

Abstract
N6-(2-Deoxy-α,β-D-erythropentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy•dG) is a major DNA lesion produced from 2’-deoxyguanosine under oxidizing conditions. Fapy•dG is produced from a common intermediate that leads to 7,8-dihydro-8-oxo-2’-deoxyguanosine (8-OxodGuo), and in greater quantities in cells. The impact of Fapy•dG on DNA structure and function is much less well understood than that of 8-OxodGuo. This is largely due to the significantly greater difficulty in synthesizing oligonucleotides containing Fapy•dG than 8-OxodGuo. We describe a synthetic approach for preparing oligonucleotides containing Fapy•dG that will facilitate intensive studies of this lesion in DNA. A variety of oligonucleotides as long as 30 nucleotides are synthesized. We anticipate that the chemistry described herein will provide an impetus for a wide range of studies involving Fapy•dG.
Keywords: DNA damage, DNA repair, DNA oxidation, oligonucleotide synthesis, formamidopyrimidines
Graphical Abstract
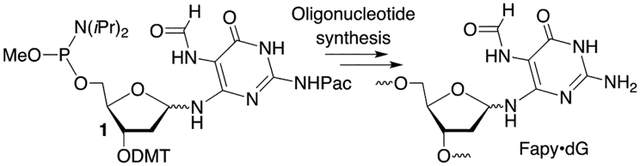
Fapy•dG is an important DNA lesion. Biochemical, cellular and structural studies on Fapy•dG have been hampered due to difficulties in synthesizing oligonucleotides containing this lesion. A robust method for synthesizing oligonucleotides containing Fapy•dG is reported that will greatly facilitate investigations on the biological and structural effects of Fapy•dG.
Introduction
DNA oxidation plays a prominent role in the etiology and treatment of diseases, as well as aging. Consequently, how individual damaged nucleotides are recognized by polymerases, glycosylases and other proteins involved in DNA repair is fundamentally important.[1] In addition, recent evidence suggests that damaged nucleotides may also play a role in regulating transcription.[2] As the most readily oxidized native nucleotide, 2’-deoxyguanosine modifications have been of significant interest.[3] Of these, 7,8-dihydro-8-oxo-2’-deoxyguanosine (8-OxodGuo), the two-electron oxidation product resulting from formal hydroxyl radical addition to the C8-position of dG (C8Add, Scheme 1) is the most well studied lesion.[4] The corresponding 2,6-diamino-4-hydroxy-5-formamidopyrimidine derived from dG (Fapy•dG) is believed to also arise from C8Add, and under some conditions is produced in greater amounts than 8-OxodGuo.[5] However, the effects of Fapy•dG on biochemical processes in the test tube and in cells, as well as nucleic acid structure, are less well understood. Obtaining knowledge about the effects of Fapy•dG is hindered due to the relative difficulty in synthesizing oligonucleotides containing this lesion. We wish to report a significant advance in synthesizing oligonucleotides containing Fapy•dG that will facilitate studies on this important DNA modification.
Scheme 1.

Formation of Fapy•dG and 8-OxodGuo.
Although formed as a consequence of oxidative stress, Fapy•dG is at the same oxidation state as dG. In fact, cyclization and dehydration of Fapy•dG is a possible biosynthetic pathway to the native nucleotide.[6] Fapy•dG is believed to result from one electron reduction of C8Add; whereas 8-OxodGuo formation requires a second oxidation step (Scheme 1).[7] The dependence of the relative levels of Fapy•dG and 8-OxodGuo on the environment in which C8Add is produced is generally consistent with this mechanism.[8] 8-OxodGuo formation is favored under O2 rich conditions, but Fapy•dG is detected at higher levels in cells. The preferential formation of Fapy•dG over 8-OxodGuo in cells is attributed to lower O2 concentration than in a test tube and the presence of reducing agents in this environment. However, a recent report described Fapy•dG formation in the test tube to require exhaustive O2 depletion in solution.[9] Fapy•dG and 8-OxodGuo have also been proposed as part of complex lesions in DNA as a result of their formation of nucleotide peroxyl radicals.[10] However, the subsequent partitioning of the peroxyl radical adduct(s) is expected to follow the same dependence as C8Add on O2.
Inferential support for the biological significance of Fapy•dG is that not only is it recognized by the proteins involved in repairing guanine oxidation (GO) products, but it is repaired in a manner to guard against introducing mutations.[3] For instance, the bacterial (Fpg) and human glycosylases (hOGG1) that selectively recognize 8-OxodGuo opposite dC versus dA act similarly on Fapy•dG. Fpg hydrolyzes the glycosidic bond of the lesion in a Fapy•dG:dC base pair almost 20-times more efficiently than when opposite dA.[11] hOGG1 is even more selective, discriminating against a Fapy•dG:dA base pair by more than 40-fold.[12] The bacterial glycosylase, MutY, incises a mismatched dA opposite Fapy•dG more rapidly than from a dG:dA mispair, but 4-fold more slowly than from a duplex containing 8-OxodGuo:dA.[11, 13] The relevance of a Fapy•dG:dA base pair is born-out by mutagenesis studies. Fapy•dG bypass in E. coli, as well as mammalian cells, gives rise to G to T transversions. The frequency of dA misincorporation opposite Fapy•dG was consistently lower in E. coli than when single-stranded plasmids containing 8-OxodGuo were replicated.[14] In mammalian cells (COS-7, HEK293T), Fapy•dG mutagenicity resulting from dA misincorporation, was much more comparable to that of 8-OxodGuo, and in some instances was greater.[15] Insight into the structural basis for Fapy•dG mutagenicity and recognition by repair enzymes have been limited to a carbacyclic analogue (carba-Fapy•dG), due to the difficulty in synthesizing oligonucleotides containing Fapy•dG.[16] Fapy•dG recognition by translesion synthesis polymerases and other repair enzymes is also not well understood. This too could be due to synthetic limitations that make it difficult for many investigators to obtain oligonucleotide substrates containing Fapy•dG.
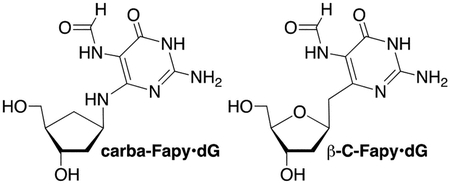
Solid-phase synthesis of oligonucleotides containing Fapy•dG is challenging due to the availability of the N6-lone pair electrons, which facilitates reversible ring opening of the deoxyribose ring (Scheme 2).[17] This results in epimerization and Fapy•dG exists as a mixture of α- and β-anomers in duplex DNA.[18] When present as a nucleoside exposure of the primary hydroxyl results in preferential isomerization to the pyranose form of Fapy•dG. Possible formation of the pyranose isomer within DNA was a major consideration when designing a method for synthesizing oligonucleotides containing Fapy•dG. Isomerization is avoided by synthesizing carbacyclic (carba-Fapy•dG) or C-nucleoside (β-C- Fapy•dG) analogues, but these model compounds may not completely recapitulate the structure of the native lesion.[19] Rizzo used conventional 3’- to 5’-solid-phase phosphoramidite chemistry to synthesize oligonucleotides containing N5-alkylated Fapy•dG variants.[20] The authors minimized isomerization to the pyranose form during acidic deprotection of the primary hydroxyl group by decreasing the detritylation reaction time. Oligonucleotides containing the pyranose isomer were removed by reverse-phase HPLC and in some instances this additional purification step was not reported. Another strategy involved post-synthetic reduction of a 5-nitro group, followed by formylation.[21] The strong electron withdrawing nitro group prevents isomerization to the pyranose isomer when the 5’-hydroxyl group is revealed during oligonucleotide synthesis. The strategy developed in our group utilizing dinucleotide phosphoramidites to prevent isomerization to the pyranose isomer also made use of the 5-nitro substituent.[22] This strategy provided oligonucleotides without any concern for pyranose formation or the need for additional purification. However, the syntheses of the dinucleotides were lengthy, coupling yields on solid-phase support were modest (50–70%) and/or required double-coupling of the Fapy•dG phosphoramidite. Finally, the ability to synthesize any oligonucleotide sequence requires preparing 4 dinucleotides, of which only two were reported.
