

Abstract
The current phase I/II clinical trial for human glycogen storage disease type-Ia (GSD-Ia) (NCT 03517085) uses a recombinant adeno-associated virus (rAAV) vector expressing a codon-optimized human glucose-6-phosphatase-α (G6Pase-α or G6PC). DNA sequence changes introduced by codon-optimization can negatively impact gene expression. We therefore generated a novel variant in which a single amino acid change, S298C, is introduced into the native human G6PC sequence. Short term gene transfer study in G6pc−/− mice showed that the rAAV-G6PC-S298C vector is 3-fold more efficacious than the native rAAV-G6PC vector. We have shown previously that restoring 3% of normal hepatic G6Pase-α activity in G6pc−/− mice prevents hepatocellular adenoma/carcinoma (HCA/HCC) development and that mice harboring <3% of normal hepatic G6Pase-α activity are at risk of tumor development. We have also shown that G6Pase-α deficiency leads to hepatic autophagy impairment that can contribute to hepatocarcinogenesis. We now undertake a long-term (66-week) preclinical characterization of the rAAV-G6PC-S298C vector in GSD-Ia gene therapy. We show that the increased efficacy of rAAV-G6PC-S298C has enabled the G6pc−/− mice treated with a lower dose of this vector to survive long-term. We further show that mice expressing ≥3% of normal hepatic G6Pase-α activity do not develop hepatic tumors or autophagy impairment but mice expressing <3% of normal hepatic G6Pase-α activity display impaired hepatic autophagy with one developing HCA/HCC nodules. Our study shows that the rAAV-G6PC-S298C vector provides equal or greater efficacy to the codon optimization approach, offering a valuable alternative vector for clinical translation in human GSD-Ia.
Keywords: recombinant adeno-associated virus vector, glucose-6-phosphatase-α variant, clinical translation, autophagy impairment
1. Introduction
Glycogen storage disease type Ia (GSD-Ia, MIM232200) is an autosomal recessive metabolic disease caused by deleterious mutations in the G6PC gene that encodes glucose-6-phosphatase-α (G6Pase-α or G6PC) [1]. Expressed primarily in the liver and the kidney, G6Pase-α catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and phosphate in the terminal step of gluconeogenesis and glycogenolysis. GSD-Ia patients manifest impaired glucose homeostasis and long-term complication of hepatocellular adenoma/carcinoma (HCA/HCC) [1]. The current dietary therapies have enabled GSD-Ia patients to attain near normal growth and pubertal development but require strong compliance to the diet. However, HCA/HCC remains in metabolically compensated GSD-Ia patients [1].
We have developed efficacious recombinant adeno-associated virus (rAAV) vectors expressing either the wild-type (rAAV-G6PC) or a codon-optimized (co) (rAAV-co-G6PC) G6Pase-α [2–5]. The codon-optimized vector, which has a higher potency, is currently being used in a phase I/II clinical trial for human GSD-Ia (NCT 03517085). Codon-optimization introduced DNA sequence changes can negatively impact gene expression and reduce potency or efficacy [6]. To explore second generation vectors with improved efficacy we identified a G6PC-S298C variant with increased catalytic activity [7]. Short-term gene therapy studies in G6pc−/− mice showed that without codon optimization, rAAV-G6PC-S298C leads to 3-fold higher expression of hepatic G6Pase-α activity compared to an identical titer of rAAV-G6PC [7]. In the current 66-week study, we examined the long-term safety and efficacy of gene therapy mediated by rAAVG6PC-S298C in G6pc−/− mice.
