

Abstract
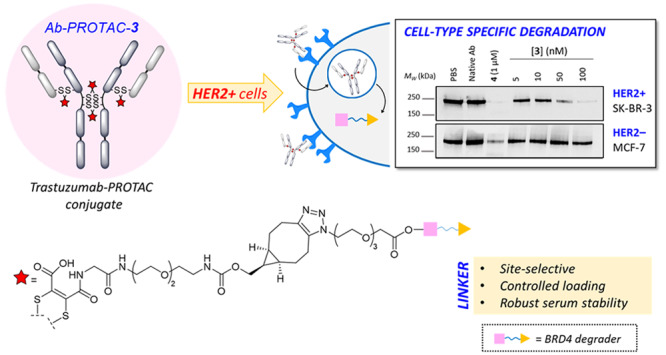
Targeting protein degradation with Proteolysis-Targeting Chimeras (PROTACs) is an area of great current interest in drug discovery. Nevertheless, although the high effectiveness of PROTACs against a wide variety of targets has been established, most degraders reported to date display limited intrinsic tissue selectivity and do not discriminate between cells of different types. Here, we describe a strategy for selective protein degradation in a specific cell type. We report the design and synthesis of a trastuzumab-PROTAC conjugate (Ab-PROTAC 3) in which E3 ligase-directed degrader activity is caged with an antibody linker which can be hydrolyzed following antibody–PROTAC internalization, releasing the active PROTAC and inducing catalytic protein degradation. We show that 3 selectively targets bromodomain-containing protein 4 (BRD4) for degradation only in HER2 positive breast cancer cell lines, while sparing HER2 negative cells. Using live cell confocal microscopy, we show internalization and lysosomal trafficking of the conjugate specifically in HER2 positive cells, leading to the release of active PROTAC in quantities sufficient to induce potent BRD4 degradation. These studies demonstrate proof-of-concept for tissue-specific BRD4 degradation, overcoming limitations of PROTAC selectivity, with significant potential for application to novel targets.
Protein degradation directed by small molecules including “molecular glues”,1−4 or Proteolysis-Targeting Chimeras (PROTACs),5−9 is among the fastest growing fields in chemical biology and drug discovery. In addition to its therapeutic potential, this technology has led to powerful chemical tools to probe protein function. PROTACs are bifunctional compounds composed of a ligand against a protein of interest (POI) connected to an E3 ligase ligand via a suitably designed linker.10 The mode of action of these molecules relies on their ability to bring the POI and E3 ligase into proximity, triggering directed polyubiquitination and subsequent proteasome-mediated degradation of the POI, in a manner that is catalytic, with respect to the PROTAC.11 PROTACs have been reported against a wide range of different targets playing important roles in biology, and particularly cancer, including Estrogen Receptor (ER),12 Androgen Receptor (AR),13,14 BET-bromodomain proteins,15,16 and various kinases.17−21 Recently disclosed safety and pharmacokinetics data for two orally bioavailable PROTAC candidates (ARV-110 targeting AR, for the treatment of prostate cancer, and ARV-471, which is an ERα degrader for breast cancer therapy) in a phase I clinical trial prefigures the potential of these compounds.22−24 Nonetheless, although numerous reported PROTACs are highly efficient degraders, they are generally not tissue-specific, since they exploit E3 ligases with broad expression profiles. Tissue-specific degradation could enable optimization of the therapeutic window and minimize side effects for broad-spectrum PROTACs, increasing their potential as drugs or chemical tools. However, PROTACs exploiting E3 ligases with restricted tissue distribution have not been reported to date, and the development of novel E3 ligase ligands remains a significant challenge.
