

SUMMARY
Zika virus (ZIKV) is an emerging, mosquito-borne flavivirus responsible for recent epidemics across the Americas, and it is closely related to dengue virus (DENV). Here, we study samples from 46 DENV-naive and 43 DENV-immune patients with RT-PCR-confirmed ZIKV infection at early-acute, late-acute, and convalescent time points from our pediatric cohort study in Nicaragua. We analyze the samples via RNA sequencing (RNA-seq), CyTOF, and multiplex cytokine/chemokine Luminex to generate a comprehensive, innate immune profile during ZIKV infection. Immunophenotyping and analysis of cytokines/chemokines reveal that CD14+ monocytes play a key role during ZIKV infection. Further, we identify CD169 (Siglec-1) on CD14+ monocytes as a potential biomarker of acute ZIKV infection. Strikingly distinct transcriptomic and immunophenotypic signatures are observed at all three time points. Interestingly, pre-existing dengue immunity has minimal impact on the innate immune response to Zika. Finally, this comprehensive immune profiling and network analysis of ZIKV infection in children serves as a valuable resource.
Graphical Abstract
In Brief
At three time points after Zika virus infection, Michlmayr et al. perform comprehensive immunoprofiling of pediatric cohort samples via RNA-seq, CyTOF, and Luminex cytokine/chemokine array, resulting in distinct temporal patterns of gene expression, cell profiles, and cytokines/chemokines. They show CD14+ monocytes play a central role, identify CD169 as a potential biomarker of acute ZIKV infection along with upregulation of CXCL10, and find no impact of prior dengue virus infection on the innate immune response to Zika.
INTRODUCTION
Zika virus (ZIKV) is a mosquito-borne virus that belongs to the Flaviviridae family and is closely related to other flaviviruses, such as dengue virus (DENV) and West Nile virus (WNV) (Barba-Spaeth et al., 2016). Because of the massive epidemic of Zika in the Americas in 2015–2016, which was associated with microcephaly and other neurological disorders in infants born of infected mothers, the World Health Organization (WHO) declared Zika a public health emergency of international concern (World Health Organization (WHO)). Profound gapsstill remain in our understanding of Zika immune responses and pathogenesis. In particular, one main concern has been whether prior exposure to DENV affects Zika outcome in areas in which these viruses co-circulate. Several recent human studies have shown that prior DENV infection results in a similar or stronger adaptive ZIKV immune response (Andrade et al., 2019; Grifoni et al., 2017) or protection against infection and/or symptomatic disease (Gordon et al., 2019; Rodriguez-Barraquer et al., 2019). However, the role of prior DENV exposure on the human innate immune responses to ZIKV remains unclear.
ZIKV can infect monocytes, macrophages, and dendritic cells (DCs) (Bowen et al., 2017; Michlmayr et al., 2017; Quicke et al., 2016). Monocytes play critical roles in the pathogenesis of many flaviviruses (Bardina et al., 2015; Lim et al., 2011; Schmid and Harris, 2014) and can be grouped into CD14hiCD16 “inflammatory,” CD14+CD16+ “intermediate,” and CD14lowCD16hi “nonclassical” monocytes (Ziegler-Heitbrock et al., 2010). Murine studies of DENV have shown that inflammatory monocytes are key targets of infection that are quickly recruited to the site of infection and express the inflammatory chemokine receptor CCR2 (Schmid and Harris, 2014). The chemokines CCL2 and CCL7, which bind to CCR2, are involved in monocytosis and monocyte recruitment into the brain during WNV encephalitis (Bardina et al., 2015). During ZIKV infection, CD14+ and CD14+CD16+ monocytes are targets of infection and play an important role in priming natural killer (NK) cells and DCs (Lum et al., 2018a; Michlmayr et al., 2017).
A key element of the innate immune response to flavivirus infection is type I interferon (IFN), which invokes a potent antiviral state in cells, resulting in upregulation of a plethora of IFN-stimulated genes (ISGs) that block viral infection (MacMicking, 2012). In vitro experiments revealed five ISGs, including RSAD2 (Viperin), OAS, PKR (RIG-1), IFITM2 and IFITM3, to be highly upregulated during flavivirus infections (Jiang et al., 2010). CD169, also called Sialoadhesin or Siglec-1, is another ISG that is important for cross-priming of CD8+ T cells during viral infection through interaction of macrophages with CD8α+ DCs (van Dinther et al., 2018). However, the extent of IFN induction and the key ISGs during the acute phase of ZIKV infection in humans are unresolved questions.
Examples of systems immunology approaches applied to infectious diseases remain relatively sparse. Most such studies of immune responses to ZIKV have concentrated on RNA sequencing (RNA-seq) and multiplex immunoassay datasets derived from primary cell culture, animal models, or small patient cohorts (Kam et al., 2017; Lum et al., 2018b; Tiwari et al., 2017; Yi et al., 2017). Here, we describe a systems immunology approach to profiling innate immune responses during acute ZIKV infection in 89 pediatric patients from a long-standing cohort study in Nicaragua. Importantly, we were able to stratify our patients into DENV-naive and DENV-immune cohorts, because of their well-characterized DENV-infection histories. We first used three high-dimensional technologies (RNA-seq, CyTOF, and Luminex cytokine/chemokine multiplex bead array) to detect globally significant changes in gene transcripts, cell populations, and serum cytokine/chemokine concentrations over three time points after infection (early acute, late acute, and convalescent). Because we observed no differences in innate immune profile due to prior DENV infection, we combined the DENV-naive and DENV-immune patient populations and built an integrative, multiscale network model based on all the datasets that revealed correlations among gene modules, cell communities, cytokines, and clinical variables, as well as key players during the immune response to Zika in these well-characterized pediatric patients.
RESULTS
Prior DENV Infection Does Not Modulate the Innate Immune Response to ZIKV
Because of the genetic similarity of ZIKV and DENV, we hypothesized that prior DENV exposure might affect the innate immune response to ZIKV. Therefore, we determined the immune signature of early-acute (1–3 days after onset of symptoms), late-acute (days 4–6), and convalescent (days 14–21) samples from DENV-naive (n = 46) and DENV-immune patients (n = 43) by RNA-seq, Luminex, and CyTOF (Figure 1). Unsupervised clustering of all samples revealed clusters based on time point, rather than on previous DENV exposure (Figures 2A and S1E). The sub-clusters identified within each time point could not separate DENV-naive and DENV-exposed groups better than random guesses because the Wilcoxon test on the ordering of the samples induced by unsupervised clustering was not different between DENV-naive and DENV-exposed patients and was not different by number of previous DENV infections (p > 0.05 for all three datasets). Thus, clustering of data within each time point is not associated with DENV serostatus or number of prior DENV infections.
Figure 1. Study Design.
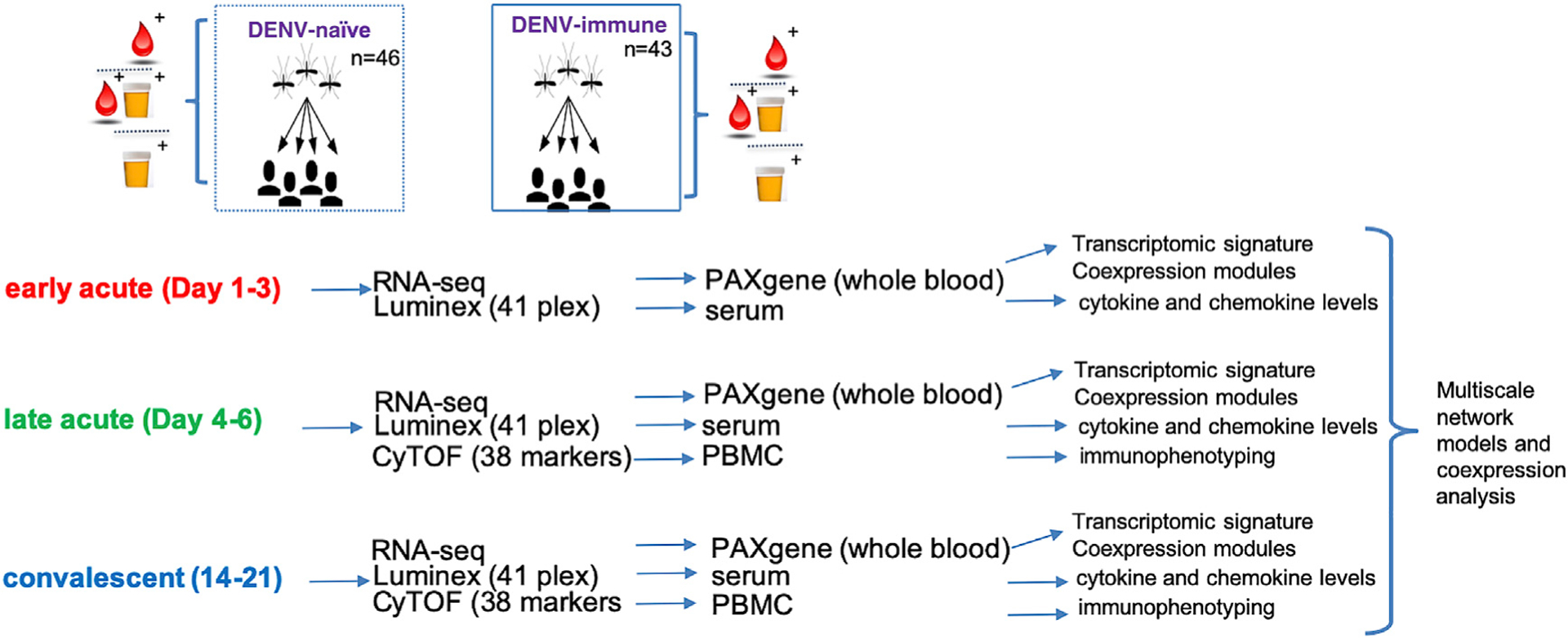
PBMC, PAXgene, and plasma samples were collected from 46 DENV-naive and 43 DENV-immune ZIKV-infected patients at three time points after symptom onset. CyTOF, RNA-seq, and cytokine/chemokine multiplex ELISA (Luminex) were performed on samples as indicated.
Figure 2. Comparison of Immune Signatures in DENV-Naive and DENV-Immune ZIKV Patients Demonstrates a Minimal Role for Prior DENV Exposure.
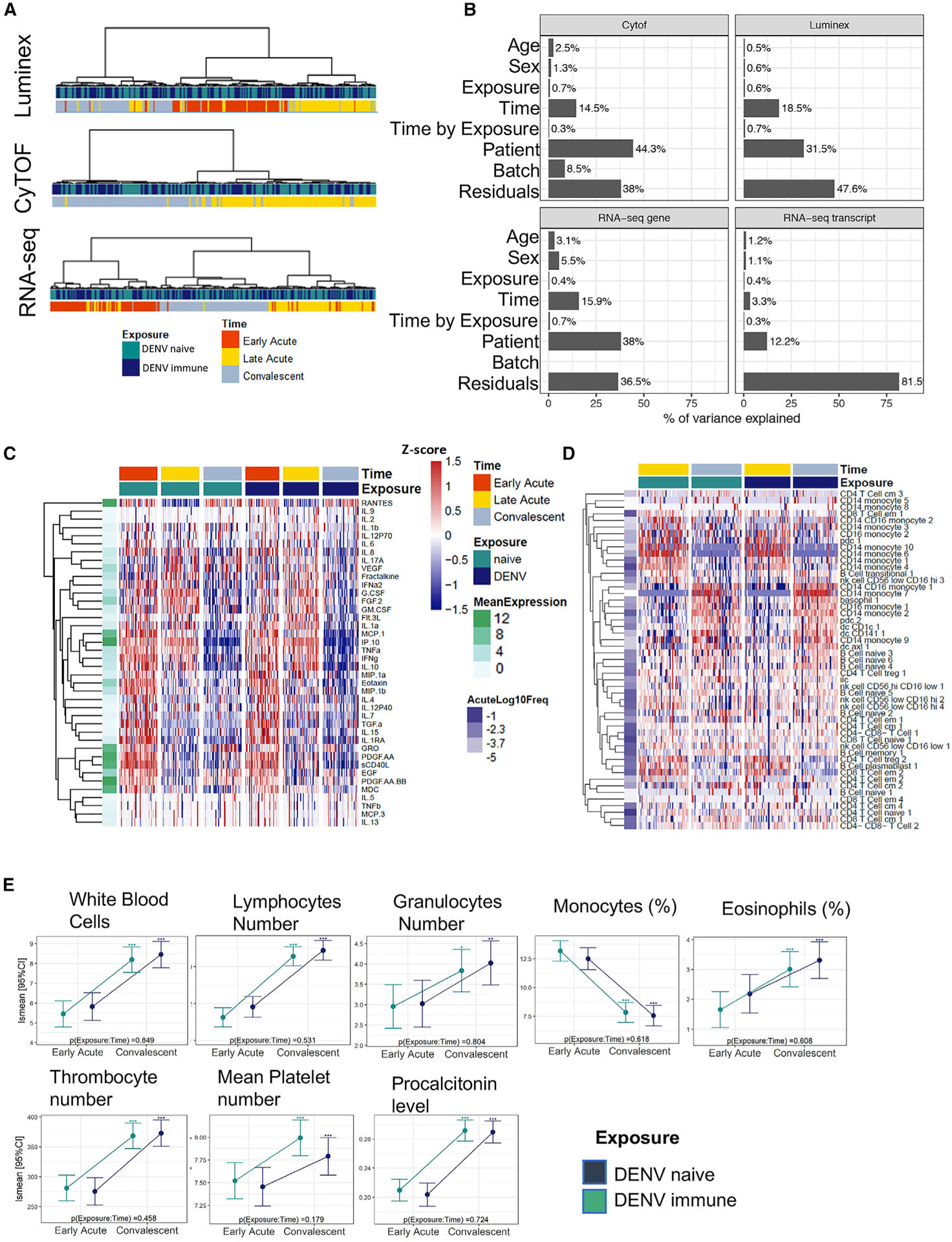
(A) Dendrogram showing unsupervised clustering of samples based on patient-normalized data for Luminex, CyTOF, and RNA-seq. Pearson’s correlation was used as a distance metric with Ward as the agglomeration procedure.