Scheme 2.
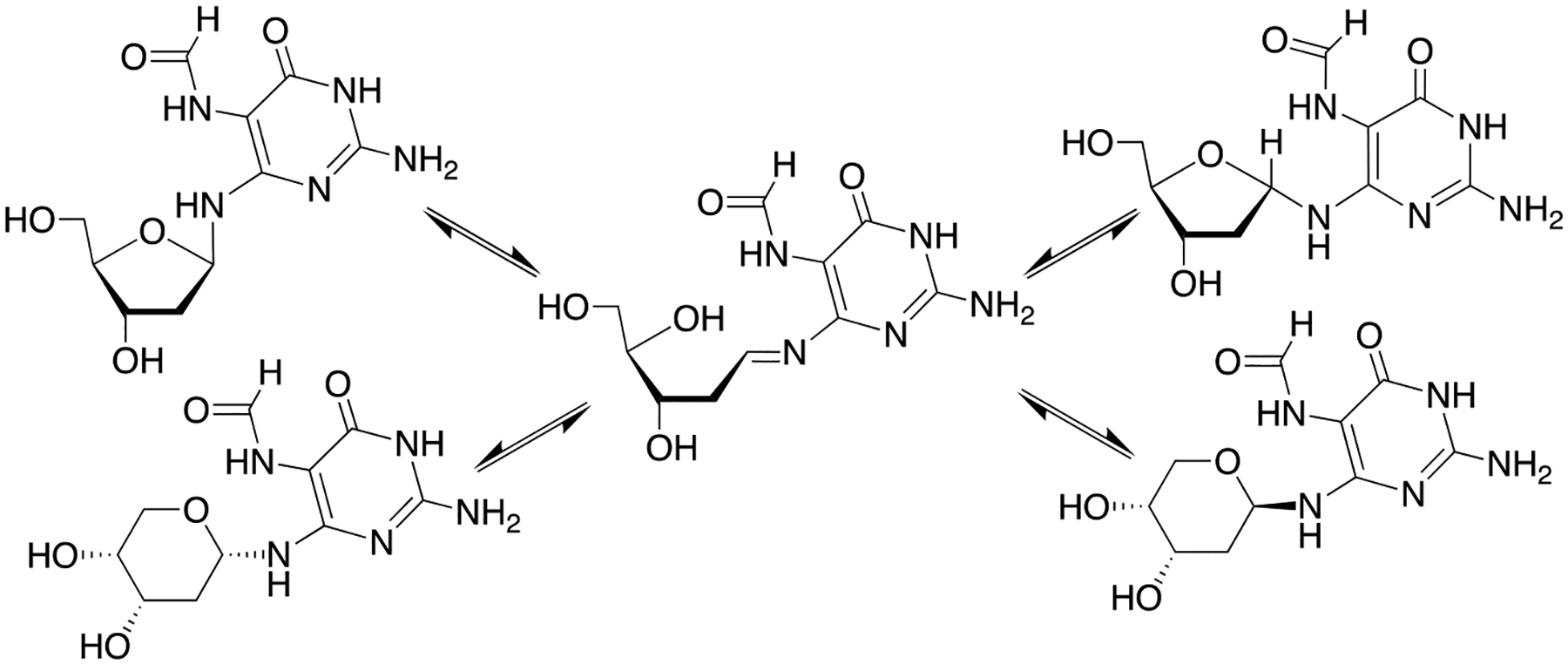
Isomerization of Fapy•dG.
Results and Discussion
Strategic overview
We postulated that oligonucleotides containing Fapy•dG could be prepared via solid-phase synthesis using a “reverse” phosphoramidite approach in which the dimethoxytrityl protecting group was at the 3’-hydroxyl position and the 5’-hydroxyl contained the phosphoramidite (1, Scheme 3). This approach avoids exposing the 5’-hydroxyl group during oligonucleotide synthesis, preventing isomerization to the pyranose isomer. The oligonucleotides would be synthesized on a solid support in the 5’- to 3’-direction, instead of the more common 3’- to 5’-direction. In addition, a single phosphoramidite could be used to synthesize any oligonucleotide sequence. The synthesis of the phosphoramidite takes advantage of the 5-nitro substituent to enable exposing the 5’-hydroxyl without incurring isomerization (Scheme 2).[22] In addition, an unusual and important aspect of this strategy includes introducing the formamide group in the presence of the phosphoramidite (2). The phosphoramidite component is usually introduced in the final step of a synthesis due to its lability. The viability of this aspect of the strategy was established by examining the compatibility of a commercially available thymidine phosphoramidite with representative hydrogenation (Pd/CaCO3, H2) and formylation (formic acetic anhydride) conditions used to convert the nitro group into a formamide prior to embarking upon the synthesis.
Scheme 3.

Strategy for the synthesis of Fapy•dG phosphoramidite 1.
Synthesis of phosphoramidite 1
The syntheses of dinucleotide phosphoramidites employed for preparing oligonucleotides containing Fapy•dG provided useful information for the synthesis of 1.[22] The current endeavor also provided an opportunity to improve transformations that were common to the two methods. For instance, carefully controlling the temperature of the electrophilic nitration reaction provided 5 in greater yield than previously obtained (90% versus 50%). The synthesis of 4 was also improved by starting from Hoffer’s sugar (6, Scheme 4), which can be prepared in large quantities.[23] The α- and β-anomers of 7 were formed in a 3:1 ratio (α:β) and were separable. However, there was no advantage to doing so and in practice 7 was used as a mixture. Following removal of the p-toluoyl groups, 8 was carried on to 9 via 3 as a mixture of anomers as previously described.[22a, 22b] Difficulty in obtaining 10 via desilylation of 9 resulted from loss of the phenoxyacetyl group (Pac) during purification. This problem was resolved by eliminating the use of triethylamine, which is often used during chromatography to guard against adventitious detritylation.
Scheme 4.