Prior to this study, we examined the relationship between hepatic levels of G6Pase-α activity and tumor development [2–5]. In studies lasting over 60-weeks we titrated the viral dose in 60 rAAV-G6PC treated G6pc−/− mice, to restore different levels of wild-type G6Pase-α activity. In 43 mice, all expressing ≥3% of normal hepatic G6Pase-α activity, no HCA/HCC developed. However, among the 17 mice expressing <3% of normal hepatic G6Pase-α activity, three (18%) developed HCA/HCC, establishing that 3% normal hepatic G6Pase-α activity is the threshold for tumor prevention [2–5]. In the current study, we again titrated the virus dose and divided the rAAV-G6PC-S298C-treated G6pc−/− mice into two groups, those expressing 3.3–35% (S298C/≥3% mice) and those expressing 0.3–2.9% (S298C/<3% mice) of normal hepatic G6Pase-α activity
Autophagy is an evolutionary conserved, degradative process that facilitates the cellular clearance or turnover of misfolded proteins, protein aggregates, and damaged organelles [8]. A deficiency in hepatic autophagy can contribute to hepatocarcinogenesis [9]. Autophagy can be positively regulated by SIRT1 (Sirtuin 1) [10, 11] and AMPK (AMP-activated protein kinase) [12, 13]. The LKB1 (liver kinase B1) activates AMPK via phosphorylation of the AMPK-α subunit [14] and the activated AMPK promotes autophagy [12, 13]. We have previously shown that hepatic G6Pase-α deficiency leads to impaired autophagy and autophagy impairment is mediated by downregulation of SIRT1 signaling [15, 16]. In the current study, we show that the S298C/<3% mice displayed impaired autophagy along with reduced SIRT1 expression.
2. Materials and methods
2.1. Infusion of G6pc−/− mice and phenotype analysis
All animal studies were conducted under an animal protocol approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Animal Care and Use Committee. The rAAV-G6PC-S298C vector was infused into 2-week-old G6pc−/− mice via the retro-orbital sinus as described previously [2]. Age-matched G6pc+/+ and G6pc+/− mice with indistinguishable phenotype were used as controls. Body composition was assessed using the Bruker Minispec NMR analyzer (Karlsruhe, Germany). Liver samples from control and rAAVG6PC-S298C-treated mice were collected at sacrifice following 12 hours or 24 hours of fast. Hepatic levels of glucose, G6P, lactate, triglyceride, and glycogen were determined as described previously [3–5]. Insulin tolerance testing of mice consisted of a 4-hour fast, prior to blood sampling, followed by intraperitoneal injection of an insulin dose of 0.25 IU/kg, and repeated blood sampling via the tail vein for 1 hour.
2.2. Phosphohydrolase assays
Liver microsome isolation and microsomal phosphohydrolase assays were performed as described previously [2]. In phosphohydrolase assays, reaction mixtures (50 μl) containing 50 mM sodium cacodylate buffer, pH 6.5, 2 mM EDTA, 10 mM G6P, and appropriate amounts of microsomal preparations were incubated at 30 °C for 10 min. Disrupted microsomal membranes were prepared by incubating intact membranes in 0.2% deoxycholate for 20 min at 4 °C. Non-specific phosphatase activity was estimated by pre-incubating disrupted microsomal preparations at pH 5 for 10 min at 37 °C to inactivate the acid-labile G6Pase-α. One unit of G6Pase-α activity represents one nmol G6P hydrolysis per minute per mg microsomal protein. The limitation of the lower level of quantitation for the microsomal G6Pase-α assay is 3 units. Enzyme histochemical analysis of G6Pase-α was performed as described previously [2].
2.3. Antibodies against human G6Pase-α
Analysis of serum antibodies against human G6Pase-α has been described [2]. A monoclonal anti-human G6Pase-α antibody [15] that also recognizes murine G6Pase-α was used as a positive control.
2.4. Statistical Analysis
The unpaired t test was performed using the GraphPad Prism Program, version 4 (GraphPad Software, San Diego, CA). Values were considered statistically significant at p< 0.05.