We considered an antibody–PROTAC conjugate as an alternative approach for selective delivery of a broad-spectrum PROTAC into specific cell types, by analogy to antibody–drug conjugates (ADCs). ADCs have gained momentum as anticancer therapeutics, since they allow delivery of a cytotoxic payload specifically to cancer cells, minimizing undesired side effects.25 ADCs can enhance therapeutic monoclonal antibodies, such as trastuzumab (Herceptin) or pertuzumab (Perjeta).26 For example, the ADC T-DM1 ado-trastuzumab emtansine (Kadcyla) has been approved by the United States Food and Drug Administration (FDA) for the treatment of metastatic HER2 positive (HER2+) breast cancer, following treatment with trastuzumab and taxanes.27,28 Nonetheless, to date, only a few ADCs have received FDA approval for commercialization, since many of these new therapeutics have failed during clinical trials, because of intrinsic limitations such as uptake into nontargeted cells. The main challenge for ADC development is related to dose-limiting toxicities (DLTs), which are frequently reported, even at suboptimal therapeutic doses, resulting in a poor balance between therapeutic efficacy and off-target toxicity of these drugs.29 Another drawback of ADCs is the low quantity of payload typically delivered into tumors, meaning that the payload must be extremely cytotoxic, which can be dose-limiting.30 We considered that a PROTAC could be an ideal ADC payload, since it benefits from catalytic degradation activity driven by substoichiometric target engagement, delivering extended and potent target degradation from a low dose of compound.31
We hypothesized that a trastuzumab-PROTAC conjugate platform could achieve selective delivery of a PROTAC and direct protein degradation specifically in HER2+ cells. Such a conjugate would bind HER2/neu receptors, inducing endosomal internalization and lysosomal release of active PROTAC (see Figure 1A). Given that bromodomain containing protein 4 (BRD4) is a potentially attractive target in inflammation and cancer, because of its role in transcriptional dysregulation,32 we were interested in exploring the potential of a trastuzumab–BRD4 degrader conjugate to achieve cell-type-specific BRD4 degradation in a breast cancer cell model. We selected PROTAC 1 for a proof-of-concept study (Figure 1B). This compound is an analogue of BRD4 degrader MZ1, which has been reported to achieve complete degradation of BRD4 at 100 nM, following 4 h of treatment.15 PROTACs of this class feature BET bromodomain ligand JQ1 as a targeting element for BET-containing proteins (such as BRD4) linked to a ligand for von Hippel–Lindau protein (VHL), which is a key component of an E3 ligase complex involved in the regulation of hypoxia.33 While our manuscript was in preparation, other examples of antibody-based targeted protein degradation were disclosed by Dragovich et al., including the delivery of Estrogen Receptor alpha (ERα) degraders selectively to targeted cells34 and selective delivery of an anti-CLL1-(GNE-987) conjugate in a model of chronic lymphocytic leukemia (CLL).35 In the latter study, the authors report a new BRD4 degrader (GNE-987) with picomolar potencies toward BRD4 degradation in EOL-1 AML cells, as well as in cell viability studies performed with different leukemia cell lines. Furthermore, with the aim of improving the pharmacokinetics properties of this compound in vivo, the derivatization of this degrader into an antibody–degrader conjugate was described. This involved the conjugation of GNE-987 to a CCL1 antibody through a disulfide-bearing linker that was attached to GNE-987 via a carbonate bond. A single IV administration of the conjugate to mice with HL-60 AML xenografts provided dose- and antigen-dependent tumor regression.
Figure 1.

(A) Proposed mode of action of an antibody–PROTAC conjugate, resulting in HER2-dependent protein degradation. (B) Structures of compounds 1 and 2. (C) Overall structure of Ab-PROTAC 3. (D) LC-MS analysis of trastuzumab-NGM-PROTAC conjugate (Ab-PROTAC 3) and the deconvoluted ion series mass spectrum. Observed mass of 152 010 corresponds to full antibody modified with four NGM-BCN-PROTAC molecules, where all NGM moieties are hydrolyzed to maleamic acids (expected: 152 008); 76 005 corresponds to the isomeric form in which the hinge cysteines are bridged in an intrachain manner (expected: 76 004). (E) Hydrolytic stability of Ab-PROTAC 3 determined by liquid chromatography–mass spectroscopy (LC-MS).
Our conjugation strategy required attachment of the PROTAC to trastuzumab through a triazole moiety. We decided to take advantage of the free hydroxyl group on the VHL ligand moiety of 1 to tether an azido-PEG linker. This hydroxyl group in 1 is essential for binding to VHL;36,37 hence, attaching an antibody linker to this position via an ester (see Figure 1B, as well as Scheme S1 in the Supporting Information) would effectively cage PROTAC activity until it is released intracellularly during lysosomal digestion of the conjugate. Thus, our tool compounds PROTAC 1 and azido-PROTAC 2 were synthesized as reported in Scheme S1.