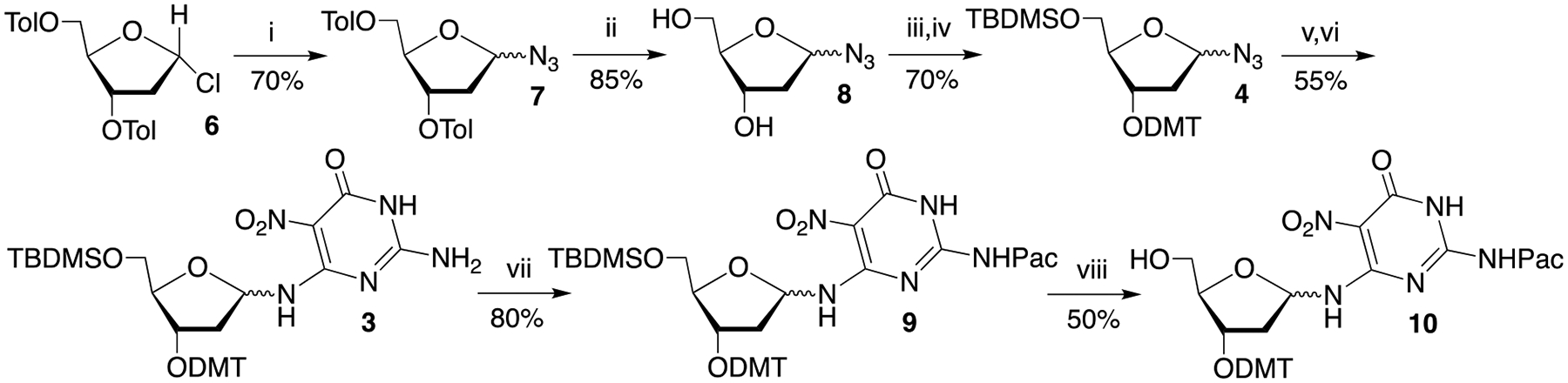
Synthesis of intermediate 9: i) BF3•Et2O, TMSN3, CH2Cl2; ii) NaOMe, MeOH; iii) TBDMSCl, pyridine; iv) DMTCl, pyridine; v) Pd/CaCO3, H2, EtOH; vi) DIPEA, 5, EtOH; vii) Phenoxyacetic acid, PyBOP, DIPEA, CH2Cl2; viii) Et3N•3HF, THF.
Retrosynthetically, 10 was only 3 transformations and 2 purifications from phosphoramidite 1 (Scheme 5). We initially tried to prepare the corresponding β-cyanoethyl phosphoramidite (12a, Scheme 5). Phosphitylation of 10 was accomplished in modest yield (50%). However, attempts to reduce and formylate 11a provided complex mixtures from which it was not possible to isolate 12a. Switching to the corresponding O-methyl phosphoramidite (11b) was less fruitful. Despite a variety of reaction conditions, 11b was contaminated with significant amounts of product(s) in which the phosphorous had undergone hydrolysis to the organophosphite.
Scheme 5.
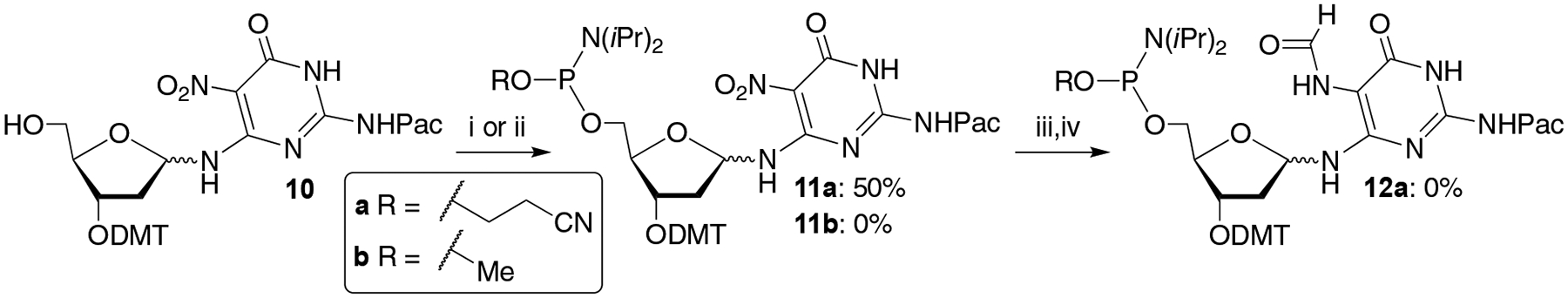
Attempts to overcome phosphoramidite instability: i) 2-Cyanoethyl-N,N,N’,N’-tetraisopropylphosphordiamidite, S-ethyltetrazole, DIPEA, CH2Cl2; ii) N,N-Diisopropylmethylphosphonamidic chloride, DIPEA, CH2Cl2; iii) Pd/C, H2, DIPEA, THF; iv) Acetic formic anhydride, pyridine, THF.
We speculated that the ability to isolate 11a in which the phosphorous is substituted with an electron withdrawing β-nitrile group but not the more electron rich O-methyl phosphorous (11b) was associated with the pyrimidine nitro-substituent. We postulated that the nitro substituent that is vital for preventing rearrangement to the pyranose, increased the acidity of the N3-proton sufficiently to facilitate hydrolysis at phosphorous in 11b. Our solution to this challenge was to remove the NH-proton by derivatizing the O4-position. Initially, a benzyl protecting group was introduced at the O4-position of 9 via a Mitsunobu reaction en route to 13 (Scheme 6). O-Methyl phosphoramidite formation proceeded in good yield (65%), consistent with the above hypothesis concerning the acidity of the N3-proton. However, subsequent reduction and formylation again yielded a complex product mixture. In addition, the benzyl group was not removed, despite experimenting with several hydrogenolysis conditions. Subsequently, the diphenyl carbamoyl (DPC) group, which has been used as an oxygen protecting group in the nitrogen heterocycles of nucleosides, was employed.[24].
Scheme 6.
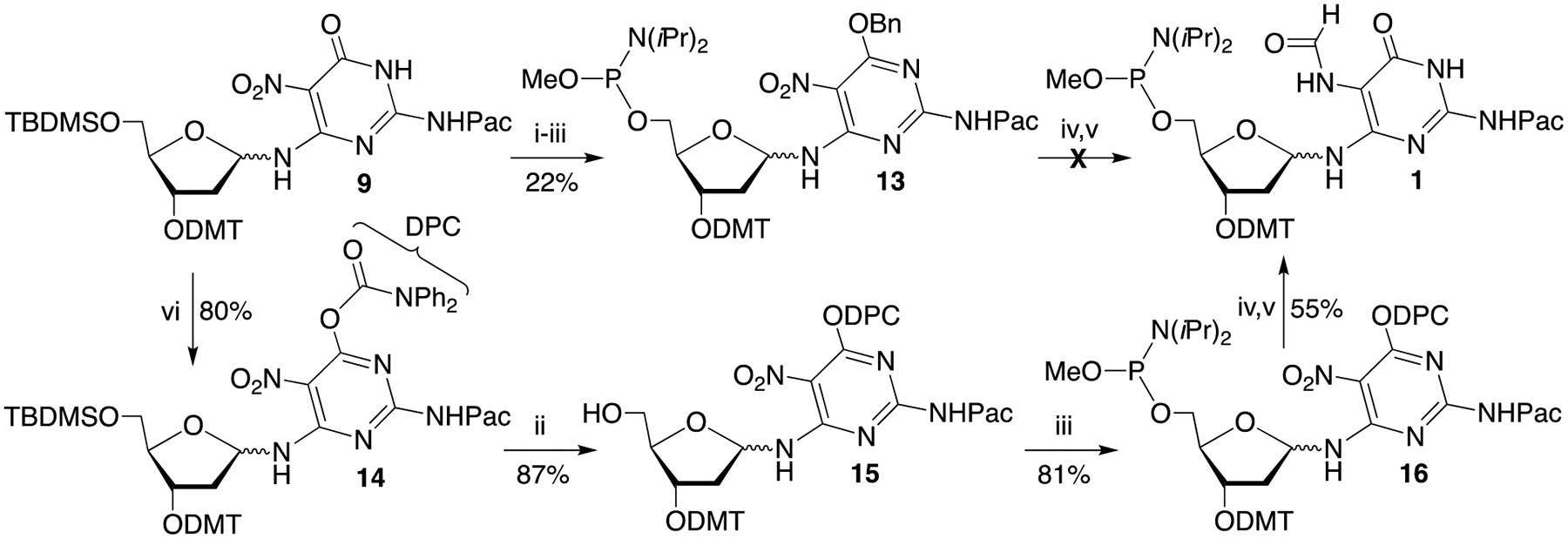
Synthesis of phosphoramidite 1 using a diphenylcarbamoyl protecting group: i) BnOH, PPh3, DEAD, THF; ii) Et3N•3HF, THF iii) N,N-Diisopropylmethylphosphonamidic chloride, DIPEA, CH2Cl2 iv) Pd/C, H2, DIPEA, ethyl acetate v) Pivalic formic anhydride, ethyl acetate vi) Diphenylcarbamoyl chloride, Et3N, pyridine.
