

Abstract
Background and Aims:
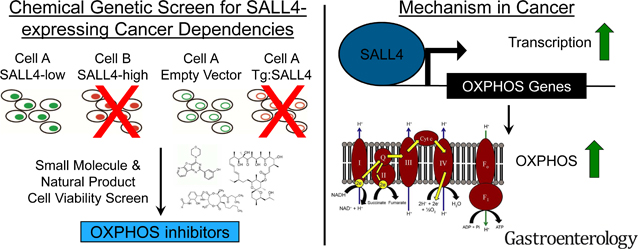
Some oncogenes encode transcription factors, but few drugs have been successfully developed to block their activity specifically in cancer cells. The transcription factor SALL4 is aberrantly expressed in solid tumor and leukemia cells. We developed a screen to identify compounds that reduce the viability of liver cancer cells that express high levels of SALL4 and we investigated their mechanisms.
Methods:
We developed a stringent high-throughput screening platform comprising unmodified SNU-387 and SNU-398 liver cancer cell lines and SNU-387 cell lines engineered to express low and high levels of SALL4. We screened 1597 pharmacologically active small molecules and 21,575 natural product extracts from plant, bacteria, and fungal sources for those that selectively reduce the viability of cells with high levels of SALL4 (SALL4hi cells). We compared gene expression patterns of SALL4hi cells vs SALL4-knockdown cells using RNA-seq and real-time PCR analyses. Xenograft tumors were grown in NOD/SCID gamma mice from SALL4hi SNU-398 or HCC26.1 cells or from SALL4lo PDX cells; mice were given injections of identified compounds or sorafenib and the effects on tumor growth were measured.
Results:
Our screen identified 1 small molecule (PI-103) and 4 natural compound analogues (oligomycin, efrapeptin, antimycin, and leucinostatin) that selectively reduced viability of SALL4hi cells. We performed validation studies, and 4 of these compounds were found to inhibit oxidative phosphorylation. The ATP synthase inhibitor oligomycin reduced the viability of SALL4hi hepatocellular carcinoma and non-small–cell lung cancer cell lines with minimal effects on SALL4lo cells. Oligomycin also reduced the growth of xenograft tumors grown from SALL4hi SNU-398 or HCC26.1 cells, to a greater extent than sorafenib, but oligomycin had little effect on tumors grown from SALL4lo PDX cells. Oligomycin was not toxic to mice. Analyses of chromatin immunoprecipitation sequencing data revealed that SALL4 binds approximately 50% of mitochondrial genes, including many oxidative phosphorylation genes, to activate their transcription. In comparing SALL4hi and SALL4-knockdown cells, we found SALL4 to increase oxidative phosphorylation, oxygen consumption rate, mitochondrial membrane potential, and utilization of oxidative phosphorylation-related metabolites to generate ATP.
Conclusions:
In a screen for compounds that reduce the viability of cells that express high levels of the transcription factor SALL4, we identified inhibitors of oxidative phosphorylation, which slowed the growth of xenograft tumors from SALL4hi cells in mice. SALL4 activates transcription of genes that regulate oxidative phosphorylation to increase oxygen consumption, mitochondrial membrane potential, and ATP generation in cancer cells. Inhibitors of oxidative phosphorylation might be used for treatment of liver tumors with high levels of SALL4.
Keywords: chemical genetic screen, HCC, metabolic vulnerability, metabolism
Lay Summary:
Liver tumors overexpress a protein called SALL4, which causes them to become dependent on specific metabolic pathways for survival. We identified a set of compounds that induce the death of these cancer cells by inhibiting this pathway.
Graphical Abstract
Introduction
Transcription factors are the second largest class of oncogenes1. However, the molecular mechanisms by which these transcription factors exert their cancer-driving effects are not well understood. There is renewed interest in phenotypic cell-based screens for studying the underlying mechanisms of various diseases, aiding in subsequent drug discovery2. Common methods for cell-based drug discovery include the screening of endogenous cell lines with and without the gene or mutation of interest, or the use of isogenic cell line systems in which the gene of interest is altered or expressed in an unaffected cell to control for genetic background2,3. In both endogenous and isogenic systems, hits are defined by their ability to selectively target cells expressing the alteration of interest, while not affecting the control cells. The disadvantage of the endogenous system is that cell lines are genetically distinct, so hits obtained may target pathways unrelated to the alteration of interest2. The isogenic system avoids the genetic complexity of the endogenous system, but suffers the drawback of compound interference with the transgene, resulting in hits that might not be biologically relevant4. To overcome these drawbacks, we developed a screening platform that encompasses both endogenous and isogenic methodologies, applying the platform to identify vulnerabilities induced by oncogene SALL4 mis-expression in hepatocellular carcinoma (HCC).
Liver cancer is the sixth most common cancer but is the second leading cause of cancer deaths worldwide owing to limited therapeutic interventions5. HCC is the predominant subtype of liver cancer, with 85% of liver cancer patients suffering from HCC. The only approved targeted therapies for treating HCC, kinase inhibitors sorafenib and regorafenib, target tumor vasculature, but they are largely ineffective and are used as a last resort6,7. There is an increased urgency to discover precision medicine interventions for this unmet need.
SALL4 (Spalt-like transcription factor 4) is an oncofetal protein essential for self-renewal and maintaining pluripotency in embryonic stem cells, and it plays a critical role in early embryonic development8–11. It is subsequently silenced in most adult tissues, but aberrantly re-expressed to drive tumorigenesis in various cancers9,12. SALL4 is highly expressed in fetal liver but is silenced in the adult liver13, and often reactivated in HCC, in which 30–50% of tumours show significant SALL4 expression14. There are two isoforms of SALL4 (SALL4A and SALL4B) that have overlapping but non-identical binding regions in the genome, and SALL4B alone can maintain pluripotency15. Both isoforms are derived from the same transcript, where SALL4A is the full length spliceoform and SALL4B lacks part of exon 29,16. It has been observed that both SALL4 isoforms are co-expressed when SALL4 is transcriptionally upregulated14. SALL4 is a C2H2 zincfinger transcription factor that can act as a transcriptional activator or repressor15,17,18. The repressive function of SALL4 is achieved through recruitment of the Nucleosome Remodelling and Deacetylase complex (NuRD)19. In cancer, SALL4 recruits NuRD to genes such as the PTEN tumour suppressor, deacetylating and silencing the locus19. The transcriptional activation function of SALL4 also plays a role in cancer. SALL4 has been shown to transcriptionally activate the c-MYC oncogene in endometrial cancer20 and HOXA9 in acute myeloid leukemia21. The in vivo tumorigenic potential of SALL4 is reflected in a mouse model of constitutive SALL4B expression, which results in the onset of acute myeloid leukemia (AML) and HCC22. Therapeutic interventions that target SALL4 and its dependencies remain elusive.