Antibody–PROTAC conjugate 3 was synthesized by rebridging the interchain disulfide bonds of trastuzumab with next-generation maleimides (NGMs), giving access to constructs with controlled PROTAC loading and robust serum stability, ensuring PROTAC release solely via hydrolysis of the cleavable ester linker.38−41 A novel dibromomaleimide-strained alkyne linker 13 (Scheme S2A in the Supporting Information) was used for convenient synthesis of 3 by copper-free strain-promoted azide–alkyne cycloaddition (SPAAC), precluding any risk of copper contamination (see Scheme S2B in the Supporting Information). Following disulfide bond reduction, trastuzumab was reacted with dibromomaleimide 13, and overnight incubation at pH 8.5 buffer facilitated quantitative hydrolysis to the desired serum stable maleamic acids (see Figures S1 and S4 in the Supporting Information). Azido-PROTAC 2 was installed via SPAAC (20 equiv 2 at 22 °C, 4 h, pH 6.9), affording Ab-PROTAC 3 with a PROTAC–antibody ratio (or drug:antibody ratio, DAR) of 4 (see Figures 1C and 1D). Denaturing LC-MS analysis shows two isomeric DAR 4 disulfide bridged conjugates, consistent with the precedent for such ADCs.39 ELISA confirmed full retention of antibody binding activity (see Figure S5 in the Supporting Information). Pleasingly, we found that 3 shows excellent stability, with 98.8% of 3 intact after 4 h of incubation (PBS, pH 7.4, 37 °C), and up to 83.5% after 24 h of incubation (see Figure 1E, as well as Figure S3 in the Supporting Information).
We examined the biological activity of 3 against two HER2 negative (HER2−) breast cancer cell lines (MCF-7 and MDA-MB-231) and two HER2+ breast cancer cell lines (SK-BR-3 and BT-474), selected based on HER2/neu receptor and BRD4 expression levels determined by Western blot and immunofluorescence (see Figures 2A and 2B) and the confirmed capacity of free PROTAC 1 to degrade BRD4 in each of these cell lines under the same conditions selected for testing Ab-PROTAC 3 (Figure 2C). The different BRD4 degradation profile of PROTAC 1 among HER2+ and HER2– phenotypes is most likely related to different expression levels of BRD4 across these cell lines.42,43 Each cell line was treated with vehicle (PBS), antibody (trastuzumab), or positive control 4 (Figure 2E), or with increasing concentrations of Ab-PROTAC 3. BRD4 degrader 4 was used as a positive control across these studies as a reference point for maximal degradation of BRD4. Cognizant of the stability of Ab-PROTAC 3 at physiological pH (above), we incubated cells either for 4 h in a serum-free medium, or for 23 h following 1-h preincubation and subsequent washout (24 h). BRD4 abundance was analyzed by SDS-PAGE, followed by Western blot analysis (Figure 2D), showing that Ab-PROTAC 3 selectively degrades BRD4 only in HER2+ cells while leaving BRD4 intact in HER2– cells. 100 nM Ab-PROTAC 3 for 4 h delivered almost-complete BRD4 depletion only in HER2+ cell lines. Significant BRD4 degradation was also observed at 50 nM, whereas no degradation was detected at any of the concentrations tested in HER2– cell lines. Furthermore, just 1 h of incubation with Ab-PROTAC 3 produced significant degradation of BRD4 24 h after treatment in HER2+ cells, suggesting that the construct achieves sufficient internalization into HER2+ cells within the 1-h treatment period to deliver a potent dose of PROTAC 1. In the case of cells lacking HER2/neu receptor surface expression, 3 is unable to undergo HER2/neu mediated internalization; hence, no degradation was observed. We note that BRD4 degradation was not observed in any experiment in which cells were treated with native trastuzumab. In the case of washout experiments with SK-BR-3 cells (Figure 2D), the minor variations in BRD4 band intensity are attributed to variation in loading, as supported by quantitation of the Western blot data (Figure S7 in the Supporting Information). The results of these experiments deliver proof of concept for selective HER2/neu-mediated PROTAC delivery.
