

Abstract
To demonstrate multifaceted contribution of aspartate β-hydroxylase (ASPH) to pancreatic ductal adenocarcinoma (PDAC) pathogenesis, in vitro metastasis assay and patient derived xenograft (PDX) murine models were established. ASPH propagates aggressive phenotypes characterized by enhanced epithelial-mesenchymal transition (EMT), 2-D/3-D invasion, extracellular matrix (ECM) degradation/remodeling, angiogenesis, stemness, transendothelial migration and metastatic colonization/outgrowth at distant sites. Mechanistically, ASPH activates Notch cascade through direct physical interactions with Notch1/JAGs and ADAMs. The ASPH-Notch axis enables prometastatic secretome trafficking via exosomes, subsequently initiates MMPs mediated ECM degradation/remodeling as an effector for invasiveness. Consequently, ASPH fosters primary tumor development and pulmonary metastasis in PDX models, which was blocked by a newly developed small molecule inhibitor (SMI) specifically against ASPH’s β-hydroxylase activity. Clinically, ASPH is silenced in normal pancreas, progressively upregulated from pre-malignant lesions to invasive/advanced stage PDAC. Relatively high levels of ASPH-Notch network components independently/jointly predict curtailed overall survival (OS) in PDAC patients (log-rank test, Ps < 0.001; Cox proportional hazards regression, P < 0.001). Therefore, ASPH-Notch axis is essential for propagating multiple-steps of metastasis and predicts prognosis of PDAC patients. A specific SMI targeting ASPH offers a novel therapeutic approach to substantially retard PDAC development/progression.
Keywords: Aspartate β-hydroxylase (ASPH), Pancreatic ductal adenocarcinoma (PDAC), Patient derived xenograft (PDX), Exosome, Notch, Small molecule inhibitor (SMI)
1. Introduction
Pancreatic cancer (PC) was responsible for 458,918 new cases and 432,240 deaths world-wide in 2018 (GLOBOCAN), with a 5-year survival rate of 5–8% [1]_ENREF_1._ENREF_10 The ASPH hydroxylizes aspartyl and asparaginyl residues in EGF-like repeats of various proteins, especially Notch receptors and ligands [2]._ENREF_2_ENREF_25 Notch pathway is critically involved in stemness, migration and invasion [3]._ENREF_31 ASPH is silenced in normal adult tissues and only upregulated during oncogenesis [4–6]. It is highly expressed in multiple tumors originated from liver (hepatocellular carcinoma; HCC), bile duct (cholangiocarcinoma) [5], pancreas [7] and nervous system [4,5,8,9]. Overexpression of ASPH or its truncated homolog Humbug confers worse prognosis of HCC [6], non-small cell lung cancer [10] and colon cancer [11]. ASPH links upstream growth factor signaling with downstream pro-oncogenic cascades [12–16]._ENREF_14 However, whether ASPH is a determinate for more aggressive/invasive phenotypes of PC remains mysterious.
Exosomes are cell-derived lipid-bilayer, membrane enclosed nanovesicles (30–100 nm) present in biofluids and cell culture supernatants [17]_ENREF_1. Exosomes locally/systemically transfer and exchange intercellular active biomolecules to reprogram biological processes in tumor development [18] and progression [19,20]. We attempt to address several critical questions whether: (a) ASPH-Notch axis is involved in exosomal cell-to-cell communication? (b) ASPH-Notch network depends on exosomal secretome to initiate PC progression? (c) inhibition of ASPH-Notch axis can reverse aggressive phenotypes and block PC development/progression?
2. Materials and methods
2.1. Tube formation
300 μl of cold (4 °C) Matrigel Matrix (10 mg/mL) was added to each well of a 24-well plate on ice. The plate was centrifuged at 300×g for 10 min at 4 °C, and quickly incubated at 37 °C for 60 min. Afterwards, 300 μl of human umbilical vein cells (HUVECs) suspension (1.2 × 105) ± exosomes (5 μg/ml) was added to each well. PC cells were incubated for 12–24 h in CO2 incubator. The formation of tube-like structures was imaged by microscope. The number of vessel joints was counted and tube area was calculated by NIH ImageJ. Results from three independent experiments were expressed as mean ± standard deviation (SD).
2.2. Pancreatosphere formation
Matrigel (300 μl) was spread evenly to each well of a 24-well plate on ice. The plate was centrifuged at 4 °C, 300×g; and immediately placed in a cell culture incubator for 30 min. Single-cells were suspended in the medium with 10% matrigel at a concentration of 2000 cells/400 μl and seeded on matrigel. PC cells were allowed to attached to the matrigel for 3 h. The medium was carefully removed and replaced with fresh medium containing 10% matrigel. After incubated for 1h, corresponding culture medium was added. Fresh medium containing 10% matrigel was changed every 2 days. The spheres formed after 5–9 days were evaluated in terms of size and number by light microscopy. All experiments were performed in triplicate wells for each condition and repeated at least twice.
2.3. ECM degradation/remodeling
Cover glass (18 mm; Fisher Scientific) was coated with pig skin Oregon green 488 conjugated Gelatin (G13186, Life Technologies). The gelatin was cross-linked with a 0.5% glutaraldehyde solution in a 12-well plate, followed by quenched with sodium borohydride (1 mg/ml) and washed 3 times with PBS. Pancreatic cancer cells (2 × 104) were seeded to each well in 2 ml of complete medium. After 18h–72h, cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, blocked with 5% bovine serum albumin (BSA), and probed for F-actin (Rhodamine phalloidin, R415, Life technologies). The coverslips were mounted over a glass slide with a drop of mounting medium containing DAPI. At least 15 fields per coverslip were imaged at all three channels (red, green and blue) under 40 × magnification. Black and white images of gelatin degradation were analyzed using NIH IMAGE J software. The degraded area was normalized to the number of nuclei in the image from the same field. Modulating agent was applied to the system to evaluate its respective impact on cellular phenotype. All experiments were performed in triplicate wells for each condition and repeated at least twice.
2.4. 3D-Embedded and 3D-On Top (co-culture layer epi-/endothelial cells on top) cultures
24-well plate was coated with 300 μl/well growth factor reduced (GFR) matrigel, and incubated (37 °C, 5% CO2) for 30 min. PC cells were harvested, counted and diluted to a concentration of 5000/ml in complete growth medium (2% GFR matrigel). 400 μl was added to each well of a matrigel pre-coated plate, and incubated (37 °C, 5% CO2) for 5–7 days. To evaluate the effects of different pharmacologic inhibitors, each compound was added to the complete medium at the time of plating followed by fresh medium changes supplemented with each compound every day while the cells grew on GFR matrigel. All experiments were performed in triplicate wells for each condition and repeated at least twice.
2.5. 3D (Spheroid) invasion
To perform 3D Culture 96-Well BME Cell Invasion Assay (Trevigen Inc. Gaithersburg, MD), PC cell monolayers were washed with PBS, dissociated by Trypsin, neutralized with complete growth medium. PC cells were counted using a hemocytometer and the cell suspension was diluted to 1 × 104 cells/ml (to obtain tumor spheroids of 300–500 μm in diameter 4 days after cell seeding). PC cell suspension was dispensed into ULA 96-well round bottom plate and centrifuged at 200×g for 5 min. The plate was transferred to CO2 incubator. After 3–5 days, PC spheroid formation was visually confirmed and proceeded to 3-D invasion assay. BMM (basement membrane matrix) was thawed on ice overnight. ULA 96-well plate containing 4-day old spheroids was placed on ice. 50 μl of BMM was gently dispensed into each U-bottom well with 6 replicates in each group. The plate was centrifuged at 300×g for 3 min at 4 °C, then transferred to CO2 incubator, allowing the BMM to solidify. After 1h, 100 μl/well of complete growth medium was gently added into each well. Invasion modulating agent was applied to the system to evaluate its respective impact on cellular phenotype. Spheroid invasion was visualized microscopically and quantitated with image analysis software NIH IMAGEJ. All experiments were performed in triplicate wells for each condition and repeated at least twice.
2.6. In Vitro Metastasis [21,22]
Matrigel invasion chambers (BD BioCoat Matrigel Invasion 24-well Chamber, 8 μm pores, BD Biosciences) were rehydrated for 2h at 37 °C with serum-free medium. HUVECs (2 × 105) in HUVEC Medium were seed in inserted chambers. After 24h, lower chambers were coated with 290 μl matrigel and filled with 500 μl HUVEC Medium (10% FBS). PC cells (1–4 × 104) stably expressing GFP in HUVECs Medium (FBS-free) were plated onto a layer of HUVECs and incubated in CO2 incubator for 3 days. Inserted 24-well chambers were removed, washed with PBS and fixed with 4% PFA for 20 min, permeabilized with Triton X-100 for 20 min, and stained with phalloidin (red) and Hoest. Transmigrated PC cells passing through HUVECs were imaged using fluorescence microscope and counted. PC cells invaded into the matrigel within the lower chambers were buried with medium containing 10% matrigel, continuously cultured in complete growth medium for 7 days to allow pancreatosphere formation. PC tumor spheres were imaged and evaluated in terms of size and number by fluorescence microscope.