(B) PVCA was performed for the unadjusted datasets indicating the percentage of variance explained by each factor.
(C and D) Heatmaps of log10-scaled cytokine levels (C) and PBMC subcommunity frequencies (D) for all samples. Markers were grouped using unsupervised hierarchical clustering, based on the distance of the time profiles between markers. Each row represents the standardized mean [(x m)/SD] profile for each marker, and as such, color scale ranges from 1.5 to 1.5 SD of the overall protein expression with red (blue) indicating overexpression (under-expression) of each group versus the rest. Mean expression of cytokines and mean cell frequency at the late-acute time point is shown to the left of each heatmap depicted in either green (Luminex) or purple (CyTOF) scales. Hierarchical clustering was applied.
(E) Selected blood variables from the acute to the convalescent time points are shown and expressed as least-square mean (lsmean) with a 95% confidence interval (CI). Asterisks above the CI bars represent significant changes from early-acute to convalescent phase, with *p < 0.05, **p < 0.01, ***p < 0.001. p values at the bottom left of each plot indicate differences between DENV-naive and DENV-immune groups.
See also Figures S1, S2, and S3.
Other analytical approaches also confirmed the lack of differences between the DENV-exposed and DENV-naive patients. Principal variation component analysis (PVCA) showed that the variance or changes in time explained by previous DENV exposure were <1% of the total variance (Figure 2B). Time profiles were similar between DENV-naive and DENV-immune patients, as shown by non-significant time by exposure interactions (false discovery rate [FDR] > 0.05, moderated F test) for all three datasets (Figures 2C, 2D, S1A, and S1B). When comparing temporal changes in RNA expression profiles, cell populations, and cytokine levels, we observed that these changes were highly correlated among groups (Pearson’s correlation coefficient, r > 0.84 for genes, r > 0.65 for transcripts, r > 0.8 for CyTOF and Luminex) (Figures S1A, S1B, S2A, and S2B) and overlapped (FDR < 0.05, fold change [FCH] > 2) between DENV-naive and DENV-immune groups (Figures S1C–S1F, S2C, S2D, and S3). However, we noted a trend that gene expression changes from early-acute to late-acute time points were greater in the DENV-naive group, albeit not significantly different.
Next, we determined whether prior exposure to DENV had an effect on complete blood cell count (CBC) and clinical signs during acute ZIKV infection. Our results showed no major differences for CBC results and clinical signs and symptoms between DENV-naive and DENV-immune groups (Figure 2E; Table 1). Therefore, moving forward, we studied the dynamics of ZIKV infection in the cohort of 89 patients based only on the time point contrast.
Table 1.
Baseline and Clinical Characteristics of All Study Participants
| Baseline Characteristics | DENV Naive | DENV Immune | p Valuea |
|---|---|---|---|
| Sex, male (%) | 22 (47.8) | 19 (46.3) | 1 |
| Age (mean [SD]) | 8.9 (3.1) | 10.2 (3.3) | 0.052 |
| Fever at early-acute | 5 (10.9) | 1 (2.4) | 0.26 |
| time point (%) | |||
| Serum (S)/Urine (U) (%)b | 0.012 | ||
| S+/U− | 22 (47.8) | 23 (56.1) | |
| S+/U+ | 22 (47.8) | 6 (14.6) | |
| S+/Unknown U status | 0 (0.0) | 7 (17.1) | |
| S−/U+ | 2 (4.3) | 5 (12.2) | |
| Symptoms | |||
| Headache (%) | 18 (39.1) | 13 (31.7) | 0.619 |
| Retro-orbital pain (%) | 7 (15.2) | 4 (9.8) | 0.659 |
| Erythema (%) | 22 (47.8) | 10 (24.4) | 0.041 |
| Cervical lymphadenopathy (%) | 5 (10.9) | 5 (12.2) | 1 |
| Reduced urine (%) | 3 (6.5) | 3 (7.3) | 1 |
| Urinary nitrate (%) | 7 (15.2) | 6 (14.6) | 1 |
| Bilirubin (%) | 7 (15.2) | 6 (14.6) | 1 |
| Arthralgia (%) | 13 (28.3) | 11 (26.8) | 1 |
| Proximal arthralgia (%) | 5 (10.9) | 4 (9.8) | 1 |
| Distal arthralgia (%) | 7 (15.2) | 5 (12.2) | 0.923 |
| Generalized rash (%) | 23 (50.0) | 17 (41.5) | 0.561 |
| Macular rash (%) | 6 (13.0) | 7 (17.1) | 0.822 |
| Papular rash (%) | 3 (6.5) | 5 (12.2) | 0.587 |
| Hemoconcentration (%) | 15 (32.6) | 14 (34.1) | 1 |
| Sample size (subjects; samples) | |||
| Luminex | 46; 138 | 43; 127 | |
| CyTOF | 46; 92 | 42; 83 | |
| RNA-seq | 46; 138 | 43; 123 | |
p values from chi-square test of independence.
Detection of ZIKV RNA by RT-PCR in serum and/or urine.
ZIKV Infection Results in an Inflammatory and Monocyte-Associated Innate Immune Response
To profile the secretion of chemokines, cytokines and growth factors during acute symptomatic ZIKV infection, we measured the serum concentration of 40 analytes by Luminex in every patient at each time point. After adjustment for plate-specific effects, we identified 32 proteins with significant changes over time (FDR < 0.05 and FCH > 1.5) with temporal dynamics grouped into four main clusters (Figures 3A and 3B; Table S1). As mentioned, no significant differences were observed between DENV-naive and DENV-immune patients, except for IFN-γ (Figure S4). Although the unadjusted p value of the mean cytokine concentration revealed differences between DENV-naive and DENV-immune patients, the result was not significant after adjustment for multiple hypotheses across cytokines (FDR > 0.05) (Table S1).
Figure 3. Plasma Cytokine and Chemokine Levels during Early Acute, Late Acute, and Convalescent ZIKV Infections Are Differentially Expressed and Reveal a Central Role for Monocytes.
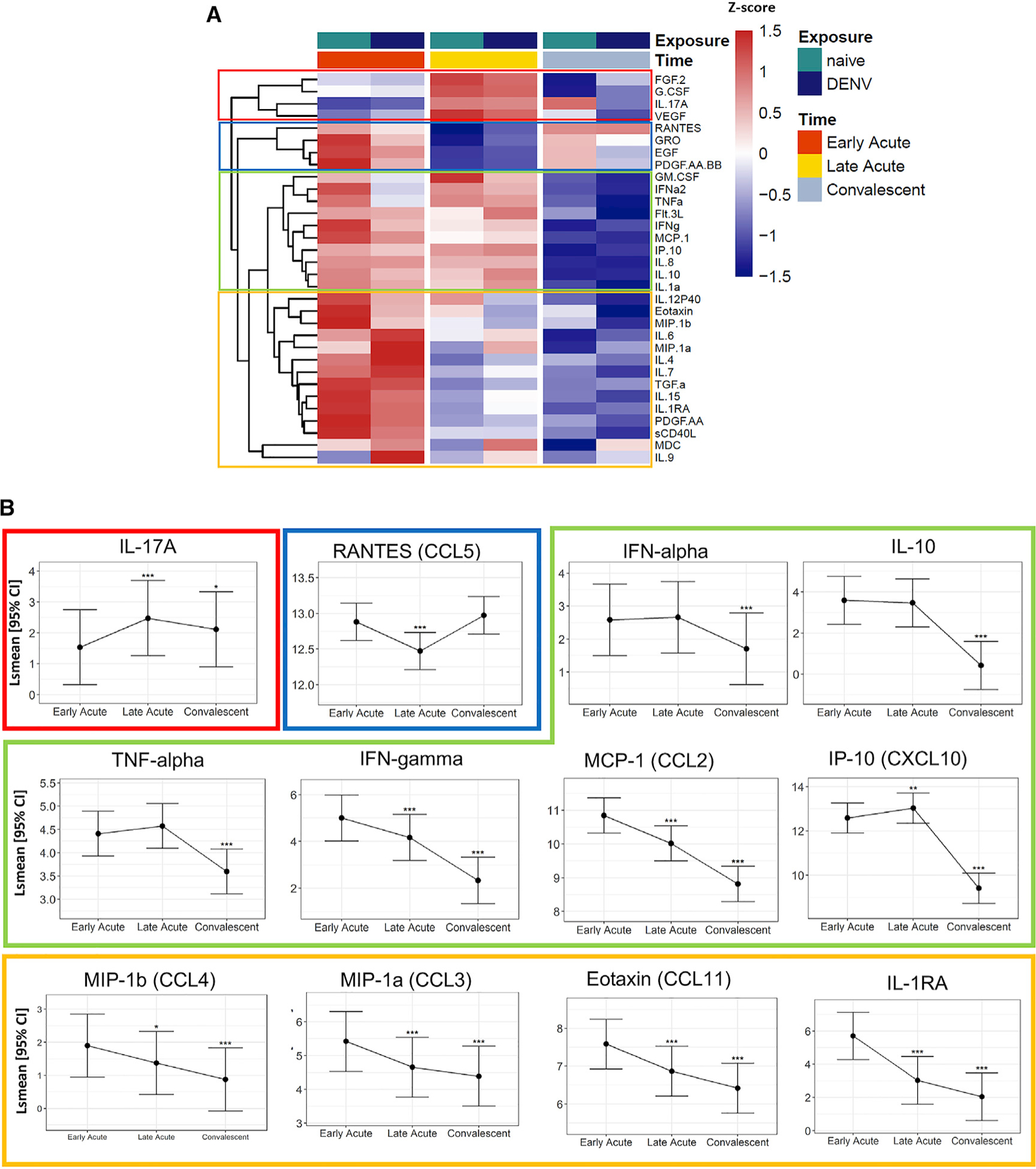
(A) Heatmap representation of the average protein expression of significant changes (FDR < 0.05) of cytokine levels (pg/mL) over time. The color of the heatmap in each row is based on Z score values calculated by centering and scaling the data by standard deviation using the formula (X m)/SD), where X is individual values, and m is the mean of the row.
(B) Estimated lsmean and 95% CI are shown for selected cytokines. Asterisks above the CI bars represent significance of the changes from late-acute or convalescent to early-acute phase (n = 89). The adjusted p value is shown as *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001.
See also Figures S4 and S5 and Table S1.
In the combined dataset (n = 89), FGF2, granulocyte-colony-stimulating factor (G-CSF), IL17A, and VEGF (red box) were significantly increased at the late-acute time point and lower at early-acute and convalescent time points (Figure 3). CCL5 (RANTES), CXCL1 (GRO), EGF, and PDGF-AA-BB (blue box) were significantly decreased at the late-acute time point compared with higher levels at early-acute and convalescent time points. Other cytokines, such as granulocyte-macro-phage-colony-stimulating factor (GM-CSF), interferon alpha (IFN-a), tumor necrosis factor alpha (TNF-α), Flt-3L, IFN-γ, CCL2 (MCP-1), CXCL10 (IP-10), interleukin-8 (IL-8), IL-10, and IL-1a were higher during both acute time points and were significantly decreased by convalescence (green box). For example, mean levels of CXCL10 and IFN-γ were 9.1- and 6.4-fold higher, respectively, at the early-acute compared with the convalescent phase of ZIKV infection (Table S1; Figure 3). The last group of cytokines, CCL3 (MIP-1α), IL-12p40, CCL11 (Eotaxin), CCL4 (MIP1β), IL-6, IL-4, IL-7, transforming growth factor alpha (TGF-α), IL-15, IL-1RA, platelet-derived growth factor-AA (PDGF-AA), sCD40L, CCL22 (MDC), and IL-9, steadily decreased from early-acute until convalescent time points (yellow box). In contrast, most interleukins, GM-CSF, TGF-α, interferons, and cell-stimulating factors exhibited low levels in the serum of all patients, regardless of time point. Further, we identified two clusters of cytokines that showed a similar pattern of up- and downregulation when comparing fold-changes between late-acute to early-acute and convalescent to late-acute phases using Pearson’s correlation: cluster 1 consisting of VEGF, IL-17A, and IL-8, and cluster 2 including sCD40L and PDGF-AA (Figure S5A). Both clusters exhibited the same degree of correlation and relatedness, suggesting that there is a relationship in their secretion during ZIKV infection. Lastly, most of the key inflammatory cytokines we identified during ZIKV infection were monocyte-associated markers.
CD14+ Monocytes Are Differentially Expanded during Acute and Convalescent Time Points of ZIKV Infection
To identify and quantify phenotypic changes within the leukocyte population at the late-acute phase and during convalescence, we performed CyTOF staining for 37 immune cell markers (Table S2A). Using a previously described computational analysis (Michlmayr et al., 2018), we defined 25 communities of canonical leukocyte populations across all samples. We also identified up to 10 different subcommunities, for a total of 51 subcommunities that reflect a detailed resolution of variation in surface-marker expression within defined cell populations at each time point (Tables S2B, S3A, and S3B).