Here, we developed a screening platform that encompasses both endogenous and isogenic methodologies, applying the platform to discover drugs targeting oncogene SALL4-induced dependencies in hepatocellular carcinoma (HCC). Our platform utilizes an endogenous pair of SALL4-expressing (SALL4hi) and SALL4 undetectable (SALL4lo) HCC cell lines, as well as isogenic SALL4 undetectable cell lines engineered to express SALL4 isoforms. We screened both synthetic and diverse natural product extract libraries to identify hit compounds that specifically decrease SALL4hi cell viability. Unexpectedly, our screen identified 4 oxidative phosphorylation inhibitors as being selective for SALL4hi cells. Our most potent and selective compound, ATP synthase inhibitor oligomycin, can selectively target a panel of SALL4hi HCC and lung cancer cell lines, over SALL4lo cells. Oligomycin also demonstrates similar in vivo tumor suppressive activity as HCC standard-of-care drug sorafenib, but at a 200 times lower dose. This in vivo efficacy is only observed in SALL4-high and not SALL4-low tumors. Analysis of SALL4 ChIP-seq data revealed SALL4 binding to a significant number of oxidative phosphorylation genes in SALL4hi HCC. SALL4 predominantly upregulates expression of these genes, as revealed by RNA-seq, mRNA expression and protein analyses. SALL4 expression functionally increases oxidative phosphorylation, as measured by cellular oxygen consumption rate, and supported by imaging and metabolite profiling. Our work demonstrates the ability of our endogenous-isogenic combination cell-based screening methodology to successfully identify a metabolic pathway vulnerability, which is therapeutically actionable with a good therapeutic index, in SALL4-expressing cancers.
Materials and Methods
Chemical genetic screen
SNU-387 empty vector, Tg:SALL4A, and Tg:SALL4B expressing isogenic cell lines were generated by transducing WT SNU-387 cells with empty vector, SALL4A or SALL4B FUWLuc-mCh-puro lentiviral constructs20. Cells were plated in 50 μl of RPMI culture media in 384well white flat-bottom plates (Corning) and incubated at 37°C in a humidified atmosphere of 5% CO2 overnight. Cell numbers per well were 1500 for SNU-398, and 750 for SNU-387 and SNU-387 isogenic lines. After overnight incubation, 0.5 μl of 100 μM drug libraries or 10 mg/ml extract libraries were added to cells with the Bravo Automated Liquid Handling Platform (Agilent). Cells were then incubated for 72 hrs at 37°C in a humidified atmosphere of 5% CO2 before 10 μl of CellTiter-Glo reagent was added to the wells with the MultiFlo Microplate Dispenser (BioTek). Cells were incubated at room temperature for a minimum of 10 minutes after which luminescence readings were recorded by an Infinite M1000 Microplate Reader (Tecan).
HCC sample collection
The collection of HCC samples from HCC patients for research is performed under Domain Specific Review Board (DSRB) protocol 2011/01580 approved by the National Healthcare Group DSRB, which governs research ethics in Singapore that involves patients, staff, premises or facilities of the National Healthcare Group as well as any other institutions under its oversight.
Results
An endogenous-isogenic chemical genetic screening platform identifies SALL4-selective compounds
Our SALL4-dependent chemical-genetic screening platform consists of a pair of endogenous HCC cell lines and a trio of isogenic cell lines (Fig. 1A). For the endogenous pair, SNU-398 expresses high levels of SALL4 protein, and its survival is dependent on SALL4 expression14. The endogenous control SNU-387 cell line has undetectable SALL4 RNA (Fig. S1A) and protein. The isogenic trio consists of lentiviral-mediated insertions into the SNU-387 SALL4 undetectable line, in which the cells are transduced with either an empty vector control, or a SALL4A or SALL4B expressing construct (Fig. 1A). The SALL4 expressing isogenic lines demonstrate SALL4 isoform-specific mRNA and protein expression (Fig. S1B, S1C and S1D) and become sensitive to SALL4 knockdown (Fig. S1D and S1E). SALL4 isoform expression in these isogenic cells does not alter their growth and proliferation rates (Fig. S1F and S1G).
Fig. 1. A chemical genetic cell-based screen to identify compounds targeting SALL4 dependencies.

(A) Schematic of screen involving the use of endogenous SALL4lo and SALL4hi HCC lines and engineered isogenic SALL4 expressing lines. (B) Venn diagram illustrating overlap of hit compounds which selectively decrease cell viability of the SALL4hi lines over their respective SALL4lo controls. (C) Workflow of natural product extract screen to identify individual compound hits from extracts containing multiple chemical entities.
The five endogenous and isogenic cell lines were screened with 1,597 pharmacologically active small molecules from the Selleck Anti-cancer and LOPAC1280 libraries, and 21,575 diverse natural product extracts of plant, fungal, and actinobacteria origin from the A*STAR Bioinformatics Institute collection23. Each natural product extract contains varying numbers of compounds, allowing multiplexing to achieve a screen with hundreds of thousands to millions of compounds efficiently. Cell viability was assessed after 72 hrs of compound or extract incubation (Fig. 1A). Extracts and compounds that reduced cell viability of the SALL4hi cell lines (SNU-398, SNU-387 Tg:SALL4A and Tg:SALL4B) by more than 1.5-fold but had minimal effect on SALL4lo (SNU-387, and SNU-387 Empty Vector) cell viability were identified as hits. The controls for the screen were proteasome inhibitor bortezomib, which significantly reduced cell viability of all cell lines, and the sole hit from the small molecule library screen, PI-103, which selectively targets the SALL4hi cells (Fig. S2A). The Z-factor of the screen was between 0.70 and 0.86.