Figure 2.

(A) Expression of HER2/neu receptor and BRD4 protein in the cell lines used in this study: Western blot analysis of HER2/neu, BRD4, and β-actin on cell lysates from MCF-7, MDA-MB-231, SK-BR-3, and BT-474 cells. (B) Representative frame from immunofluorescence microscopy images of HER2– MCF-7 and HER2+ SK-BR-3 cells labeled with a primary anti-HER2 antibody and stained with a secondary AF488-conjugated antibody (green). Nuclei were stained with DAPI (blue). (C) Monitoring of BRD4 degradation properties of PROTAC 1 in MCF-7, MDA-MB-231, SK-BR-3 and BT-474 cells. Western blot analysis of BRD4 and β-actin after treatment of cells with PROTAC 1, or control compounds. Cells were incubated with PROTAC 1 for either 4 h in the medium before harvesting cell lysates (“4 h incubation”), or for 1 h followed by washout (“washout”), which consisted of removal of the medium, washing with PBS, the addition of fresh medium, and harvesting 23 h later. (D) Ab-PROTAC 3 induces selective degradation of BRD4 in HER2+ cell lines, in a dose-dependent manner: Western blot analysis of BRD4 and β-actin following treatment of MCF-7, MDA-MB-231, SK-BR-3, or BT-474 cells with Ab-PROTAC 3, or control compounds. Cells were incubated with Ab-PROTAC 3 for either 4 h in the medium before harvesting cell lysates (“4 h incubation”), or for 1 h followed by washout (“washout”), consisting of removal of the medium, washing with PBS, the addition of fresh medium, and harvesting 23 h later. (E) Structure of BRD4 degrader 4 used as a positive control to gauge BRD4 degradation. (F) BRD4 degradation by Ab-PROTAC 3 is proteasome-dependent: Western blot analysis of BRD4 and β-actin after treatment of SK-BR-3 and BT-474 cells with Ab-PROTAC 3 or control compounds in the presence or absence of proteasome inhibitor bortezomib (BTZ). Cells were incubated for 2 h with BTZ or PBS (pH 6.9), followed by 1 h of treatment with Ab-PROTAC (3). A washout was performed on the wells treated with Ab-PROTAC 3, and cells were harvested 23 h after washout.
We next determined whether BRD4 degradation induced by Ab-PROTAC 3 is proteasome-dependent, as expected for PROTAC-mediated degradation. Cells were incubated with vehicle (PBS), positive control 4, or 100 nM Ab-PROTAC 3 in the presence or absence of proteasome inhibitor Bortezomib (BTZ). Western blot analysis of cell lysates confirmed that proteasome inhibition prevents Ab-PROTAC 3-mediated BRD4 degradation in both HER2+ cell lines (Figure 2F).
Receptor-mediated antibody internalization has been extensively studied through different approaches, including radiolabeling, fluorescence microscopy, flow cytometry, or cytotoxicity assays.44−47 Among these, confocal microscopy with fluorescently tagged antibodies allows antibody localization upon internalization across different cellular compartments, such as endosomes or lysosomes, and, hence, is a commonly used technique. Trastuzumab internalization and intracellular compartmentalization using this method has been previously studied, evidencing endosomal internalization of the HER2/trastuzumab complex.48 Taking these precedents into account, we aimed to investigate the trafficking of Ab-PROTAC 3 in live cells by confocal microscopy to confirm internalization and further analyze the fate of our conjugate within the intracellular environment. For these studies, we used a fluorescently labeled AlexaFluor488-Ab-PROTAC (AF488-3) (see Scheme S2 and Figure S4 in the Supporting Information) synthesized by treating construct 3 with AlexaFluor488-NHS ester (10 equiv, 22 °C, 2 h, pH 7.4), resulting in conjugate AF488-3 with an average fluorophore:antibody ratio (FAR) of 3.8 (see Figure S6 in the Supporting Information). HER2+ SK-BR-3 cells were incubated with 100 nM AF488-3 for 1 h, and cells were then washed and imaged at 1, 4, 8, and 24 h time points to study conjugate trafficking (see Figure 3A). At 1 h, AF488-3 was observed on the cell surface, whereas after 4 h of incubation, AF488-3 was already partially internalized into distinct intracellular compartments, presumably early and late endosomes upstream from lysosome trafficking for degradation, and partially colocalized with LysoTracker DeepRed labeled lysosomes (see Figures 3A and 3C). After 8 h, AF488-labeled compartments colocalized extensively with lysosomes (Figures 3A and 3C); similar results were seen without conjugate washout (see Figure S8 in the Supporting Information). Note that, upon lysosomal digestion of Ab-PROTAC 3, the free active PROTAC 1 must be released and then access the nucleus to trigger BRD4 degradation. MCF-7 cells expressing very low levels of HER2 receptor were unable to uptake AF488-3 under the same conditions (Figure 3B), consistent with resistance of these cells to Ab-PROTAC-mediated BRD4 degradation.