We identified 20 subcommunities with significant changes post-ZIKV infection (Figure 4A). Of these, 10 were significantly more abundant in the acute phase, whereas 10 were more so in the convalescent phase. In particular, specific CD14+ monocyte subsets changed across time. Specifically, subcommunities 7 and 2 of CD14+ monocytes (FDR = 7.16 × 10−40 and FDR = 1.46 × 10−22, respectively) were the most significantly overrepresented monocyte subcommunities during convalescence and were absent or low during the acute phase (Figure 4B). Other cell communities elevated at convalescence were one CD14+CD16+ and one CD16+ monocyte subcommunity, basophils, CD141+ DCs, Axl+ DCs, a subset of pDCs, and certain groups of B cells. In contrast, subcommunities 1, 4, 6, and 10 of CD14+ monocytes (FDR = 1.82 × 10−36, FDR = 2.06 × 10−15, FDR = 6.18 × 10−23, and FDR = 3.91 × 10−5, respectively) were the most significantly overrepresented monocyte populations during the late-acute phase and were low or absent during convalescence (Figure 4B). Thus, acute symptomatic ZIKV infection was strongly associated with an abundance of specific monocyte subcommunities at certain time points, reflecting distinct monocytic patterns that could be useful for defining biomarkers of disease progression for Zika. Other subcommunities that were more abundant at the late-acute time point at FDR < 0.05 were a subset of pDCs, subsets of CD4+ and CD8+ T cells, plasmablasts, and a subset of CD16+ NK cells.
Figure 4. Acute Symptomatic ZIKV Infection Results in Changes in Immunophenotypic Signatures.
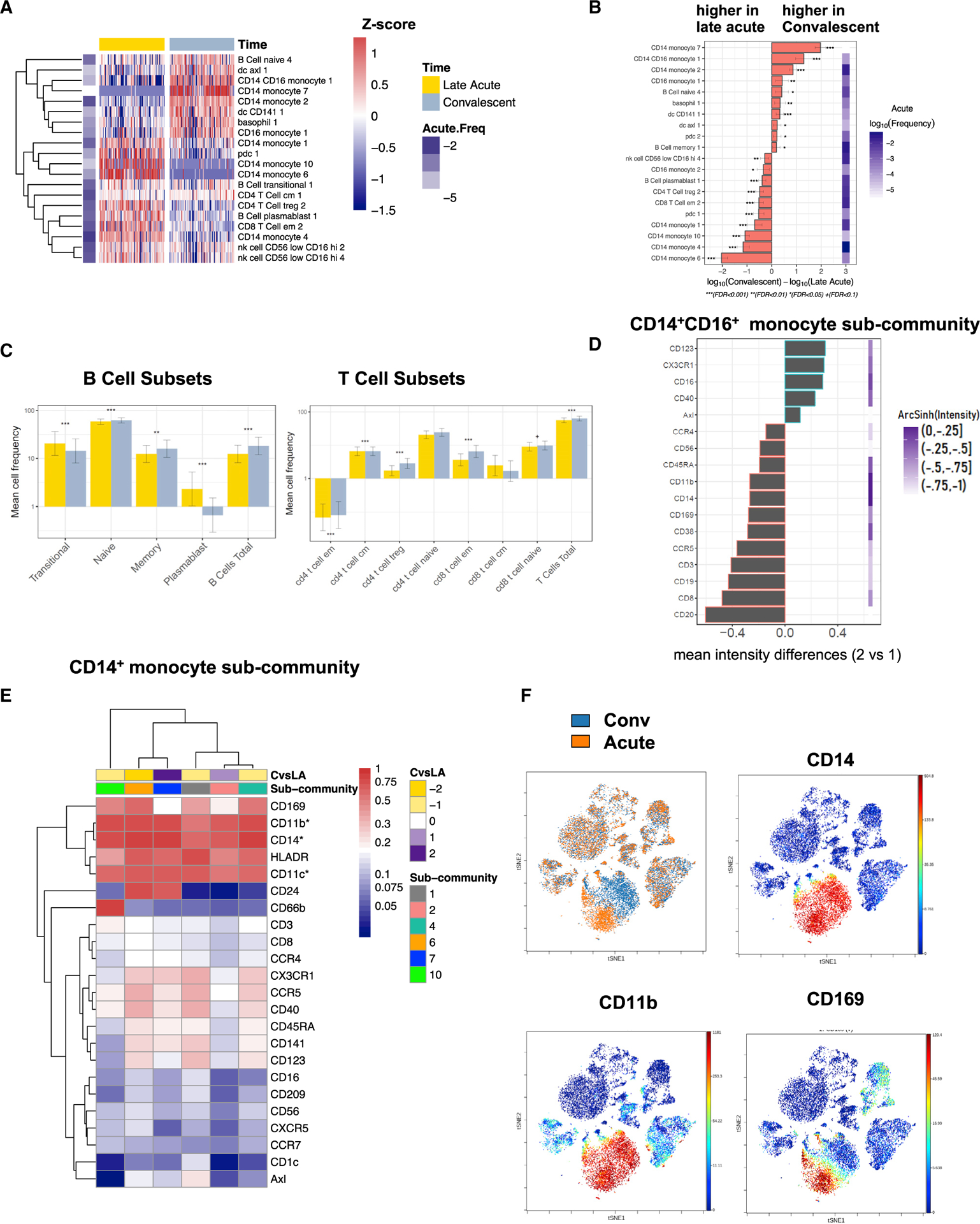
(A) Heatmap representation of the average log10-scaled cell frequency profiles in ZIKV-infected patients for cell subcommunities (FDR < 0.05, FCH > 1.5). Values were standardized for each cell population. The color key to the left of the heatmap indicates the average abundance of that subcommunity at the late-acute phase.
(B) Changes in cell subcommunities from acute to convalescent time points (FDR < 0.05, FCH > 1.5).
(C) Mean cell frequency for B and T cell subsets at the late-acute (yellow) and convalescent (gray) phase, with asterisks representing significance among time points. Error bars are 95% CI; *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001.
(D) Differences in marker expression estimated as the median intensity across CD14+CD16+ populations (subcommunity 2 vs subcommunity 1) with a mean arcsinh (intensity) greater than −1.
(E) Mean marker expression for subcommunities of CD14+ monocytes that are significantly changed during ZIKV infection (FDR < 0.05, FCH > 1.3).
(F) Representative viSNE plots of a DENV-naive patient (patient ID 4515). The top-left panel depicts acute (orange) and convalescent (blue) PBMCs. The top-right, bottom-right, and left panels depict intensity of expression of CD14, CD11b, and CD169, respectively, in the same patient. Intensity of expression is expressed as color ranging from red (high expression) to blue (no expression).
Next, we performed a subset analysis of B and T cells across time. Among B cells, we found that plasmablasts and transitional B cells were significantly more frequent during the late-acute phase of infection, whereas naive and memory B cells were more frequent during convalescence (Figure 4C, left panel). In fact, plasmablasts were almost entirely absent in the convalescent-phase samples, whereas all other B cell subcommunities showed similar levels of mean cell frequency across time points. The frequency of CD4+ and CD8+ effector memory T cell subcommunities was greater during the convalescent time point (Figure 4C, right panel). Overall, the mean cell frequency of T cells was greater during convalescence (FDR < 0.001).
For CD14+CD16+ monocytes, we identified subcommunities 1 and 2, which were significantly changed between late-acute and convalescent ZIKV infection (Figure 4B and 4D). Specifically, CD14+CD16+ monocyte subcommunity 1 was present at a greater frequency at convalescence (FDR p = 8.6 × 10 12). Among CD14+ monocytes, we identified six different monocyte subcommunities that exhibited a specific pattern of cell-surface expression (Figure 4E; Table S4). Furthermore, we noticed a marked shift within the monocyte population from acute to convalescent time points within all patient samples (Figure 4F; Table S3). In particular, CD169 (Siglec-1) was highly expressed on the surface of acute-phase-associated subcommunities (Figures 4E and 4F) and could serve as an immune marker. Representative visualization of t-Distributed Stochastic Neighbor Embedding (viSNE) plots of one patient are depicted in Figure 4F, which show the differential expansion of monocytes across time point and their expression of CD169. Several other markers also exhibited a higher expression profile on acute-phase CD14+ monocytes, including activation markers CD40, CD86, and HLA-DR and homing/adhesion molecules, such as CCR5, CD209, CD11c, and CD11b (Figure 4E). Differential expression of the genes for these surface markers across all time points was confirmed by RNA-seq. Given the strong associations of these distinct subcommunities with time points after ZIKV infection, these data suggest that unappreciated heterogeneity within CD14+ monocyte phenotypes may enable different roles for subcommunities of these monocytes during ZIKV infection. Within the CD16+ monocytes, we detected a significant expansion of CD16+ monocyte 1 subcommunity in convalescent samples and expansion of CD16+ monocyte subcommunity 2 during the acute phase of ZIKV infection (Figure 4B).
To determine whether cytokine levels are associated with changes in cell communities identified by CyTOF, we correlated changes in Luminex protein concentrations with changes in subcommunity frequencies using Spearman’s correlation and hierarchical clustering of the samples (Figure S5B). No significant correlations were observed between the late-acute and the convalescent time point between Luminex and CyTOF data.
Transcriptomic Signatures of ZIKV Infection Exhibit Temporal Patterns
To measure global transcriptional changes during the three sampled phases of ZIKV infection, we employed RNA-seq and transcript-level quantification, followed by differential expression analysis (Table S5). A strong transcriptional signature changing across the three time points was obtained, with 6,350 differentially expressed transcripts (DETs) at FDR < 0.01 and FCH > 2 (Figures 5A–5F and S6A–S6C). Among these top DETs, hierarchical clustering differentiated transcripts into transcripts that were elevated during the late-acute time point (top cluster), transcripts that decreased expression over time (middle cluster), or transcripts that were elevated during the early-acute and convalescent time points (bottom cluster) (Figure 5A). Seeking to quantify the relative number of DETs that fit into distinct temporal patterns, we performed unsupervised hierarchical clustering of all 6,350 DETs and discovered four main temporal dynamic patterns of ZIKV infection (Figures 5B–5F). We then analyzed the biological categories and pathways they represent (Figure S6D; Table S6).
Figure 5. ZIKV Infection Leads to Distinct Temporal Patterns of Top Gene Transcripts, DETs, and CCL2 Isoforms.
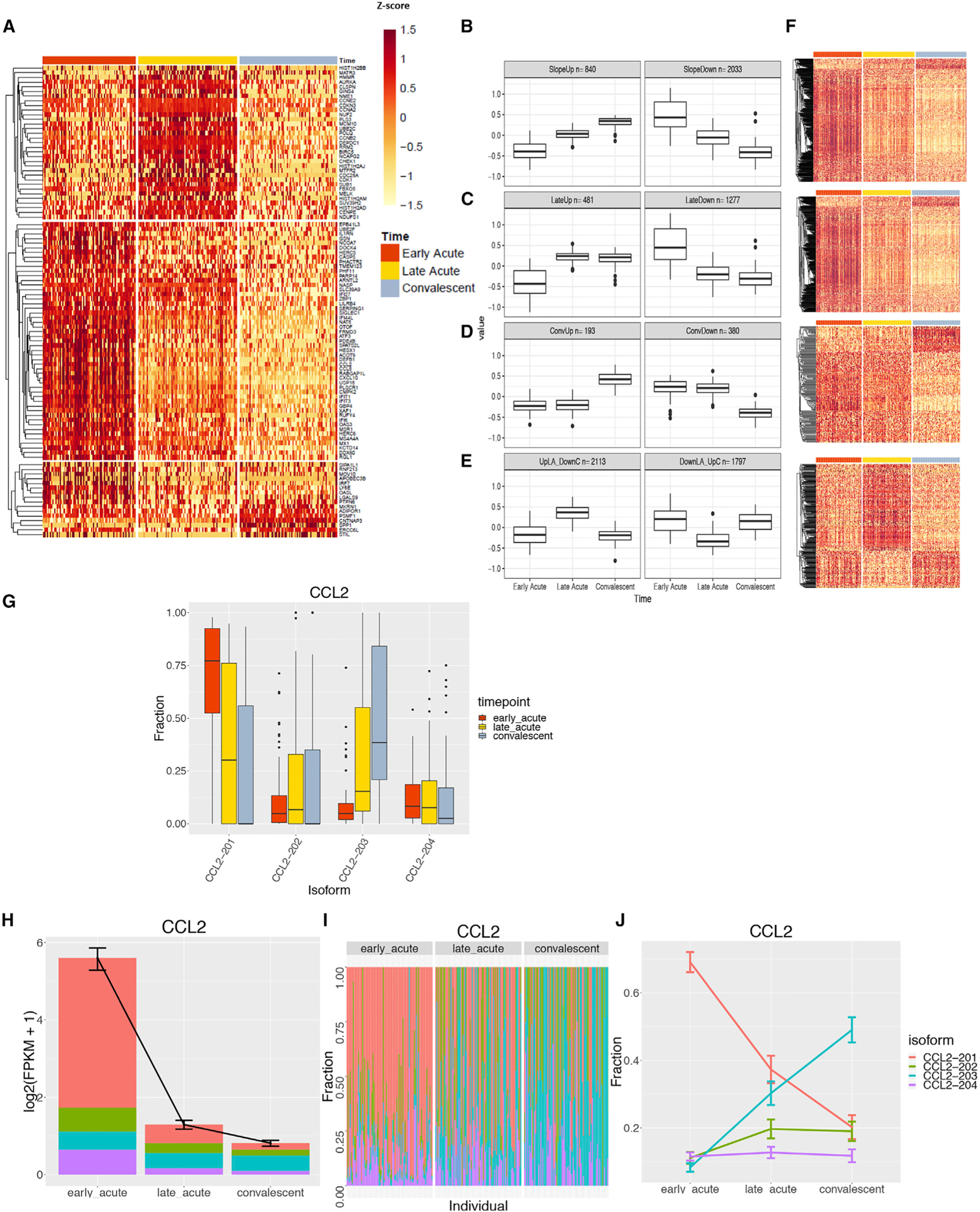
(A) Top protein-coding transcripts with significant changes over time (FCH > 10, FDR < 0.001) filtered to the 100 transcripts with the largest overall changes seen among all time points.