We obtained three categories of hits from the screen: compounds/extracts that selectively targeted endogenous SALL4hi SNU-398 over SALL4lo control SNU-387 (117 hits), compounds/extracts that selectively targeted Tg:SALL4A cells over Empty Vector control (420 hits), and compounds/extracts that selectively targeted Tg:SALL4B cells over control (960 hits) (Fig. 1B). Each category gave at least 100 hits but taken together, the overlapping results gave only 17 hits (1 small molecule and 16 natural product extract hits). Our combined screening methodology yields a small number of hits that conform to stringent SALL4-specificity requirements, decreasing the time and cost for further validation and work-up of hits.
Since each natural product extract we screened is a mixture of compounds, we determined the specific active components responsible for the SALL4hi response. 31 natural product extract hits from the Tg:SALL4A-SNU-398 overlap (3 hits), Tg:SALL4B-SNU-398 overlap (12 hits), and all three cell line overlap (16 hits) were retested in the screening assay, and only 18 were reproducible (Fig. 1C). These 18 hits were then validated with dose response curves, where only 12 hits from the all three cell line overlap category were validated (Fig. 1C). No hits from the Tg:SALL4A-SNU-398 or Tg:SALL4B-SNU-398 categories passed through this validation step. Next, we fractionated the 12 validated hit extracts into 38 fractions each. Fractions were then screened to identify 9 discrete fractions that were selective for SALL4-high cells, and positive fractions were subjected to Q-TOF mass spectrometry and nuclear magnetic resonance analysis to identify active components (Fig. 1C).
Oxidative phosphorylation inhibitors target SALL4-dependent cell viability
Overall, the screen identified one small molecule hit, PI-103, and 4 natural compound analogues of oligomycin, efrapeptin, antimycin, and leucinostatin as being selective for SALL4hi cells (Fig. 2A and S2A), with a hit rate of 0.02%. Oligomycin and leucinostatin are known inhibitors of the F0 ATP synthase subunit, efrapeptin inhibits the F1 ATP synthase subunit, and antimycin targets cytochrome c reductase in Complex III of oxidative phosphorylation24,25 (Fig. 2B). PI-103 has been shown to induce mitochondrial apoptosis in acute myeloid leukemia cells26. Since the CellTiter-Glo reagent we used for the screen quantifies ATP levels as a measure of cell viability, and our hits target oxidative phosphorylation and the mitochondria, which is a major source of cellular ATP, we further validated our hits with the CyQUANT DNA dye as an alternative measure of cell viability. The dose response curves for the 5 hits using either CellTiter-Glo or CyQUANT were highly comparable (Fig. S2B and S2C). We also tested various analogues of oligomycin and efrapeptin in our cell-based assay (Table S1A). The 4 natural compounds and their analogues demonstrated potent IC50 values in the 0.1 to 10 nM range for the endogenous SALL4hi SNU-398 line and partial cell viability decreases in the SALL4hi isogenic lines, with selectivity ratios ranging from 200 to 20,000 fold compared to the IC50 values in the SALL4lo control cells (Fig. 1A and S1C, Table S1A). In SALL4-high cells, oligomycin A seems to induce cell death through apoptosis, as suggested by the presence of cleaved caspase-3 with oligomycin treatment in a dose response manner (Fig. S2D).
Fig. 2. SALL4-dependent cells are susceptible to mitochondrial oxidative phosphorylation inhibitors.

(A) Cell viability dose-response curves for cells treated for 96 hrs with hit compounds from the natural product extract screen, oligomycin, efrapeptin, antimycin, and leucinostatin, measured with CellTiter-Glo reagent and normalized to untreated cell viability (mean of 3 replicates ± SD). (B) Diagram indicating oxidative phosphorylation targets of validated hit compounds.
Oligomycin A suppresses SALL4-dependent tumorigenesis
We selected oligomycin A for downstream tumor-suppression and mechanistic studies since it had the most potent SALL4hi cell IC50 of 0.5 nM and the highest selectivity of 20,000 fold over the SALL4lo cells. Oligomycin A is also readily available commercially. To determine if oligomycin A could selectively target other SALL4hi cell lines, we performed dose response cell viability experiments on a panel of HCC cell lines. This panel includes two patient-derived primary cell lines, HCC9.2 and HCC26.1, from two Singapore HCC cases, and an immortalized normal liver cell line THLE-3 (Fig. 3A and S3A). We also tested oligomycin A in a pair of nonsmall cell lung cancer (NSCLC) cell lines, in which the SALL4hi H661 line was previously shown to be dependent on SALL4 expression, while the SALL4lo H1299 line was not27 (Fig. S3B and S3C, Table S1B). Our data suggests that oligomycin A is potent and selective against SALL4hi expressing HCC and NSCLC cell lines (Fig. 3A and S3A-C, Table S1A and B).
Fig. 3. Oligomycin A suppresses SALL4-dependent HCC.

(A) Cell viability dose-response curves for a panel of HCC cell lines treated with oligomycin A for 72 hrs, measured with CellTiter-Glo reagent and normalized to untreated cell viability (mean of 3 replicates ± SD). (B) Tumor size plot of SALL4-high SNU-398 mouse xenografts injected (intraperitoneal) with vehicle, sorafenib, or oligomycin A (mean ± SD). (C) Plot of tumor size at day 13 of the xenograft experiments in (B) (mean ± SD). (D) Tumor size plot of SALL4-high PDX HCC26.1 mouse xenografts injected (intraperitoneal) with vehicle, sorafenib, oligomycin A, or a combination of 20 mg/kg sorafenib and 0.1 mg/kg oligomycin (mean ± SD). (E) Plot of tumor size at day 25 of the xenograft experiments in (D) (mean ± SD). (F) Tumor size plot of SALL4-low PDX1 mouse xenografts injected (intraperitoneal) with vehicle or oligomycin A (mean ± SD). (G) Plot of tumor size at day 32 of the xenograft experiments in (F) (mean ± SD). (H-J) Mouse weight quantification plot from the respective mouse xenograft experiments in (B-G) (mean ± SD).
