

Abstract
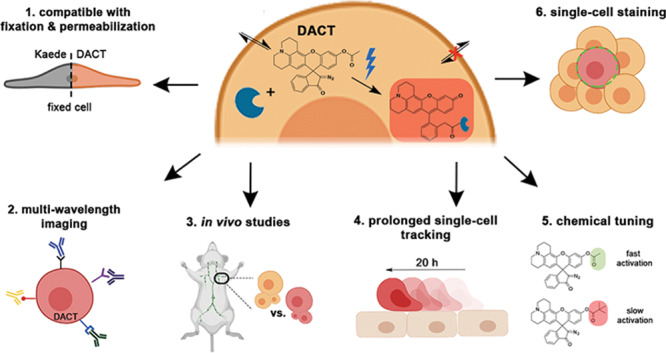
Cell trackers are fluorescent chemical tools that facilitate imaging and tracking cells within live organisms. Despite their versatility, these dyes lack specificity, tend to leak outside of the cell, and stain neighboring cells. Here, we report a dual-activatable cell tracker for increased spatial and temporal staining control, especially for single-cell tracking. This probe overcomes the typical problems of current cell trackers: off-target staining, high background signal, and leakage from the intracellular medium. Staining with this dye is not cytotoxic, and it can be used in sensitive primary cells. Moreover, this dye is resistant to harsh fixation and permeabilization conditions and allows for multiwavelength studies with confocal microscopy and fluorescence-activated cell sorting. Using this cell tracker, we performed in vivo homing experiments in mice with primary splenocytes and tracked a single cell in a heterogeneous, multicellular culture environment for over 20 h. These experiments, in addition to comparative proliferation studies with other cell trackers, demonstrated that the signal from this dye is retained in cells for over 72 h after photoactivation. We envision that this type of probes will facilitate the analysis of single-cell behavior and migration in cell culture and in vivo experiments.
Fluorescent cell trackers are powerful tools that enable direct visualization of biological processes like cell proliferation, cell migration, and cell–cell interactions.1,2 Fluorescent markers can be either small-molecule dyes or fluorescent proteins. Specific cell types can be labeled with genetically encoded fluorescent proteins (e.g., GFP and YFP).3 Additional control over labeling can be achieved using fluorescent proteins like photoactivatable GFP4 and photoconvertible Kaede1,5 and Kikume.6 These photoconvertible proteins are green fluorescent but undergo a bathochromic shift in emission wavelength upon irradiation with UV light.7 The ability to control the emission of the protein by light irradiation allows for advanced applications, for example, in kinetic studies of cellular migration.8
Despite the usefulness of photoconvertible fluorescent proteins, their application is limited by the availability of the transgenic organism, and gene knockout comparative studies increase substantially the complexity of the experimental setup. Furthermore, these proteins often lose their fluorescence following fixation and permeabilization protocols.9 Compared to genetically encoded fluorescent proteins,10 small-molecule dyes benefit from high quantum yields,11 photostability,12 and chemical tuning of their properties.13 On the other hand, they must also fulfill multiple requirements, including membrane permeability, uniform cellular staining, fluorogenic response upon intracellular targeting, intracellular retention, and low cytotoxicity.14,15
Commercial cell proliferation dyes, such as eFluor670 and CellTrace Violet (CTV), are used to quantify cell divisions and track cell populations in live animals.16 Their chemical structures contain functional groups that can form irreversible bonds with reactive nucleophiles. Nevertheless, eFluor670 and CTV are always in a fluorescent state. This feature can cause high background fluorescence and false positive signals if the molecule accumulates in an off-target location.17 In contrast, other cell trackers like carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) react with intracellular carboxylesterases (CEs) to give a bright fluorescein reaction product.18 Succinimidyl groups can covalently bind to intracellular nucleophiles; therefore, cell trackers like CFDA-SE can be retained inside cells, even after cellular division.19,20 Chloromethyl containing probes, like chloromethyl fluorescein diacetate (CMFDA), also form irreversible bonds to intracellular targets containing free thiols, including glutathione.21 Even though this strategy enhances selectivity toward specific intracellular nucleophiles, irreversible cross-linking to reduced glutathione can affect the redox balance of the cell and affect its function.22,23 Both of these fluorescein derivatives are protected with acetyl groups, which makes them very labile toward active CEs. Particularly under inflammatory conditions, immune cells secrete abundant cytotoxic products that can lead to cell death with the subsequent release of intracellular components, including CEs, to the extracellular space.24,25 This condition may lead to the hydrolysis of acetyl-containing dyes and contribute to unspecific fluorescence in the tissue.