It was important to utilize well purified 9 to obtain good yields of 14, but the hydrophobicity of the DPC group also improved the ease of its chromatographic purification. The protected pyrimidine (14) was carried on to the nitro phosphoramidite (16), with the desilylation (15) and phosphitylation steps proceeding in good yield. Anomers of 15 were separated but only the major isomer was carried forward. The target phosphoramidite (1) was obtained, albeit in low yield (20%) when using the previously reported hydrogenation and formylation conditions.[22] Fortuitously, the DPC protecting group was cleaved during hydrogenation.
The low yield of the one pot hydrogenation and formylation reaction is attributed to proclivity of the phosphoramidite in 1 to hydrolyze during reaction and even flash chromatography on silica gel. Hence, the difficulty of preparing acetic formic anhydride (AFA) free of acetic acid posed a significant problem when preparing 1.[25] Attempts to prepare and distill pure AFA yielded inconsistent results. Consequently, a variety of formylation methods were examined using model compound 9. Transfer hydrogenation and formylation using ammonium formate was unsuccessful.[26] In addition, formylation using formic acid and carbodiimide or formic acid, imidazole and oxalyl chloride did not provide the expected formamide product. The overall process was significantly improved by carrying out the hydrogenation reaction in the more hydrophobic ethyl acetate instead of THF, and substituting pivalic formic anhydride (PFA) for AFA. PFA was prepared under solvent free condition and distilled under high vacuum at 0 ⁰C.[27] The obtained anhydride product contains only trace of acids even stored for weeks. Reactions using PFA proved to be more reproducible than those with AFA. We attributed this to the greater ease of purification of PFA by distillation, as well as by deleting pyridine from the reaction mixtures.[22a, 22b] These conditions provided less complicated reaction mixtures, which facilitated separation and purification of the anomers of 1. The Fapy•dG phosphoramidite (1) is very sensitive to the acidity of silica gel. However, 1 could be purified by conventional silica gel flash chromatography using a mixture of acetone and dichloromethane containing 0.1% triethylamine. This proved to be more reliable in our hands than an automated flash chromatography system (Combiflash Rf+) equipped with a less reactive cyano end-capped silica gel column, from which 1 could be eluted using ethyl acetate/hexane (50%−100%).
Characterization of the anomers of 1 was challenging. The overall yield of 1 from 16 was 55% and the more polar isomer was favoured ~2-fold over the less polar anomer. The diagnostic formamide proton was evident as a mixture of rotamers in the 1H NMR at δ 8.65–8.69 for the minor anomer, which is consistent with reported Fapy·dG derivatives and their restricted rotation.[22] However, the corresponding proton for the major, more polar isomer was less evident in the 1H NMR spectrum. A peak accounting for ~0.7 protons was evident further upfield at δ 7.66, which is close to the region at which the aromatic protons resonate. One possibility was that the resonance for the formamide proton of other rotamers of this anomer were obscured by the large number of aromatic protons. Although mass spectrometry was compatible with its identification as an isomer of 1, confirmatory evidence for the presence of the formamide group was obtained via a 1H-13C HSQC experiment, which showed the expected correlation between the formamide proton (δ 7.66) and the formyl carbon signal at δ158 (Figure S35). A similar correlation was observed for the formamide proton of the minor anomer (Figure S32). If the major isomer is α−1, the upfield shift of the formyl proton in the major isomer could be attributed to shielding by the π-electrons of the aromatic ring(s). However, NOE experiments aimed at providing confirmation of this hypothesis were ambiguous.
Synthesis and deprotection of oligonucleotides containing Fapy•dG using 1
Initial conditions for synthesizing oligonucleotides containing Fapy•dG using 1 utilized previous reports as a starting point.[22a, 22b] Detritylation and oxidation were carried out using trichloroacetic acid and t-BuOOH, respectively. Following coupling of 1 (0.1 M), capping with phenoxyacetic anhydride and 1-methylimidazole was replaced with pivalic anhydride/lutidine/THF. Coupling conditions of 1 using 4,5-dicyanoimidazole as activator were optimized by synthesizing a 15mer consisting of thymidines and a single Fapy•dG (17) on a (“reverse”) 3’-dimethoxytrityl thymidine support containing a succinate linkage between the 5’-hydroxyl and long chain alkylamine. On-line dimethoxytrityl cation monitoring indicated that 1 coupled from 70–80% when a 15 min reaction time was used. Separate anomers of 1 were initially used, but no difference in coupling yields was detected (data not shown). Consequently, an anomeric mixture of 1 is routinely used in oligonucleotide synthesis, which also facilitates purifying the phosphoramidite. The Fapy•dG phosphoramidite (1) was much more sensitive to water in the acetonitrile used to dissolve it than commercially available phosphoramidites. To avoid hydrolysis, activated molecular sieves were added to the solution of 1 after filtering and were present throughout oligonucleotide synthesis. After demethylating the Fapy•dG phosphate triester with disodium 2-carbamoyl-2-cyanoethylene-1,1-dithiolate trihydrate (0.2 M, 30 min, 25 °C),[28] 17 was deprotected and cleaved from the solid-phase support using K2CO3 (50 mM) in MeOH (25 °C, 4 h). The gel purified product was characterized by ESI-MS (Figure S1). The coupling yield of 1, as well as its synthesis was a marked improvement over the corresponding dinucleotide phosphoramidites employed previously.[22]

Having established satisfactory coupling conditions for 1, conditions for synthesizing oligonucleotides containing Fapy•dG and all 4 native nucleotides using reverse phosphoramidites were explored. To make the synthesis of Fapy•dG containing oligonucleotides accessible to the greatest number of scientists possible, we sought to maximize the use of commercially available phosphoramidites and solid-phase synthesis supports. Reverse dT and fast deprotecting dCAc, dAPac and dGiPrac phosphoramidites are compatible with the K2CO3 conditions used to deprotect 17.[22a, 22b] However, the latter two reverse phosphoramidites are not commercially available. Reverse dimethylformamidine protected dG (dGdmf) and the respective solid-phase support are commercially available. This required establishing mutually compatible conditions for Fapy•dG and native phosphoramidite deprotection, as well as oligonucleotide cleavage from the solid-phase supports.