2.7. Patient’s tissue procurement for PDX model
Tumor tissues were collected from 6 patients (Table S1) with primary PDAC who had undergone surgical resection at Rhode Island Hospital of Brown University. This study was approved by the Institutional Review Board of Rhode Island Hospital and conducted in accordance with all current ethical guidelines.
2.8. Establishment of F1 generation patient-derived xenograft (PDX) murine models
PDAC tumor specimens were transferred to the animal procedure room within 0.5–1h after surgical resection, washed by DMEM (1% penicillin/streptomycin), and diced into 5 × 5 × 5 mm3 fragments. Female (5–6-week-old; n = 6/group) NSG mice (Jackson Laboratory) were anesthetized with isoflurane (4% induction, 2% maintenance). A small incision was made on the lower back; 1–2 tumor fragments subcutaneously implanted. The residual tumor fragments were formalin-fixed or stored in liquid nitrogen for IHC or RT-PCR. Tumor was measured 3 times/week. Tumor volume=(length × width2)/2. All animal procedures were approved by IACUC at Rhode Island Hospital.
2.9. Establishment of F2 through F7 PDX murine models
When reached approximately 500 mm3, F1 tumors were excised, washed, diced into 5 × 5 × 5 mm3 fragments, transplanted into 5–6-week-old female NSG mice under anesthesia, and serially passaged to F7 generation. Necropsy was performed when reached approximately 800–900 mm3. PDX derived tumors, liver and lymph nodes were fixed for histology/IHC, and lungs immersed in Bouin’s solution (HT10132; Sigma-Aldrich, St. Louis, MO) to determine macro-/micro-metastases.
2.10. Anti-tumor effects of SMI in vivo
Potential anti-tumor effect of small molecule inhibitor (SMI) [14] on PDAC was analyzed in F5–F7 generation PDX mice harboring tumors derived from Patient B. When tumors reached 100 mm3 (grown subcutaneously on the back of NSG mice for 4–5 weeks after transplantation), mice were randomized into experimental or control group treated with MO-I-1182 or DMSO, respectively. Torpac mouse capsule filled with lactose and MO-I-1182 (or DMSO) was administered by dosing syringe injection into mouse’s stomach through mouth and esophagus. MO-I-1182 was administered orally at 10 mg/kg daily for 5 weeks. Mice were sacrificed at 5 weeks. Necropsy was performed and primary tumors, lungs, liver and lymph nodes surgically removed. The lungs were immersed in Bouin’s solution and metastatic nodules counted.
3. Statistics
Data were analyzed with SPSS and GraphPad software packages. Nonparametric data were analyzed with Kruskal-Wallis two-way ANOVA and Tamhane’s post hoc test. Normally distributed data were analyzed using two-way ANOVA and Bonferroni post hoc. Spearman’s rank correlation coefficient (ρ) and Pearson’s correlation coefficient (r) were used to evaluate the relationship of ASPH expression with other components levels in tumor tissue by IHC. OS time was calculated from the date of diagnosis to the date of death or last follow-up. Median survival time was estimated using Kaplan-Meier plot and compared with log-rank test. Univariate explanatory variables and multivariate Cox proportional hazards regression models were applied to evaluate individual and combined contribution of ASPH network components on OS, adjusting for clinical factors. A p < 0.05 (2-tailed) was considered statistically significant.
4. Results
4.1. ASPH physically interacts with Notch receptors, ligand, and regulators to activate Notch cascade in PC
ASPH expression in PC cell lines has been evaluated previously [7]. MIA-Paca2 cells stably expressing empty vector vs. ASPH using lenti-virus transfection [7]; whereas AsPC-1 and HPAFII stably expressing CRISPR-vector vs. ASPH knock-our (KO) using CRISPR-CAS9 were established.
ASPH physically interacts with Notch receptors/ligands (JAGs) and regulators (ADAM10/ADAM17). Aspartyl/Asparaginyl β-hydroxylase (HydroX) domain is required for ASPH-Notch extracellular domain (ECD) interaction. ASPH amplifies Notch-JAG interaction, facilitates ligand-receptor binding and thus stimulates S2 cleavage of Notch receptors executed by ADAM10/17. In both ligand-dependent and ligand-independent (ADAM10/17 dependent) manners, Notch signaling is activated by ASPH [7,22]. Since ASPH’s function depends on β-hydro-xylase activity [7], SMIs against enzymatic activity of ASPH were characterized. MO-I-1182 exerted a dose-dependent effect on cell viability [7]. ASPH mediated Notch signaling activation was eliminated by SMI based on Luciferase Reporter (Fig.S1AC) and Western blot assays (Fig. 1A and B; Fig. S1D). Notch signal inhibitor DAPT efficiently reduced migration/invasion (Fig. S1E–F), EMT (upregulation of Vimentin, Figs. S1K) and 3D invasion (Fig. 1C), ECM degradation/remodeling (Fig. 1D) and stemness (Fig.1E; Fig. S2A).
Fig. 1.

ASPH activates Notch signaling pathway in PC.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A-B) Expression profile of Notch signaling major components in response to SMI and DAPT, respectively.
(C) 3D tumor spheroid invasion in response to DAPT.
(D) ECM degradation/remodeling in response to DAPT.
(E) 3D pancreatosphere formation in response to DAPT.
(F) Scheme of In Vitro Metastasis assay of PC cells, which mimics local invasion (penetration through basement membrane) at the primary site, intravasation/extravasation, invasion into distant tissue and eventual metastatic colonization/outgrowth at distant sites.
(G) Transendothelial migration and intravasation/extravasation.
(H) Invasion through basement membrane and subsequent pancreatosphere formation in response to DAPT.
We newly developed in vitro metastasis assay (Fig. 1F) to mimic how PC cells invade through basement membrane at primary site, subsequently intravasate into/extravasate out of vasculature system (transendothelial migration), consequently invade through basement membrane and form metastatic colonization/outgrowth (pancreatosphere formation) at distant sites [21,22]. Endogenous ASPH induced migration/invasion (Fig. S1G–J), EMT (downregulation of E-Cadherin and upregulation of Vimentin, Fig.S1L–N), 3D invasion (Fig. S1O), ECM degradation/remodeling (Fig. S1P–Q), stemness (upregulation of cancer stem cell marker CD44 or EpCAM [Fig. S2B–D] and enhanced tumor sphere formation [Fig. S2E–F]) and in vitro metastasis (Fig. 1G and H; Fig. S2G–H) were impaired by ASPH KO or DAPT.