To test the in vivo efficacy of oligomycin A in suppressing HCC tumors, we utilized a SALL4-high mouse xenograft model of SALL4-dependent SNU-398 cells, a SALL4-high patient-derived xenograft model derived from the HCC26.1 patient primary cell line expressing high levels of SALL4 (Fig. S3A), and a SALL4-low patient-derived xenograft model of a tumor named PDX1. In the SALL4-high SNU-398 cell line model, oligomycin A was able to suppress tumor size to a similar degree to the standard-of-care drug in HCC, sorafenib, but at a 200 times lower dose of 0.1 mg/kg compared to 20 mg/kg for sorafenib (Fig. 3B, 3C and S3D). Similarly, oligomycin A or sorafenib treatment was able to suppress tumors in our SALL4-high PDX model with tumor suppression synergy observed in the sorafenib-oligomycin combination treatment (Fig. 3D, 3E and S3E). The PDX1 tumors, which showed very low SALL4 protein levels (Fig. S3F), did not respond to oligomycin treatment (Fig. 3F, 3G and S3G). Mouse weight was not significantly affected by oligomycin treatment in all models (Fig. 3H-J). We examined the known oligomycin side effects of muscle weakness, respiratory depression, and convulsions28,29 in mice treated with vehicle or oligomycin over 3 weeks. To assess muscle weakness, we carried out the open field test, grip strength test, and rotarod test. In the open field test, the distance travelled by the mice in 30 mins was not significantly affected, while their average velocity of movement was slightly decreased with oligomycin treatment (Fig. S3H). In the grip strength test, the normalized full body force was not significantly affected, while the forepaw force was slightly decreased with oligomycin treatment (Fig. S3I). In the rotarod test, the latency to fall of the mice was not significantly affected by oligomycin treatment (Fig. S3J). We did not observe any respiratory depression or convulsions in the mice. Our data suggest that the drug was not highly toxic to the mice at this therapeutic dose.
To examine a potential correlation of oxidative phosphorylation inhibition in patients, we reexamined a HCC patient dataset that we previously published for SALL4 expression14,30. The first-line treatment for Type II diabetes is the biguanide drug metformin, which has been shown to inhibit oxidative phosphorylation31,32. We previously observed that 60% of HCC patient tumors had detectable levels of SALL4, but when we stratified patients with and without diabetes, we noticed a significant difference (Fig. S3K). Non-diabetic patients showed the same trend of 60% SALL4 positivity as all patients combined, however, the trend was reversed in diabetic patients with only 40% having SALL4 positive tumors (Fig. S3K). Patient information on the type of diabetes and metformin use is unavailable so more clinical work is needed to validate this correlation. We tested phenformin, an analogue of metformin with known oxidative phosphorylation inhibition activity32, in our SALL4 isogenic cell lines. We observed partial sensitivity to phenformin in the SALL4-expressing cells compared to the parental SALL4 low line, but the effect was not as prominent as that of oligomycin A (Fig. S3L). The lower effectiveness of phenformin is expected since it is a less potent inhibitor of oxidative phosphorylation (mM IC50)32 compared to oligomycin A (nM IC50)33. Our data suggests the possibility that oxidative phosphorylation inhibition by metformin treatment in diabetic patients suppresses SALL4-positive tumorigenesis.
Oncogenic SALL4 binds oxidative phosphorylation genes and predominantly upregulates them
Since the hits from our screen predominantly target oxidative phosphorylation, we examined our previous SALL4 and acetylated H3K27 chromatin immunoprecipication sequencing (ChIP-seq) data in the SNU-398 cells34. We found that SALL4 binds up to 45% of mitochondrial genes, as defined by the MitoCarta 2.0 gene list, and gene ontology analysis revealed that a significant number of these genes are involved in oxidative phosphorylation (Fig. 4A, Table S2). Gene meta analysis of SALL4 and H3K27ac occupancy at these mitochondrial genes revealed that SALL4 binds predominantly at the promoter region, between the H3K27ac double peaks35 (Fig. 4B and 4C).
Fig. 4. SALL4 binds and upregulates oxidative phosphorylation gene expression.

(A) Venn diagram of mitochondrial genes from the MitoCarta 2.0 dataset bound by SALL4 from our prior SALL4 ChIP-seq experiment performed on SNU-398 cells. Selected significant pathways from Gene Ontology analysis of the SALL4 bound genes are shown. (B) ChIP-seq region plots of the SALL4 bound mitochondrial genes in (A), representing the regions bound by SALL4 and marked by H3K27ac in SNU-398 cells (from analysis of prior data), −3 kb upstream of the transcription start site (TSS) and +3 kb downstream of the transcription end site (TES). (C) Representative ChIP-seq input, H3K27ac, and SALL4 peaks for control gene SUMO1 and electron transport chain genes ATP5D, ATP5E, and NDUFA3. (D) RNA-seq expression level fold change for a panel of mitochondrial genes from the SALL4 bound list in (A), in the SALL4 expressing cell lines, normalized to expression levels in the empty vector control, performed in singlet. (E) Western blots for SALL4-bound oxidative phosphorylation genes and ACTB loading control in the cell lines used in the screen. Bands were quantified by densitometry with SNU-387 and EV bands as references. (F) Western blots for the genes in (E) with SALL4 knockdown for 72 hrs in the SNU-398 cell line. Bands were quantified by densitometry with sh-scr bands as reference.