Figure 3.

(A) HER2+ SK-BR-3 cells were treated with Alexa488-conjugated Ab-PROTAC (AF488-3) (100 nM) for 1, 4, 8, and 24 h (all post-1-h washout). Cells were then labeled with LysoTracker DeepRed (50 nM, 30 min). Shown is a representative frame from live imaging of cells via confocal microscopy. Scale bar = 20 μm. (B) HER2– MCF-7 cells were treated with AF488-3 (100 nM) for 30 min, followed by incubation with LysoTracker DeepRed (50 nM) for an additional 30 min. Shown is a representative frame from live imaging of cells via confocal microscopy. (C) Quantification of AF488-3 colocalization with LysoTracker DeepRed. The Pearson’s correlation coefficient is shown for various incubation time points in SK-BR-3 cells; data are mean + the standard error of measurement (SEM); n = 6 cells. Statistical analysis was performed by one-way ANOVA, followed by Tukey’s multiple comparison test (triple asterisk symbol (***) denotes P < 0.0001; “ns” = nonsignificant).
In summary, we have developed a new example of antibody–PROTAC conjugate by linking azido-PROTAC 2 to dibromomaleimide-strained alkyne-functionalized trastuzumab through a triazole moiety leading to Ab-PROTAC 3 bearing four molecules of PROTAC. This site-selective conjugate directs proteasomal BRD4 degradation in HER2+ cells, following HER2-dependent internalization, lysosomal trafficking, and active PROTAC release. Our studies demonstrate a new antibody–PROTAC system for antigen-specific targeted protein degradation through antigen-dependent delivery of a PROTAC, broadening the scope of the Ab-PROTAC technology and providing the basis for the development of antibody–PROTACs for the degradation of a variety of different targets. We conclude that Ab-PROTACs have the potential to overcome traditional limitations of targeted degraders and ADCs by combining the key advantages of each modality: the catalytic potency of PROTACs with the tissue specificity of ADCs.
Methods
Experimental procedures are described in detail in the Supporting Information.
Acknowledgments
The authors thank GlaxoSmithKline Research and Development, Ltd. (Stevenage, U.K.) for providing pan-bromodomain inhibitor JQ1 and Von Hippel–Lindau ligand (VHL), and The Francis Crick Institute (London, U.K.) for providing the cell lines used in this study. M.M. thanks the Xunta de Galicia for a Postdoctoral Fellowship. This work was funded by the Engineering and Physical Sciences Research Council (EPSRC) (Grant No. EP/R512540/1, Project No. 2100261 to E.W.T. and Grant No. EP/R034621/1 to J.R.B. and V.C.) and GlaxoSmithKline Research and Development, Ltd.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00285.
Supplementary figures and schemes; materials and methods for chemical synthesis and bioconjugation, cell-based experiments, and microscopy studies; compound characterization data and NMR spectra; Ab-PROTACs_Suppl_info (PDF)
The authors declare the following competing financial interest(s): E.W.T. is a founder, Director, and shareholder of Myricx Pharma, Ltd. The other authors declare no competing financial interests.