In this study, we report a probe that combines the best features of small-molecule dyes and photoconvertible fluorescent proteins. This dual-activatable cell tracker (DACT) relies on activation by both intracellular CEs and light. The fluorescent product is bright and binds covalently to intracellular nucleophiles, giving a highly durable signal that is resistant toward harsh fixation and permeabilization conditions. DACT is simple to use and does not require any genetic manipulation. Moreover, it is compatible with highly sensitive primary cells without inducing considerable cytotoxicity. Finally, we demonstrate the benefits of spatiotemporal control of DACT by implementing inverted fluorescence recovery after photobleaching (iFRAP) to stain single splenocytes selectively in heterogeneous cell mixtures and track their location over a long time.26
Results and Discussion
Design, Synthesis, and Properties of DACTs
Seeking to create a probe that could stain the intracellular region of cells selectively and robustly, we designed a library of DACTs taking into consideration five stages of a live-cell labeling experiment (Figure 1). First, we envisioned that membrane permeability could be facilitated by an initial nonfluorescent form of the DACT that is stable and relatively lipophilic, but noncytotoxic (Figure 1A, I). Upon accumulation of this DACT within the intracellular space, active CEs present in the cell would hydrolyze the ester moiety of DACT to give a nonemissive, preactivated form (Figure 1A, II).27 The main difference between the nonhydrolyzed and hydrolyzed DACT is the electronic character of the xanthene core (Figure 1B). Nonhydrolyzed DACT in the extracellular space has an electron poor xanthene core, and photolysis with UV light induces an intramolecular rearrangement that produces a nonfluorescent photoproduct. Hydrolyzed DACT in the intracellular space has an electron-rich xanthene core, and photolysis with UV light induces the formation of a bright fluorescent product that forms a cross-link with intracellular nucleophiles (Figure 1A, III).27 Therefore, preactivating DACT with intracellular CEs is a crucial step that determines the outcome of the photoreaction and provides selectivity for intracellular labeling. Using the emission of the cross-linked photoproduct (λmax = 560 nm), cells can be detected and tracked with confocal microscopy (Figure 1A, IV) or quantified and categorized by multiwavelength fluorescence assisted cell sorting (FACS; Figure 1A, V).
Figure 1.

Labeling experiment and mechanism of DACT. (A) Five stages of experimental design and how DACTs respond to the stimulus of each step. (B) Structures and photochemical mechanism of activation of DACT probes. PE = Phycoerythrin.
We envisioned that we could modulate the reactivity of the probe toward CEs by variation of the bulkiness of the acyl group R (Figure 1B). Therefore, we prepared a small library of DACTs 1–4 of increasing steric bulk. Each of these compounds was obtained in a one-step synthetic procedure starting from the diazoindanone rhodol precursor 5 and the corresponding acyl chlorides under basic conditions (Figure S1).27 The relative reactivity of DACTs 1–4 toward intracellular CEs was tested in HeLa cells using confocal microscopy (Figure S2 A,B). The susceptibility of DACTs toward hydrolysis by CEs correlated well with the bulkiness of the ester group (Figure S2), similarly to what has been observed for the hydrolysis of other esters in live cells and tissues.28 This trend was further confirmed by FACS (Figure S3).
DACTs are diazoindanone xanthene dyes that are transformed into a ketene intermediate that can be trapped by intracellular nucleophiles.27,29 To prove that DACTs also label intracellular structures covalently, we tested whether DACTs diffuse out of the cell after photoactivation. Even after 27 h of incubation, the intracellular fluorescent signal was detectable for all DACT derivatives (Figure S4). Moreover, we irradiated a small intracellular region of interest (ROI) in an iFRAP experiment and measured a line profile of fluorescence intensity within the irradiated ROI (Figure S5). No significant change in the position of intracellular fluorescence was observed over time for DACTs. In addition, DACTs stayed attached to their intracellular targets after the photoirradiation step (Figure S6). To determine whether cell staining with DACT-1 (Figure 2A) is as homogeneous and persistent as with commonly used dyes, we conducted imaging experiments with cell trackers CTV and CFDA-SE and diffusion experiments with eFluor670 (Figure 2C and Figure S6). Dyes CTV and CFSE showed homogeneous labeling of the cell, but the intensity of CTV decreased significantly after more than 12 h (Figure 2C). DACT-1, in contrast, did not decrease in fluorescence intensity significantly over this period. In contrast, confocal microscopy images of cells stained with eFluor670 revealed a punctate pattern typical of endosomal uptake and/or exocytosis even after short incubation times (Figure S6). In general, DACT-1 performs similarly or better than established cell trackers in terms of homogeneity of staining (Figure 2C).
Figure 2.