Nucleobase deprotection conditions were tested using 17 and an oligonucleotide containing dA, dC, dG and dT (18). Oligonucleotide 18 was prepared on commercially available UnyLinker™ solid-phase synthesis support. Use of this support obviates the need for users to independently synthesizing LCAA-CPG containing reverse dAPac. Furthermore, any conditions sufficient for cleaving oligonucleotides from UnyLinker™ will be sufficient for removing oligonucleotides linked via the standard, more labile succinate group. dGdmf was incompletely deprotected using K2CO3 in MeOH, and remnants of the UnyLinker™ were retained at the 5’-terminus. Saturated aqueous ammonia solution in ethanol (3:1, 15 °C, 10 h), which is used to deprotect oligonucleotides containing carba-Fapy•dG completely deprotected dGdmf within 18 but cleaved at Fapy•dG in 17.[16a] Oligonucleotide 17 was also cleaved at Fapy•dG when treated with concentrated NH4OH at room temperature overnight or with ammonia/methyl amine (1:1, v/v, 55 °C, 1 h).[29] Fapy•dG also underwent cleavage when subjected to diisopropylethylamine (10% by volume) and β-mercaptoethanol (0.25 M) in MeOH (25 °C, 2 h).
Treating 17 with t-butylamine in H2O (1:3 v/v) under the recommended conditions (60 °C, 4 h) produced significant amounts of cleavage at Fapy•dG.[30] However, subjecting 17 and 18 to these conditions at 40 °C for 8 h provided good yields of completely deprotected oligonucleotides. The general utility of this method was demonstrated by synthesizing a series of oligonucleotides on UnyLinker™ support that contain Fapy•dG and the 4 native nucleotides (20-26) (Figure S4–10). In addition, 27-29 (Figure S11–13) were synthesized on supports containing the appropriate 5’-terminal reverse nucleoside. However, Fapy•dG was cleaved during the deprotection when it was present at the 3’-terminus in 19. In addition, we also determined that conditions reported for the deprotection and cleavage of oligonucleotides containing N5-methyl Fapy•dG (0.1 M NaOH, 25 °C, 12 h) were compatible Fapy•dG, the phosphoramidites employed, as well as UnyLinker™ support. Oligonucleotides 18, 19 and 26-29 were successfully obtained in comparable yields as when deprotected with t-butylamine. However, 26 required 16 h to achieve complete deprotection of the 12 dGdmf groups, while complete deprotection of 26 was achieved with t-butylamine at 40 °C for 8 h. Finally, it should be noted that the deprotection methods employed here were unable to deprotect commercially available dABz.
Conclusion
Fapy•dG is a biologically interesting DNA lesion formed from a common intermediate as the most well studied DNA lesion, 8-OxodGuo. Moreover, Fapy•dG is formed in greater quantities than the latter under some O2 deficient conditions. Structural and biochemical knowledge of Fapy•dG, as well as its effects on replication and transcription in cells is limited due to difficulties in synthesizing oligonucleotides containing it. Consequently, research on the formamidopyrimidines has lagged behind that of 8-OxodGuo. We describe the implementation of a synthetic strategy using reverse phosphoramidites that is a marked improvement over previously reported methods for synthesizing oligonucleotides containing Fapy•dG. The process is robust, enabling the synthesis of oligonucleotides containing Fapy•dG suitable for biochemical and structural studies. The approach should also be compatible with synthesizing oligonucleotides containing N5-alkylated formamidopyrimidines. The synthesis of phosphoramidite 1 is a significant improvement over previous methods, in terms of its effectiveness and ease of implementation. The availability of 1 and its compatibility with readily available solid-phase oligonucleotide synthesis reagents will facilitate research on this biologically significant family of DNA lesions by the research community.
Experimental Section
General Methods
Triethylamine, pyridine, diisopropylamine, t-butylamine, 2,6-lutidine, ethyl acetate and dichloromethane were distilled from CaH2 under Ar. Methanol was dried over molecular sieves 3 Å and distilled under Ar. THF was distilled from Na under Ar. Pivalic anhydride was distilled under vacuum. All other reagents were purchased from commercial sources and used without further purification unless otherwise stated. Nuclear magnetic resonance spectra were acquired on Bruker 400 MHz for 1H, 101 MHz for 13C and 162 MHz for 31P. High resolution ESI mass spectra for HRMS of synthesized molecules were recorded on a Waters Acquity / Xevo-G2 UPLC-MS system in positive mode. MALDI-TOF spectra were recorded on a Bruker AutoFlex III MALDI-TOF/TOF mass spectrometry in negative mode. Low resolution ESI mass spectrometry was carried out on a Thermo Finnigan Surveyor LCQ Fleet instrument. Oligonucleotide synthesis was carried out on an ABI-394 synthesizer. N-Phenoxyacetyl protected 3’-dimethoxytrityl 5’-β-cyanoethyl 2’-deoxyadenosine phosphoramidite (dAPac) was synthesized as previously described (41). The requisite “reverse” 5’-β-cyanoethyl phosphoramidites for thymidine (dT) and N,N-dimethylformamidine 2’-deoxyguanosine (dGdmf) were obtained from Glen Research. Reverse phosphoramidite, N-acetyl 2’-deoxycytidine (dCAc) as well as Universal UnyLinker™ Support were purchased from Chemgenes. All other commercially available oligonucleotide synthesis reagents were purchased from Glen Research. HP Cyano RediSep Rf gold chromatography column (15.5 g) was purchased from Teledyne ISCO.
Preparation of 2-Deoxy-3,5-bis[O-(p-toluoyl)]-α,β-D-erythro-pentofuranosyl azide (7).
[31] Hoffer’s chloro sugar[23] (6, 10 g, 25.7 mmol) was suspended in DCM (50 mL) added with BF3∙Et2O (360 mg, 2.6 mmol) and trimethylsilyl azide (3.8 g, 31 mmol) at 0 °C. After 30 min, the reaction was brought to RT and stirred for 5 h. The reaction mixture was diluted with DCM (200 mL), washed with H2O, brine and dried over Na2SO4. The organic solution was removed by evaporation and residue containing both α- and β-anomers was directly used for next step without purification. For characterization purposes, the mixture was purified by flash chromatography (5% −10% EtOAc in Hexane) to afford 7 (9 g, α/β = 3:1, 70%) as a white solid. α-anomer: 1H NMR (400 MHz, CDCl3) δ7.98 (d, J= 8.4 Hz, 2H), 7.91 (d, J= 8.4 Hz, 2H), 7.22–7.27 (m, 4H), 5.70–5.71 (m, 1H), 5.48–5.51 (m, 1H), 4.71 (m, 1H), 4.50–4.63 (m, 2H), 2.52–2.59 (m, 1H), 2.41 (d, J= 3.2 Hz, 6H), 2.21–2.42 (m, 1H); β-anomer: 1H NMR (400 MHz, CDCl3) 7.99 (d, J= 8.4 Hz, 2H), 7.89 (d, J= 8.4 Hz, 2H), 7.20–7.24 (m, 4H), 5.70 (t, J= 5.2 Hz, 1H), 5.56–5.59 (m, 1H), 4.52–4.60 (m, 3H), 2.38–2.42 (m, 8H). The spectra of the α- and β-isomers are consistent with the reported data.
Preparation of 2-Deoxy-α,β-D-[erythro-]pentofuranosyl azide (8).