Supplementary Material
References
- Uehara T.; Minoshima Y.; Sagane K.; Sugi N. H.; Mitsuhashi K. O.; Yamamoto N.; Kamiyama H.; Takahashi K.; Kotake Y.; Uesugi M.; Yokoi A.; Inoue A.; Yoshida T.; Mabuchi M.; Tanaka A.; Owa T. (2017) Selective Degradation of Splicing Factor CAPERα By Anticancer Sulfonamides. Nat. Chem. Biol. 13, 675–680. 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- Han T.; Goralski M.; Gaskill N.; Capota E.; Kim J.; Ting T. C.; Xie Y.; Williams N. S.; Nijhawan D. (2017) Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 356, eaal3755 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Faust T. B.; Yoon H.; Nowak R. P.; Donovan K. A.; Li Z.; Cai Q.; Eleuteri N. A.; Zhang T.; Gray N. S.; Fischer E. S. (2020) Structural Complementarity Facilitates E7820-Mediated Degradation of RBM39 by DCAF15. Nat. Chem. Biol. 16, 7. 10.1038/s41589-019-0378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X.; Volkov O. A.; Czerwinski R. M.; Tan H. L.; Huerta C.; Morton E. R.; Rizzi J. P.; Wehn P. M.; Xu R.; Nijhawan D.; Wallace E. M. (2019) Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 27, 1625–1633. 10.1016/j.str.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P.; Lithgow H.; Churcher I. (2016) Small Molecule-Mediated Protein Knockdown as a New Approach to Drug Discovery. MedChemComm 7, 2206–2216. 10.1039/C6MD00347H. [DOI] [Google Scholar]
- Ottis P.; Crews C. M. (2017) Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 12, 892–898. 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- Churcher I. (2018) PROTAC-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?. J. Med. Chem. 61, 444–452. 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. (2019) Proteolysis TArgeting Chimeras (PROTACs) — Past, Present and Future. Drug Discovery Today: Technol. 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. (2019) Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discovery 18, 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Paiva S. L.; Crews C. M. (2019) Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. (2015) Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Hu B.; Wang M.; Xu F.; Miao B.; Yang C. Y.; Wang M.; Liu Z.; Hayes D. F.; Chinnaswamy K.; Delproposto J.; Stuckey J.; Wang S. (2019) Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 62, 1420–1442. 10.1021/acs.jmedchem.8b01572. [DOI] [PubMed] [Google Scholar]
- Han X.; Wang C.; Qin C.; Xiang W.; Fernandez-Salas E.; Yang C. Y.; Wang M.; Zhao L.; Xu T.; Chinnaswamy K.; Delproposto J.; Stuckey J.; Wang S. (2019) Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 62, 941–964. 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- Salami J.; Alabi S.; Willard R. R.; Vitale N. J.; Wang J.; Dong H.; Jin M.; McDonnell D. P.; Crew A. P.; Neklesa T. K.; Crews C. M. (2018) Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Commun. Biol. 1, 100. 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli (2015) A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. (2015) Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W. D.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. (2018) Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 115, E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A. P.; Raina K.; Dong H.; Qian Y.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferraro C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. (2018) Identification and Characterization of von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 61, 583–598. 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Song J.; Chen X.; Hines J.; Crews C. M. (2018) Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 140, 16428–16432. 10.1021/jacs.8b10320. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Lan T.; Su S.; Rao Y. (2019) Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem. Commun. 55, 369–372. 10.1039/C8CC07813K. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Burgess K. (2019) PROTACs Suppression of CDK4/6, Crucial Kinases for Cell Cycle Regulation in Cancer. Chem. Commun. 55, 2704–2707. 10.1039/C9CC00163H. [DOI] [PubMed] [Google Scholar]
- Arvinas, Inc . A Phase 1 Clinical Trial of ARV-110 in Patients With Metastatic Castration-resistant Prostate Cancer; available via the Internet at: https://www.clinicaltrials.gov/ct2/show/NCT03888612?term=ARV-110 (accessed December 2, 2019).
- Arvinas, Inc . Clinical Trial of ARV-471 in Patients with ER+/HER2– Locally Advanced or Metastatic Breast Cancer (mBC); available via the Internet at: https://clinicaltrials.gov/ct2/show/NCT04072952 (accessed April 4, 2020).
- Mullard A. (2019) Arvinas’s PROTACs pass first safety and PK analysis. Nat. Rev. Drug Discovery 18, 895. 10.1038/d41573-019-00188-4. [DOI] [PubMed] [Google Scholar]
- Walko C. M.; West H. J. (2018) Antibody Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 69, 191–207. [DOI] [PubMed] [Google Scholar]
- Kadcyla Seems Better Than Herceptin for Early-Stage, HER2-Positive Residual Breast Cancer; available via the Internet at: https://www.breastcancer.org/research-news/kadcyla-vs-herceptin-for-early-stage-her2-pos (accessed Dec. 2, 2019).