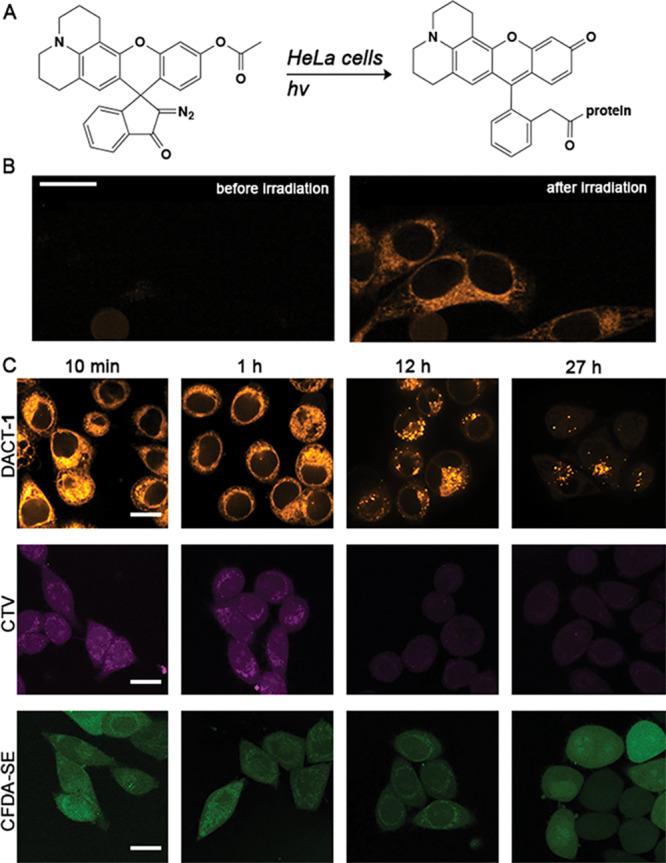
Fluorescent signal durability of cells stained with DACT-1, CTV, and CFDA-SE with varying incubation times after staining protocol. (A) Schematic representation of the intracellular reaction of DACTs. (B) Confocal images obtained from treating HeLa cells with DACT-1 (10 μM) for 10 min. Photoactivation and read-out were achieved using 405 nm (1 s, 30 mW) and 561 nm lasers (0.5 s, 120 mW), respectively. (C) Comparison of fluorescence images of cells incubated with DACT-1 (10 μM for 30 min and photoirradiated), CTV, or CFDA-SE (in a solution of 5 μM dye in FluoroBright, for 30 min) individually, washed and imaged after 10 min and 1, 12, and 27 h using confocal microscopy. Representative images from three independent experiments are displayed. Scale bars = 10 μm.
Cell Tracking and Profiling Using DACT-1 in Vivo
After demonstrating the performance of DACTs as cell trackers in HeLa cells, we focused on their application in primary cells and whole animals. We chose to work with primary murine splenocytes, which are highly sensitive cells. As the fluorescent marker, we decided to use DACT-1 because it produced the brightest intracellular signal after short incubation times. To assess the toxicity induced by irradiation and the dye itself, freshly isolated murine splenocytes were subjected to different UV irradiation times (t = 0, 1, 2, 3, 5 min) and DACT-1 concentrations (1, 10, 20 μM). Toxicity was assessed by staining with Zombie Aqua, a dye that detects membrane permeability, and Annexin V, an indicator of apoptosis, using FACS (Figures S7 and S8). Ten micromoles of DACT-1 and 1.5 min of UV exposure resulted in optimal staining with minimal toxicity (Figures S9 and S10), and these conditions were selected for all subsequent experiments.
Because splenocytes comprise several cell types, we examined whether DACT-1 would stain the whole population of splenocytes homogeneously (Figure S11). For this purpose, splenocytes were incubated with DACT-1 for 30 min at 37 °C and irradiated (405 nm, 1.5 min). The stained splenocytes were mixed in a ratio of 1:1 with unstained splenocytes, and the mixtures were incubated with the corresponding antibodies (30 min at 4 °C) to distinguish dendritic cells and CD4 and CD8 T cells by multicolor FACS analysis. This experiment revealed that cells stained with DACT-1 were consistently distinguishable from nonlabeled cells. Moreover, the fact that in all cell types analyzed the ratio of DACT-1+ to DACT-1– cells remained approximately 1:1 suggests no immediate toxicity of the labeling procedure in primary cells (Figure S11).
To evaluate further the innocuousness of DACT-1 in primary cells, we performed an in vivo homing study to assess cell functionality. A 1:1 mixture of singly (eFluor670) and doubly (eFluor670 and DACT-1) labeled splenocytes was injected into the tail vein of a mouse (Figure 3A). Splenocytes primarily consist of T cells and B cells. The latter are well-known to recirculate constantly between blood and secondary lymphoid organs, such as lymph nodes (LNs), which they rapidly enter by transmigrating through high endothelial venules.30 To analyze the presence of the adoptively transferred cells in lymphoid organs, auricular, axillary and inguinal LNs were harvested after 19 h, processed, and analyzed by FACS. This analysis detected a sizable population of the transferred cells in all LNs and revealed that the 1:1 input ratio of eFluor670+DACT-1– and eFluor670+DACT-1+ was maintained in all harvested LNs (Figure 3B,C). This result demonstrates that DACT-1 staining does not interfere with the active migration of splenocytes in a live animal.
Figure 3.

In vivo homing assay confirmation of no functional differences in splenocytes labeled with DACT-1 compared to splenocytes labeled with eFluor670. (A) Representative FACS analysis of eFluor670+DACT-1– and eFluor670+DACT-1+ splenocytes (1:1 ratio). (B) Representative FACS analysis of LN cell suspensions showing percentages of labeled cells. (C) Analysis of ratios of DACT-1+ to DACT-1– cells in LNs; values were determined using the gating strategies displayed in B. Pooled data from three independent experiments are displayed. Results were normalized to the input and displayed as individual data points (n = 3) and mean (line).