[21, 22b] Sodium methoxide (1.2 g, 22.7 mmol) was added to a solution of 7 (9 g, 22.7 mmol) in dry MeOH (70 mL). The reaction was stirred at 40 °C for 2 h, at which time Amberlite GC50-H+ ion exchange resin (3.5 g) was added. The reaction mixture was stirred for another 30 min at room temperature. The resin was filtered off and the solvent was removed under vacuum. The residue was purified by flash chromatography (5% −10% MeOH in DCM) to afford 8 (3.1 g, 80%, α/β = 3:2) as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.68 (dd, J= 3.4, 1.0 Hz, 1H), 5.59 (t, J= 4.6 Hz, 0.5H), 4.51–4.67 (m, 0.5H), 4.30–4.25 (m, 2H), 4.51–4.00 (q, J= 4.0 Hz, 0.5H), 3.82–3.65(m, 3H), 2.23–1.93(m, 3H). The chemical shifts of β-isomers are consistent with the reported data.
Preparation of 6-[2’-Deoxy-3’-O-dimethoxytrityl-5’-O-tert-butyl(dimethyl)silyl-α/β-D-ribofuranose-1’-yl]amino-4-(diphenylcarbamoyl)oxy-5-nitro-2-(phenoxyacetyl)aminopyrimidine (14).
Compound 9[22b] (1 g, 1.2 mmol) was coevaporated with pyridine (3 × 3 mL) and then dissolved in pyridine (5 mL). Diphenylcarbamoyl chloride (340 mg, 1.4 mmol. 1.2 eq.) was added, followed by Et3N (183 mg, 1.8 mmol) The reaction was stirred in the dark for 40 min and then partitioned into a mixture of 5% NaHCO3/ EtOAc (1:1, 50 mL). The organic phase was washed with brine, dried over Na2SO4, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Hexane/EtOAc = 4:1–3:1) to provide 14 as a mixture of anomers (1 g, more polar isomer/less polar isomer = 5:1, 80%) as a foam. (The 1H NMR integration of the anomeric mixture follows. The integration is reported such that a single proton for the major anomer is normalized to 1.) 1H NMR (400 MHz, CDCl3): δ 9.20–9.22 (d, J= 8 Hz, 1H), 8.66 (br s, 1H), 6.81–7.46 (m, 35H), 6.34–6.36 (m, 0.2H), 6.17 (t, J= 8 Hz, 1H), 4.77–4.79 (m, 2.4H), 4.33–4.35 (m, 1H), 4.27(s, 1H), 3.77 (s, 8H), 3.32–3.36 (m, 2.4H), 1.93 (m, 1H), 0.78 (s, 12H), −0.09 - −0.06(d, J= 12 Hz, 8H). 13C-NMR (101 MHz, CDCl3): δ 166.8, 161.6, 158.6, 157.5, 157.1, 155.3, 149.6, 145.0, 136.4, 136.2, 130.3, 130.2, 130.1, 129.8, 129.2, 128.3, 128.2, 128.1, 127.9, 126.9, 122.3, 116.9, 114.9, 114.8, 113.4, 113.3, 113.2, 88.0, 87.2, 83.2, 77.4, 77.3, 77.1, 76.8, 75.5, 68.1, 63.6, 55.2, 38.7, 31.6, 25.3, 20.7, 18.3, 1.0, −5.3, −5.6. HRMS (ESI-TOF) m/z [M+Na]+ calcd. for C57H60N6O11SiNa 1055.3982, found: 1055.3960.
Preparation of 6-(2’-Deoxy-3’-O-dimethoxytrityl-α,β-D-ribofuranose-1’-yl)amino-4-(diphenylcarbamoyl)oxy-5-nitro-2-(phenoxyacetyl)aminopyrimidine (15).
Compound 14 (870 mg, 0.84 mmol) was coevaporated with toluene/DCM (1:1, 2 × 2 mL) and then dissolved in THF (10 mL). Et3N∙3HF (1.5 mL, 8.4mmol,) was added and the reaction was stirred at room temperature overnight. The mixture was diluted with ethyl acetate and was washed with saturated NaHCO3, and brine. The organic layer was dried over Na2SO4, filtered and evaporated to dryness under vacuum. The residue was purified by flash chromatography (Hexane/EtOAc = 2:1–1:1) to provide two isomers of 15 (670 mg, 87%, more polar isomer/less polar isomer = 4:1) as a foam. Less polar anomer: 1H NMR (400 MHz, CDCl3) δ 8.93–8.91 (d, J= 8 Hz, 1 H), 7.18 (br s, 1H), 6.81–7.46 (m, 28H), 6.34–6.36 (m, 1H), 4.80–4.81 (m, 2H), 4.35–4.36 (m, 1H), 4.11–4.13 (m, 1H), 3.94 (s, 6H), 3.15–3.53 (m, 2H). 13C NMR (101 MHz, CDCl3): δ: 167.2, 161.5, 158.7, 157.4, 155.4, 149.5, 145.3, 136.5, 136.4, 130.3, 130.2, 129.8, 129.3, 128.3, 128.0, 127.0, 122.2, 116.7, 114.9, 113.3, 87.1, 86.2, 82.9, 77.5, 77.2, 76.8, 74.8, 68.2, 62.7, 55.2, 40.9, 21.1, 14.2; More polar anomer 1H NMR (400 MHz, CDCl3) δ 9.23 (d, J= 8 Hz, 1 H), 8.77 (br s, 1H), 6.82–7.46 (m, 28H), 6.24 (t, J= 7.2 Hz, 1H), 4.67–4.77 (m, 2H), 4.32 (d, J= 6.8 Hz, 1H), 4.23 (s, 1H), 3.77 (s, 6H), 3.21–3.53 (m, 2H), 1.88–1.91 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 176.0, 172.0, 166.3, 163.5, 162.5, 161.8, 160.1, 154.4, 149.7, 141.1, 140.9, 135.0, 134.9, 134.5, 133.0, 132.9, 131.8, 127.1, 121.7, 119.6, 118.2, 91.8, 87.9, 82.3, 81.9, 81.6, 80.3, 72.8, 67.5, 65.2, 60.0, 43.9, 25.8, 19.0. HRMS (ESI-TOF) m/z [M+Na]+ calcd. for C51H46N6NaO11 941.3117, found: 941.3099 (less polar), 941.3095 (more polar).
Preparation of 6-(2’-Deoxy-3’-O-dimethoxytrityl-α,β-D-ribofuranose-1’-yl)amino-4-(diphenylcarbamoyl)oxy-5-nitro-2-(phenoxyacetyl)aminopyrimidine 5’-methyl-N,N’-diisopropyl phosphoramidite (16).
The major isomer of compound 15 (270 mg, 0.29 mmol) was coevaporated with toluene (2 × 10mL) and dried in vacuo for 2 h before it was dissolved DCM (5 mL) at 0°C. DIPEA (185 mg, 1.5 mmol) was then added, followed by diisopropylmethylphosphonamidic chloride (110 mg, 0.53 mmol). After 10 min, the reaction was brought to room temperature and stirred for 1.5 h, at which time it was diluted with DCM (2 mL). The resulting solution was washed with 5% NaHCO3 and brine, dried over Na2SO4, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Hexane/EtOAc = 2:1) to provide 16 (250 mg, 80%) as a foam. 1H NMR (400 MHz, CDCl3): δ 9.20–9.22 (d, J= 8 Hz, 1 H), 6.82–7.38 (m, 28H), 6.16–6.22 (m, 1H), 4.77–4.80 (d, J= 8 Hz, 2H), 4.43–4.37 (dd, J= 16, 8 Hz, 1H), 4.33–4.28 (m, 1H), 3.74 (s, 6H), 3.42–3.46 (m, 2H), 3.26–3.31 (m, 3H), 1.12–1.13 (m, 14H). 31P NMR (162 MHz, CDCl3): δ 149.0, 149.1. HRMS (ESI-TOF) m/z [M+H]+ calcd. for C58H63N7O12P 1080.4267, found: 1080.4252.