(B–E) Unsupervised clustering of all DETs (FCH > 2, FDR < 0.01) identify four main clusters with different temporal profiles: transcripts that change monotonically over time (B), transcripts that change at late-acute and convalescent time points (C), transcripts that change only at the convalescent time point (D), and transcripts that change only at the late-acute time point (E). (B–E) For each row, the box plot on the left shows the expression patterns for the top cluster per time point (EA, LA, and Conv) and the right box plot corresponds to the bottom cluster of the heatmaps in (F). Boxplots of transcript expression (adjusted by mean expression per gene) represent the average time profile for each cluster. Lower and upper hinges of the boxplot correspond to the first and third quartiles; whiskers extend to the most distant values no further than 1.5× the interquartile range from the hinge.
(F) Transcripts are grouped based on increase/decrease in expression over time. Heatmaps of all DETs corresponding to the DETs shown in (B)–(E).
(G) Four isoforms of CCL2 transcripts and fraction of expression level of each isoform per time point are shown as example of DETs. Box hinges and whiskers are calculated as in (B-E)
(H) The total expression level of CCL2 isoforms in log2 (fragments per kilobase of transcript per million [FPKM]).
(I) Isoform usage across each individual, grouped by the three time points
(J) Change in isoform usage across three time points. (H and J) Shown are the mean plus SEM for each data point.
Within the first group of transcripts—which monotonically change over time—we identified 840 and 2,033 transcripts that either increased or decreased, respectively, from the early-acute to convalescent time point (Figure 5B). DETs that decreased over time belonged to the group of ISGs, such as OAS1, 2 and 3; OASL; DDX58 (RIG-I); IFIH1 (MDA-5); and IFIT5; pattern recognition receptors, including TLR4 and TLR7; apoptosis pathway genes, such as caspases (CASP1 and CASP3); and viral entry receptors (Axls) or inhibitory receptors, including CD300A, which is highly expressed on myeloid cells. The time profile of these transcripts suggests that they can be used to differentiate the phase of ZIKV infection. As such, we used random forest, a broadly used machine learning algorithm (Breiman, 2001), on patient-adjusted gene expression data to predict the infection phase. When genes were ranked by their ability to predict infection phase (quantified by the Gini coefficient), the top 25 genes we found included ISGs, such as USP18, IFI27, HERC6, OAS1, IFIT, and STAT2 (Figure S6E). The Gini coefficient allowed us to rank genes based on their ability to predict the time point of infection with high accuracy. A significant drop in the mean Gini coefficient value was observed after the top 25 genes. These 25 transcripts represent a potential biomarker signature in the RNA-seq data to predict ZIKV infection phase, with an area under the curve (AUC) = 0.94 and an accuracy of 0.99 in testing and 0.86 in cross-validation.
The second pattern encompasses gene transcripts that either increased (481 DETs) or decreased (1,277) in expression between the early- and late-acute time points without further changes thereafter (Figure 5C). The DETs decreasing over time included STAT1; CD169 (Siglec-1); Siglec 7, 10, and 14; CXCL10; and transcripts that belong to the scavenger receptor family MARCO, typically expressed on monocytes. A third pattern included transcripts that did not change their expression from early- to late-acute phase but increased (193 DETs) or decreased (380) their expression levels between the late-acute and convalescent time points, including DDX17 and the translation factor EIF4A1 (Figure 5D). Finally, 2,133 and 1,797 DETs reached highest or lowest levels of expression, respectively, in the late-acute phase compared with other time points (Figure 5E). Heatmaps depicting DETs for these patterns are shown in Figure 5F. The top five significant pathways identified using the Gene Ontology (GO) Consortium and Wikipathway (WP) databases included genes such as IFITM1, 2 and 3; IFIT5; OASL; OAS1; OAS3; MX-1; and RSAD2, which are ISGs and have a pivotal role in innate immunity (Table S6). Again, there were no significant differences between samples from DENV-naive versus DENV-immune patients.
In eukaryotes, alternative splicing and differential isoform usage expands the transcriptome complexity and functional diversity in response to infection. Analysis of alternatively spliced (AS) genes showed a signature of 4,693 AS genes (FDR < 0.05 and FCH > 2) changing across time. CCL2 is a key chemokine associated with CD14+ monocytes; therefore, we investigated the differential isoform expression level of CCL2 transcripts (Figures 5G–5J). Four different CCL2 isoforms (CCL2–201 to −204) exhibited a temporal expression profile (Figure 5G). The isoform CCL2–201 was most abundant at the early-acute phase, whereas alternative isoforms had a much lower expression level at that time point (Figures 5G–5J). In contrast, isoform CCL2–203 was most abundantly expressed during convalescence.
A Multiscale Interaction Network Reveals a Roadmap of the Innate Immune Response to ZIKV Infection
To discover relationships between Luminex, CyTOF, and RNA-seq datasets during ZIKV infection, we integrated all data into a multiscale network analysis. We first performed co-expression clustering using weighted gene co-expression network analysis (WGCNA) of all 46 DENV-naive and 43 DENV-immune patient samples across all time points. Coexpression modules for the DENV-naive samples were strongly preserved in the DENV-immune samples, with a Zsummary > 10 for all modules (Langfelder et al., 2011). To increase the statistical power with a larger sample pool, we combined the DENV-naive and DENV-immune patients into a single WGCNA analysis. The WGCNA analysis of all pooled samples produced 10 coexpression network modules (coEMs) (Figure 6A), named after arbitrary colors. The association of the coEM profiles (defined through the eigenvectors of each module) with clinical variables showed a strong and significant association (FDR < 0.05) only between all coEMs and age (first column) and time point (second column) (Figure 6B).
Figure 6. Multiscale Network Analysis Highlights the Temporal Dynamics during the Innate Immune Response to ZIKV Infection and Identifies Key Markers at the Protein and RNA Level.
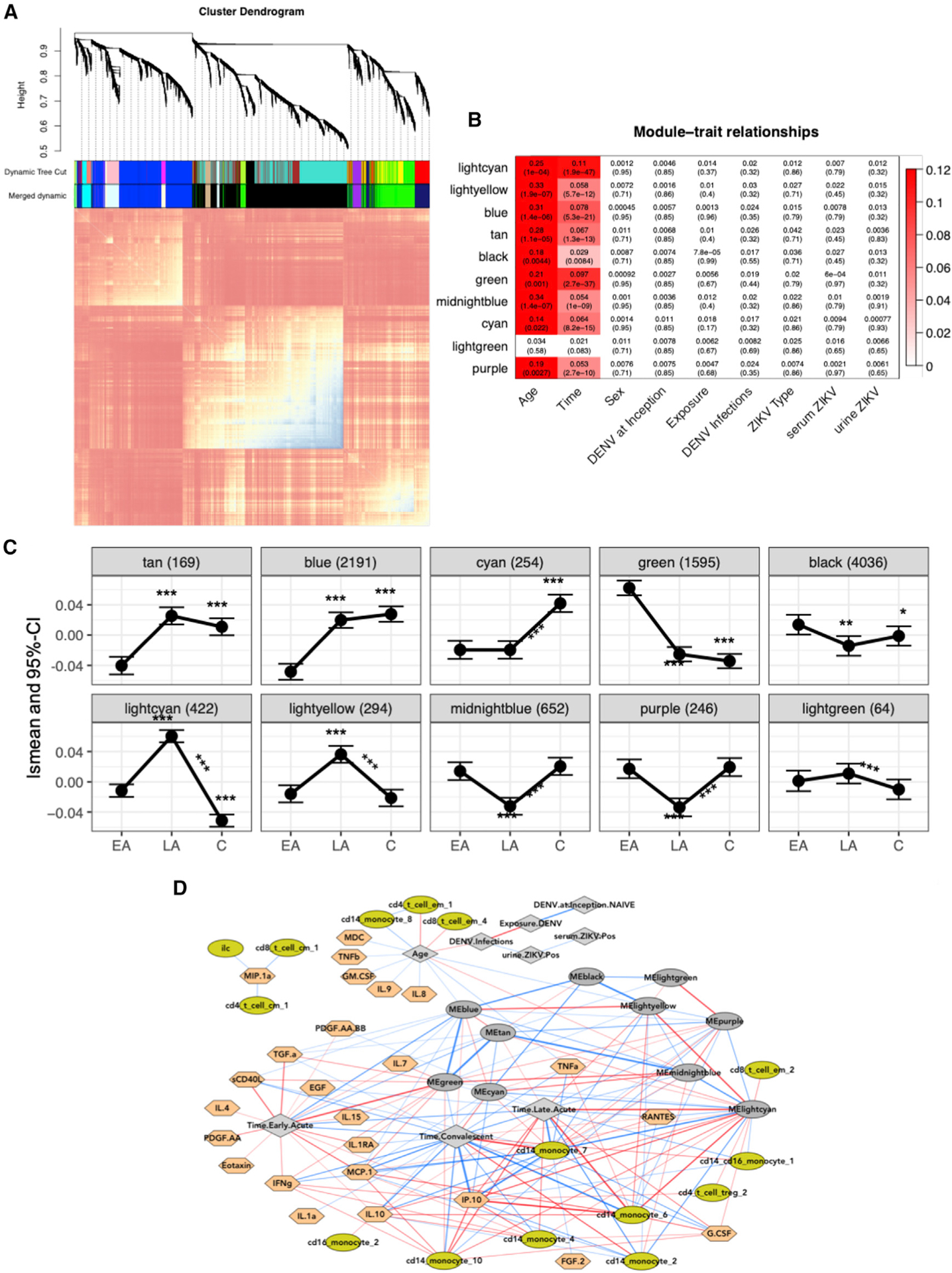
(A) Topological overlap matrix (TOM) plot of coexpression networks created from gene expression profiling of all samples across all time points. At the top, a dendrogram of the hierarchical clustering of the matrix that undergoes a dynamic tree-cut operation to form gene co-expression network modules (coEMs) is shown by the colored bars on the edges of the TOM plot (color assignment is arbitrary).
(B) Association between modules and important covariates by module. FDR-adjusted p values are displayed in parentheses. Colors represent the effect size of significant associations and are shown in the legend on the right (FDR < 0.05).
(C) Estimated lsmean and 95% CI are shown at each time point for each selected gene module. Asterisks above and below the CI bars represent significant changes from late-acute or convalescent to early-acute time points. Asterisks above the lines connecting the time points represent significant changes between the late-acute and convalescent time points. *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001.
(D) Multiscale network of all cell subcommunities, cytokines, and coEM eigengenes are depicted with a force-directed layout. Light green, cell subcommunities; dark gray oval, coEM eigengenes; gray diamond, clinical variables; peach, cytokines; red and blue lines (edges) are positive or negative correlations, respectively. Lines are filtered to FDR < 0.01, and the thickness of the line corresponds to the square of the correlations.
See also Table S7.
We then examined more closely the temporal dynamics of each coEM (Figure 6C). Transcripts in the light green module remained relatively unchanged across the three time points, but different temporal trends across time emerged for the other nine coEMs (tan, blue, cyan, green, black, light cyan, light yellow, midnight blue, and purple). The green coEM is of particular interest because it contains all transcripts with elevated expression at the early-acute time point and significant downregulation thereafter (Figure 6C). Genes in this module include OASL, STAT1, and STAT2, all of which have a protective role in the innate immune response (Table S7), as well as Toll-like receptor (TLR) genes and genes involved in TNF signaling, including TNFSF 10 and 13, as activity hubs. Of note, the tan coEM is enriched in ribosomal proteins. The light cyan and light yellow coEMs follow patterns for genes peaking in the late-acute phase (Figure 5E), with light cyan mostly containing genes involved in cell cycle, mitosis, and replication (Table S7) and light yellow containing basal transcription factors. In line with these two modules, genes in the cyan module were also involved in tissue repair, meiosis, and mitosis but were significantly greater only at the convalescent time point. The blue module contains genes that belong mostly to the zinc-finger antiviral protein family, which are important for RNA degradation and translation inhibition and are significantly greater at late-acute and convalescence compared with early-acute phase. The purple module contains DETs, such as YBX3 and SPTA1, that have been described as binding to flaviviruses. Along with the purple module, the midnight blue and black gene modules were also lower at the late-acute time point and returned to levels similar to the early-acute time point by convalescence. Among all temporal trends and within each time point, none of the coEMs had significantly different overall profiles between DENV-naive and DENV-immune patients (ANOVA FDR > 0.08).
Integrating cell subcommunity frequencies, cytokine profiles, and clinical outcomes with the network co-expression modules produced a global multiscale network (Figure 6D). This global network visualizes all interactions in this study, except CyTOF-CyTOF and Luminex-Luminex, in a compact force-directed layout. As seen previously (Figure S5B), CyTOF and Luminex measurements are positively correlated within each assay, thus the associated interactions were excluded from the network visual. However, in the top center of the network layout, aspects of the innate immune signature most strongly associated with time are drawn out of these clusters and toward the late-acute and early-acute nodes (gray diamonds with blue lines). These variables may be considered the most globally contrasting features of the ZIKV immune signature across time points. The secretion of inflammatory cytokines, including CCL2 (MCP-2), TGF-α, sCD40L, IL-1RA, IL-4, IL15, PDGF.AA, Eotaxin, and IFN-γ, were strongly correlated with the early-acute time point and the green gene module, which contains mainly ISGs (Figure 6D; Table S7). During the late-acute time point, 3 CD14+ monocyte populations identified during acute ZIKV infection by CyTOF (CD14+ monocytes 4, 6, and 10) were highly correlated with CXCL10 (IP-10) and CCL2 (MCP-2) and the light cyan, light yellow, and tan gene modules. This again verifies a central role of the monocyte-heavy signature during the acute phase of infection. In contrast, CD14+ monocytes 2 and 7 were highly correlated with the convalescent time point and were negatively correlated with the inflammatory chemokines CCL2 and CXCL10 and the green gene module (Figure 6D). Of note, CXCL10 (IP-10) and CCL2 (MCP1), as well as RANTES and IL-10, are the central nodes connecting cell population and gene expression variables. In the global multiscale network model, the interactions between DENV exposure and the measured immune signature variables are not strong enough to emerge as edges, and therefore, DENV immunity remains isolated at the top center of the layout, only connected to the rest of the network via the “Age” variable, thus confirming our finding that previous DENV exposure has a minimal role during acute, symptomatic ZIKV infection.