- Lambert J. M.; Chari R. V. J. (2014) Ado-Trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 57, 6949–6964. 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- Amiri-Kordestani L.; Blumenthal G. M.; Xu Q. C.; Zhang L.; Tang S. W.; Ha L.; Weinberg W. C.; Chi B.; Candau-Chacon R.; Hughes P.; Russell A. M.; Miksinski S. P.; Chen X. H.; McGuinn W. D.; Palmby T.; Schrieber S. J.; Liu Q.; Wang J.; Song P.; Mehrotra N.; Skarupa L.; Clouse K.; Al-Hakim A.; Sridhara R.; Ibrahim A.; Justice R.; Pazdur R.; Cortazar P. (2014) FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 20, 4436–4441. 10.1158/1078-0432.CCR-14-0012. [DOI] [PubMed] [Google Scholar]
- Mahalingaiah P. K.; Ciurlionis R.; Durbin K. R.; Yeager R. L.; Philip B. K.; Bawa B.; Mantena S. R.; Enright B. P.; Liguori M. J.; Van Vleet T. R. (2019) Potential Mechanisms of Target-Independent Uptake and Toxicity of Antibody-Drug Conjugates. Pharmacol. Ther. 200, 110–125. 10.1016/j.pharmthera.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Teicher B. A.; Chari R. V. J. (2011) Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 17, 6389–6397. 10.1158/1078-0432.CCR-11-1417. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. (2017) Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discovery 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. (2012) BET domain co- regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12, 465–477. 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. L.; Jiang B. H.; Rue E. A.; Semenza G. L. (1995) Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. U. S. A. 92, 5510–5514. 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillow T. H.; Adhikari P.; Blake R. A.; Chen J.; Del Rosario G.; Deshmukh G.; Figueroa I.; Gascoigne K. E.; Kamath A. V.; Kaufman S.; Kleinheinz T.; Kozak K. R.; Latifi B.; Leipold D. D.; Sing Li C.; Li R.; Mulvihill M. M.; O’Donohue A.; Rowntree R. K.; Sadowsky J. D.; Wai J.; Wang X.; Wu C.; Xu Z.; Yao H.; Yu S.-F.; Zhang D.; Zang R.; Zhang H.; Zhou H.; Zhu X.; Dragovich P. S. (2020) Antibody Conjugation of a Chimeric BET Degrader Enables in Vivo Activity. ChemMedChem 15, 17–25. 10.1002/cmdc.201900497. [DOI] [PubMed] [Google Scholar]
- Dragovich P. S.; Adhikari P.; Blake R. A.; Blaquiere N.; Chen J.; Cheng Y.-X.; den Besten W.; Han J.; Hartman S. J.; He J.; He M.; Rei Ingalla E.; Kamath A. V.; Kleinheinz T.; Lai T.; Leipold D. D.; Li C. S.; Liu Q.; Lu J.; Lu Y.; Meng F.; Meng L.; Ng C.; Peng K.; Lewis-Phillips G.; Pillow T. H.; Rowntree R. K.; Sadowsky J. D.; Sampath D.; Staben L.; Staben S. T.; Wai J.; Wan K.; Wang X.; Wei B.; Wertz I. E.; Xin J.; Xu K.; Yao H.; Zang R.; Zhang D.; Zhou H.; Zhao X. (2020) Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα) Bioorg. Med. Chem. Lett. 30, 126907–126914. 10.1016/j.bmcl.2019.126907. [DOI] [PubMed] [Google Scholar]