Preparation of 6-(2’-Deoxy-3’-O-dimethoxytrityl-α,β-D-ribofuranose-1’-yl)amino-5-formamido-2-(phenoxyacetyl)amino pyrimidine-3H-4-one 5’-methyl-N,N’-diisopropyl phosphoramidite (1).
Phosphoramidite 16 (200 mg, 0.17 mmol), DIPEA (180 mg, 1.3 mmol) and 10% palladium on carbon (160 mg) in ethyl acetate (4 mL) were placed in a pressure tube. The reaction mixture was pressurized with H2 and vented ten times and then stirred under H2 (60 psi) for 3 h. The reaction atmosphere is exchanged with Ar by freeze-pump-thaw degassing. The solution was cooled to 0 ⁰C. Pivalic formic anhydride (50 mg, 0.36 mmol) was added and the reaction mixture was stirred at 0 ⁰C for 1 h. The reaction mixture was diluted with 5 mL of EtOAc and centrifuged at 10000 RPM for 3 min. The solution was carefully removed and the solid was washed with 5 mL EtOAc. After centrifuging, the EtOAc solutions were combined, washed with 5% NaHCO3, brine and dried over Na2SO4. The solution was filtered and evaporated to dryness under vacuum. The residue was purified by flash chromatography using DCM/Acetone = 5:1 and 0.1 % Et3N until the less polar isomer eluted, then 2:1 DCM/Acetone to elute the more polar isomer to provide the individual anomers of 1 (90 mg, 55%, more polar/less polar=2:1). Note: Because of tailing, fractions eluted from the column were dilute. Column fractions (~6 mL) were concentrated to ~1 mL prior to TLC analysis. Less polar isomer: 1H NMR (400 MHz, CD3CN) δ: 8.65–8.69 (m, 1H), 8.55–8.58 (m, 1H), 7.44–7.46 (m, 2H), 7.33–7.36 (m, 10H), 7.24–7.27 (m, 1H), 7.00–7.05 (m, 3H), 6.89 (d, J= 8.4 Hz, 5H), 6.03–6.04 (m, 1H), 5.96 (q, J= 8.4 Hz, 1H), 4.72 (s, 2H), 4.24–4.30 (m, 1H), 4.12 (,m, 1H), 3.78 (s, 6H), 3.32–3.49 (m, 4H), 1.72–1.84 (m, 2H), 0.98–1.10 (m, 12H), 31P NMR (162 MHz, CD3CN) δ: 148.92, 148.60. More polar isomer: 1H NMR (400 MHz, CD3CN) δ: 7.66 (s, 0.7H), 7.46–7.51 (m, 2H), 7.30–7.40 (m, 10H), 7.21–7.26 (m, 2H), 7.00–7.05 (m, 3H), 6.87–6.90 (m, 5H), 6.36–6.41 (m, 1H), 5.93–6.01 (m, 1H), 4.79 (s, 2H), 4.19–4.31 (m, 2H), 3.77 (s, 6H), 3.32–3.51 (m, 4H), 3.21–3.27 (m, 3H), 2.76–2.84 (m, 1H), 1.69–1.81 (m, 1H), 0.98–1.12 (m, 12H). 31P NMR (162 MHz, CD3CN) δ: 148.86, 148.64. HRMS (ESI-TOF) m/z [M+H]+ calcd. for C40H43N5O11P 800.2691, found: 800.2684 (less polar isomer), 800.2678 (more polar isomer). (Please note that the mass corresponds to the hydrolyzed organophosphite product)
Oligonucleotide Synthesis, Deprotection, and Characterization.
The incorporation of Fapy•dG (1) into oligonucleotides with reverse β-cyanoethyl phosphoramidites was carried out on 1 μmol scale using 4,5-dicyanoimidazole (0.25 M in acetonitrile) as activating agent. Acetonitrile solutions of phosphoramidites synthesized in our laboratory (1, dAPac) were filtered using 0.45 μm syringe filters. The coupling of 1 is extremely sensitive to water. Thus, molecular sieves were added to the solution after filtering and kept in the bottle throughout oligonucleotide synthesis. The wait time for the coupling of Fapy•dG phosphoramidite was extended to 900 s, while for other phosphoramidites the wait time was 180 s. After coupling 1, commercially available phenoxyacetic anhydride (Cap A mix) and N-methyl imidazole (Cap B mix) solutions were replaced by pivalic anhydride/2, 6-lutine/THF (1:1:8, v/v/v). Capping was carried out 60 s at all steps during oligonucleotide synthesis. t-BuOOH (1 M in toluene) was used for oxidation (40 s). Following solid phase synthesis, the methyl group was removed from the Fapy•dG phosphate triester using disodium 2-carbamoyl-2-cyanoethylene-1,1-dithiolate trihydrate (0.2 M in DMF, 0.3 mL) for 30 min at room temperature [22a, 22b]. The resin was then rinsed successively with MeOH, THF and CH2Cl2, vacuum dried and treated with aq. NaOH (0.1 M) at room temperature for 12 h. The deprotection time was extended to 16 h for oligonucleotides containing large numbers of dG (e.g. 28). The solution was neutralized (per pH paper) with AcOH (5% in MeOH, ~0.5 vol. of the NaOH solution), concentrated to dryness and purified using 20% denaturing polyacrylamide gel electrophoresis. Alternatively, the resin was incubated with tBuNH2/H2O (1:3, v/v) at 40 ⁰C for 8 h and neutralized (per pH paper) with glacial acetic acid (~0.1 vol. of the tBuNH2 solution). The isolated oligonucleotides were characterized by MALDI-TOF MS or ESI-MS (Figure S1–13).