One important limitation of some commercially available dyes and most fluorescent proteins is the loss of signal when cells are subjected to fixation and permeabilization protocols.9,14 These procedures are not only essential for sample preservation but also for other applications, such as electron microscopy31 or intracellular staining of proteins using antibodies.32 Therefore, we tested the robustness of DACT-1 under these conditions. Splenocytes labeled with DACT-1 were mixed with unlabeled cells in a 1:1 ratio. The resulting mixture was fixed and permeabilized using a kit for staining nuclear transcription factors, a particularly harsh protocol. After FACS analysis, the ratio of DACT-1+ to DACT-1– was maintained, indicating that DACT-1 was retained and that the fluorescent signal was not lost during fixation (Figure S12). These results were further confirmed and contrasted by comparing the median fluorescence intensity (MFI) of DACT-1, CTV, CFDA-SE, and eFluor670 before and after fixation and permeabilization of the cells (Figure S13). Notably, nearly 80% of the fluorescence signal in cells stained with CFDA-SE was lost after the fixation and permeabilization protocol. For cells stained with DACT-1, CTV, and eFluor670, no significant change was observed (Figure S13), demonstrating that these dyes are better suited for this kind of experiment.
DACT-1 Is Not Transferred between Cells after Photoactivation
Although some commercially available cell trackers are retained inside the cell, noticeable intercellular transfer occurs with some dyes, eroding their specificity in cell tracking experiments.18 Therefore, we constructed a cell interaction model to compare dye leakage of splenocytes labeled with either DACT-1 or the popular cell tracking dye eFluor670. The model consisted of a conditionally immortalized lymphatic endothelial cell (imLEC) monolayer to which splenocytes were added to study cell crawling under diverse conditions.33 Splenocytes were stained separately with eFluor670 (5 μM), DACT-1 (10 μM), or a 1:1 mixture of eFluor670 and DACT-1. After irradiation and incubation for 19 h at 37 °C on confluent imLEC monolayers, the cells were analyzed by confocal microscopy (Figure 4 and Figure S14). The dyes eFluor670 and DACT-1 were detected in the 647 and 561 nm channels, respectively, and no bleed-through between imaging channels was observed (Figure 4A,B). The signal measured in the imLEC layer at 647 nm revealed that tracker eFluor670 had leaked significantly from splenocytes into the imLEC monolayer (Figure 4A, white arrow). In contrast, imaging at 561 nm gave a signal only in splenocytes and not in the imLEC monolayer (Figure 4B). The same result was observed in splenocytes that had been treated with both dyes simultaneously (Figure 4C). Additional analysis using FACS revealed that 14% of the imLEC population contained eFluor670 dye, whereas no DACT-1 dye was found in imLECs (Figure 4D,E). Similar leakage values were obtained for conditions incubated with a 1:1 mixture of stained splenocytes (eFluor670+ and DACT-1+). This result confirmed that, whereas eFluor670 leaked significantly from stained splenocytes, DACT-1 displayed no detectable leakage (Figure 4E and Figure S14). Similar FACS experiments revealed that DACT-1 was comparable to CTV and slightly superior to CFDA-SE (Figure S15). These results demonstrate that DACT-1 performs as well as CTV and better than eFluor607 or CFDA-SE in terms of nonspecific dye transfer between different cell types.
Figure 4.

Minimal DACT-1 intercellular transfer in coculture studies. Representative images of splenocytes labeled with DACT-1 (A), eFluor670 (B), and mixed in a 1:1 ratio (C), incubated on imLECs for 19 h. Examples of splenocytes are indicated with yellow arrows, and examples of leakage to ECs are shown with white arrows. Scale bars = 50 μm in A, 5 μm in C. (D) Representative FACS analysis of coculture studies of CD31+ imLECs (Figures S14, S15). (E) Analysis of the total percentage of imLECs stained with DACT-1 and eFluor670 caused by intercellular transfer of dyes from splenocytes. Data are plotted as mean ± SEM (n = 3 independent experiments are pooled).
DACT-1 Signal Is Retained in Cells for a Long Time
To evaluate further the durability of the signal in primary cells, we performed proliferation assays with labeled T cells.18 To this end, we performed experiments with bone marrow derived dendritic cells (BM-DCs), which when presenting a peptide derived from ovalbumin (OVA) on MHCII are able to stimulate the proliferation of T cells expressing a transgenic OVA-specific αβ-T cell receptor (OT-II T cells).34 Assuming that the T cells are evenly labeled initially and the dye is efficiently retained, FACS analysis would be expected to reveal proliferation peaks over several T cell division cycles, corresponding to successive reductions of fluorescence by 50% due to cell divisions. OT-II T cells were labeled with DACT-1, CTV, CFDA-SE, or eFluor670 and subsequently incubated for 72 h with BM-DCs and an MHCII-binding peptide derived from OVA. Four cell generations were clearly distinguished for all nonphotoactivatable dyes (CTV, CFDA-SE, and eFluor670). For cells stained with DACT-1, peaks corresponding to the different cell generations were less clearly resolved, presumably because of the low number of fluorescent molecules obtained from the photoactivation step (Figure S16). Nevertheless, we could still distinguish four generations of cells employing DACT-1 staining, demonstrating that the dye signal was retained and not lost in these cells. During these experiments, we noticed that the percentage of proliferated cells stained with CTV was slightly lower than for other staining conditions (Figure S17).