To assess gene expression changes caused by SALL4 activity, we performed RNA-seq on the isogenic SALL4 expressing cells and SNU-398 SALL4-high cells with SALL4 knockdown (Fig. S4A). We observed that oxidative phosphorylation and other mitochondrial genes with SALL4-bound promoters show increased mRNA expression with SALL4 expression, particularly with the SALL4B isoform (Fig. 4D). In addition, SALL4 knockdown downregulates the expression of these genes (Fig. S4B). We validated the observed RNA-seq expression patterns of some of these genes by qRT-PCR (Fig. S4C and S4D). Gene Set Enrichment Analysis (GSEA)36 of the RNA-seq data revealed significant enrichment of oxidative phosphorylation genes in the SNU-398 control compared to SALL4 knockdown, and in the SALL4B expressing isogenic cell line compared to empty vector control (Fig. S4E, Table S3A-F). This suggests that the binding of SALL4, to oxidative phosphorylation and other mitochondrial gene promoters, predominantly activates transcription of these genes. Genes that are not bound by SALL4 such as SUMO1 are unaffected (Fig. 4C, 4D and S4B). Western blots of SALL4-bound oxidative phosphorylation genes ATP5D, ATP5E, ATP5G2, and NDUFA3, and other SALL4-bound mitochondrial genes ARG2, MRPL24, and SLC25A23, show similar trends in gene expression data, in which SALL4 expression (predominantly SALL4B) upregulates their protein levels while SALL4 knockdown downregulates these levels (Fig. 4E, 4F, S4F and S4G).
SALL4 expression functionally increases oxidative phosphorylation
Since SALL4 expression in our HCC cell lines enhances oxidative phosphorylation gene mRNA and protein expression, we examined if these changes would result in functional alterations in oxidative phosphorylation. We first measured the oxygen consumption rate (OCR) of the SALL4hi and SALL4lo cells used in the screen, since oxidative phosphorylation requires oxygen. We observed that the OCR is significantly increased in the SNU-398 SALL4hi line and by expressing either SALL4A or SALL4B in the isogenic lines (Fig. 5A). The opposite occurs with SALL4 knockdown in SNU-398 cells, in which OCR decreases proportionally with decreasing SALL4 protein levels, as shSALL4–2 reduces SALL4 protein level to a greater degree than shSALL4–1 (Fig. 5B and S4G). This suggests that SALL4 expression increases oxidative phosphorylation-dependent OCR.
Fig. 5. SALL4 expression upregulates oxidative phosphorylation.

(A) OCR measurements of SALL4 endogenous and isogenic lines used in the screen, normalized to DNA content measured by CyQUANT reagent (mean of 3 replicates ± SD). (B) OCR measurements for SALL4 knockdown in SNU-398 endogenous SALL4-high cells, normalized to DNA content measured by CyQUANT reagent (mean of 3 replicates ± SD). (C) Representative images of SALL4 endogenous and isogenic cell lines stained with DAPI nuclear dye, Mitotracker Red mitochondrial membrane potential dye, and immunostained with cytochrome c antibody. Scale bars are 20 μm in length. (D) Quantification of cytochrome c fluorescence signal per cell, normalized to DAPI signal (median, quartile and range). (E) Quantification of MitoTracker fluorescence signal per cell, normalized to DAPI signal (median, quartile and range). (F) ATP levels per DNA content for the SALL4 isogenic cell lines measured by CellTiter-Glo ATP detection reagent values normalized to CyQUANT DNA quantification reagent values (mean of 3 replicates ± SD). (G) NADH/NAD+ values measured by HPLC-mass spectrometry metabolite profiling of the SALL4 isogenic cell lines (mean of 3 replicates ± SD).
To assess mitochondrial localization and the mitochondrial membrane potential gradient generated by oxidative phosphorylation, we performed immunofluorescence imaging of the SALL4 endogenous and isogenic cell lines with oxidative phosphorylation membrane protein Cytochrome c and MitoTracker dye, a dye which localizes to the mitochondrial membrane in a membrane potential-dependent manner (Fig. 5C). Quantification of the fluorescence signals per cell revealed that Cytochrome c is significantly upregulated in the SALL4A expressing cells (Fig. 5D). In addition, the MitoTracker signal is significantly increased in the SNU-398 and both SALL4A and SALL4B expressing cells (Fig. 5E). These results suggest that SALL4 expression increases oxidative phosphorylation-dependent mitochondrial membrane potential.
Since oxidative phosphorylation is functionally increased by SALL4 expression, we analysed the levels of oxidative phosphorylation-related metabolites. We first measured ATP levels normalized to DNA content in the SALL4 expressing cells and found that ATP levels are significantly increased in both the SALL4A and SALL4B expressing lines (Fig. 5F). We also performed metabolite profiling on the SALL4 expressing lines and through Metabolite Set Enrichment Analysis (MSEA)37, observed that electron transport chain (oxidative phosphorylation) and malate-aspartate shuttle metabolites are significantly altered in both SALL4A and SALL4B expression (Fig. S5A and S5B). The malate-aspartate shuttle facilitates the transfer of electrons from membrane impermeable NADH generated during glycolysis in the cytosol to mitochondrial oxidative phosphorylation38. NADH levels are significantly lower in the SALL4 expressing lines while NAD+ levels are significantly higher, implying that there is an increased conversion of NADH into NAD+ by oxidative phosphorylation Complex I (Fig. 5G). Malate-aspartate shuttle metabolites are also significantly increased, suggesting an increase in the transfer of electrons (NADH) generated in glycolysis to oxidative phosphorylation (Fig. S5C). Our metabolite profiling data implies that SALL4 expression increases the utilization of oxidative phosphorylation-related metabolites to generate more ATP.
Many cancers demonstrate the Warburg effect, where glycolysis is upregulated by the PI3K/mTOR signalling pathway39. Our small molecule SALL4-selective hit from the screen, PI-103, is a pan PI3K inhibitor (Fig S2A). We therefore examined the effects of SALL4 expression on glycolysis in our oxidative phosphorylation-dependent model. From our metabolite profiling data, glycolytic metabolites are primarily downregulated with SALL4 expression (Fig. S5D). The levels of L-lactate, the end product of anaerobic respiration, were unchanged with SALL4 expression (Fig. S5E). Further, we measured the extracellular acidification rate (ECAR) of the SALL4 isogenic cell lines, which measures lactate being secreted into the extracellular environment, and observed a slight decrease in the ECAR with SALL4 expression (Fig. S5F). In the glycolysis stress test, we observed a marked decrease in glycolytic rate and a slight decrease in glycolytic capacity in the SALL4 expressing cells (Fig. S5G). To ascertain if PI3K inhibition is important for SALL4-selectivity, we tested a number of PI3K isoform-specific and mTOR inhibitors in our endogenous and isogenic cell lines. However, most of these inhibitors did not recapitulate the specificity for SALL4-expressing lines observed with PI-103 treatment (Fig. S6A). The SALL4-selectivity of PI-103 could be due to an off-target effect, rather than by modulating the PI3K pathway. From these experiments, it is likely that SALL4 expression in cancer neither initiates the Warburg effect nor creates a dependency on glycolysis.