Supplementary Material
Acknowledgements
We are grateful for support of this research from the National Institute of Environmental Health Studies (ES-027558). We thank Professor Carmelo Rizzo for helpful discussions and Professor Rebekka Klausen for providing access to the Combiflash Rf+ apparatus.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Cadet J, Davies KJA, Medeiros MHG, Di Mascio P, Wagner JR, Free Rad. Biol. Med 2017, 107, 13–34; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dizdaroglu M, Mutat. Res. Rev. Mutagen 2015, 763, 212–245. [DOI] [PubMed] [Google Scholar]
- [2].a) Fleming AM, Ding Y, Burrows CJ, Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Redstone SCJ, Fleming AM, Burrows CJ, Chem. Res. Toxicol 2019, 32, 437–446; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu J, Fleming AM, Burrows CJ, ACS Chem. Biol 2018, 13, 2577–2584; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK, Hegde ML, Radak Z, Bacsi A, Brasier AR, Ba X, Boldogh I, J. Biol. Chem 2016, 291, 25553–25566; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Allgayer J, Kitsera N, Bartelt S, Epe B, Khobta A, Nucleic Acids Res. 2016, 44, 7267–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shafirovich V, Geacintov NE, Free Rad. Biol. Med 2017, 107, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Amente S, Di Palo G, Scala G, Castrignanò T, Gorini F, Cocozza S, Moresano A, Pucci P, Ma B, Stepanov I, Lania L, Pelicci PG, Dellino GI, Majello B, Nucleic Acids Res. 2019, 47, 221–236; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Freudenthal BD, Beard WA, Perera L, Shock DD, Kim T, Schlick T, Wilson SH, Nature 2015, 517, 635–639; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Menoni H, Shukla MS, Gerson V. r., Dimitrov S, Angelov D, Nucleic Acids Res. 2012, 40, 692–700; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) van der Kemp PA, de Padula M, Burguiere-Slezak G, Ulrich HD, Boiteux S, Nucleic Acids Res. 2009, 37, 2549–2559; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA, Nucleic Acids Res. 2009, 37, 2830–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Greenberg MM, Acc. Chem. Res 2012, 45, 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Becker S, Thoma I, Deutsch A, Gehrke T, Mayer P, Zipse H, Carell T, Science 2016, 352, 833–836. [DOI] [PubMed] [Google Scholar]
- [7].Dizdaroglu M, Kirkali G, Jaruga P, Free Rad. Biol. Med 2008, 45, 1610–1621. [DOI] [PubMed] [Google Scholar]
- [8].a) Jaruga P, Kirkali G, Dizdaroglu M, Free Rad. Biol. Med 2008, 45, 1601–1609; [DOI] [PubMed] [Google Scholar]; b) Douki T, Martini R, Ravanat J-L, Turesky RJ, Cadet J, Carcinogenesis 1997, 18, 2385–2391; [DOI] [PubMed] [Google Scholar]; c) Pouget JP, Frelon S, Ravanat JL, Testard I, Odin F, Cadet J, Radiat. Res 2002, 157, 589–595; [DOI] [PubMed] [Google Scholar]; d) Cadet J, Wagner JR, Cold Spring Harbor Perspect. Biol 2013, 5, A012559/012551-A012559/012518; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xue L, Greenberg MM, J. Am. Chem. Soc 2007, 129, 7010–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Alshykhly OR, Fleming AM, Burrows CJ, J. Org. Chem 2015, 80, 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Douki T, Riviere J, Cadet J, Chem. Res. Toxicol 2002, 15, 445–454; [DOI] [PubMed] [Google Scholar]; b) Bergeron F, Auvré F, Radicella JP, Ravanat J-L, Proc. Nat. Acad. Sci. USA 2010, 107, 5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wiederholt CJ, Delaney MO, Pope MA, David SS, Greenberg MM, Biochemistry 2003, 42, 9755–9760. [DOI] [PubMed] [Google Scholar]
- [12].Krishnamurthy N, Haraguchi K, Greenberg MM, David SS, Biochemistry 2008, 47, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Chmiel NH, Livingston AL, David SS, J. Mol. Biol 2003, 327, 431–443; [DOI] [PubMed] [Google Scholar]; b) Porello SL, Leyes AE, David SS, Biochemistry 1998, 37, 14756–14764. [DOI] [PubMed] [Google Scholar]
- [14].Patro JN, Wiederholt CJ, Jiang YL, Delaney JC, Essigmann JM, Greenberg MM, Biochemistry 2007, 46, 10202–10212. [DOI] [PubMed] [Google Scholar]
- [15].a) Kalam MA, Haraguchi K, Chandani S, Loechler EL, Moriya M, Greenberg MM, Basu AK, Nucleic Acids Res. 2006, 34, 2305–2315; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pande P, Haraguchi K, Jiang Y-L, Greenberg MM, Basu AK, Biochemistry 2015, 54, 1859–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Gehrke TH, Lischke U, Gasteiger KL, Schneider S, Arnold S, Müller HC, Stephenson DS, Zipse H, Carell T, Nature Chem. Biol 2013, 9, 455–461; [DOI] [PubMed] [Google Scholar]; b) Coste F, Ober M, Carell T, Boiteux S, Zelwer C, Castaing B, J. Biol. Chem 2004, 279, 44074–44083. [DOI] [PubMed] [Google Scholar]
- [17].Berger M, Cadet J, Z. Naturforsch 1985, 40b, 1519–1531. [Google Scholar]
- [18].Patro JN, Haraguchi K, Delaney MO, Greenberg MM, Biochemistry 2004, 43, 13397–13403. [DOI] [PubMed] [Google Scholar]
- [19].a) Burgdorf LT, Carell T, Chem. Eur. J 2002, 8, 293–301; [DOI] [PubMed] [Google Scholar]; b) Delaney MO, Greenberg MM, Chem. Res Toxicol 2002, 15, 1460–1465. [DOI] [PubMed] [Google Scholar]
- [20].a) Christov PP, Brown KL, Kozekov ID, Stone MP, Harris TM, Rizzo CJ, Chem. Res. Toxicol 2008, 21, 2324–2333; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Christov PP, Angel KC, Guengerich FP, Rizzo CJ, Chem. Res. Toxicol 2009, 22, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lukin M, Minetti CASA, Remeta DP, Attaluri S, Johnson F, Breslauer KJ, de los Santos C, Nucleic Acids Res. 2011, 39, 5776–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM, J. Am. Chem. Soc 2002, 124, 3263–3269; [DOI] [PubMed] [Google Scholar]; b) Haraguchi K, Greenberg MM, J. Am. Chem. Soc 2001, 123, 8636–8637; [DOI] [PubMed] [Google Scholar]; c) Jiang YL, Wiederholt CJ, Patro JN, Haraguchi K, Greenberg MM, J. Org. Chem 2005, 70, 141–147. [DOI] [PubMed] [Google Scholar]
- [23].Rolland V, Kotera M, Lhomme J, Syn. Comm 1997, 27, 3505–3511. [Google Scholar]
- [24].a) Robins MJ, Zou R, Guo Z, Wnuk SF, J. Org. Chem 1996, 61, 9207–9212; [Google Scholar]; b) Kamimura T, Tsuchiya M, Urakami K, Koura K, Sekine M, Shinozaki K, Miura K, Hata K, J. Am. Chem. Soc 1984, 106, 4552–4557. [Google Scholar]
- [25].Cai H, Guengerich FP, Chem. Res. Toxicol 2000, 13, 327–335. [DOI] [PubMed] [Google Scholar]
- [26].Pratap TV, Baskaran S, Tetrahedron Lett. 2001, 42, 1983–1985. [Google Scholar]
- [27].a) Schijf R, Stevens W, Recl. Trav. Chim. Pays-Bas 1966, 85, 627–628; [Google Scholar]; b) Vlietstra EJ, Zwikker JW, Nolte RJM, Drenth W, Recl. Trav. Chim. Pays-Bas 1982, 101, 460–461. [Google Scholar]
- [28].Scaringe SA, Wincott FE, Caruthers MH, J. Am. Chem. Soc 1998, 120, 11820–11821. [Google Scholar]
- [29].Reddy MP, Hanna NB, Farooqui F, Tetrahedron Lett. 1994, 35, 4311–4314. [Google Scholar]
- [30].The Glen Report (Glen Research), Sterling, Virginia: 20164, 2013. [Google Scholar]
- [31].Bag SS, Talukdar S, Matsumoto K, Kundu R, J. Org. Chem 2013, 78, 278–291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.