3. Results
3.1. rAAV-G6PC-S298C directs long-term hepatic G6Pase-α expression
We examined the safety and efficacy of rAAV-G6PC-S298C-mediated gene transfer over a 66-week study by infusing 2-week-old G6pc−/− mice with 1013 (n = 2), 3 × 1012 (n = 6), and 1012 (n = 9) vp/kg of rAAV-G6PC-S298C. Based on previous titration data these were expected to reconstitute a range from <3% to 40% of normal hepatic G6Pase-α activity. The mean hepatic G6Pase-α activity of 62–66-week-old control mice (n = 15) was 240.2 ± 15.8 units, representing 100% normal hepatic G6Pase-α activity. The limitation of the lower level of quantitation for the microsomal G6Pase-α assay is 3 units, equivalent to 1.3% of normal hepatic G6Pase-α activity. Hepatic G6Pase-α activities in rAAV-G6PC-S298C-treated mice were 64.4 to 85.7 (1013 vp/kg), 8.0 to 30.6 (3 × 1012 vp/kg), and 0.8 to 9.0 (1012 vp/kg) units, respectively (Fig.1A). Given the previous extensive observations [2–5] that had established 3% of normal hepatic G6Pase-α activity is required to prevent HCA/HCC development, we grouped the resulting mice for analysis based on their hepatic G6Pase-α activity levels. Among the nine G6pc−/− mice treated with 1012 vp/kg of rAAV-G6PC-S298C, eight had <7.0 units of G6Pase-α activity (<2.9% of normal hepatic G6Pase-α activity) and were named as S298C/<3% mice. The ninth mouse expressing 9 units (3.8%) of G6Pase-α activity was grouped with G6pc−/− mice treated with 3 ×1012 vp/kg and 1013 vp/kg of rAAV-G6PC-S298C expressing 3.3–13% and 27–35% of normal hepatic G6Pase-α activity, respectively. These nine mice were named as S298C/≥3% mice. As expected, none of the control or S298C/≥3% mice developed HCA/HCC over the 66-week study. One of the eight S298C/<3% mice restoring 1.7 unit of hepatic G6Pase-α activity developed hepatic tumors (Fig.1A), an incidence consistent with the larger studies we have reported previously [2–5]. Pathological analysis showed that this mouse harbored two HCA and one HCC nodules.
Fig. 1.
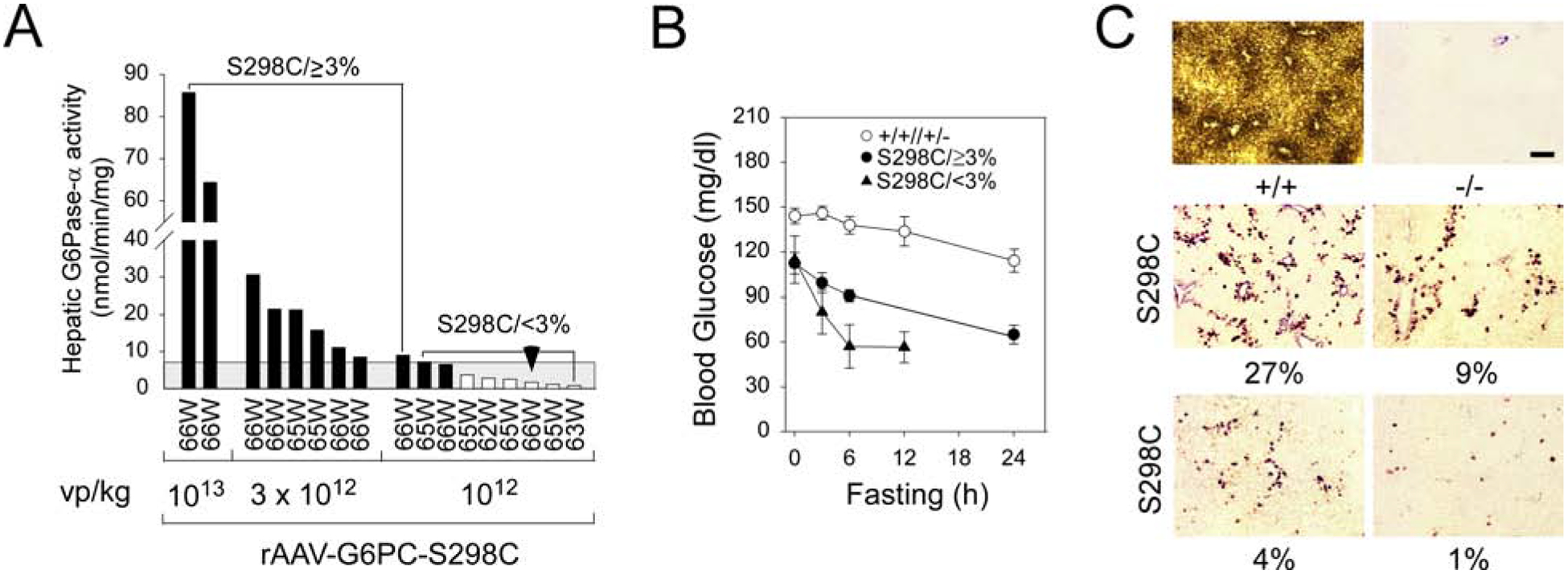
Biochemical analysis of 62–66-week-old rAAV-G6PC-S298C-treated G6pc−/− mice. (A) Hepatic microsomal G6Pase-α activity in rAAV-G6PC-S298C-treated G6pc−/− mice is shown at the indicated ages in weeks (W). The mice were grouped based on viral dosages: 1013 vp/kg (n = 2), 3 × 1012 vp/kg (n = 6), and 1012 vp/kg (n = 9). Two major subgroups emerge for mice restoring 3.3–35% (S298C/≥3% mice, n = 9) and 0.3–2.9% (S298C/<3% mice, n = 8), respectively of normal hepatic G6Pase-α activity. Hepatic microsomal G6Pase-α activity in 62–66-week-old wild-type mice (n = 15) averaged 240.2 ± 15.8 units, representing 100% normal hepatic G6Pase-α activity. The grey area denotes 3% of normal hepatic G6Pase-α activity. (B) Fasting blood glucose tolerance profiles. (C) Histochemical analysis of hepatic G6Pase-α activity. Each image represents an individual mouse. (+/+), wild-type, (−/−), untreated G6pc−/−, and (S298C), rAAVG6PC-S298C-treated mice. Scale bar = 200μm. The numbers in percentage represent hepatic G6Pase-α activity restored in the rAAV-G6PC-S298C-treated mice.