Interestingly, the top altered metabolic pathway due to SALL4 expression was the urea cycle (Fig. S5A and S5B). We observed significant upregulation of urea cycle metabolites, particularly in the SALL4B expressing cells, in our metabolite profiling data (Fig. S7A). When we examined our ChIP-seq data for urea cycle genes, we only observed SALL4 binding at the promoter region of ARG2 (Fig. S7B). This suggests a possible coupling of oxidative phosphorylation and the urea cycle through ARG2 regulation by SALL4. However, since SALL4 binds only one gene in the urea cycle, it is unlikely that the urea cycle plays a direct role in SALL4-dependent cancer.
We also examined mitochondrial DNA (mtDNA) copy number through qRT-PCR analysis with mtDNA gene-specific primers40 and found that the examined mtDNA regions are significantly amplified in SNU-398 SALL4hi cells and SALL4 expressing isogenic lines (Fig. S7C). This suggests that SALL4 expression promotes an increase in mtDNA copy number in relation to increased oxidative phosphorylation functionality in the mitochondria. We also examined the expression of mitochondrial biogenesis regulators PGC-1α, PGC-1β, TFAM, NRF1, and NRF241–43, in our SALL4-expressing isogenic lines. Only PGC-1α was significantly upregulated in the SALL4B-expressing line while there were no appreciable alterations in TFAM, NRF1, and NRF2 (Fig. S7D). PGC-1β expression was not detected in these lines. In our ChIP-seq data, SALL4 binding was only observed at the promoters of NRF2 and TFAM (Fig. S7E). Our data suggests that SALL4 does not directly regulate the expression of mitochondrial biogenesis genes.
Conclusions:
A combined chemical-genetic screening to discover oncogenic transcription factor vulnerabilities as precision medicine
Our chemical genetic screening platform with endogenous and isogenic SALL4 expressing HCC cell lines allows for the efficient and stringent identification of a small number of hits that target both the endogenous and isogenic SALL4hi lines, increasing the likelihood that these hits are specifically affecting SALL4-related biology. The endogenous pair gives biological relevance while the isogenic trio controls for genetic background. Our combination endogenous-isogenic screen is therefore able to identify compounds that target SALL4-specific biology in a biologically relevant fashion. The 4 natural compound hits identified target different oxidative phosphorylation components and by doing so, they potently and selectively target SALL4 expressing cells in both HCC and NSCLC systems. We demonstrate that ATP synthase inhibitor oligomycin A effectively targets SALL4hi cells in a panel of HCC cell lines and can suppress tumors in vivo to a similar degree as the current standard-of-care drug sorafenib. Oligomycin and sorafenib also act in synergy to suppress tumorigenesis when combined. This suggests that our system can identify tool compounds that are specific to transcription factor cancer biology efficiently and effectively. Our proof-of-concept screen could have important implications for future academic studies of oncogenic transcription factor downstream pathways, and potential precision medicine applications.
A previously unknown metabolic role of SALL4 in tumorigenesis
From prior work, the widely accepted role of transcription factor SALL4 in cancer has been to modulate the expression of both pro- and anti-cancer genes, such as by recruiting the NuRD complex to chromatin to silence PTEN, or by directly upregulating oncogene MYC levels.
Our screening results and subsequent investigation into the altered processes in SALL4-dependent tumorigenesis reveals a previously unknown metabolic reprogramming function of SALL4. We demonstrate that SALL4 binds a significant number of oxidative phosphorylation and other mitochondrial genes at their promoters and predominantly upregulates their mRNA expression. This gene expression upregulation ultimately leads to increased protein levels of these genes. SALL4 expression also leads to a functional increase in oxidative phosphorylation, with increased cellular OCR, mitochondrial membrane potential, oxidative phosphorylation-related metabolites and mtDNA copy number. Since SALL4 expression in our isogenic cell lines does not affect cell proliferation, we believe that oxidative phosphorylation is specifically coopted by SALL4 mis-expression in cancer, and not as a result of increased proliferation rate upregulating non-specific housekeeping processes. Our work proposes that SALL4 expression in cancer confers a dependency on oxidative phosphorylation through direct gene expression regulation, although the underlying preference for this metabolic reprogramming in tumorigenesis is still unclear.
We did not observe the Warburg effect, the preference for cancers to upregulate anaerobic glycolysis for energy, in our SALL4-expressing cancer cell models. Recent studies have challenged the hypothesis that the Warburg effect is cancer specific, suggesting that the effect is a result of metabolic changes associated with a proliferative state, rather than a unique feature of malignancy44. Many non-malignant cells utilize the Warburg effect to proliferate. There are many advantages of non-Warburg aerobic respiration to proliferating cells, such as the supply of large quantities of anabolic precursors such as nucleotides, proteins, and lipids. Many tumor cells have been shown to utilize the TCA cycle and oxidative phosphorylation to generate ATP and balance reactive oxygen species45. Tumorigenesis has also been shown to be dependent on mitochondrial function. Cancer cells can use fatty acids and amino acids, rather than glucose, to supply intermediates for the TCA cycle and maintain mitochondrial respiration, particularly during changes in the tumor microenvironment30,46. This might explain why SALL4-expressing cells upregulate oxidative phosphorylation to become tumorigenic, thereby becoming sensitive to oxidative phosphorylation inhibitors, rather than demonstrating the Warburg effect.
Other than being a potent oncogene, SALL4 is an important developmental gene in the fetal liver and in stem cells. It would be interesting to determine if oxidative phosphorylation and other metabolic processes are similarly regulated by homeostatic SALL4 expression in the developing embryo or the stem cell compartment during liver regeneration post injury. The role of SALL4 in liver regeneration is poorly understood and future studies are prudent for dissecting this role in greater detail.