4.2. ASPH potentiates PC cells to secrete exosomes delivering prooncogenic/pro-invasive cargoes
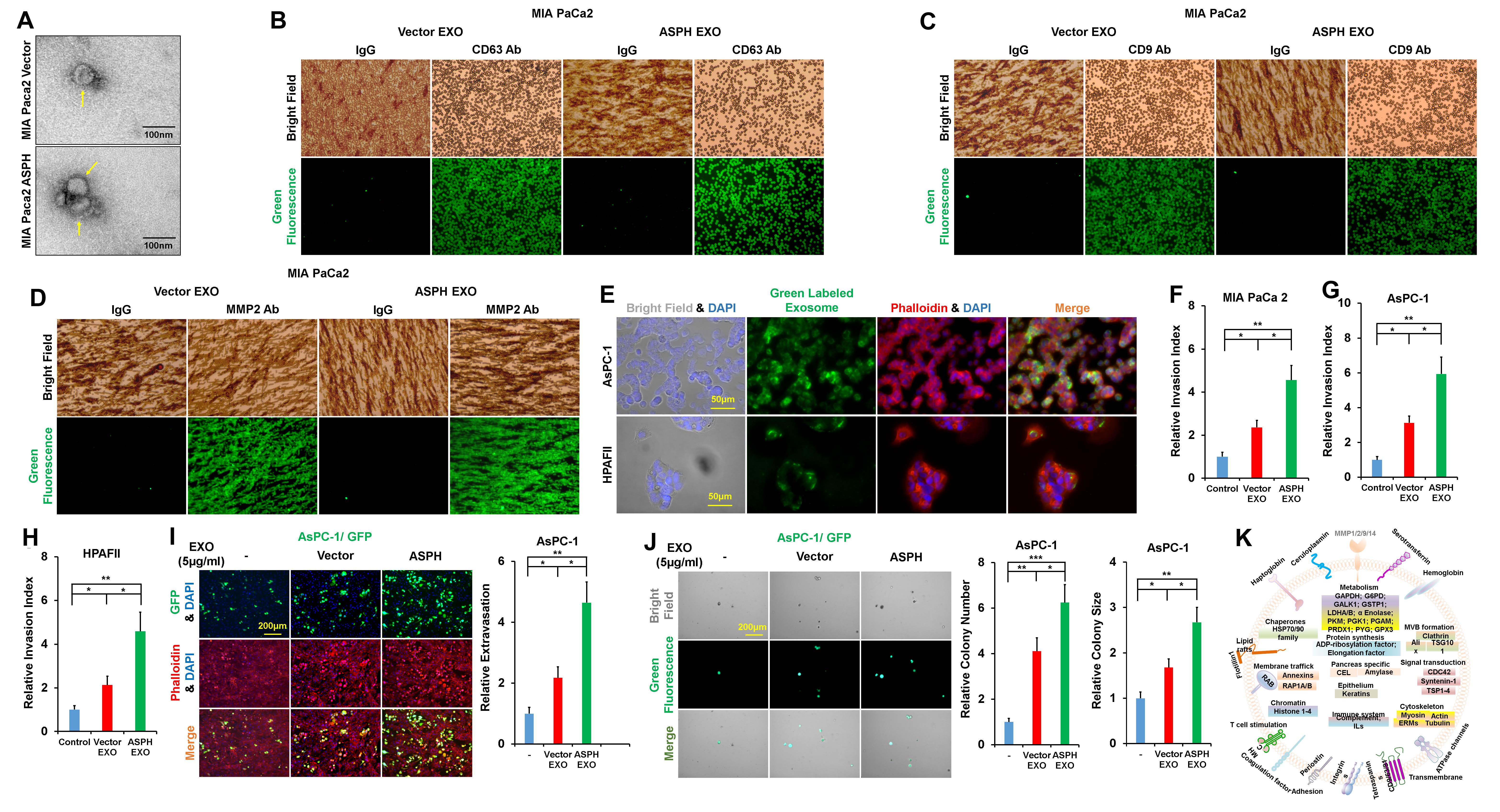
Notch signaling pathway is pivotal for exosomes secretion/biological activity [23,24]._ENREF_23_ENREF_60 We hypothesize ASPH-Notch axis stimulates PC cells to generate pathologic exosomes and thus reprogram cellular invasiveness. Exosomes were extracted and purified from PC cell lines with ultracentrifugation and characterized by density at 1.10–1.12 g/mL on sucrose gradients, morphology under transmission electron microscopy (Fig. S3A), and expression of CD9/CD63, MMP2/MMP9 (Fig. S3B–D).
Exosomes secreted by donors were avidly taken up by recipients (Fig. S3E). Docking station of exosomes was substantiated to ECM degradation sites under immunofluorescent microscopy as demonstrated by co-localization of exosome marker CD63 with (matrix metalloproteinase 2) MMP2 (Fig. 2A). Donors transferred specific exosomes to recipients, to render them with aggressive phenotypes: invasion (Fig. S3F–H), ECM degradation and pancreatosphere formation. Exosomes secreted by MIA-Paca2 overexpressing ASPH dramatically strengthened tube formation (in vitro analysis of angiogenesis) (Fig. 2B) in vitro metastasis (Fig. 2C and D; Fig. S3I–J) of parental PC cells.
Fig. 2.
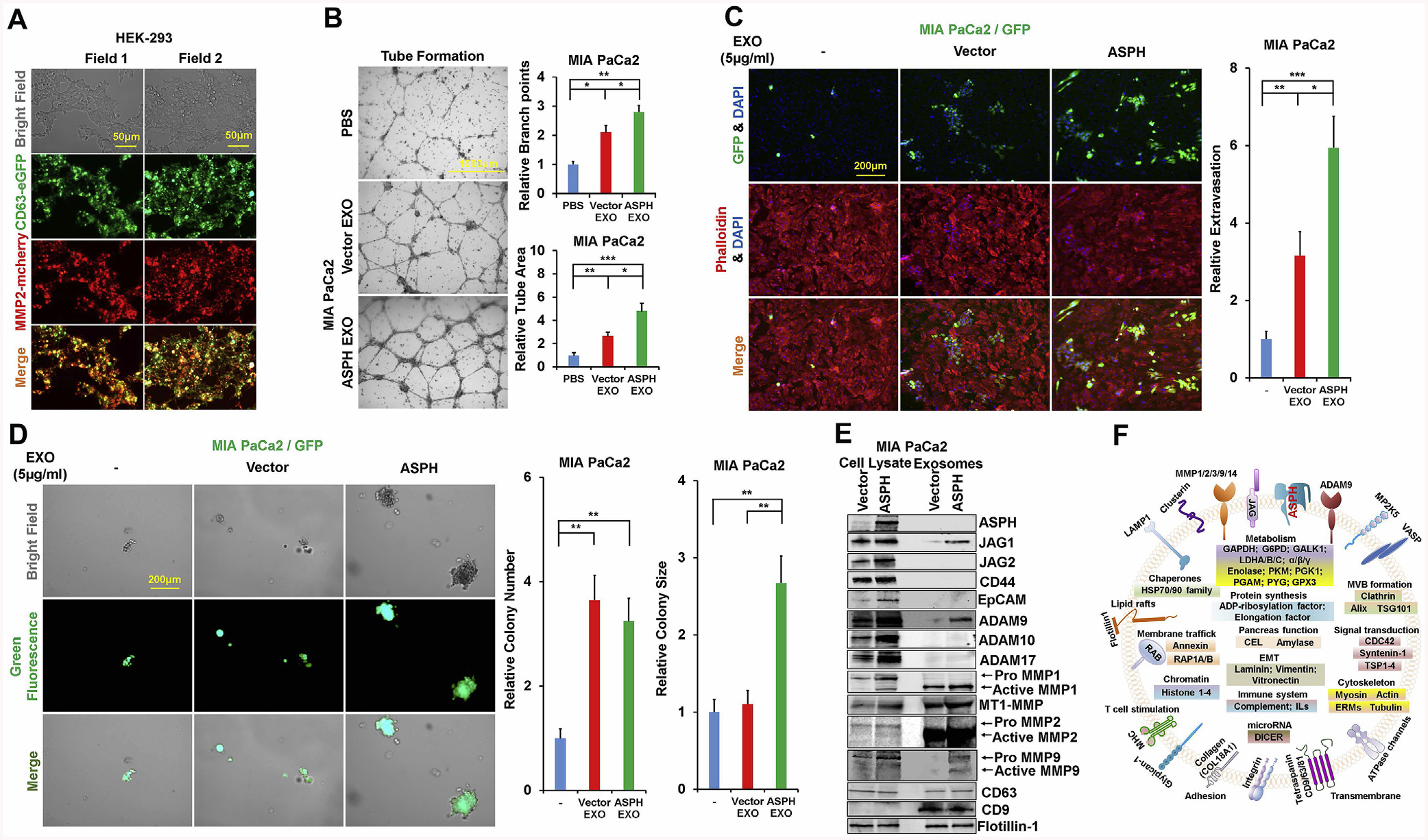
ASPH guides PC cells to secrete pro-oncogenic/pro-metastatic exosomes and acquire more malignant phenotypes.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A) Co-localization of exosomal marker CD63 and ECM degradation machinery MMP2.
(B) Tube formation of parental PC cells incubated with exosomes secreted by MIA PaCa2 cells overexpressing empty vector and ASPH, respectively.
(C) Transendothelial migration and extravasation.
(D) Invasion through basement membrane and subsequent pancreatosphere formation of parental PC cells incubated with exosomes secreted by MIA Paca2 cells overexpressing empty vector and ASPH, respectively.
(E) Compared to empty vector, the exosomes released by MIA Paca2 cells stably expressing ASPH exhibited enrichment of Notch signaling components.
(F) Representative protein cargoes carried by exosomes released by MIA Paca2 stably expressing ASPH as deciphered with proteomics using Mass Spectrometry.
Compared to empty vector (Fig. S3K), ASPH promoted MIA-Paca2 to secrete exosomes carrying pro-invasive/pro-metastatic (Fig. 2E) and immunosuppressive elements (Fig. 2F). Differential gene expression (protein levels) between MIA-ASPH and MIA-Vector has been deciphered, compared and annotated. Representative examples of major components possibly involved in ASPH-mediated aggressive malignant phenotypes are presented as a clue for further investigation. Neutral sphingomyelinase 2 (N-SMase 2) is essential for exosomes synthesis/release [25]. ASPH guided exosomes may confer invasiveness depending partially on N-SMase activity [26]. ASPH mediated migration and/or invasion (Fig. S4A–C) and ECM degradation (Fig. 3A; Figs. S4D and F) were reduced by GW4869 (specific non-competitive inhibitor of N-SMase). Notably, this inhibitory effect of GW4869 was rescued by re-addition of exosomes secreted by MIA-Paca2 expressing ASPH (Fig.3B; Figs. S4E and G). ASPH mediated 3D invasion (Fig.3C; Fig. S4H), stemness (tumor sphere formation Fig.3D; Fig. S4I–J) and in vitro metastasis (Fig. 3E and F; Fig. S4K–L) were also attenuated by GW4869. Notably, synthesis/release of exosomes is undermined by GW4869 treatment (Fig. 3G).