DISCUSSION
In this study, we comprehensively analyzed the innate immune profile of symptomatic ZIKV infection in children using Luminex, CyTOF, and RNA-seq on samples from patients who were either DENV-naive or DENV-immune, identified through our long-term cohort study in Nicaragua. Although other Zika studies have attempted to identify biomarkers based on a single methodology, this study provides a high-resolution view of the immune response to Zika in pediatric patients at three time points after infection that provides distinct molecular and cellular signatures. Based on analysis of all three datasets at each time point, we identified key players during acute ZIKV infection, including CD14+ monocytes, CD169, CCL2, CXCL10, and several ISGs. These results represent a highly comprehensive immune characterization of acute ZIKV infection in humans.
Because of the antigenic similarity between ZIKV and DENV, it has been hypothesized that prior immunity to DENV could affect subsequent Zika outcome, but it remains unclear how and to what extent. One study from our cohort in Nicaragua showed that prior DENV infection is protective against symptomatic ZIKV infection (Gordon et al., 2019), whereas another study in Brazil concluded that prior DENV infection reduced both ZIKV infection and disease risk (Rodriguez-Barraquer et al., 2019). Regarding the adaptive immune response, previous DENV immunity resulted in a more-rapid T cell response of greater magnitude after ZIKV infection (Grifoni et al., 2017) but only minimally influenced the breadth and magnitude of the post-ZIKV B cell and serum-neutralizing antibody response against ZIKV (Andrade et al., 2019; Collins et al., 2017). To determine the effect of prior DENV immunity on the innate immune response to acute ZIKV infection, we stratified our data into DENV-naive and DENV-immune Zika patients. Our results showed no differences between these groups based on the innate immune profile, suggesting that prior DENV infection has a minimal role in innate immunity to ZIKV infection.
Cytokines and chemokines are key players during viral infection and can be either detrimental or protective. Four studies in adults during the acute phase of ZIKV infection show strong upregulation of monocyte-associated chemokines (Foo et al., 2018; Kam et al., 2017; Lum et al., 2018b; Naveca et al., 2018), consistent with our findings, which also revealed a monocyte-associated cytokine profile. This is not surprising because monocytes are the primary targets of ZIKV infection in human blood (Michlmayr et al., 2017). Acute ZIKV infection resulted in high levels of chemokines, whereas interleukins were secreted at low levels. This suggests that the leukocyte recruitment axis is most affected during ZIKV infection, likely resulting in altered migration of immune cells into tissues for viral clearance. The secretion of cytokines followed four temporal patterns from early-to late-acute phase and to convalescence. Another study of early-acute ZIKV infection showed a similar cytokine/chemokine profile that was associated with viremia during Zika (Naveca et al., 2018).
We identified CCL2 and CXCL10 as key cytokines during acute, symptomatic ZIKV infection, both abundantly secreted by inflammatory CD14+ monocytes. High levels of CCL2 correspond with expansion of CD14+ and CD14+CD16+ monocytes. The CCL2-CCR2 axis is pivotal for monocyte recruitment to the brain and viral clearance during other flavivirus infections (Lim et al., 2011). Similar to our results, recent studies showed that CCL2 levels are high during the acute phase of ZIKV infection (Naveca et al., 2018). However, increased CCL2 levels have also been linked with severe DENV infection, including neurological complications and apoptosis of neuronal cells (de-Oliveira-Pinto et al., 2012; King et al., 2002). CCL2 was particularly high in ZIKV-infected pregnant women who delivered babies with congenital birth defects, and CCL2 was identified as a potential prognostic biomarker for neurological complications in infants (Kam et al., 2017). Different isoforms of CCL2 are secreted to varying degrees by different cells and have higher or lower affinity to the cognate chemokine receptor CCR2 (Carpenter et al., 2014; Leeman and Gilmore, 2008). Here, we identified four CCL2 isoforms, of which CCL2–201 was abundantly expressed and associated with early acute ZIKV infection. The functional implications of each CCL2 isoform in disease outcome are not yet well understood.
CXCL10 is an IFN-stimulated chemokine identified as a potential biomarker in several flaviviral studies (Kam et al., 2017; Naveca et al., 2018). CXCL10 binds to the chemokine receptor CXCR3 (Michlmayr and McKimmie, 2014) and was one of the top DETs in the acute phase of Zika in this study. Furthermore, CXCL10 was highly secreted during acute ZIKV infection and was correlated with acute-phase CD14+ monocytes (Foo et al., 2017; Michlmayr and McKimmie, 2014). High CXCL10 levels during the acute phase of ZIKV infection correlate with viremia and clinical symptoms of Zika, including neuronal damage in the developing baby (Kam et al., 2017; Naveca et al., 2018). High CXCL10 levels have been associated with severe manifestations of DENV and yellow fever virus infection (Chaudhary et al., 2017; Singh et al., 2017). Our results also demonstrate strong upregulation and secretion of CXCL10 during the acute phase of symptomatic ZIKV infection, as shown by Luminex and RNA-seq. However, we were unable to evaluate the role of CXCL10 or CCL2 in relation to disease severity because our study population experienced mild disease overall.
We found that certain CD14+ and CD14+CD16+ monocyte subsets are highly expanded during the acute phase of infection, in accordance with previous findings (Lum et al., 2018a; Michlmayr et al., 2017). We also show that in acute-phase, but not in convalescent-phase, CD14+ monocytes, CD169 is one of the top DETs and is also highly expressed on the surface of CD14+ monocytes. Thus, CD169 could potentially be a biomarker of acute ZIKV infection. CD169 is an interesting protein because it is critical for bridging the innate and adaptive immune responses. CD169 is an ISG, and levels of expression correlate with HIV viral load (Kim et al., 2015a). Additionally, murine studies with vaccinia virus have shown that CD169 has a pivotal role in priming adaptive immunity via cross-presentation of antigens to DCs, which, in turn, activate T cells (van Dinther et al., 2018). However, more studies are necessary to understand the functional role of CD169 on monocytes during acute ZIKV infection.
Most RNA-seq studies of ZIKV to date have been performed in vitro in murine and human cells. These studies show that ZIKV, and the closely related DENV, lead to temporal gene expression differences that underscore the highly dynamic nature of infection (Carlin et al., 2018; Popper et al., 2012). Here, we show striking differences in transcriptomic patterns by time point, including days 1–3 versus days 4–6 versus days 14–21, and we identify 25 transcripts that can serve to determine the stage of disease progression. At present, this cannot be defined clearly or accurately in clinical practice.
Many genes upregulated during the early-acute phase were involved in interferon pathways, pattern-recognition factors, and ISG expression, consistent with other RNA-seq studies of ZIKV-infected cells in vitro (Carlin et al., 2018; Lum et al., 2018a; Sun et al., 2017; Tiwari et al., 2017; Yi et al., 2017). Moreover, 2′−5′-oligoadenylate synthetase (OAS) and 2′−5′-oligoadenylate synthase-like protein (OASL) genes, among the top DEGs upregulated in RNA-seq, have been shown to reduce DENV and WNV replication, and polymorphisms in these genes in humans are associated with WNV encephalitis and dengue hemorrhagic fever/dengue shock syndrome (Lim et al., 2009; Lin et al., 2009). STAT-2, which was also elevated in our study, is involved in the IFN pathway and is specifically targeted by the ZIKV NS5 protein, which antagonizes IFN signaling and restricts the innate immune response (Chaudhary et al., 2017), analogous to DENV NS5 (Ashour et al., 2009; Grant et al., 2016). Interferon-induced transmembrane (IFITM), another well-characterized group of ISGs upregulated during ZIKV infection, restricts WNV infection and pathogenesis and prevents ZIKV replication and ZIKV-induced cell death (Gorman et al., 2016). IFIT1, 3, and 5 have an important role during many flavivirus infections and were in the top DETs in all our patients (Bowen et al., 2017; Hsu et al., 2013; Zhang et al., 2016). In particular, they have a beneficial role in DENV infection, preventing apoptosis and reducing DENV titers (Hsu et al., 2013). For ZIKV, high levels of gene expression of IFIT1 and 3 are important for controlling viral replication in vitro (Bowen et al., 2017). In summary, the ISGs that we identified have critical roles during many flavivirus infections, including ZIKV infection.
Top gene pathways enriched at the early-acute time point were associated with regulation of viral genome replication, type II IFN signaling pathways, apoptosis, IFN cellular response, regulation of IFN, and TLR signaling pathways. We created a multiscale network model that integrates changes in cell communities, cytokines, and gene transcript expression into a comprehensive map of innate immunity to ZIKV infection, leveraging modularity, rather than traditional interference testing. This network is valuable for the discovery of biomarkers for acute symptomatic ZIKV infection and improves our understanding of key players in the innate immune response in humans.
The strength of our study is that we comprehensively characterized ZIKV infection in humans in many children (n = 89) at two acute and one convalescent time point after onset of illness. One limitation of our study is that we were not able to stratify by severity of ZIKV infection; however, Zika is characterized by mild illness in children (Burger-Calderon et al., 2020), and our study population experienced insufficient severe symptoms to allow stratification by severity. Additionally, we could not stratify the DENV-exposed patients by serotype to determine whether DENV serotype affects clustering of RNA-seq, Luminex, and CyTOF data because this would have required many more patient samples. However, DENV serotype is unlikely to have a critical role because the comparison of DENV-naive and DENV-immune Zika patients did not reveal differences in the innate immune response. Furthermore, although we were not able to include markers of viral infection in our CyTOF panel to identify infected cell types during ZIKV infection and how infected cells change during acute illness, we have previously characterized ZIKV infection of peripheral blood mononuclear cells (PBMCs) in Nicaraguan patients (Michlmayr et al., 2017).
In conclusion, we anticipate that the comprehensive immune and molecular profiling of 89 pediatric Zika cases at three time points that we report here will be an important resource for interpreting future experiments of Zika biology in cultured cells and other systems by providing the context of immune profiling in human patients with acute symptomatic ZIKV infection. Our study answered a long-standing question in the field and definitively showed that prior DENV exposure does not modulate the innate immune response to ZIKV infection. Our immune profiling revealed an innate response largely centered on changes in CD14+ monocyte subpopulations and monocyte-related cytokines and genes. In particular, CD14+ monocytes during the acute but not convalescent phase all expressed CD169. Furthermore, we identified CCL2 and CXCL10 as key cytokines secreted during the early- and late-acute phase. Our RNA-seq analysis demonstrated significant upregulation of ISGs and other IFN pathway genes and revealed a clear temporal pattern of gene expression during ZIKV infection differentiating early-acute from late-acute from convalescent phases. Overall, these findings provide a unique global perspective on the biomolecular and immunological landscape of ZIKV infection and spark new hypotheses for future studies to further discern the mechanisms of ZIKV pathogenesis and immunity in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Eva Harris, PhD (eharris@berkeley.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the RNA-seq, CyTOF and Luminex data sets reported in this paper is Immport: SDY1476. The raw RNA-seq data is additionally deposited in “GeoOmnibus:GSE12982”. The code generated in this study is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
As part of the Pediatric Dengue Cohort Study (PDCS) in Managua, Nicaragua, blood and urine samples collected from a total of 46 DENV-naive and 43 previously DENV-infected (DENV-immune) children presenting to the study health center in July and August 2016 were included in this study. Information about sex, gender, and age for all study participants is shown in Table 1.
Samples were collected at three time points: early acute (day 1–3 post-symptom onset [p.s.o]), late acute (day 4–6 p.s.o) and convalescent (day 14–21 p.s.o) (Figure 1). We used PAXgene and plasma samples for RNA-seq and Luminex, respectively, for all three time points and PBMCs for CyTOF at the late-acute and convalescent time points, when they were available (Figure 1). Suspected Zika cases presented with rash, fever, or one or more of the following: conjunctivitis, arthralgia, myalgia, and/or peri-articular edema, regardless of fever. ZIKV infection was confirmed by real-time RT-PCR in acute-phase serum and/or urine samples performed at the National Virology Laboratory of the Ministry of Health in Managua using either of two triplex assays that simultaneously detect ZIKV, CHIKV and DENV infections: the ZCD assay (Waggoner et al., 2016a; 2016b) or the CDC Trioplex assay (Centers for Disease Control and Prevention (CDC), 2017), followed in some cases by Zika virus isolation. In addition, seroconversion by ZIKV IgM capture ELISA in paired acute and convalescent sera was evaluated (Balmaseda et al., 2018). Confirmed ZIKV-positive cases were classified as DENV-naive if they entered the cohort study with no detectable anti-DENV antibodies, as measured by DENV inhibition ELISA (IE) assay (Balmaseda et al., 2006; Katzelnick et al., 2017), and had no documented DENV infections (symptomatic or inapparent) during their time in the cohort. Confirmed ZIKV-positive cases were classified as DENV-immune if they entered the cohort study with no detectable anti-DENV antibodies and had ≥ 1 documented DENV infections during their time in the cohort or if they entered the cohort with detectable anti-DENV antibodies.