Long-Term, Single-Cell Tracking
Tracking single cells in complex heterogeneous mixtures enables the characterization of cell–cell interactions and cell migration with increased temporal dynamics. These experiments, however, are often limited by leakage, photobleaching, or toxicity of the fluorescent marker. To test DACT-1 in a long-term, single-cell tracking experiment, we prepared a confluent monolayer of imLECs and added unstained splenocytes. This heterogeneous mixture of cells was incubated with DACT-1 (10 μM). Without any further washing step, we selected a single splenocyte and irradiated it in an iFRAP experiment to photoconvert DACT-1 only in that selected cell (Figure 5A). A 24 h time-lapse acquisition revealed the displacement of the splenocyte throughout the field of view (Figure 5B and Movie 1). We were able to monitor up to 12 splenocytes simultaneously in different fields of view over that time frame. We observed splenocytes crawling on the surface of the imLECs and detected a selected single splenocyte in the presence of other nonfluorescent cells (Figure 5C, Movie 2). These results demonstrate the suitability of DACT-1 staining for tracking single cells over time without any need to genetically modify the organism or isolate cells for independent staining.
Figure 5.

Long-term tracking of a single splenocyte on an imLEC monolayer. (A) Brightfield image of a splenocyte (dashed green line) on an imLEC monolayer. Fluorescent intensity in the 561 nm channel (5 s, 120 mW) recorded before and after iFRAP (405 nm, 1 s, 30 mW). (B) Time lapse of the ROI (magenta dotted square) displayed in panel A of the same lymphocyte over the course of 20 h. (C) Example of a splenocyte selected in brightfield mode, activated with iFRAP and tracked over 24 h (same irradiation conditions). Crawling of the splenocyte is observed between 16 and 24 h (yellow arrows). Representative data from two out of 12 experiments are shown. Scale bars = 5 μm.
Summary and Conclusions
In this report, we described DACTs, a family of photoactivatable, photo-cross-linking cell trackers. These probes combine the best features of photoconvertible proteins and small-molecule dyes, including a bright fluorescent signal, low cytotoxicity, intracellular dye retention, and simple application that does not require complicated genetic engineering approaches.
We demonstrated that these DACT probes are preactivated by intracellular CEs and that the kinetics of this preactivation can be tuned. This way, an appropriate DACT could even be selected for experimental conditions in which substantial CEs might be present. Active esterases can be found throughout an entire organism,35 and studies have detected the presence of active CEs both in human and animal plasma.36 It is believed that secretion of active CEs is mainly caused by liver microsomes,37 and recent studies have revealed the unique overexpression of hCEII in cancerous tissue,38,39 a characteristic feature used for pro-drug activation. We therefore propose that adapting the protecting group of DACT can increase resistance toward CE hydrolysis in tissue and circulating blood, enhance the selectivity toward specific substrates, and prevent preactivation of the probe in nondesired locations. After preactivation by CEs in the intracellular compartment, photoactivation of DACTs generates a photo-cross-linked fluorescent product. This covalently bound fluorophore is bright and does not leave the cell even after many hours. Moreover, DACT staining was compatible with harsh fixation and permeabilization procedures often necessary for staining intracellular proteins and profiling cells by multiwavelength FACS.
Importantly, even in combination with photoactivation, DACT probes were not cytotoxic to the tested HeLa cells or the much more sensitive primary murine splenocytes. Moreover, DACT-1 labeling did not compromise cellular function, as evidenced by homing experiments in which DACT-1-labeled splenocytes migrated as avidly as eFluor670-labeled cells from blood into LNs of mice. Cellular coculture studies further confirmed that DACT-1 was not transferred between cells, in contrast to the commercial dye eFluor670, which displayed considerable leakage from dye-labeled splenocytes to unlabeled imLECs. These data illustrate the specificity problem inherent to some fluorescent dyes available on the market18 and confirm that DACT-1 labeling is not jeopardized by costaining with other dyes. Moreover, proliferation assays revealed that DACT-1 was retained in primary cells for up to 72 h. We therefore suggest that DACT-1 can be used as a reliable photoactivatable cell tracker because it displays high retention within the intracellular space, minimal intercellular transfer, and long durability of the signal.