Fig. 3.
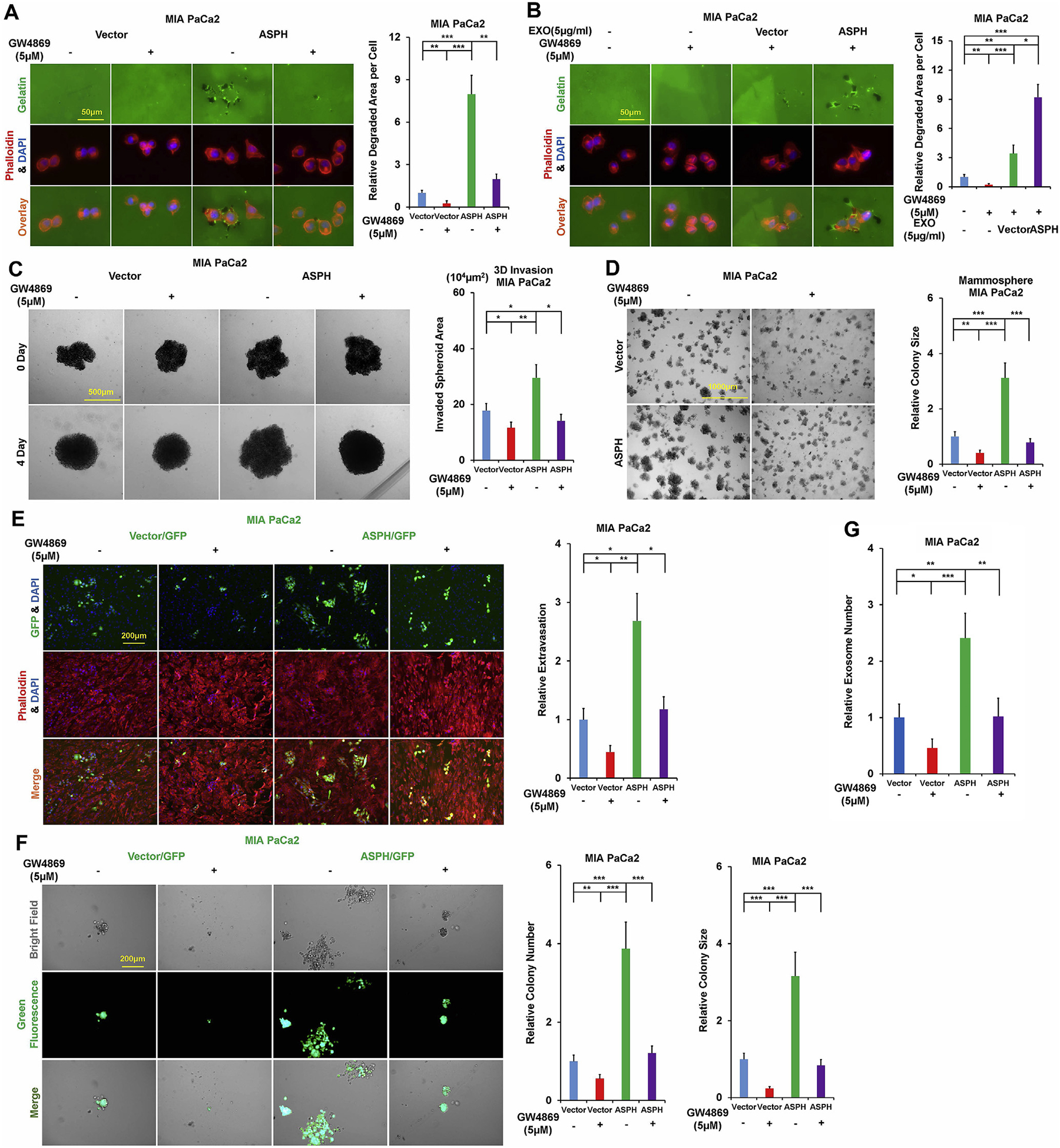
Exosomes contribute to ASPH mediated aggressive malignant phenotypes in PC, which are significantly attenuated in vitro by N-SMase inhibitor GW4869.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A) ECM degradation/remodeling in response to GW4869.
(B) GW4869 blocks ECM degradation/remodeling of parental PC cells, which could be completely rescued by addition of exosomes secreted by MIA Paca2 cells stably expressing ASPH (vector to a much less extent).
(C) 3D tumor spheroid invasion in response to GW4869.
(D) 3-D pancreatosphere formation in response to GW4869.
(E) Transendothelial migration and intravasation/extravasation.
(F) Invasion through basement membrane and subsequent pancreatosphere formation in response to GW4869.
(G) Synthesis/release of exosomes in response to GW4869.
4.3. Exosomes act as outlets of MMPs in PC
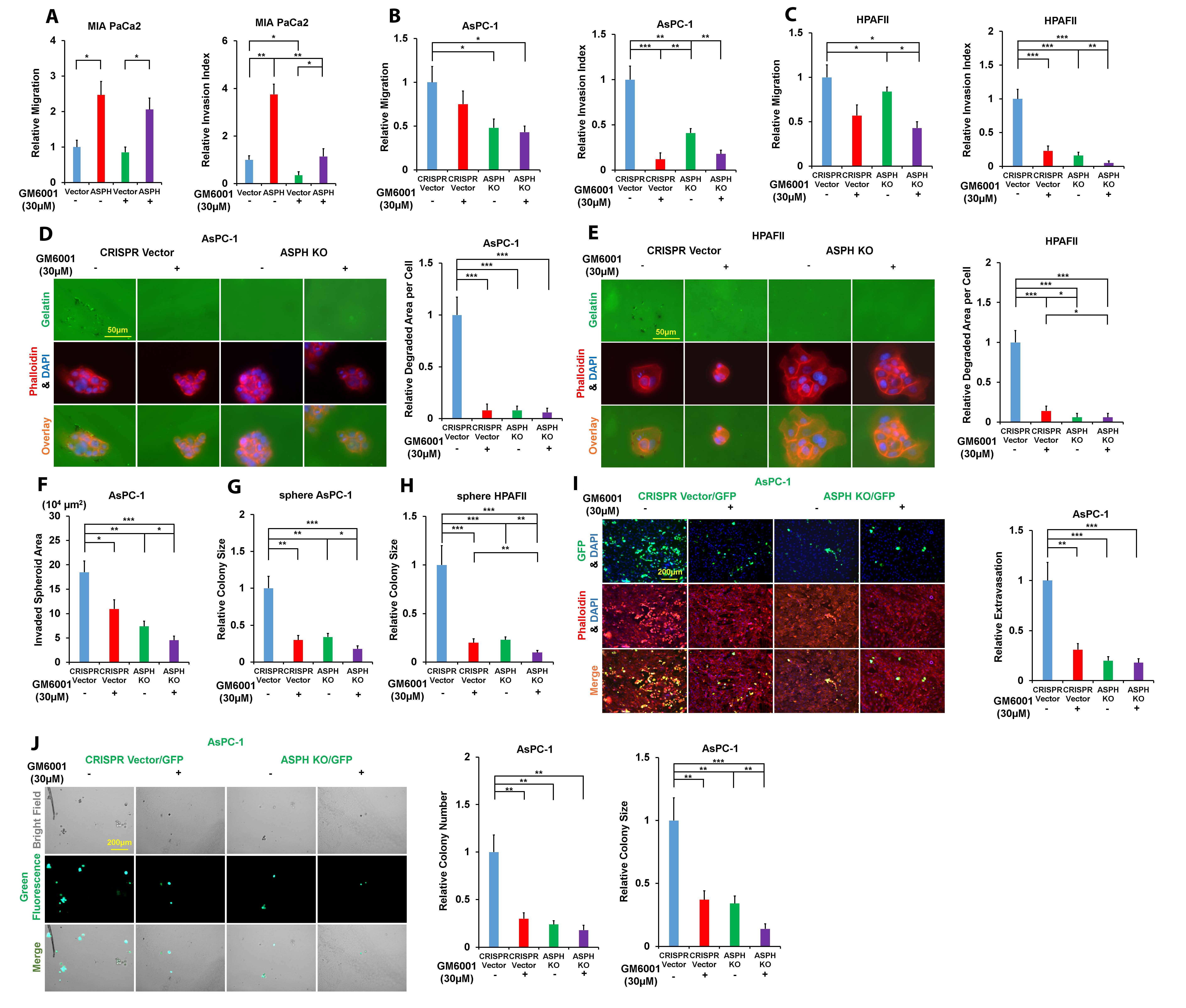
ASPH activates Notch signaling to stabilize ADAMs and upregulate downstream MMPs, with an outlet of being packaged into exosomes. We hypothesize exosomal MMPs are executors for ASPH mediated malignant phenotype. ASPH enhanced migration/invasion (Fig. S5A–C), ECM degradation (Fig. 4A; Fig. S5D–E), 3D invasion (Fig.4B; Fig. S5F), stemness (Fig.4C; Fig. S5G–H) and in vitro metastasis (Fig. 4D and E; Fig. S5I–J) were inhibited by GM6001, a broad-spectrum MMPs family inhibitor.