SALL4 as a potential biomarker for oxidative phosphorylation precision medicine in cancer
Clinical trials have been conducted to assess the effectiveness of oxidative phosphorylation inhibitors as effective cancer therapies47. However, the direct molecular mechanisms of oxidative phosphorylation phosphorylation upregulation in cancer are not well understood, particularly in liver cancer. This, coupled with toxicity associated with targeting a ubiquitous cellular pathway, currently make these inhibitors less appealing as cancer drugs.
Our study demonstrates the possibility of SALL4 to be used as a companion biomarker to select cancer patients who may benefit from oxidative phosphorylation inhibitors in the clinic. Mechanistically, we propose a direct link between SALL4 upregulation and an increase in oxidative phosphorylation, where SALL4 binds and transcriptionally activates oxidative phosphorylation genes during tumorigenesis. Tumors that express significant levels of SALL4 are more sensitive to oxidative phosphorylation inhibition at very low doses, as we have demonstrated both in vitro and in vivo. A larger therapeutic window for clinical oxidative phosphorylation inhibitors is therefore possible in patients harboring SALL4-expressing tumors. Targeting SALL4-dependent cancer with oxi oxidative phosphorylation inhibitors could lead to an effective suppression of tumorigenesis with minimal toxicity. The patient data we examined shows promise for precision medicine use of oxidative phosphorylation inhibitors in SALL4 patients, but the limitations of the annotated patient bio-data, the small samples size, and the low potency of biguanides as oxidative phosphorylation inhibitors, means that more clinical studies are needed to confirm the clinical utility of our findings.
A limitation of our study is that we did not obtain SALL4A- and SALL4B-specific hits. Further studies are needed to determine the unique mechanisms by which each isoform drives cancer. A confounding issue is that SALL4A and B are co-expressed from the same gene locus as splice isoforms, and from prior work, are always co-expressed in the same cell line or tumor tissue. Our study demonstrates that both SALL4 isoforms can functionally upregulate, and thus create a dependency on, oxidative phosphorylation. Targeting this pathway shared by both isoforms with oxidative phosphorylation inhibitors is therefore a viable therapeutic option for SALL4-expressing cancers. We have observed that SALL4 is upregulated in about 20–30% of all solid tumors12,14, so the potential clinical utility of oxidative phosphorylation inhibitors with a companion SALL4 diagnostic is highly significant.
Our study demonstrates that a SALL4 biomarker can be used in conjunction with oligomycin, a highly potent oxidative phosphorylation inhibitor that has not yet been tested extensively in clinical trials to our knowledge. The LD33 (lethal dose that kills 33%) of oligomycin in rats is 0.5 mg/kg (1 mg/kg in mice), while 100% of rats survived with 0.1 mg/kg of drug (0.2 mg/kg in mice)28,48. Our study doses mice at the sub-lethal dose of 0.1 mg/kg oligomycin, which is 10 times less than the LD33, and we observe significant and selective tumor size suppression in SALL4-high tumors with low toxicity. It might be worthwhile to explore the clinical use of oligomycin in SALL4-expressing tumors.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT: The transcription factor SALL4 is mis-expressed in cancer cells. We developed a screen to identify compounds that reduce the viability of liver cancer cells that mis-express SALL4 and the mechanisms by which these compounds act.
NEW FINDINGS: We identified a metabolic vulnerability in liver (and possibly lung) cancer cells, due to overexpression of SALL4, which can be targeted by natural product oxidative phosphorylation inhibitors.
LIMITATIONS: This was a chemical screen for compounds that affect the viability of a small number of cell lines in culture and growth as xenograft tumors in mice. Additional studies in other animal models of liver cancer, and on other cell lines, are needed.
IMPACT: We developed a screen to identify compounds that kill cancer cells that overexpress or underexpress a specific protein. This screen can be used to identify compounds with toxicity to cells with other alterations in gene expression and identify the mechanisms regulated by these alterations.
Acknowledgments:
We thank Min Yuan for her assistance with the metabolite profiling experiments.
Grant support: This work was supported by the Genome Institute of Singapore Innovation Fellow Award (J.L.T.); A*STAR A*ccelerate Gap Funding ETPL/18-GAP018-R20H (J.L.T.); Singapore Ministry of Health’s National Medical Research Council (Singapore Translational Research (STaR) Investigator Award, D.G.T.; NMRC/OFIRG/0064/2017, W.L.T; LCG17MAY004, W.L.T.; NMRC/TCR/015-NCC/2016, W.L.T.); National Research Foundation Singapore (NRF-NRFF2015-04 Grant, W.L.T.) and the Singapore Ministry of Education under its Research Centres of Excellence initiative; Singapore Ministry of Education Academic Research Fund Tier 3, grant number MOE2014-T3-1-006 (D.G.T); NIH/NCI Grant R35CA197697 (D.G.T); NIH/NHLBI Grant P01HL095489 (L.C.); LLS Grant P-TRP-5854-15 (L.C). This work was supported in part by the JSPS KAKENHI (Grant Numbers: JP17H05623, JP17K19603, JP18H05102, JP19H01177, and JP19H05267) (A.S.).
Disclosures: J.L.T., Y.K., S.B.N., W.L.T., L.C., and D.G.T. are co-inventors on a patent application filed by A*STAR A*ccelerate, the National University of Singapore, and Brigham & Women’s Hospital relating to work in this manuscript.
Footnotes
Data and materials availability: All sequencing data have been deposited in the NCBI Gene Expression Omnibus databases GSE114808 and GSE112729.