Finally, by tracking the migration of single splenocytes on imLEC monolayers, we could demonstrate that it is possible to specifically irradiate single cells in a complex 3D environment and to track such a cell for long time periods (>20 h). This type of experiment requires a robust photoactivatable dye that does not leak out of the cell of interest and would have been impossible using currently available dyes. This technique could therefore be useful for fate-mapping and tracking of cells of interest in in vitro experiments40 On the other hand, DACT-1-based labeling could also be combined with ex vivo or in vivo time-lapse in tissues of either wild-type or genetically modified mice using intravital microscopy.41 In this case, iFRAP photoactivation could be implemented to select a ROI or single cells for studying cellular migration or for labeling cells for subsequent ex vivo isolation, in analogy to experiments that have thus far been performed with transgenic mice expressing photoconvertible proteins.42,43 For such experiments, the use of DACT dyes could represent a versatile and cost-effective alternative.
Methods
Chemical Synthesis and Compound Characterization
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Varian Gemini 300, Varian Mercury 300, Bruker ARX 300, Bruker DRX 400, Bruker AV 400, and Bruker AV-NEO 500 spectrometers at 300, 400, or 500 MHz (1H) and 75 MHz, 100 MHz, or 125 MHz (13C), respectively. Chemical shifts (δ) are reported in parts per million downfield from tetramethylsilane using the residual deuterated solvent signals as an internal reference. For 1H NMR, coupling constants J are given in hertz, and the resonance multiplicity is described as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). All spectra were recorded at 25 °C. Mass spectrometry (MS) and high-resolution mass spectrometry (HR-MS) were performed by the MS-service of the Laboratory for Organic Chemistry at the ETH Zürich on a Waters Micromass AutoSpec-Ultima spectrometer (EI), on a Bruker maXis spectrometer (ESI), or on a Varian IonSpec FT-ICR spectrometer (MALDI). For MALDI measurements, the matrix was 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) or 3-hydroxypyridine-2-carboxylic acid (3-HPA). Masses are reported in m/z units for the molecular ion M+ for the exact (ChemDraw) and the detected mass. NMR spectra can be obtained upon request.
Reactions
All reactions that were conducted under exclusion of air and water were performed in oven-dried glassware and under a N2 atmosphere. Flash column chromatography (FC) was carried out using silica gel (particle size: 40–63 μm, 230–400 mesh ASTM; Silicycle) employing a Büchi Reveleris PREP purification system with HPLC-grade solvents. Analytical thin layer chromatography (TLC) was performed on aluminum sheets or glass plates coated with silica gel 60 F254 (Merck); visualization with a UV lamp (254 and 366 nm). Evaporation under reduced pressure was performed at 45–60 °C and 900–10 mbar with the use of a rotary evaporator (Heidolph).
Cell Lines
HeLa cells (ATCC CCL2) were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with fetal bovine serum (FBS, 10%) and penicillin-streptomycin (0.1%), referred to as growth medium, at 37 °C in a 95% humidity atmosphere under a 5% CO2 environment. For imaging, HeLa cells were grown to 90% confluence and seeded onto an eight-well Nunc Lab-Tek II chambered cover glass plates (50 000 cells per well) for confocal microscopy or onto Ibidi μ-slide eight-well plates (50 000 cells per well) a day prior to imaging experiments. To generate imLECs, conditionally immortalized lymphatic endothelial cells were isolated from Immorto mice44 and kept in liquid nitrogen. To expand the cells, imLECs were seeded on collagen and fibronectin coated dishes (both 10 μg mL–1) and cultured at 33 °C in a medium containing 40% DMEM (low glucose), 40% F12-Ham, 20% FCS, 56 μg mL–1 heparin, 10 μg mL–1 endothelial cell mitogen, antibiotic antimycotic solution, and l-glutamine. Additionally, murine interferon-γ (IFNγ; 1U mL–1) was added to induce large T-antigen expression. Forty-eight hours before functional assays, imLECs were cultured without IFNγ at 37 °C in the same medium. For functional assays, the cells were cultured for 48 h at 37 °C without IFNγ prior the experiments,44 and the monolayer formation was confirmed under the microscope.
Animals
C57BL/6 mice and OT-II mice34 were bred under specific pathogen free (SPF) conditions in the ETH Rodent Center HCI facility. In vivo homing studies and splenocyte and bone marrow isolation were performed in accordance with protocols approved by the Cantonal Veterinary Office Zurich.
Acknowledgments
This work was funded by an ETH research grant (ETH-02 16-1) and the European Research Council (grant 801572, HDPROBES) to P.R.-F. Microscopy experiments were carried out at the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich or the Bioimaging and Optics (BIOP) core facility of EPF Lausanne. We thank J. Nguyen for performing some confocal microscopy experiments. Some figures were created using Biorender.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00208.