Fig. 4.

Exosomes function as outlets of MMPs production; MMPs act as executors for exosomes attributed to ASPH-induced cellular behaviors, which are undermined in vitro by pan-MMPs inhibitor GM6001.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A) ECM degradation/remodeling in response to GM6001.
(B) 3D tumor spheroid invasion in response to GM6001.
(C) 3D pancreatosphere formation in response to GM6001.
(D) Transendothelial migration and extravasation.
(E) Invasion through basement membrane and subsequent pancreatosphere formation in response to GM6001.
4.4. ASPH promotes PC metastasis in vivo
The demographic/pathologic characteristics of selected patients with PDAC have been detailed recently [21]. Three representative PDX tumors from F0 generation NSG mice derived from 3 PDAC patients were serially propagated from F1 to F7 generation. Case #3 (Patient B) had spontaneously developed pulmonary metastasis whereas Case #1 (Patient A) and Case #6 (Patient C) were free from detectable metastases. The original histologic architecture of PDAC was bona fide preserved in F1–F4 PDX mice, where transformed glandular epithelium was partially enclosed by a dense desmoplastic stroma (Fig. 5A; Fig. S6A–D). Glandular cellularity was gradually increased in PDX mice following serial passages. ASPH expression in the original tumor derived from PDAC patients was continuously recapitulated for > 56 weeks (Fig.5B; Fig. S6A–D). Patient B with pulmonary metastasis transmitted this phenotype faithfully to subsequent PDX model from F1 to F7 generation (Fig. 5A and B; Fig. S6E–F). 100% of PDX mice spontaneously developed macro-/micro-pulmonary metastases, where ASPH expression was authentically maintained. No pulmonary metastasis was detected in F1–F7 PDX mice derived from Patient A and C.
Fig. 5.
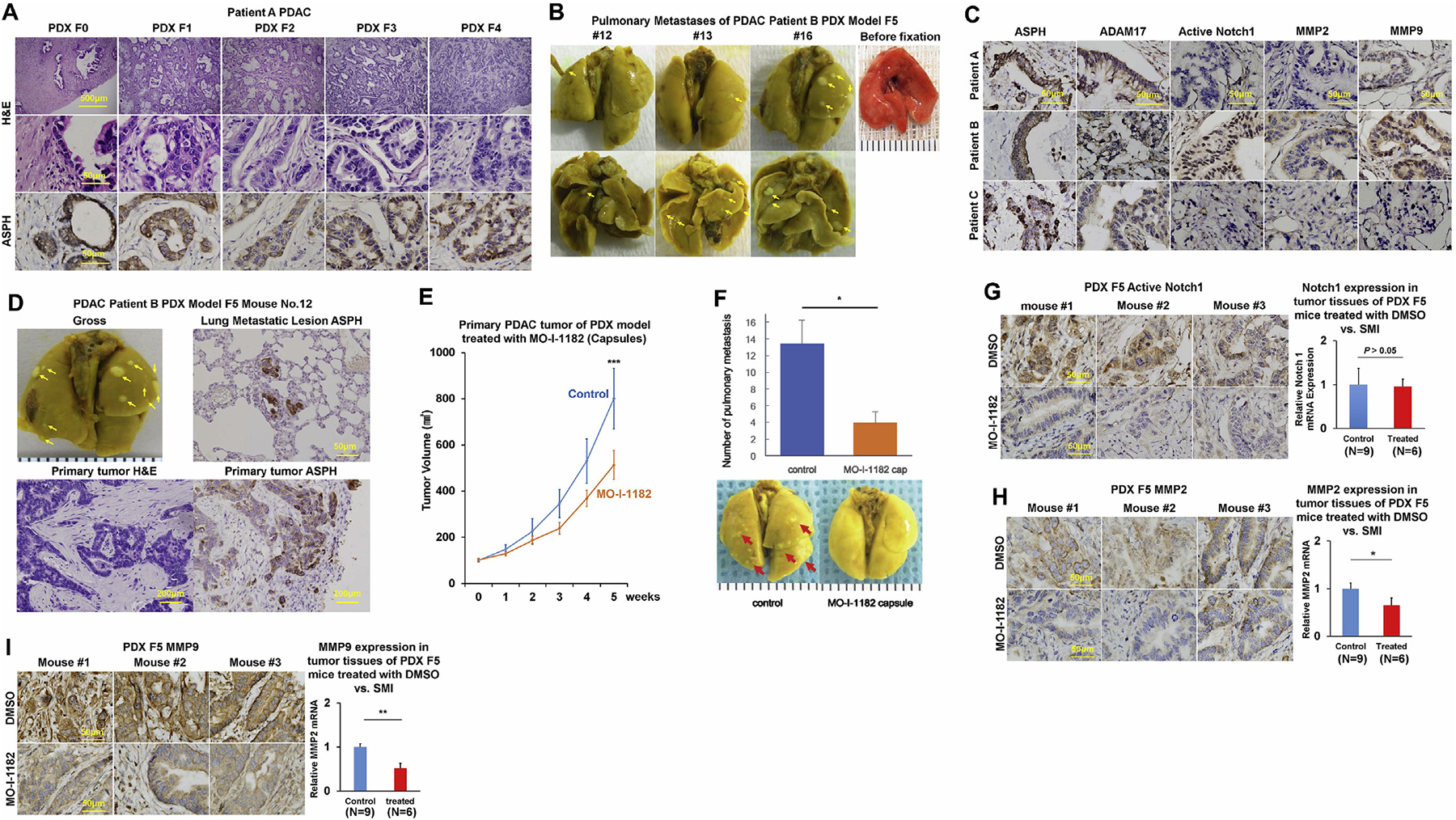
In vivo antitumor effects of a 3rd generation SMI (MO-I-1182) targeting ASPH enzymatic activity on PDX murine models of human PDAC.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A) Histopathologic characteristics (H&E) of original tumors (F0) derived from a representative PDAC Patient A and xenografted tumors in representative mice of F1 through F4 generation PDX model.
(B) Pulmonary macro-metastases (arrows) in representative F5 PDX mice (n = 16) derived from Patient B. 100% (16/16) of the F5 generation PDX mice derived from Patient B had spontaneously developed pulmonary metastases.
(C) Expression profiling of ASPH-Notch components in resected primary PDAC tumor specimens derived from Patients A, B and C.
(D) Gross appearance of the involved lungs, histopathologic characteristics (H&E) and expression profiling of ASPH in transplanted primary tumors as well as pulmonary macro-metastases (arrows) in a representative mouse of F5 generation PDX model derived from PDAC Patient B.
(E) Transplanted primary tumor growth in mice of F5 generation PDX model in response to orally formulated SMI vs. DMSO control. It took 4–5 weeks for the transplanted tumors to grow up to 100 mm3, when treatment with MO-I-1182 (10 mg/kg, orally, every other day) was initiated. The mice were followed up for 5 weeks until the tumors grew up to 1000 mm3.
(F) Antitumor effects of the SMI on pulmonary metastasis administered with orally formulated (capsules) MO-I-1182 blocks pulmonary micro-/macro-metastases (arrows) in mice of F5 generation PDX derived from Patient B.
(G-I) Expression profiling of ASPH, Active Notch1 and MMPs detected by IHC and qRT-PCR in representative mice of F5 generation PDX model treated with SMI vs. DMSO control.
4.5. ASPH activates Notch signaling pathway in vivo
To evaluate if ASPH activates Notch cascade in vivo, expression profiling of ASPH network was measured in F4–F7 generation PDX tumors. All 3 patients exhibited ASPH positive PDX tumors. Only Case #3 (Patient B) with transmissible spontaneous pulmonary metastasis displayed markedly activated Notch1, upregulation of regulators ADAM17 and downstream MMPs (Fig. 5C and D; Fig. S6G). In Patient A and C, the full-length Notch1 was not present. Thus, no active form of Notch1 was detected and downstream target genes of Notch1 were not upregulated, either, as expected. Tumor development/progression were accelerated with serial passages in PDX model. Spontaneous pulmonary metastasis is attributable to activation of Notch cascade in primary PDX tumor [7,27–29]. Notch signaling components were consistently upregulated or downregulated in ASPH positive vs. negative tumors in 10 PDAC specimens randomly selected from pathology archives at Rhode Island Hospital.
4.6. A SMI specifically targeting ASPH enzymatic activity inhibits primary tumor growth and pulmonary metastasis
When sacrificed at 5 weeks later, the animals from DMSO group developed significantly larger tumor volume (at the back) and 3.5-fold more metastatic lesions in their lungs than those in SMI-treated group. Thus, both primary PDAC tumor growth and pulmonary metastases were substantially inhibited by orally formulated preparation of SMI (Fig. 5E–F).