- Min J. H.; Yang H.; Ivan M.; Gertler F.; Kaelin W. G.; Pavietich N. P. (2002) Structure of a HIF-1α-PVHL Complex: Hydroxyproline Recognition in Signaling. Science 296, 1886–1889. 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Hon W. C.; Wilson M. I.; Harlos K.; Claridge T. D. W.; Schofield C. J.; Pugh C. W.; Maxwell P. H.; Ratcliffe P. J.; Stuart D. I.; Jones E. Y. (2002) Structural Basis for the Recognition of Hydroxyproline in HIF-1α by PVHL. Nature 417, 975–978. 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Nunes J. P. M.; Morais M.; Vassileva V.; Robinson E.; Rajkumar V. S.; Smith M. E. B.; Pedley R. B.; Caddick S.; Baker J. R.; Chudasama V. (2015) Functional Native Disulfide Bridging Enables Delivery of a Potent, Stable and Targeted Antibody-Drug Conjugate (ADC). Chem. Commun. 51, 10624–10627. 10.1039/C5CC03557K. [DOI] [PubMed] [Google Scholar]
- Morais M.; Nunes J. P. M.; Karu K.; Forte N.; Benni I.; Smith M. E. B.; Caddick S.; Chudasama V.; Baker J. R. (2017) Optimisation of the Dibromomaleimide (DBM) Platform for Native Antibody Conjugation by Accelerated Post-Conjugation Hydrolysis. Org. Biomol. Chem. 15, 2947–2952. 10.1039/C7OB00220C. [DOI] [PubMed] [Google Scholar]
- Robinson E.; Nunes J. P. M.; Vassileva V.; Maruani A.; Nogueira J. C. F.; Smith M. E. B.; Pedley R. B.; Caddick S.; Baker J. R.; Chudasama V. (2017) Pyridazinediones Deliver Potent, Stable, Targeted and Efficacious Antibody-Drug Conjugates (ADCs) with a Controlled Loading of 4 Drugs per Antibody. RSC Adv. 7, 9073–9077. 10.1039/C7RA00788D. [DOI] [Google Scholar]
- Forte N.; Chudasama V.; Baker J. R. (2018) Homogeneous Antibody-Drug Conjugates via Site-Selective Disulfide Bridging. Drug Discovery Today: Technol. 30, 11–20. 10.1016/j.ddtec.2018.09.004. [DOI] [PubMed] [Google Scholar]
- Sahni J. M.; Keri R. A. (2018) Targeting Bromodomain and Extraterminal Proteins in Breast Cancer. Pharmacol. Res. 129, 156–176. 10.1016/j.phrs.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.proteinatlas.org/ENSG00000141867-BRD4/cell (accessed Apr. 6, 2019).
- Casalini P.; Caldera M.; Canevari S.; Mrnard S.; Mezzanzanica D.; Tosi E.; Gadina M.; Colnaghi M. I. (1993) A Critical Comparison of Three Internalization Assays Applied to the Evaluation of a Given mAb as a Toxin-Carrier Candidate. Cancer Immunol. Immunother. 37, 54–60. 10.1007/BF01516942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazin J.; Moldenhauer G.; Altevogt P.; Brady N. R. (2015) A Novel Method for Measuring Cellular Antibody Uptake Using Imaging Flow Cytometry Reveals Distinct Uptake Rates for Two Different Monoclonal Antibodies Targeting L1. J. Immunol. Methods 423, 70–77. 10.1016/j.jim.2015.04.024. [DOI] [PubMed] [Google Scholar]
- Isa M.; Asanuma D.; Namiki S.; Kumagai K.; Kojima H.; Okabe T.; Nagano T.; Hirose K. (2014) High-Throughput Screening System to Identify Small Molecules That Induce Internalization and Degradation of HER2. ACS Chem. Biol. 9, 2237–2241. 10.1021/cb500654q. [DOI] [PubMed] [Google Scholar]
- Sorkin A., and Duex J. E. (2010) Quantitative Analysis of Endocytosis and Turnover of Epidermal Growth Factor (EGF) and EGF Receptor. Curr. Protoc. Cell Biol. 46, 10.1002/0471143030.cb1514s46 (Chapter 15: p. Unit 1514). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin C. D.; De Maziere A. M.; Pisacane P. I.; van Dijk S. M.; Eigenbrot C.; Sliwkowski M. X.; Klumperman J.; Scheller R. H. (2004) Endocytosis and Sorting of ErbB2 and the Site of Action of Cancer Therapeutics Trastuzumab and Geldanamcin. Mol. Biol. Cell 15, 5268–5282. 10.1091/mbc.e04-07-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.