PBMCs were isolated from whole blood as described previously (Zompi et al., 2012). Sampling times closely adhered to the targeted late-acute (standard deviation [SD] = 0.5 days) and convalescent (SD = 0.6 days) timepoints. Clinical information was collected during medical consults and was digitized by double-data entry with quality control checks performed daily and weekly. This study was conducted as a collaboration between the Nicaraguan Ministry of Health and the University of California, Berkeley, and was reviewed and approved by the Institutional Review Boards (IRBs) of the University of California, Berkeley, and the Nicaraguan Ministry of Health. Parents or legal guardians of all subjects provided written informed consent, and subjects 6 years of age and older provided verbal assent as approved by the IRBs.
METHODS DETAILS
PBMC isolation
For PBMC preparation, blood samples were collected in Vacutainer tubes (Becton-Dickenson) with EDTA anticoagulant reagent. Upon receipt in the Nicaraguan National Virology Laboratory, 4–5 mL of blood was transferred into a Leucosep tube (Greiner Bio-One) containing 3 mL of Ficoll Histopaque (Sigma) and centrifuged at 500 g for 20 min at room temperature. The PBMC fraction was collected and transferred to a 15 mL conical tube containing 9 mL of PBS with 2% fetal bovine serum (FBS; Denville Scientific) and 1% penicillin/streptomycin (Sigma). Cells were washed three times in this solution by centrifugation at 500 g for 10 min and resuspended in 10 mL of complete medium. Before the third wash, an aliquot was used to obtain a cell count using a hematology analyzer (Sismex XS-1000i). After the third wash, cells were resuspended at a concentration of 107 cells per ml in freezing medium consisting of 90% FBS and 10% dimethyl sulfoxide, and aliquoted. Cryovials containing the cell suspension were placed in isopropanol containers (Mr. Frosty, Nalgene) at −80°C overnight and transferred to liquid nitrogen for storage.
CyTOF sample processing and acquisition
CyTOF uses metal-labeled reagents and inductively coupled plasma mass spectrometry to overcome the limits of fluorescence spectral overlap in flow cytometry, allowing measurement of up to 41 analytes at single-cell resolution. Cryopreserved PBMC samples from the late-acute and convalescent phases of infection were thawed and stained with Rhodium (Rh)103 nucleic acid intercalator (Fluidigm) as a viability marker. The viability of cells as determined by Rh103 staining and the count of cells per sample are displayed in Table S8. PBMC samples that showed viable cell frequency of < 30%, or those for which fewer than 50,000 events were recovered were excluded from downstream analyses. Paired PBMC samples from each time point were first barcoded using a CD45 antibody-based barcoding approach (Mei et al., 2016), and each early and late-acute and convalescent paired sample was pooled as a single patient sample for subsequent processing to minimize technical variability and potential batch effects. The pooled patient samples were then stained with a previously tested 37-marker CyTOF antibody panel for 30 min on ice. The samples were then fixed, permeabilized barcoded using commercial palladium (Pd) barcoding kits (Fluidigm) and pooled as sets of 20 samples. These pooled samples were then incubated with Iridium (Ir) nucleic acid intercalator (Fluidigm) in freshly diluted 2% formaldehyde. The samples were then stored at 4C in PBS containing 0.1% BSA until acquisition. Immediately prior to CyTOF acquisition, the samples were washed with deionized water (diH20), counted and resuspended in diH20 containing a 1/20 dilution of Equation 4 Element beads (Fluidigm). Following routine auto-tuning, the samples were acquired on a CyTOF2 mass cytometer (Fluidigm) equipped with a SuperSampler fluidics system (Victorian Airships) at an event rate of < 400 Hz. Each composite barcoded sample required approximately 20 h of acquisition time.
CyTOF data analysis
Following data acquisition, the FCS files were normalized using the bead-based normalization algorithm in the CyTOF control software. Cell events associated with specific patients were first debarcoded based on Pd barcodes using the Fluidigm software, and uploaded to Cytobank for initial data processing. Normalization beads were excluded based on Cerium (Ce)140 signal, and cell events were identified based on Ir191/193 DNA signal. A conservative doublet exclusion gate was applied based on DNA and event length, and Rh103+ dead cells were also excluded. The cell events associated with the late-acute and convalescent samples were then manually de-barcoded based on CD45–194Pt and CD45–198Pt expression, respectively, and were split and exported as separate samples for subsequent analyses using a semi-supervised computational analysis pipeline. Potential intra-individual batch effects and PVCA were carried out as previously described (Michlmayr et al., 2018). Traditional hierarchical gating was applied to a subset of 20 samples to identify 8 major immune compartments: T cells, B cells, NK cells, NKT cells, monocytes, mDCs, pDCs, and basophils. These manually gated data were used to train a logistic regression classifier (Nod label), which was then applied to identify these populations in all the samples. We tested the logistic regression model performance over the cell subsets, both by manually examining the prediction and determining the F1 score. In general, our logistic regression model showed acceptable performance for all cell subsets as measured by both precision/recall and F1 score, with larger subsets showing better performance due to the increased availability of training data. Next, we applied Phenograph (Levine et al., 2015) as an unbiased approach to define the phenotypic heterogeneity within each of these compartments (HybridLouvain). The cell clusters identified in each single sample were then meta-clustered across all samples to identify phenotypically similar communities that were reproducibly present across multiple samples (MetaHybridLouvain) (Michlmayr et al., 2018). These meta-clusters were then manually annotated based on overall marker expression profiles and their association with known immune cell subsets, allowing for the presence of additional phenotypically distinct sub-clusters within these known subsets. These annotations were mapped back to the individual samples, and the relative frequency and median marker expression patterns of these consistently annotated clusters were then exported for further statistical analyses. Meta-clusters that were characterized by protein expression patterns that did not correspond to any known cell subsets, including those that appeared to be cell-cell doublets, were annotated as “undefined” and not included in subsequent statistical or multiscale network analyses.
Multiplex ELISA
Cytokines and chemokines were measured using a multiplex ELISA-based assay (Luminex). Each sample was run in duplicate in a 96-well micro titer plate using 25 μL serum from each patient from early- and late-acute and convalescent time points using the multiplex cytokine panels (Multiplex High Sensitivity Human Cytokine Panel, Millipore Corp.). Forty analytes (cytokines and chemokines) were measured using a Luminex-200 system and the XMap Platform (Luminex Corporation). Acquired mean fluorescence data were analyzed and calculated by the xPONENT software. The lower and upper detection limits for these assays are 3.0pg/mL and 15,000 pg/mL, respectively. Quality control of each sample was performed, and a bead count of < 50 was not used for analysis. The experimental design for the Luminex assay was carried out using PlateDesigner (platedesigner.net) with samples from the same patient being randomly allocated to a plate and well, guaranteeing that if technical confounders exist they can properly be adjusted for in the analysis steps (Suprun and Suaréz Fariñas, 2019).
Preparation of RNA sequencing libraries
Total RNA was extracted from PAXgene RNA blood solution with the PAXgene Blood RNA Kit (QIAGEN) by following manufacturers’ instructions including DNase digestion and an additional clean-up using RNeasy MinElute kit (QIAGEN). Purified RNA samples were quantified by Qubit 3.0 fluorometer with RNA High Sensitivity Assay kit (Thermo-Fisher). We confirmed the quality of the RNA with the RNA High Sensitivity ScreenTape using the TapeStation 2200 (Agilent Technologies). Sample libraries were then prepared from the libraries of the 267 samples (from three time points of 89 paired patient samples). First, ribosomal RNA (rRNA) and globin mRNA were removed from 200 ng total RNA, and the remaining RNA was fragmented and primed for cDNA synthesis using TruSeq Total Stranded RNA HT kit with Ribo-Zero Globin on a Microlab STAR automated liquid handling system (Hamilton). The libraries were barcoded with TruSeq HT indices to allow for multiplexing, and ligation-mediated PCR was performed to enrich barcoded libraries for 15 cycles, then purified with the Agencourt AMPure XP beads system (Beckman Coulter). The libraries were assessed for quality with the high-sensitivity DNA chip in a TapeStation 2200 (Agilent) and quantified with KAPA Library Quantification Kits for Illumina platforms (Kapa Biosystems). The libraries were diluted to 2nM and combined equimolarly in pools of 12. These pools were then clustered using a cBot (Illumina) with a HiSeq 3000/4000 paired-end cluster kit on a patterned flow cell, one pool per lane. The flow cell was sequenced on a HiSeq4000 using a HiSeq 3000/4000 SBS kit (300 cycles, Illumina). Two technical replicates were sequenced per biological sample, for a total of 2 sequencing runs.
Pre-processing of RNA-seq data
Sequencer-generated base call (BCL) files were converted to FASTQ files, and the multiplexed samples were separated using bcl2fastq, which was then assessed for sequencing quality using FastQC (version 0.11.4, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The fastq files were quality filtered by using FASTAX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) with the invocation fastq quality filter -q 30 -p 50 -v -Q 33, and only the sequencing reads that met all quality control requirements were aligned to the latest human reference genome (GRCh38) using HISAT2 (version 2.0.4) (Kim et al., 2015b). SAMtools (version 0.1.19) was used to sort and convert the SAM files to BAM (Li et al., 2009). Aligned sequences were assembled into potential transcripts, and gene expression in units of FPKM was quantified using StringTie (version 1.2.2) (Pertea et al., 2015). To generate the count matrices for genes and transcripts of reads mapped to particular genomic features (genes and transcripts), Python script prepDE.py was used to extract the read count information directly from the files generated by StringTie.
Gene set enrichment analyses
The acute-convalescent and viral titer DET signatures were analyzed for enrichment of Gene Ontology (GO) biological process (The Gene Ontology Consortium, 2015), Panther (2016), and Reactome (2016) terms using the Enrichr platform (Chen et al., 2013; Fabregat et al., 2016; Mi et al., 2013). DETs were selected based on varying FDR and FCH thresholds to create sets of transcripts and mapped to unique gene symbols, which all produced qualitatively similar results for the top enriched terms; representative results for those DETs are presented in this study. Enrichr improves on the typical method of ranking term significance with one-sided Fisher’s exact tests by multiplying their log-scaled p values by a Z-score of the deviation from the expected rank for each term, which decreases the bias of the Fisher’s exact method toward terms with few gene assignments (Chen et al., 2013). Enrichment of WGCNA coEMs for terms from GO biological process (2015), Reactome (2016), and WikiPathways (2016) was similarly calculated using Enrichr without ranking or cutoffs (Kutmon et al., 2016). Enrichment of DET signatures within each coEM was calculated using one-sided Fisher’s exact tests and a Benjamini-Hochberg adjustment.
Differential expression analyses
For differential expression analysis at the transcript level we use R packages (version 3.4.3). Transcripts where pseudo-alignment counts in units of counts per million (CPM) were <1 in >10 samples were removed, and the remaining 58,650 transcripts were analyzed. Transcripts per million (TPM) values for transcript quantification and overlapping read counts for gene level quantification were normalized using the voom transformation, which models the variance based on abundance and heterogeneity of the samples and typically achieves better control over FDR than other RNA-seq methods (Law et al., 2014). Additionally, it converts the data to a linear, normally distributed scale allowing the use of classical linear models, including addition of covariates and extensions to models for longitudinal data. Expression profiles were modeled using mixed-effects models in the limma framework to account for the paired structure of the data. Limma uses an empirical Bayes moderation of the standard errors toward the prior transcript variances, which was fitted using an intensity-dependent trend. All models included age and gender as covariates. The following calculation was used for the mixed model to evaluate DEGs: (l Y~Age+Gender+Time). For the model evaluating changes by DENV exposure (see first result section), An interaction of exposure-by-time was also included in the model, but DENV exposure was not included in the model of Figure 5 since it did not show an effect. Of note, supplementary tables include the results of both models. p values from the moderated (paired) t test were adjusted for multiple hypotheses using the Benjamini-Hochberg (FDR-controlling) procedure. Pathway-based visualization of differentially expressed genes was performed with the pathview R package and KEGG annotations (Luo and Brouwer, 2013; Ogata et al., 1999). For alternative splicing (AS) analysis in ZIKV infection, the diffSplice function in limma was applied to the dataset using both F-test and Simes-correction (Hu et al., 2013).
Construction of gene co-expression networks and co-expression modules
Gene co-expression networks were constructed from the gene-level expression data for all samples using the WGCNA (version 1.51) and coexpp (version 0.1.0, https://bitbucket.org/multiscale/coexpp) R packages (Zhang and Horvath, 2005). WGCNA leverages natural variance in expression between sampled individuals and time points to build a network structure from the Pearson correlations for all gene-gene pairs (Zhang and Horvath, 2005). WGCNA converts the gene-gene correlation matrix into an adjacency matrix using a power function that optimizes for scale-free topology, and adjacencies are then transformed into a topological overlap matrix (TOM) that represents normalized counts of neighbors that are shared between the nodes on either side of each edge. Genes were grouped using average-linkage hierarchical clustering of the TOM, followed by a dynamic cut-tree algorithm that divides the dendrogram branches into gene coexpression network modules (coEMs) (Langfelder and Horvath, 2008). coexpp is a specialized implementation of WGCNA that optimizes memory and multicore usage. Gene expression data were preprocessed for WGCNA by applying a log2 transformation to the FPKM quantification and removing the lowest-variance quartile of genes. Relationships among coEMs and the other data were evaluated using eigengenes (the first principal component of each coEM), calculating the Pearson correlations for all possible pairings of the coEM eigengenes, clinical variables, and cell subpopulations (Langfelder and Horvath, 2007). Network layout was performed using the ForceAtlas2 algorithm in Gephi (version 0.9.1) followed by visualization in Cytoscape (version 3.4.0) (Bastian and Heymann, 2009; Smoot et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
R (R-project.org) version 3.5.0 and available packages were used for all analyses. Luminex data were analyzed as log2 of the protein concentration obtained from the standard curve interpolation. IL-13 was eliminated because the value was under LOD for >90% of the samples. CyTOF population frequencies were log10 transformed prior to analysis. Batch effect for Luminex and CyTOF were visually ascertained using Principal component analysis (PCA) and the percentage (%) of variance explained by the batch was estimated by Principal variation Component analysis (PVCA). Luminex and CyTOF data were adjusted for each batch using a linear model with factors sex, gender, time and DENV pre-exposure status before ZIKV infection. Changes in laboratory markers, cell population frequency, marker intensity within each cell subpopulation and protein expression were modeled using linear mixed-effect models. The models considered fixed effects gender, age, and time, and when modeling by prior DENV exposure, also time by DENV exposure interaction, a random intercept for each patient. Hypotheses of interest were tested from the fitted model using contrasts and adjustment for multiple hypotheses across markers within each technology were carried out using the Benjamini-Hochberg procedure, which controls the FDR.