4.7. Downregulation of Notch signaling pathway in response to SMI treatment
To validate molecular mechanisms discovered from in vitro system, tumor tissue derived from F5 generation PDX model of Patient B treated with DMSO (control) vs. SMI was examined for expression profiling of ASPH network. ASPH levels are comparable because the SMI inhibits its enzymatic activity but not transcription. Likewise, Notch1 mRNA was unchanged. Significant reduction in activated Notch1 protein was observed only in the SMI treated tumors. As downstream target genes regulated by Notch1, MMPs expression was reduced at both mRNA and protein levels (Fig. 5G–I). Therefore, inhibition of ASPH’s enzymatic activity reduces activation of Notch cascade, and downstream MMPs gene expression.
4.8. Expression profiling of ASPH network predicts clinical outcome of PDAC patients
ASPH-Notch axis functions in PDAC patients, as illustrated by differential expression between tumor and adjacent non-malignant pancreatic tissue. The demographic/clinical features of study population (N = 166) have been summarized recently [21]. ASPH was detected in 97.6% of PDAC patients, with a negative, low, moderate, high or very high expression rate of 2.4%, 22.9%, 23.5%, 27.7%, and 23.5%, respectively. ASPH was undetectable in adult normal pancreas (Fig. 6A), inflammatory diseases (acute/chronic pancreatitis) or pancreatic neuroendocrine tumor [7]; upregulated at early stage preinvasive pancreatic neoplasm including PanIN (pancreatic intraepithelial neoplasia), IPMN (intraductal papillary mucinous neoplasm) and MCN (mucinous cystic neoplasm) (Fig. 6B); whereas markedly expressed in invasively advanced/spontaneously metastatic PC (Fig. 6C), compared to adjacent non-malignant pancreas (Fig. 6D).
Fig. 6.
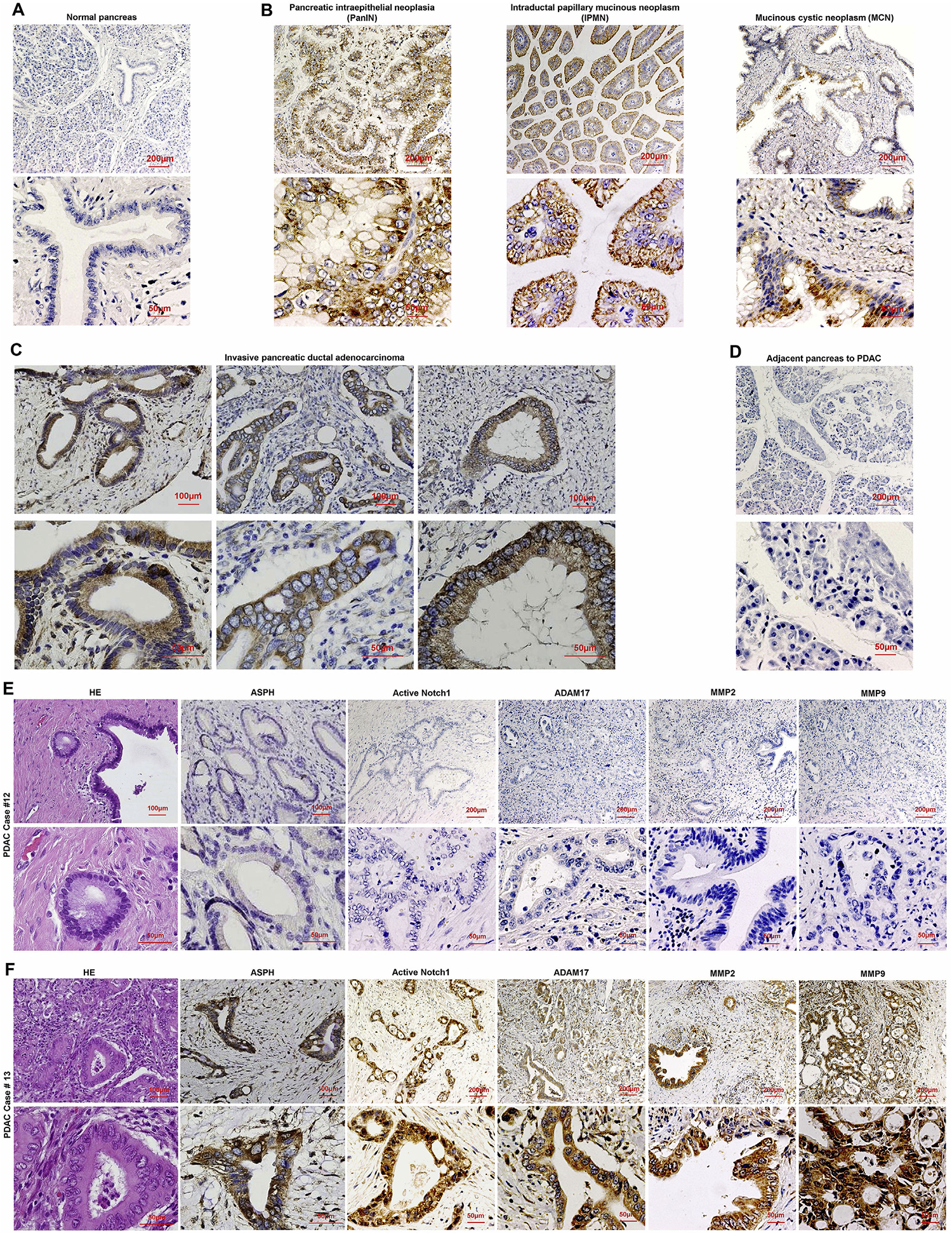
Major elements of ASPH-Notch axis act synergistically in PC pathogenesis.
(A-D) Expression of ASPH in (A) normal pancreas; (B) precursor lesions for sporadic PC: pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN); (C) invasive PDAC; (D) adjacent non-malignant pancreas.
(E-F) Histopathologic characteristics and ASPH network expression profiling in 2 representative tumors derived from PDAC patients. Notch pathway was consistently (E) downregulated/silenced vs. (F) upregulated/activated Notch pathway in ASPH negative (Patient #12) vs. positive (Patient #13) PDAC.
Notch network components were consistently downregulated or upregulated in ASPH negative vs. positive PDAC patients. ASPH was moderately-(very) strongly expressed in poorly differentiated more aggressively tumors, whereas negatively-weakly expressed in moderately-well differentiated less invasive tumors (Fig. 6E–F; Fig. S7A–F). ASPH level positively correlated with active Notch1, MMPs, and ADAM17 (r ≥ 0.6, p < 0.001, 2-sided) (Fig. 7A–D). Active Notch1 expression positively correlated with ADAM17 and MMPs (Fig. 7E–G).
Fig. 7.

Expression levels of ASPH-Notch axis elements predict clinical outcome of PDAC patients.
*p < 0.05; **p < 0.01; ***p < 0.001.
(A-D) ASPH expression level positively correlated with Notch signaling pathway components levels.
(E-G) Active Notch1 expression level positively correlated with MMPs and ADAM17 levels.
(H-L) Compared to a negative-low level, a moderate-high level of ASPH; active Notch1; ADAM17; MMP2/9 expression conferred reduced OS of PC patients (log-rank test, Ps < 0.001).
(M-N) Combined effects of 4 (Notch axis) or 5 (ASPH-Notch network) molecules on OS of PC patients using Kaplan-Meier method. The numbers from 0 to 12 indicate the total expression scores of at-risk proteins (log-rank test, p < 0.001).
(O) Working hypothesis for ASPH guided exosomes as an executor for initiating aggressive malignant phenotypes of pancreatic cancer.
ASPH network level predicts prognosis of PDAC patients based on Kaplan–Meier plots and Cox proportional hazards regression models. Compared to a negative-low level, a moderate-very high level of ASPH, activated Notch1, ADAM17, MMPs correlated with curtailed overall survival (OS) of PC patients (log-rank test, Ps < 0.001) (Fig. 7H–L). Individual molecules jointly undermined OS when combing the unfavorable expression scores significantly associated with reduced survival in multivariate models. Increased numbers/expression levels of 5 unfavorable molecules conferred reduced OS [21] (Fig. 7M–N; Table S1). Patients with 0–1, 2–4, 5–7, and 8–12 unfavorable scores had median survival time of 38.0, 19.7, 10.4, and 5.0 months, respectively (P < 0.001). Compared to 0–1, for patients carrying 2–4, 5–7, or 8–12 unfavorable scores, adjusted hazard ratio (HR) (95% confidence interval) was 2.94 (1.90–4.56), 7.76 (4.72–12.77), and 14.29 (5.94–34.41), respectively (P < 0.001).