Author Contributions
∥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Tomura M.; Yoshida N.; Tanaka J.; Karasawa S.; Miwa Y.; Miyawaki A.; Kanagawa O. (2008) Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 105, 10871–10876. 10.1073/pnas.0802278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clerck L. S.; Bridts C. H.; Mertens A. M.; Moens M. M.; Stevens W. J. (1994) Use of fluorescent dyes in the determination of adherence of human leucocytes to endothelial cells and the effect of fluorochromes on cellular function. J. Immunol. Methods 172, 115–124. 10.1016/0022-1759(94)90384-0. [DOI] [PubMed] [Google Scholar]
- Day R. N.; Davidson M. W. (2009) The fluorescent protein palette: tools for cellular imaging. Chem. Soc. Rev. 38, 2887–2921. 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson G. H.; Lippincott-Schwartz J. (2002) A photoactive GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877. 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Ando R.; Hama H.; Yamamoto-Hino M.; Mizuno H.; Miyawaki A. (2002) An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99, 12651–12656. 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H.; Karasawa S.; Shimizu H.; Nukina N.; Miyawaki A. (2005) Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 6, 233–238. 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov A. M.; Mishin A. S.; Yampolsky I. V.; Belousov V. V.; Chudakov D. M.; Subach F. V.; Verkhusha V. V.; Lukyanov S.; Lukyanov K. A. (2009) Green fluorescent proteins are light-induced electron donors. Nat. Chem. Biol. 5, 459–461. 10.1038/nchembio.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shand F. H. W.; Ueha S.; Otsuji M.; Koid S. S.; Shichino S.; Tsukui T.; Kosugi-Kanaya M.; Abe J.; Tomura M.; Ziogas J.; Matsushima K. (2014) Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc. Natl. Acad. Sci. U. S. A. 111, 7771–7776. 10.1073/pnas.1402914111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Segala M. G.; Sun M. G.; Shtengel G.; Viswanathan S.; Baird M. A.; Macklin J. J.; Patel R.; Allen J. R.; Howe E. S.; Piszczek G.; Hess H. F.; Davidson M. W.; Wang Y.; Looger L. L. (2015) Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat. Methods 12, 215–218. 10.1038/nmeth.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemet I.; Ropelewski P.; Imanishi Y. (2015) Applications of phototransformable fluorescent proteins for tracking the dynamics of cellular components. Photochem. Photobiol. Sci. 14, 1787–1806. 10.1039/C5PP00174A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavis L. D.; Raines R. T. (2008) Bright ideas for chemical biology. ACS Chem. Biol. 3, 142–155. 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. (2015) A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–253. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Muthusamy A. K.; Liang Y.; Brown T. A.; Lemon W. C.; Patel R.; Lu R.; Macklin J. J.; Keller P. J.; Ji N.; Lavis L. D. (2017) A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace P. K.; Muirhead K. A. (2007) Cell tracking 2007: a proliferation of probes and applications. Immunol. Invest. 36, 527–561. 10.1080/08820130701812584. [DOI] [PubMed] [Google Scholar]
- Parish C. R. (1999) Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 77, 499–508. 10.1046/j.1440-1711.1999.00877.x. [DOI] [PubMed] [Google Scholar]
- Begum J.; Day W.; Henderson C.; Purewal S.; Cerveira J.; Summers H.; Rees P.; Davies D.; Filby A. (2013) A method for evaluating the use of fluorescent dyes to track proliferation in cell lines by dye dilution. Cytometry, Part A 83, 1085–1095. 10.1002/cyto.a.22403. [DOI] [PubMed] [Google Scholar]
- Flannagan R. S.; Heinrichs D. E. (2018) A fluorescence based-proliferation assay for the identification of replicating bacteria within host cells. Front. Microbiol. 9, 1–13. 10.3389/fmicb.2018.03084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah B. J. C.; Parish C. R. (2012) New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods 379, 1–14. 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Tario J. D. J.; Conway A. N.; Muirhead K. A.; Wallace P. K. (2018) Monitoring cell proliferation by dye dilution: considerations for probe selection. Methods Mol. Biol. 1678, 249–299. 10.1007/978-1-4939-7346-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filby A.; Begum J.; Jalal M.; Day W. (2015) Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods 82, 29–37. 10.1016/j.ymeth.2015.02.016. [DOI] [PubMed] [Google Scholar]
- Poot M.; Pierce R. H. (2001) Analysis of mitochondria by flow cytometry. Methods Cell Biol. 64, 117–128. 10.1016/S0091-679X(01)64009-3. [DOI] [PubMed] [Google Scholar]
- Lulevich V.; Shih Y. P.; Lo S. H.; Liu G. Y. (2009) Cell tracing dyes significantly change single cell mechanics. J. Phys. Chem. B 113, 6511–6519. 10.1021/jp8103358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz R. C.; Lemus R.; Lange R. W.; Karol M. H. (2001) Rapid reduction of intracellular glutathione in human bronchial epithelial cells exposed to occupational levels of toluene diisocyanate. Toxicol. Sci. 60, 348–355. 10.1093/toxsci/60.2.348. [DOI] [PubMed] [Google Scholar]