Visualization of small correlation matrices was performed with the corrplot R package. Quantitative measures of external cluster validity were calculated susing the clusterCrit R package (version 1.2.7). Dendogram analysis was conducted using the Dendextend R package. Statistical significance, error bars, heatmap scale bars, confidence intervals, and the exact value of n for each dataset are described in each Figure Legend and the experimental approach is explained in the Methods Details section.
Statistical analyses of all datasets
For all 89 patient samples quantitative measures of external cluster validity were calculated using the clusterCrit R package (version 1.2.7). Elastic net regularized regression, which fits a logistic regression model with L1 and L2 penalties (the elastic net penalty), was performed with the glmnet R package (version 2.0–5) (Friedman et al., 2010). Elastic net hyper-parameters α and λ were both selected empirically per model by a grid search that maximized AUC under five-fold nested cross validation. 100 bootstrap resampling runs were used to estimate the 95% confidence interval for the AUC.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Antibodies used for CyTOF are summarized in Table S2 | ||
| Anti-human CCR5 (clone NP-6G4) | Fluidigm | Cat# 3156015A; RRID:AB_2661814 |
| Anti-human SIGLEC1(clone 7–239) | Biolegend | Cat# 346002; RRID:AB_2189031 |
| Anti-human CXCR3 (cloneG025H7) | Biolegend | Cat# 353702; RRID:AB_353702 |
| Anti-human CD45RA (clone HI130) | Biolegend | Cat# 304002; RRID:AB_2661811 |
| Anti-human Eotaxin | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human Flt.3L | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL5 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human MCP1 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD19 (clone HIB19) | Biolegend | Cat# 302202; RRID:AB_2661817 |
| Anti-humanBDCA1 (clone L161) | Biolegend | Cat# 331502; RRID:AB_2661820 |
| Anti-humanPD1(clone EH12.2H7) | Biolegend | Cat# 329902; RRID:AB_940488 |
| Anti-human CD24 (clone ML5) | Biolegend | Cat# 311102; RRID:AB_314851 |
| Anti-human CCR4 (clone 205410) | R&D Systems | Cat# MAB1567; RRID:AB_2074395 |
| Anti-human GCSF | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human MCP3 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL1A | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human TNFa | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human VEGF | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD11c (clone Bu15) | Biolegend | Cat# 301616; RRID:AB_439736 |
| Anti-human CD45RA (clone HI100) | Biolegend | Cat# 304102; RRID:AB_314406 |
| Anti-human CD127 (clone A019D5) | Biolegend | Cat# 351302; RRID:AB_10718513 |
| Anti-human CEACAM8 (clone G10F5) | Biolegend | Cat# 305102; RRID:AB_2661823 |
| Anti-human IFNg | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL12p70 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL1RA | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL9 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL4 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IP10 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD56 (clone B159) | BD Biosciences | Cat# 555513; RRID:AB_2661829 |
| Anti-human CD161(clone HP-3G10) | Biolegend | Cat# 339902; RRID:AB_2661837 |
| Anti-human EGF | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human TGFa | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human GRO | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human MDC | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL13 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL6 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human MIP1a | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human MIP1b | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human TNFb | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD4 (clone RPA-T4) | Biolegend | Cat# 300502; RRID:AB_314070 |
| Anti-human CD8 (clone RPA-T8) | Biolegend | Cat# 301002; RRID:AB_2661818 |
| Anti-human CXCR5 (clone J252D4) | Biolegend | Cat# 356902; RRID:AB_2561811 |
| Anti-human CD38 (clone HB-7) | Biolegend | Cat# 356602; RRID:AB_2661836 |
| Anti-human IFNa | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human sCD40L | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human RANTES | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD27 (clone O323) | Biolegend | Cat# 302802; RRID:AB_2661825 |
| Anti-human CCR6 (clone G034E3) | Biolegend | Cat# 353402; RRID:AB_10918625 |
| Anti-human Fractalkine | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL10 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL12p40 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human PDGF AA BB | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL17 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL8 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human THBD (clone M80) | Biolegend | Cat# 344102; RRID:AB_2201808 |
| Anti-human IL2RA (clone M-A251) | Biolegend | Cat# 356102; RRID:AB_2661833 |
| Anti-human CD3 (clone UCHT1) | Biolegend | Cat# 300402; RRID:AB_2661835- |
| Anti-human CXCR3 (clone 2A9–1) | Biolegend | Cat# 341602; RRID:AB_1595422 |
| Anti-human AXL (clone 108724) | R&D Systems | Cat# MAB154; RRID:AB_2062558 |
| Anti-human GMCSF | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human PDGF AA | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human CD57 (clone HCD57) | Biolegend | Cat# 322302; RRID:AB_2661815 |
| Anti-human CD20 (clone 2H7) | Fluidigm | Cat# 3147001B |
| Anti-human CD123 (clone 6H6) | Biolegend | Cat# 306002; RRID:AB_2661822 |
| Anti-human FGF2 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL15 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL1b | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL2 | Millipore Corp | Cat# HSTCMAG-28SK |
| Anti-human IL7 | Millipore Corp | Cat# HSTCMAG-28SK |
| Biological Samples | ||
| PAXgene samples from humans | Dengue pediatric cohort study in Nicaragua | Accession number: SDY1476 https://www.immport.org/home |
| Serum samples from humans | Dengue pediatric cohort study in Nicaragua | Accession number: SDY1476 https://www.immport.org/home |
| PBMCs samples from humans | Dengue pediatric cohort study in Nicaragua | Accession number: SDY1476 https://www.immport.org/home |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Histopaque-1077 | Sigma | Cat# 10771 |
| Fetal bovine serum Premium US source | Thomas scientific | Cat# 1005837 |
| Phosphate buffered saline (PBS) GIBCO | Sigma | Cat# P5119 |
| 10,000 U Penicillin-Streptomycin | Sigma | Cat# P4333 |
| Dimethyl sulfoxide | Sigma | Cat# C6164 |
| Cell-ID Intercalator-Rh—500 μM | Fluidigm | Cat# 201103A |
| EDTA | Sigma | Cat# E6758 |
| Cell-ID Intercalator-Ir—125 μM | Fluidigm | Cat# 201192A |
| EQ Four Element Calibration Beads— 100 mL | Fluidigm | Cat# 201078 |
| Formaldehyde solution | Sigma-Aldrich | Cat# 252549 |
| Molecular grade water | Thermo Fisher | Cat# Am9935 |
| RPMI-1640 | Sigma | Cat# R7388 |
| Benzonase Nuclease | Millipore | Cat# E1014 |
| Critical Commercial Assays | ||
| MILLIPLEX MAP Human High Sensitivity Cytokine Panel | Millipore Corp | HSTCMAG-28SK |
| TrueSeq RNA CD Index Plate (96 Indexes, 96 samples) | Illumina | Cat# 20019792 |
| TruSeq Total Stranded RNA HT kit (w/Ribo-Zero Globin) | Illumina | Cat# 20020612 |
| Paxgene Blood RNA Kit | QIAGEN | Cat# 762164 |
| RNeasy MinElute Cleanup kit | QIAGEN | Cat# 74204 |
| Qubit RNA High Sensitivity Assay kit | Thermo Fisher | Cat# Q32852 |
| RNA ScreenTapes | Agilent | Cat# 5067-5576 |
| D1000 ScreenTapes | Agilent | Cat# 5067-5582 |
| Superscript II | Invitrogen | Cat# 18-064-022 |
| KAPA Library Quantification Kit Illumina | Kapa Biosystems | Cat# 79960298001 |
| HiSeq 3000/4000 paired-end cluster kit | Illumina | Cat# PE410-1001 |
| HiSeq 3000/4000 SBS kit | Illumina | Cat# FC410-1003 |
| Agencourt AMPure XP beads system | Beckman Coulter | Cat# A63881 |
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | Cat# 201060 |
| MAXPAR X8 Multimetal labeling kit | Fuidigm | Cat# 201300 |
| Deposited Data | ||
| RNA-seq transcript and gene data | This study | https://www.ncbi.nlm.nih.gov/geo Accession number: GEO: GSE12982 and https://www.immport.org/home “Immport: SDY1476” |
| Luminex data | This study | https://www.immport.org/home “Immport: SDY1476” |
| CyTOF data | This study | https://www.immport.org/home “Immport : SDY1476” https://flowrepository.org/: FR-FCM-Z2HQ |
| Software and Algorithms | ||
| CyTOF analysis | Cytobank | https://www.cytobank.org/ |
| XMap Platform | Luminex Corporation | https://www.luminexcorp.com/xmap-technology |
| PlateDesigner | Suprun and Suaréz Fariñas, 2019 | platedesigner.net |
| xPONENT Multiplex analysis software | Luminex corporation | https://www.luminexcorp.com/xponent/ |
| RNA -preprocessing reads FASTQC | FASTQC | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| RNA-preprocessing tool FASTAX Toolkit | Kim et al., 2015a | (http://hannonlab.cshl.edu/fastx_toolkit/ version 0.11.4 |
| HISAT 2 | Kim et al., 2015b | http://daehwankimlab.github.io/hisat2/ (version 2.0.4) |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ (version 0.1.19) |
| StringTie | Pertea et al., 2015 | https://ccb.jhu.edu/software/stringtie/ (version 1.2.2) |
| Python | prepDE.py | https://www.python.org/ |
| EnrichR platform | Chen et al., 2013; Fabregat et al., 2016; Mi et al., 2013) | https://amp.pharm.mssm.edu/Enrichr/ |
| Gene Ontology (GO) biological process | The Gene Ontology Consortium, 2019; Ashburner et al., 2000 | http://geneontology.org/ |
| Panther | Panther, 2016 | http://www.pantherdb.org/ |
| Reactome | Reactome, 2016 | https://reactome.org/ |
| WikiPathways | WikiPathways, 2016 | https://www.wikipathways.org/index.php/WikiPathways |
| R studio | R project | https://www.r-project.org/ (version 3.5.0) |
| voom | Law et al., 2014 | https://rdrr.io/bioc/limma/man/voom.html |
| KEGG | Luo and Brouwer, 2013; Ogata et al., 1999 | https://www.genome.jp/kegg/annotation/ |
| limma | Smyth et al., 2005 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| WGCNA | Zhang and Horvath, 2005 | https://cran.r-project.org/web/packages/WGCNA/index.html (version 1.51) |
| coexpp | Zhang and Horvath, 2005 | (version 0.1.0, https://bitbucket.org/multiscale/coexpp) |
| ForceAtlas2 algorithm | Bastian and Heymann, 2009, Int. Assoc. Adv. Artif. Intell. Conf. Weblogs Soc. Media, conference | https://github.com/gephi/gephi/wiki/Force-Atlas-2 Gephi (version 0.9.1) |
| Cytoscape | Bastian and Heymann, 2009, Int. Assoc. Adv. Artif. Intell. Conf. Weblogs Soc. Media, conference; Smoot et al., 2011 | https://cytoscape.org/ (version 3.4.0) |
| corrplot | R-project | https://cran.r-project.org/web/packages/corrplot/vignettes/corrplot-intro.html |
| clusterCrit R | R-project | https://cran.r-project.org/web/packages/clusterCrit/index.html (version 1.2.7). |
| Dendextend R package | R-project | https://cran.r-project.org/web/packages/dendextend/index.html |
| glmnet R package | Friedman et al., 2010 | https://cran.r-project.org/web/packages/glmnet/index.html (version 2.0-5) |
Highlights.
CD14+ monocytes play a central role during ZIKV infection and express CD169
CD169 is a biomarker of acute ZIKV infection and correlates with CXCL10 levels
ZIKV infection leads to distinct temporal patterns of gene and protein expression
Pre-existing dengue immunity has minimal impact on the innate immune response to Zika
ACKNOWLEDGMENTS
This work was supported by grants U19AI118610 (E.H.) and P01AI106695 (E.H.) from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH). We thank Michael Stewart for help with RNA isolation and sequencing and Theodore Pak and Li Wang for their constructive comments on the manuscript and data analysis. We also thank study personnel at the Centro de Salud Sócrates Flores Vivas, the National Virology Laboratory of the Nicaraguan Ministry of Health, and the Sustainable Sciences Institute in Managua, Nicaragua. Finally, we are grateful to the study participants and their families.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107569.
DECLARATION OF INTEREST
The authors declare no competing interests.