5. Discussion
ASPH promotes PC progression through activating Notch signaling pathway [7,21]. ASPH-Notch axis guides tumor cells to secrete prooncogenic/pro-metastatic exosomes [22], to strengthen ECM degradation, invasion, stemness, angiogenesis (tube formation), transendothelial migration (intravasation/extravasation), and metastatic colonization/outgrowth at distant sites (e.g., lungs) (Fig. 8). At-risk ASPH-Notch axis elements independently/synergistically confer worse clinical outcome of PC patients in dose-/intensity-dependent manners. Patient’s OS was decreased with their increased numbers/expression levels. Thus, ASPH-Notch network components serve as prognostic factors for PDAC.
Fig. 8.

Hypothesized mechanisms underlying how ASPH-Notch axis is critically involved in pathogenesis of pancreatic cancer.
ASPH stabilizes Notch receptors [12,13], ligands JAGs and regulators ADAMs, enhances Notch receptors-ligands interactions, activates Notch cascade and upregulates downstream target genes responsible for EMT, angiogenesis, stemness, invasion and metastasis. ASPH mediated aggressive phenotype is attenuated by inhibiting Notch signaling with DAPT.
Exosomes secreted autocrinally/paracrinally by tumor and microenvironment spur tumor-induced immunosuppression, angiogenesis and pre-metastatic niche formation [30,31]. ASPH enhanced bio-synthesis/release of characteristic exosomes probably through upregulating RAB proteins as indicated by proteomics. RAB10 regulates tubule formation, fusion events [32],_ENREF_35 and exocytosis (intracellular vesicle docking, trafficking and transport). RAB11 spurs docking and fusion of MVBs to cell membrane in response to increased cytosolic calcium [33]. ASPH may guide the more malignant donor cells to secrete exosomes delivering designated cargos, and thus to reprogram biological processes of recipient cells. Notably, ASPH secreted by cancer cells and delivered by exosomes can be detected in biofluids (e.g., plasma/serum). A highly sensitive immunoassay using serum samples has been developed, with an accuracy of 93.8%, demonstrating that ASPH/HAAH is a biomarker for early diagnosis of multiple-cancer [34]. As a potential pan-cancer biomarker, ASPH has been enriched in exosomes derived from cancer patients. This novel assay is performing in a large patient population to establish ASPH as a biomarker for pancreatic cancer diagnosis and prognosis.
ASPH mediates ECM degradation and altered morphology in 3-D culture. MMPs upregulated by Notch signaling and ADAMs stabilized by ASPH are critical elements for pro-oncogenic/pro-metastatic exosomes secretome_ENREF_33. MMPs act as outlets of Notch cascade and executioners for ECM degradation to facilitate invasion and metastasis of PC cells. Exosomes are proposed to educate microenvironment to promote PC pulmonary metastatic colonization/outgrowth.
It’s challenging to develop drugs targeting metastasis due to lack of suitable animal models that realistically mimic human tumor histopathology. We have established a PDX model of human PDAC that spontaneously metastasizes to murine lungs from subcutaneous xeno-graft. This phenotype faithfully recapitulated histopathological/clinical characteristics of original tumor derived from a specific patient; and was serially propagated in 100% of mice from F2 to F7 generation. ASPH activates Notch1 cascade [35,36] in the primary PDAC tumor, upregulates downstream target genes (e.g., MMPs) and drives multiple-steps of metastasis. Specific SMI inhibiting ASPH enzymatic activity substantially disrupts primary tumor growth and impedes pulmonary metastasis.
ASPH-Notch axis functions in randomly selected PDAC patients. Expression levels of ASPH-Notch axis components independently predict prognosis of PDAC. Carrying more unfavorable molecules confers poorer OS. ASPH is silenced in adult normal pancreas, whereas upregulated at premalignant pancreatic lesions and highly expressed in advanced or spontaneously metastatic PC. The Notch signaling elements are consistently downregulated or upregulated in ASPH negative vs. positive PC patients. Individual components of ASPH-Notch axis exert joint effects on reduced OS in a dose-/intensity-dependent manner. ASPH may serve as a biomarker for early diagnostics and prognostics of PC.
ASPH may play preeminent roles in different events of PC metastasis, from local invasion, intravasation, survival in circulation, extra-vasation, to metastatic colonization/outgrowth. The SMIs bind to the catalytic site of ASPH, significantly decrease β-hydroxylase activity [7,14,37–39], subsequently block pathobiological processes as acterized by diminished activation of Notch signaling pathway. When ASPH turns on Notch signal, designated exosomes are released to pare pre-metastatic niche; upon treatment with SMIs, exosomal chinery is shut-down and pro-oncogenic/pro-metastatic properties versed. This study together with previous findings [4,5,7,8,10,11,16,38–44] establish ASPH as a therapeutic target for PC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Pharmaceutical Sciences, College of Pharmacy-Glendale, Midwestern University, Glendale (Arizona 85308, USA) for synthesizing, structural characterization and providing data on the purity of these small molecule inhibitors (SMIs) of ASPH (including MO-I-1182); Laboratory for Molecular Biology and Cytometry Research at the University of Oklahoma Health Sciences Center for the use of the Core Facility which provided Mass Spectrometry/Proteomics service; Dr. Iannis Aifantis at NYU School of Medicine for providing with pcDNA3 NOTCH1 full-length plasmid.
Funding resources
This work was supported in part by National Institutes of Health (NIH) grant CA123544, P30GM110759 and Institutional Funds of United States (to J.R.W and X.Q.D.); National Natural Science Foundation of China (Grant No. 81670583 to B.S.).
Abbreviations
- 2-D
two-dimensional
- 3-D
three-dimensional
- ADAM
A Disintegrin and Metalloprotease Domain
- ASPH
Aspartate β-hydroxylase
- BSA
bovine serum albumin
- co-IP
co-immunoprecipitation
- DLL
Delta-like
- ECD
extracellular domain
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EMT
epithelial–mesenchymal transition
- FBS
fetal bovine serum
- FFPE
formalin-fixed paraffin embedded
- GFR
growth factor reduced
- HCC
hepatocellular carcinoma
- H&E
hematoxylin and eosin
- HR
hazard ratio
- HUVEC
human umbilical vein/vascular endothelium
- HydroX
Aspartyl/Asparaginyl β-hydroxylase domain
- IACUC
institutional animal care and use committee
- IHC
immunohistochemistry
- IPMN
intraductal papillary mucinous neoplasm
- JAG
Jagged
- KO
knockout
- ICD
intracellular domain
- MCN
mucinous cystic neoplasm
- MMP
matrix metalloproteinase
- MVBs
multivesicular bodies
- NSG
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; NOD-scid IL2Rgnull
- N-SMase
Sphingomyelin phosphodiesterase
- OS
overall survival
- PanIN
pancreatic intraepithelial neoplasia
- PC
pancreatic cancer
- PDAC
pancreatic ductal adenocarcinoma
- PDX
patient-derived xenograft
- PFA
paraformaldehyde
- RAB
Member RAS Oncogene Family
- SMI
small molecule inhibitor
Footnotes
Declarations of competing interest
The authors declare no competing interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2020.02.039.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics CA A Cancer J. Clin 69 (2019) 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- [2].Dinchuk JE, Henderson NL, Burn TC, Huber R, Ho SP, Link J, O’Neil KT, Focht RJ, Scully MS, Hollis JM, Hollis GF, Friedman PA, Aspartyl beta -hydroxylase (Asph) and an evolutionarily conserved isoform of Asph missing the catalytic domain share exons with junctin, J. Biol. Chem 275 (2000) 39543–39554. [DOI] [PubMed] [Google Scholar]
- [3].Artavanis-Tsakonas S, Rand MD, Lake RJ, Notch signaling: cell fate control and signal integration in development, Science 284 (1999) 770–776. [DOI] [PubMed] [Google Scholar]
- [4].Ince N, de la Monte SM, Wands JR, Overexpression of human aspartyl (asparaginyl) beta-hydroxylase is associated with malignant transformation, Canc. Res 60 (2000) 1261–1266. [PubMed] [Google Scholar]
- [5].Lavaissiere L, Jia S, Nishiyama M, de la Monte S, Stern AM, Wands JR, Friedman PA, Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma, J. Clin. Invest 98 (1996) 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang K, Liu J, Yan ZL, Li J, Shi LH, Cong WM, Xia Y, Zou QF, Xi T, Shen F, Wang HY, Wu MC, Overexpression of aspartyl-(asparaginyl)-beta-hydroxylase in hepatocellular carcinoma is associated with worse surgical outcome, Hepatology 52 (2010) 164–173. [DOI] [PubMed] [Google Scholar]