- Nathan C. (2006) Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182. 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- Andersen M. H.; Schrama D.; thor Straten P.; Becker J. C. (2006) Cytotoxic T cells. J. Invest. Dermatol. 126, 32–41. 10.1038/sj.jid.5700001. [DOI] [PubMed] [Google Scholar]
- Hauke S.; von Appen A.; Quidwai T.; Ries J.; Wombacher R. (2017) Specific protein labeling with caged fluorophores for dual-color imaging and super-resolution microscopy in living cells. Chem. Sci. 8, 559–566. 10.1039/C6SC02088G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabi E. A.; Thiel Z.; Trapp N.; Pinotsi D.; Rivera-Fuentes P. (2017) A photoactivatable probe for super-resolution imaging of enzymatic activity in live cells. J. Am. Chem. Soc. 139, 13200–13207. 10.1021/jacs.7b07748. [DOI] [PubMed] [Google Scholar]
- Tian L.; Yang Y.; Wysocki L. M.; Arnold A. C.; Hu A.; Ravichandran B.; Sternson S. M.; Looger L. L.; Lavis L. D. (2012) Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc. Natl. Acad. Sci. U. S. A. 109, 4756–4761. 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel Z.; Rivera-Fuentes P. (2019) Single-molecule imaging of active mitochondrial nitroreductases using a photo-crosslinking fluorescent sensor. Angew. Chem., Int. Ed. 58, 11474–11478. 10.1002/anie.201904700. [DOI] [PubMed] [Google Scholar]
- von Andrian U. H.; Mackay C. R. (2000) T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343, 1020–1034. 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- Graham L.; Orenstein J. M. (2007) Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat. Protoc. 2, 2439–2450. 10.1038/nprot.2007.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisman A., and Becher B. (2014) T-Helper Cells (Waisman A., and Becher B., Eds.), Springer, New York. [Google Scholar]
- Teijeira A.; Hunter M. C.; Russo E.; Proulx S. T.; Frei T.; Debes G. F.; Coles M.; Melero I.; Detmar M.; Rouzaut A.; Halin C. (2017) T cell migration from inflamed skin to draining lymph nodes requires intralymphatic crawling supported by ICAM-1/LFA-1 interactions. Cell Rep. 18, 857–865. 10.1016/j.celrep.2016.12.078. [DOI] [PubMed] [Google Scholar]
- Barnden M. J.; Allison J.; Heath W. R.; Carbone F. R. (1998) Defective TCR expression in transgenic mice constructed using cDNA- based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76, 34–40. 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- Wang D.; Zou L.; Jin Q.; Hou J.; Ge G.; Yang L. (2018) Human carboxylesterases: a comprehensive review. Acta Pharm. Sin. B 8, 699–712. 10.1016/j.apsb.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Sedlacek M.; Manoharan I.; Boopathy R.; Duysen E. G.; Masson P.; Lockridge O. (2005) Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 70, 1673–1684. 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Yan B.; Yang D.; Bullock P.; Parkinson A. (1995) Rat serum carboxylesterase. J. Biol. Chem. 270, 19128–19134. 10.1074/jbc.270.32.19128. [DOI] [PubMed] [Google Scholar]
- Xu G.; Zhang W.; Ma M. K.; McLeod H. L. (2002) Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 8, 2605–2611. [PubMed] [Google Scholar]
- Yano H.; Kayukawa S.; Iida S.; Nakagawa C.; Oguri T.; Sanda T.; Ding J.; Mori F.; Ito A.; Ri M.; Inagaki A.; Kusumoto S.; Ishida T.; Komatsu H.; Inagaki H.; Suzuki A.; Ueda R. (2008) Overexpression of carboxylesterase-2 results in enhanced efficacy of topoisomerase I inhibitor, irinotecan (CPT-11), for multiple myeloma. Cancer Sci. 99, 2309–2314. 10.1111/j.1349-7006.2008.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piltti K. M.; Cummings B. J.; Carta K.; Manughian-Peter A.; Worne C. L.; Singh K.; Ong D.; Maksymyuk Y.; Khine M.; Anderson A. J. (2018) Live-cell time-lapse imaging and single-cell tracking of in vitro cultured neural stem cells – Tools for analyzing dynamics of cell cycle, migration, and lineage selection. Methods 133, 81–90. 10.1016/j.ymeth.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson A. L.; Fujisaki J.; Wu J.; Runnels J. M.; Turcotte R.; Celso C. L.; Scadden D. T.; Strom T. B.; Lin C. P. (2013) Tracking single cells in live animals using a photoconvertible near-infrared cell membrane label. PLoS One 8, e69257 10.1371/journal.pone.0069257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medaglia C.; Giladi A.; Stoler-Barak L.; De Giovanni M.; Salame T. M.; Biram A.; David E.; Li H.; Iannacone M.; Shulman Z.; Amit I. (2017) Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 358, 1622–1626. 10.1126/science.aao4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora G. D.; Schwickert T. A.; Fooksman D. R.; Kamphorst A. O.; Meyer-Hermann M.; Dustin M. L.; Nussenzweig M. C. (2010) Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143, 592–605. 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigl B.; Aebischer D.; Nitschké M.; Iolyeva M.; Röthlin T.; Antsiferova O.; Halin C. (2011) Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 118, 205–215. 10.1182/blood-2010-12-326447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.