REFERENCES
- Andrade P, Gimblet-Ochieng C, Modirian F, Collins M, Cárdenas M, Katzelnick LC, Montoya M, Michlmayr D, Kuan G, Balmaseda A, et al. (2019). Impact of pre-existing dengue immunity on human antibody and memory B cell responses to Zika. Nat. Commun 10, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour J, Laurent-Rolle M, Shi P-Y, and García-Sastre A (2009). NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol 83, 5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmaseda A, Hammond SN, Tellez Y, Imhoff L, Rodriguez Y, Saborío SI, Mercado JC, Perez L, Videa E, Almanza E, et al. (2006). High seroprevalence of antibodies against dengue virus in a prospective study of schoolchildren in Managua, Nicaragua. Trop. Med. Int. Health 11, 935–942. [DOI] [PubMed] [Google Scholar]
- Balmaseda A, Zambrana JV, Collado D, García N, Saborío S, Elizondo D, Mercado JC, Gonzalez K, Cerpas C, Nuñez A, et al. (2018). Comparison of four serological methods and two reverse transcription-PCR assays for diagnosis and surveillance of Zika virus infection. J. Clin. Microbiol 56, e01785–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba-Spaeth G, Dejnirattisai W, Rouvinski A, Vaney M-C, Medits I, Sharma A, Simon-Lorière E, Sakuntabhai A, Cao-Lormeau V-M, Haouz A, et al. (2016). Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 536, 48–53. [DOI] [PubMed] [Google Scholar]
- Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, Charo IF, Lu W, Pletnev AG, and Lim JK (2015). Differential roles of chemokines CCL2 and CCL7 in monocytosis and leukocyte migration during West Nile virus infection. J. Immunol 195, 4306–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian Mathieu, and Heymann Sebastien (2009). Gephi: An Open Source Software for Exploring and Manipulating Networks. Proceedings of the Third International ICWSM Conference. https://gephi.org/publications/gephi-bastian-feb09.pdf. [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, and Suthar MS (2017). Zika virus antagonizes type i interferon responses during infection of human dendritic cells. PLoS Pathog. 13, e1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L (2001). Random Forests. Machine Learning 45, 5–32. https://link.springer.com/content/pdf/10.1023/A:1010933404324.pdf. [Google Scholar]
- Burger-Calderon R, Bustos Carrillo F, Gresh L, Ojeda S, Sanchez N, Plazaola M, Katzelnick L, Mercado BL, Monterrey JC, Elizondo D, et al. (2020). Age-dependent manifestations and case definitions of paediatric Zika: a prospective cohort study. Lancet Infect. Dis 20, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin AF, Vizcarra EA, Branche E, Viramontes KM, Suarez-Amaran L, Ley K, Heinz S, Benner C, Shresta S, and Glass CK (2018). Deconvolution of pro- and antiviral genomic responses in Zika virus-infected and bystander macrophages. Proc. Natl. Acad. Sci. USA 115, E9172–E9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Ricci EP, Mercier BC, Moore MJ, and Fitzgerald KA (2014). Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol 14, 361–376. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) (2017). Trioplex Real-time RT-PCR Assay. https://www.cdc.gov/zika/pdfs/trioplex-real-time-rt-pcr-assay-instructions-for-use.pdf.
- Chaudhary V, Yuen K-S, Chan JF-W, Chan C-P, Wang P-H, Cai J-P, Zhang S, Liang M, Kok K-H, Chan C-P, et al. (2017). Selective activation of type II interferon- signaling by Zika virus NS5 protein. J. Virol 91, e00163–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MH, McGowan E, Jadi R, Young E, Lopez CA, Baric RS, Lazear HM, and de Silva AM (2017). Lack of durable cross-neutralizing antibodies against Zika virus from Dengue virus infection. Emerg. Infect. Dis 23, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de-Oliveira-Pinto LM, Gandini M, Freitas LP, Siqueira MM, Marinho CF, Setúbal S, Kubelka CF, Cruz OG, and Oliveira SA (2012). Profile of circulating levels of IL-1Ra, CXCL10/IP-10, CCL4/MIP-1b and CCL2/ MCP-1 in dengue fever and parvovirosis. Mem. Inst. Oswaldo Cruz 107, 48–56. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, et al. (2016). The reactome pathway knowledgebase. Nucleic Acids Res. 44 (D1), D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo S-S, Chen W, Chan Y, Bowman JW, Chang L-C, Choi Y, Yoo JS, Ge J, Cheng G, Bonnin A, et al. (2017). Asian Zika virus strains target CD14+ blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat. Microbiol 2, 1558–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo S-S, Chen W, Chan Y, Lee W-S, Lee S-A, Cheng G, Nielsen-Saines K, Brasil P, and Jung JU (2018). Biomarkers and immunoprofiles associated with fetal abnormalities of ZIKV-positive pregnancies. JCI Insight 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, and Tibshirani R (2010). Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Gresh L, Ojeda S, Katzelnick LC, Sanchez N, Mercado JC, Chowell G, Lopez B, Elizondo D, Coloma J, et al. (2019). Prior dengue virus infection and risk of Zika: A pediatric cohort in Nicaragua. PLoS Med. 16, e1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman MJ, Poddar S, Farzan M, and Diamond MS (2016). The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J. Virol 90, 8212–8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, and García-Sastre A (2016). Zika virus targets human STAT2 to inhibit Type I interferon signaling. Cell Host Microbe 19, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A, Pham J, Sidney J, O’Rourke PH, Paul S, Peters B, Martini SR, de Silva AD, Ricciardi MJ, Magnani DM, et al. (2017). Prior Dengue virus exposure shapes T cell immunity to zika virus in humans. J. Virol 91, 601–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y-L, Shi S-F, Wu W-L, Ho L-J, and Lai J-H (2013). Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PLoS ONE 8, e79518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Huang Y, Du Y, Orellana CF, Singh D, Johnson AR, Monroy A, Kuan P-F, Hammond SM, Makowski L, et al. (2013). DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 41, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Weidner JM, Qing M, Pan X-B, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi P-Y, et al. (2010). Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J. Virol 84, 8332–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam Y-W, Leite JA, Lum F-M, Tan JJL, Lee B, Judice CC, Teixeira DAT, Andreata-Santos R, Vinolo MA, Angerami R, et al. ; Zika-Unicamp Network (2017). Specific biomarkers associated with neurological complications and congenital central nervous system abnormalities from Zika virus-infected patients in Brazil. J. Infect. Dis 216, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, Balmaseda A, and Harris E (2017). Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W-K, McGary CM, Holder GE, Filipowicz AR, Kim MM, Beydoun HA, Cai Y, Liu X, Sugimoto C, and Kuroda MJ (2015a). Increased expression of CD169 on blood monocytes and its regulation by virus and CD8 T cells in macaque models of HIV infection and AIDS. AIDS Res. Hum. Retroviruses 31, 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015b). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CA, Anderson R, and Marshall JS (2002). Dengue virus selectively induces human mast cell chemokine production. J. Virol 76, 8408–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutmon M, Riutta A, Nunes N, Hanspers K, Willighagen EL, Bohler A, Mélius J, Waagmeester A, Sinha SR, Miller R, et al. (2016). WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res. 44 (D1), D488–D494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2007). Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, and Horvath S (2011). Is my network module preserved and reproducible? PLoS Comput. Biol 7, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman JR, and Gilmore TD (2008). Alternative splicing in the NF-kB signaling pathway. Gene 423, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, Johnson H, Pape J, Foster GA, Krysztof D, et al. (2009). Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 5, e1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, and Murphy PM (2011). Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J. Immunol 186, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Yu HP, Chang BL, Tang WC, Liao CL, and Lin YL (2009). Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against Dengue virus infection. J. Immunol 183, 8035–8043. [DOI] [PubMed] [Google Scholar]
- Lum F-M, Lee D, Chua T-K, Tan JJL, Lee CYP, Liu X, Fang Y, Lee B, Yee W-X, Rickett NY, et al. (2018a). Zika virus infection preferentially counterbalances human peripheral monocyte and/or NK cell activity. MSphere 3, e00120–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum F-M, Lye DCB, Tan JJL, Lee B, Chia P-Y, Chua T-K, Amrun SN, Kam Y-W, Yee W-X, Ling W-P, et al. (2018b). Longitudinal study of cellular and systemic cytokine signatures to define the dynamics of a balanced immune environment during disease manifestation in Zika virus-infected patients. J. Infect. Dis 218, 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, and Brouwer C (2013). Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol 12, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei HE, Leipold MD, and Maecker HT (2016). Platinum-conjugated antibodies for application in mass cytometry. Cytometry A 89, 292–300. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, and Thomas PD (2013). PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlmayr D, Andrade P, Gonzalez K, Balmaseda A, and Harris E (2017). CD14+CD16+ monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol 2, 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlmayr D, and McKimmie CS (2014). Role of CXCL10 in central nervous system inflammation. Int. J. Interferon, Cytokine, Mediat. Res 6, 1–18. [Google Scholar]
- Michlmayr D, Pak TR, Rahman AH, Amir ED, Kim EY, Kim-Schulze S, Suprun M, Stewart MG, Thomas GP, Balmaseda A, et al. (2018). Comprehensive innate immune profiling of chikungunya virus infection in pediatric cases. Mol. Syst. Biol 14, e7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveca FG, Pontes GS, Chang AY-H, Silva GAVD, Nascimento VAD, Monteiro DCDS, Silva MSD, Abdalla LF, Santos JHA, Almeida TAP, et al. (2018). Analysis of the immunological biomarker profile during acute Zika virus infection reveals the overexpression of CXCL10, a chemokine linked to neuronal damage. Mem. Inst. Oswaldo Cruz 113, e170542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, and Kanehisa M (1999). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popper SJ, Gordon A, Liu M, Balmaseda A, Harris E, and Relman DA (2012). Temporal dynamics of the transcriptional response to dengue virus infection in Nicaraguan children. PLoS Negl. Trop. Dis 6, e1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, et al. (2016). Zika virus infects human placental macrophages. Cell Host Microbe 20, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Barraquer I, Costa F, Nascimento EJM, Nery N, Castanha PMS, Sacramento GA, Cruz J, Carvalho M, De Olivera D, Hagan JE, et al. (2019). Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363, 607–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MA, and Harris E (2014). Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog. 10, e1004541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Guest J-M, Kanwar M, Boss J, Gao N, Juzych MS, Abrams GW, Yu F-S, and Kumar A (2017). Zika virus infects cells lining the blood-retinal barrier and causes chorioretinal atrophy in mouse eyes. JCI Insight 2, e92340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang P-L, and Ideker T (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G, Gentleman R, Carey V, Dudoit S, Irizarry R, and Hubert W (2005). Limma: linear models for microarray data In Bioinformatics and Computational Biology Solutions using R and Bioconductor, First Edition, Gentleman R, Carey V, Huber W, Irizarry R, and Dudoit S, eds. (Springer; ), pp. 397–420. [Google Scholar]
- Sun X, Hua S, Chen H-R, Ouyang Z, Einkauf K, Tse S, Ard K, Ciara-nello A, Yawetz S, Sax P, et al. (2017). Transcriptional Changes during Naturally Acquired Zika Virus Infection Render Dendritic Cells Highly Conducive to Viral Replication. Cell Rep. 21, 3471–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprun M, and Suaréz Fariñas M (2019). PlateDesigner: a web-based application for the design of microplate experiments. Bioinformatics 35, 1605–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium (2019). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari SK, Dang J, Qin Y, Lichinchi G, Bansal V, and Rana TM (2017). Zika virus infection reprograms global transcription of host cells to allow sustained infection. Emerg. Microbes Infect 6, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinther D, Veninga H, Iborra S, Borg EGF, Hoogterp L, Olesek K, Beijer MR, Schetters STT, Kalay H, Garcia-Vallejo JJ, et al. (2018). Functional CD169 on macrophages mediates interaction with dendritic cells for CD8+ T cell cross-priming. Cell Rep. 22, 1484–1495. [DOI] [PubMed] [Google Scholar]
- Waggoner JJ, Gresh L, Mohamed-Hadley A, Ballesteros G, Davila MJV, Tellez Y, Sahoo MK, Balmaseda A, Harris E, and Pinsky BA (2016a). Single-reaction multiplex reverse transcription PCR for detection of Zika, Chikungunya, and Dengue viruses. Emerg. Infect. Dis 22, 1295–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JJ, Gresh L, Vargas MJ, Ballesteros G, Tellez Y, Soda KJ, Sahoo MK, Nuñez A, Balmaseda A, Harris E, and Pinsky BA (2016b). Viremia and clinical presentation in Nicaraguan patients infected with Zika virus, Chikungunya virus, and Dengue virus. Clin. Infect. Dis 63, 1584–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L, Pimentel H, and Pachter L (2017). Zika infection of neural progenitor cells perturbs transcription in neurodevelopmental pathways. PLoS ONE 12, e0175744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, article 17. [DOI] [PubMed] [Google Scholar]
- Zhang J, Sze DM-Y, Yung BY-M, Tang P, Chen W-J, Chan K-H, and Leung PH-M (2016). Distinct expression of interferon-induced protein with tetratricopeptide repeats (IFIT) 1/2/3 and other antiviral genes between subsets of dendritic cells induced by dengue virus 2 infection. Immunology 148, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJM, Liu YJ, MacPherson G, Randolph GJ, et al. (2010). Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–e80. [DOI] [PubMed] [Google Scholar]
- Zompi S, Montoya M, Pohl MO, Balmaseda A, and Harris E (2012). Dominant cross-reactive B cell response during secondary acute dengue virus infection in humans. PLoS Negl. Trop. Dis 6, e1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO) Zika Virus and Complications: 2016. Public Health Emergency of International Concern. (WHO Press), https://www.who.int/emergencies/zika-virus/en/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq, CyTOF and Luminex data sets reported in this paper is Immport: SDY1476. The raw RNA-seq data is additionally deposited in “GeoOmnibus:GSE12982”. The code generated in this study is available upon request.