- [7].Dong X, Lin Q, Aihara A, Li Y, Huang CK, Chung W, Tang Q, Chen X, Carlson R, Nadolny C, Gabriel G, Olsen M, Wands JR, Aspartate beta-Hydroxylase expression promotes a malignant pancreatic cellular phenotype, Oncotarget 6 (2015) 1231–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, de la Monte SM, Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) beta-hydroxylase suppress migration of cholangiocarcinoma cells, J. Hepatol 38 (2003) 615–622. [DOI] [PubMed] [Google Scholar]
- [9].Sepe PS, Lahousse SA, Gemelli B, Chang H, Maeda T, Wands JR, de la Monte SM, Role of the aspartyl-asparaginyl-beta-hydroxylase gene in neuroblastoma cell motility, Laboratory investigation, J. Tech. Methods Pathol 82 (2002) 881–891. [DOI] [PubMed] [Google Scholar]
- [10].Luu M, Sabo E, de la Monte SM, Greaves W, Wang J, Tavares R, Simao L, Wands JR, Resnick MB, Wang L, Prognostic value of aspartyl (asparaginyl)-beta-hydroxylase/humbug expression in non-small cell lung carcinoma, Hum. Pathol 40 (2009) 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang J, de la Monte SM, Sabo E, Kethu S, Tavares R, Branda M, Simao L, Wands JR, Resnick MB, Prognostic value of humbug gene overexpression in stage II colon cancer, Hum. Pathol 38 (2007) 17–25. [DOI] [PubMed] [Google Scholar]
- [12].Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, Wands JR, Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms, Hepatology 44 (2006) 446–457. [DOI] [PubMed] [Google Scholar]
- [13].Chung W, Kim M, de la Monte S, Longato L, Carlson R, Slagle BL, Dong X, Wands JR, Activation of signal transduction pathways during hepatic oncogenesis, Canc. Lett 370 (2016) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aihara A, Huang CK, Olsen MJ, Lin Q, Chung W, Tang Q, Dong X, Wands JR, A cell-surface beta-hydroxylase is a biomarker and therapeutic target for hepatocellular carcinoma, Hepatology 60 (2014) 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wands JR, Kim M, WNT/beta-catenin signaling and hepatocellular carcinoma, Hepatology 60 (2014) 452–454. [DOI] [PubMed] [Google Scholar]
- [16].Tomimaru Y, Koga H, Yano H, de la Monte S, Wands JR, Kim M, Upregulation of T-cell factor-4 isoform-responsive target genes in hepatocellular carcinoma, Liver Int. : Off. J. Int. Assoc. Study Liver 33 (2013) 1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, Reissfelder C, Pilarsky C, Fraga MF, Piwnica-Worms D, Kalluri R, Glypican-1 identifies cancer exosomes and detects early pancreatic cancer, Nature 523 (2015) 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C, Microvesicles: mediators of extracellular communication during cancer progression, J. Cell Sci 123 (2010) 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, Ostrowski M, Thery C, Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression, Canc. Res 72 (2012) 4920–4930. [DOI] [PubMed] [Google Scholar]
- [20].Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D, Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET, Nat. Med 18 (2012) 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ogawa K, Lin Q, Li L, Bai X, Chen X, Chen H, Kong R, Wang Y, Zhu H, He F, Xu Q, Liu L, Li M, Zhang S, Nagaoka K, Carlson R, Safran H, Charpentier K, Sun B, Wands J, Dong X, Aspartate beta-hydroxylase promotes pancreatic ductal adenocarcinoma metastasis through activation of SRC signaling pathway, J. Hematol. Oncol 12 (2019) 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin Q, Chen X, Meng F, Ogawa K, Li M, Song R, Zhang S, Zhang Z, Kong X, Xu Q, He F, Bai X, Sun B, Hung MC, Liu L, Wands J, Dong X, ASPH-notch Axis guided Exosomal delivery of Prometastatic Secretome renders breast Cancer multi-organ metastasis, Mol. Canc 18 (2019) 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ, Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways, Cell 159 (2014) 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D, Tumour exosome integrins determine organo-tropic metastasis, Nature 527 (2015) 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shamseddine AA, Airola MV, Hannun YA, Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes, Adv. Biol. Regul 57 (2015) 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ogretmen B, Hannun YA, Biologically active sphingolipids in cancer pathogenesis and treatment, Nat. Rev. Canc 4 (2004) 604–616. [DOI] [PubMed] [Google Scholar]
- [27].Brown GT, Murray GI, Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis, J. Pathol 237 (2015) 273–281. [DOI] [PubMed] [Google Scholar]
- [28].Qiu H, Tang X, Ma J, Shaverdashvili K, Zhang K, Bedogni B, Notch1 auto-activation via transcriptional regulation of furin, which sustains Notch1 signaling by processing Notch1-activating proteases ADAM10 and membrane type 1 matrix metalloproteinase, Mol. Cell Biol 35 (2015) 3622–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ma J, Tang X, Wong P, Jacobs B, Borden EC, Bedogni B, Noncanonical activation of Notch1 protein by membrane type 1 matrix metalloproteinase (MT1-MMP) controls melanoma cell proliferation, J. Biol. Chem 289 (2014) 8442–8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hoshino D, Kirkbride KC, Costello K, Clark ES, Sinha S, Grega-Larson N, Tyska MJ, Weaver AM, Exosome secretion is enhanced by invadopodia and drives invasive behavior, Cell Rep. 5 (2013) 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mathivanan S, Ji H, Simpson RJ, Exosomes: extracellular organelles important in intercellular communication, J. Proteom 73 (2010) 1907–1920. [DOI] [PubMed] [Google Scholar]
- [32].English AR, Voeltz GK, Rab10 GTPase regulates ER dynamics and morphology, Nat. Cell Biol 15 (2013) 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Savina A, Fader CM, Damiani MT, Colombo MI, Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner, Traffic 6 (2005) 131–143. [DOI] [PubMed] [Google Scholar]
- [34].Semenuk MA, Cifuentes AS, Ghanbari ER, Lebowitz MS, Ghanbari HA, Improved detection of cancer specific serum exosomal aspartyl (asparaginyl) beta hydroxylase (HAAH), Canc. Res 77 (2017) Abstract #723. [Google Scholar]
- [35].Akinleye A, Iragavarapu C, Furqan M, Cang S, Liu D, Novel agents for advanced pancreatic cancer, Oncotarget 6 (2015) 39521–39537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM, Australian I Pancreatic Cancer Genome, Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UM, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV, Grimmond SM, Genomic analyses identify molecular subtypes of pancreatic cancer, Nature 531 (2016) 47–52. [DOI] [PubMed] [Google Scholar]
- [37].Huang CK, Iwagami Y, Aihara A, Chung W, de la Monte S, Thomas JM, Olsen M, Carlson R, Yu T, Dong X, Wands J, Anti-tumor effects of second generation beta-hydroxylase inhibitors on cholangiocarcinoma development and progression, PloS One 11 (2016) e0150336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Iwagami Y, Huang CK, Olsen MJ, Thomas JM, Jang G, Kim M, Lin Q, Carlson RI, Wagner CE, Dong X, Wands JR, Aspartate beta-hydroxylase modulates cellular senescence through glycogen synthase kinase 3beta in hepatocellular carcinoma, Hepatology 63 (2016) 1213–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tomimaru Y, Mishra S, Safran H, Charpentier KP, Martin W, De Groot AS, Gregory SH, Wands JR, Aspartate-beta-hydroxylase induces epitope-specific T cell responses in hepatocellular carcinoma, Vaccine 33 (2015) 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shimoda M, Tomimaru Y, Charpentier KP, Safran H, Carlson RI, Wands J, Tumor progression-related transmembrane protein aspartate-beta-hydroxylase is a target for immunotherapy of hepatocellular carcinoma, J. Hepatol 56 (2012) 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Noda T, Shimoda M, Ortiz V, Sirica AE, Wands JR, Immunization with aspartate-beta-hydroxylase-loaded dendritic cells produces antitumor effects in a rat model of intrahepatic cholangiocarcinoma, Hepatology 55 (2012) 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].de la Monte SM, Tamaki S, Cantarini MC, Ince N, Wiedmann M, Carter JJ, Lahousse SA, Califano S, Maeda T, Ueno T, D’Errico A, Trevisani F, Wands JR, Aspartyl-(asparaginyl)-beta-hydroxylase regulates hepatocellular carcinoma invasiveness, J. Hepatol 44 (2006) 971–983. [DOI] [PubMed] [Google Scholar]
- [43].Maeda T, Taguchi K, Aishima S, Shimada M, Hintz D, Larusso N, Gores G, Tsuneyoshi M, Sugimachi K, Wands JR, de la Monte SM, Clinicopathological correlates of aspartyl (asparaginyl) beta-hydroxylase over-expression in cholangiocarcinoma, Canc. Detect. Prev 28 (2004) 313–318. [DOI] [PubMed] [Google Scholar]
- [44].Palumbo KS, Wands JR, Safran H, King T, Carlson RI, de la Monte SM, Human aspartyl (asparaginyl) beta-hydroxylase monoclonal antibodies: potential bio-markers for pancreatic carcinoma, Pancreas 25 (2002) 39–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.