To review GEO accession GSE112729:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112729 Enter token gdyfgeoapjevdql into the box
To review GEO accession GSE114808:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114808 Enter token ajudymeanhstnqd into the box
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes:
- 1.Patel MN, Halling-Brown MD, Tym JE, et al. Objective assessment of cancer genes for drug discovery. Nat Rev Drug Discov 2013;12:35–50. [DOI] [PubMed] [Google Scholar]
- 2.Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today 2013;18:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res 2014;74:2377–2384. [DOI] [PubMed] [Google Scholar]
- 4.Janzen WP. Screening Technologies for Small Molecule Discovery: The State of the Art. Chem Biol 2014;21:1162–1170. [DOI] [PubMed] [Google Scholar]
- 5.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: ources, methods and major patterns in GLOBOCAN 2012. Int J Cancer J Int Cancer 2015;136:E359–386. [DOI] [PubMed] [Google Scholar]
- 6.Bruix J, Raoul J-L, Sherman M, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol 2012;57:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebocontrolled, phase 3 trial. The Lancet 2017;389:56–66. [DOI] [PubMed] [Google Scholar]
- 8.Celis JF de Barrio R. Regulation and function of Spalt proteins during animal development. Int J Dev Biol 2009;53:1385–1398. [DOI] [PubMed] [Google Scholar]
- 9.Tatetsu H, Kong NR, Chong G, et al. SALL4, the missing link between stem cells, development and cancer. Gene 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Tam W-L, Tong GQ, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol 2006;8:1114–1123. [DOI] [PubMed] [Google Scholar]
- 11.Lim CY, Tam W-L, Zhang J, et al. Sall4 Regulates Distinct Transcription Circuitries in Different Blastocyst-Derived Stem Cell Lineages. Cell Stem Cell 2008;3:543–554. [DOI] [PubMed] [Google Scholar]
- 12.Nicolè L, Sanavia T, Veronese N, et al. Oncofetal gene SALL4 and prognosis in cancer: A systematic review with meta-analysis. Oncotarget 2017;8:22968–22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oikawa T, Kamiya A, Kakinuma S, et al. Sall4 regulates cell fate decision in fetal hepatic stem/progenitor cells. Gastroenterology 2009;136:1000–1011. [DOI] [PubMed] [Google Scholar]
- 14.Yong KJ, Gao C, Lim JSJ, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med 2013;368:2266–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao S, Zhen S, Roumiantsev S, et al. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol Cell Biol 2010;30:5364–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohlhase J, Chitayat D, Kotzot D, et al. SALL4 mutations in Okihiro syndrome (Duaneradial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum Mutat 2005;26:176–183. [DOI] [PubMed] [Google Scholar]
- 17.Elling U, Klasen C, Eisenberger T, et al. Murine inner cell mass-derived lineages depend on Sall4 function. Proc Natl Acad Sci U S A 2006;103:16319–16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Gao C, Chai L, et al. A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PloS One 2010;5:e10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Jeong H-W, Jeong H, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PloS One 2009;4:e5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li A, Jiao Y, Yong KJ, et al. SALL4 is a new target in endometrial cancer. Oncogene 2015;34:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li A, Yang Y, Gao C, et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J Clin Invest 2013;123:4195–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Cui W, Yang J, et al. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood 2006;108:2726–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng SB, Kanagasundaram Y, Fan H, et al. The 160K Natural Organism Library, a unique resource for natural products research. Nat Biotechnol 2018;36:570–573. [DOI] [PubMed] [Google Scholar]
- 24.Slater EC. The mechanism of action of the respiratory inhibitor, antimycin. Biochim Biophys Acta BBA - Rev Bioenerg 1973;301:129–154. [DOI] [PubMed] [Google Scholar]
- 25.Hong S, Pedersen PL. ATP Synthase and the Actions of Inhibitors Utilized To Study Its Roles in Human Health, Disease, and Other Scientific Areas. Microbiol Mol Biol Rev MMBR 2008;72:590–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park S, Chapuis N, Bardet V, et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008;22:1698–1706. [DOI] [PubMed] [Google Scholar]
- 27.Yong KJ, Li A, Ou W-B, et al. Targeting SALL4 by entinostat in lung cancer. Oncotarget 2016;7:75425–75440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramar R, Hohenegger M, Srour AN, et al. Oligomycin toxicity in intact rats. Agents Actions 1984;15:660–663. [DOI] [PubMed] [Google Scholar]
- 29.Smith RM, Peterson WH, McCoy E. Oligomycin, a new antifungal antibiotic. Antibiot Chemother 1954;4:962–70. [PubMed] [Google Scholar]
- 30.Lu G-D, Ang YH, Zhou J, et al. CCAAT/enhancer binding protein α predicts poorer prognosis and prevents energy starvation–induced cell death in hepatocellular carcinoma. Hepatol Baltim Md 2015;61:965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu 2007:13–20. [PubMed] [Google Scholar]
- 32.Bridges HR, Jones AJY, Pollak MN, et al. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 2014;462:475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang M, Qu Y, Gao B, et al. Oligomycin, a Complex V Inhibitor, Decreases Left Ventricular Contractility in Isolated Rabbit Heart. FASEB J 2011;25:lb362–lb362. [Google Scholar]
- 34.Liu BH, Jobichen C, Chia CSB, et al. Targeting cancer addiction for SALL4 by shifting its transcriptome with a pharmacologic peptide. Proc Natl Acad Sci U S A 2018;115:E7119–E7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008;40:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledgebased approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia J, Wishart DS. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res 2010;38:W71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu M, Zhou L, Stanley WC, et al. Role of the Malate-Aspartate Shuttle on the Metabolic Response to Myocardial Ischemia. J Theor Biol 2008;254:466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courtnay R, Ngo DC, Malik N, et al. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep 2015;42:841–851. [DOI] [PubMed] [Google Scholar]
- 40.Phillips NR, Sprouse ML, Roby RK. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: A multiplex real-time PCR assay. Sci Rep 2014;4:3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendham AE, Duffield R, Coutts AJ, et al. Similar mitochondrial signaling responses to a single bout of continuous or small-sided-games-based exercise in sedentary men. J Appl Physiol Bethesda Md 1985 2016;121:1326–1334. [DOI] [PubMed] [Google Scholar]
- 42.Eisele PS, Salatino S, Sobek J, et al. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J Biol Chem 2013;288:2246–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992–1003, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017;168:657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palm W, Thompson CB. Nutrient acquisition strategies of mammalian cells. Nature 2017;546:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashton TM, McKenna WG, Kunz-Schughart LA, et al. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res 2018;24:2482–90. [DOI] [PubMed] [Google Scholar]
- 48.Freireich EJ, Gehan EA, Rall DP, et al. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep 1966;50:219–244. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.