j.molonc.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152 1173–1183. 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Radisky D. C., Kenny P. A., Bissell M. J. (2007). Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J. Cell. Biochem. 101 830–839. 10.1002/jcb.21186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringnér M., Fredlund E., Häkkinen J., Borg Å., Staaf J. (2011). GOBO: gene expression-based outcome for breast cancer online. PLoS One 6:e17911. 10.1371/journal.pone.0017911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson E. J., Khaled W. T., Abell K., Watson C. J. (2006). Epithelial-to-mesenchymal transition confers resistance to apoptosis in three murine mammary epithelial cell lines. Differentiation 74 254–264. 10.1111/j.1432-0436.2006.00075.x [DOI] [PubMed] [Google Scholar]
- Sanli I., Lalevee S., Cammisa M., Perrin A., Rage F., Lleres D., et al. (2018). Meg3 non-coding RNA expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 23 337–348. 10.1016/j.celrep.2018.03.044 [DOI] [PubMed] [Google Scholar]
- Scheel C., Weinberg R. A. (2012). Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol. 22 396–403. 10.1016/j.semcancer.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J. M., Panzilius E., Bartsch H. S., Irmler M., Beckers J., Kari V., et al. (2015). Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 10 131–139. 10.1016/j.celrep.2014.12.032 [DOI] [PubMed] [Google Scholar]
- Sheng X., Li J., Yang L., Chen Z., Zhao Q., Tan L., et al. (2014). Promoter hypermethylation influences the suppressive role of maternally expressed 3, a long non-coding RNA, in the development of epithelial ovarian cancer. Oncol. Rep. 32 277–285. 10.3892/or.2014.3208 [DOI] [PubMed] [Google Scholar]
- Shimono Y., Zabala M., Cho R. W., Lobo N., Dalerba P., Qian D., et al. (2009). Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138 592–603. 10.1016/j.cell.2009.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson V., Hilmarsdottir B., Sigmundsdottir H., Fridriksdottir A. J., Ringner M., Villadsen R., et al. (2011). Endothelial induced EMT in breast epithelial cells with stem cell properties. PLoS One 6:e23833. 10.1371/journal.pone.0023833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Apostolou E., Akutsu H., Fukuda A., Follett P., Natesan S., et al. (2010a). Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465 175–181. 10.1038/nature09017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Maherali N., Borkent M., Hochedlinger K. (2010b). A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat. Methods 7 53–55. 10.1038/nmeth.1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Li Y., Yang B. (2016). Downregulated long non-coding RNA MEG3 in breast cancer regulates proliferation, migration and invasion by depending on p53’s transcriptional activity. Biochem. Biophys. Res. Commun. 478 323–329. 10.1016/j.bbrc.2016.05.031 [DOI] [PubMed] [Google Scholar]
- Sun L., Luo H., Liao Q., Bu D., Zhao G., Liu C., et al. (2013). Systematic study of human long intergenic non-coding RNAs and their impact on cancer. Sci. China Life Sci. 56 324–334. 10.1007/s11427-013-4460-x [DOI] [PubMed] [Google Scholar]
- Sun M., Xia R., Jin F., Xu T., Liu Z., De W., et al. (2014). Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol. 35 1065–1073. 10.1007/s13277-013-1142-z [DOI] [PubMed] [Google Scholar]
- Tabar L., Dean P. B., Yen A. M., Tarjan M., Chiu S. Y., Chen S. L., et al. (2014). A proposal to unify the classification of breast and prostate cancers based on the anatomic site of cancer origin and on long-term patient outcome. Breast Cancer 8 15–38. 10.4137/bcbcr.S13833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam W. L., Weinberg R. A. (2013). The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 19 1438–1449. 10.1038/nm.3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima M., Tange S., Ishimura A., Suzuki T. (2017). MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial-mesenchymal transition in lung cancer cell lines. J. Biol. Chem. 292 82–99. 10.1074/jbc.m116.750950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J. P. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2 442–454. 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- Tierling S., Dalbert S., Schoppenhorst S., Tsai C. E., Oliger S., Ferguson-Smith A. C., et al. (2006). High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 87 225–235. 10.1016/j.ygeno.2005.09.018 [DOI] [PubMed] [Google Scholar]
- Uroda T., Anastasakou E., Rossi A., Teulon J. M., Pellequer J. L., Annibale P., et al. (2019). Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol. Cell 75 982–995.e9. 10.1016/j.molcel.2019.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdmanis P. N., Roy-Chaudhuri B., Kim H. K., Sayles L. C., Zheng Y., Chuang C. H., et al. (2015). Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene 34 94–103. 10.1038/onc.2013.523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Ren Z., Sun P. (2012). Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J. Cell. Biochem. 113 1868–1874. 10.1002/jcb.24055 [DOI] [PubMed] [Google Scholar]
- Wang Y., Kong D. (2018). Knockdown of lncRNA MEG3 inhibits viability, migration, and invasion and promotes apoptosis by sponging miR-127 in osteosarcoma cell. J. Cell. Biochem. 119 669–679. 10.1002/jcb.26230 [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhou B. P. (2013). Epithelial-mesenchymal transition—a hallmark of breast cancer metastasis. Cancer Hallm. 1 38–49. 10.1166/ch.2013.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. D., Gao D., Redfern A., Thompson E. W. (2019). Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 19 716–732. 10.1038/s41568-019-0213-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Zhang T., Qi L., Zhou C., Wei J., Feng F., et al. (2019). Integrated analysis of co-expression and ceRNA network identifies five lncRNAs as prognostic markers for breast cancer. J. Cell. Mol. Med. 23 8410–8419. 10.1111/jcmm.14721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yersal O., Barutca S. (2014). Biological subtypes of breast cancer: prognostic and therapeutic implications. World J. Clin. Oncol. 5 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D. D., Liu Z. J., Zhang E., Kong R., Zhang Z. H., Guo R. H. (2015). Decreased expression of long noncoding RNA MEG3 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer. Tumour Biol. 36 4851–4859. 10.1007/s13277-015-3139-2 [DOI] [PubMed] [Google Scholar]
- Zaravinos A. (2015). The regulatory role of microRNAs in EMT and cancer. J. Oncol. 2015:865816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M., Kalluri R. (2004). The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 82 175–181. 10.1007/s00109-003-0517-9 [DOI] [PubMed] [Google Scholar]
- Zhang X., Rice K., Wang Y., Chen W., Zhong Y., Nakayama Y., et al. (2010). Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions. Endocrinology 151 939–947. 10.1210/en.2009-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhou Y., Mehta K. R., Danila D. C., Scolavino S., Johnson S. R., et al. (2003). A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 88 5119–5126. 10.1210/jc.2003-030222 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhong Y., Wang Y., Zhang X., Batista D. L., Gejman R., et al. (2007). Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 282 24731–24742. 10.1074/jbc.m702029200 [DOI] [PubMed] [Google Scholar]
- Zhu W., Botticelli E. M., Kery R. E., Mao Y., Wang X., Yang A., et al. (2019). Meg3-DMR, not the Meg3 gene, regulates imprinting of the Dlk1-Dio3 locus. Dev. Biol. 455 10–18. 10.1016/j.ydbio.2019.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data for this article has been submitted to GEO, with the GEO accession number GSE142268, see here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142268