At age 62–66 weeks, the fasting blood glucose profiles of S298C/≥3% mice paralleled those of the control mice, but blood glucose levels were consistently lower (Fig.1B). The fasting blood glucose levels of the S298C/<3% mice were low, and fasting was terminated at 12 hours (Fig. 1B) when most mice had blood glucose levels below 60 mg/dl. In summary, the rAAVG6PC-S298C-treated G6pc−/− mice could all tolerate over 12 hours of fasting.
Enzyme histochemical analysis showed that G6Pase-α in wild-type mice was distributed throughout the liver with significantly higher levels in proximity to blood vessels (Fig. 1C). There was no detectable G6Pase-α activity in the liver sections of untreated G6pc−/− mice (Fig. 1C). G6Pase-α in the rAAV-G6PC-S298C-treatedG6pc−/− mice was also distributed throughout the liver but with foci containing markedly higher levels of enzymatic activity with a substantial proportion of hepatocytes harbored little or no G6Pase-α (Fig. 1C), suggesting that uniform hepatic G6Pase-α expression is not required for rescuing the GSD-Ia phenotype.
3.2. The phenotype of 62–66-week-old rAAV-G6PC-S298C-treated G6pc−/− mice
At age 62–66 weeks, the rAAV-G6PC-S298C-treated G6pc−/− mice exhibited no hepatic histological abnormalities except increased glycogen storage (Fig. 2A). Compared to control mice, hepatic glycogen contents were markedly higher in rAAV-G6PC-S298C-treated mice and glycogen levels were inversely correlated to hepatic G6Pase-α activity restored (Fig. 2A). Pathological analysis showed that the tumor-bearing S298C/<3% mouse had two HCA nodules of 16 and 4 mm in diameter characterized by a lack of portal tracts and compressed adjacent parenchyma, and one HCC nodule of 3 mm in diameter consisting of anisocytotic and anisokaryotic hepatocytes (Fig. 2A). The average body weight (BW) and body fat of the rAAVG6PC-S298C-treated mice were 72% and 68%, respectively of their age-matched control mice (Fig. 2B), indicating that the mice with lower than wild-type hepatic G6Pase-α activity were protected against age-related obesity, as had been noted previously [3]. The liver weight (LW) values of the 62–66-week-old rAAV-G6PC-S298C-treated G6pc−/− mice were inversely correlated to the hepatic G6Pase-α activity restored (Fig. 2C). When LW was expressed as percent of BW, the treated G6pc−/− mice had significantly higher values primarily because of their lower BW.
Fig. 2.
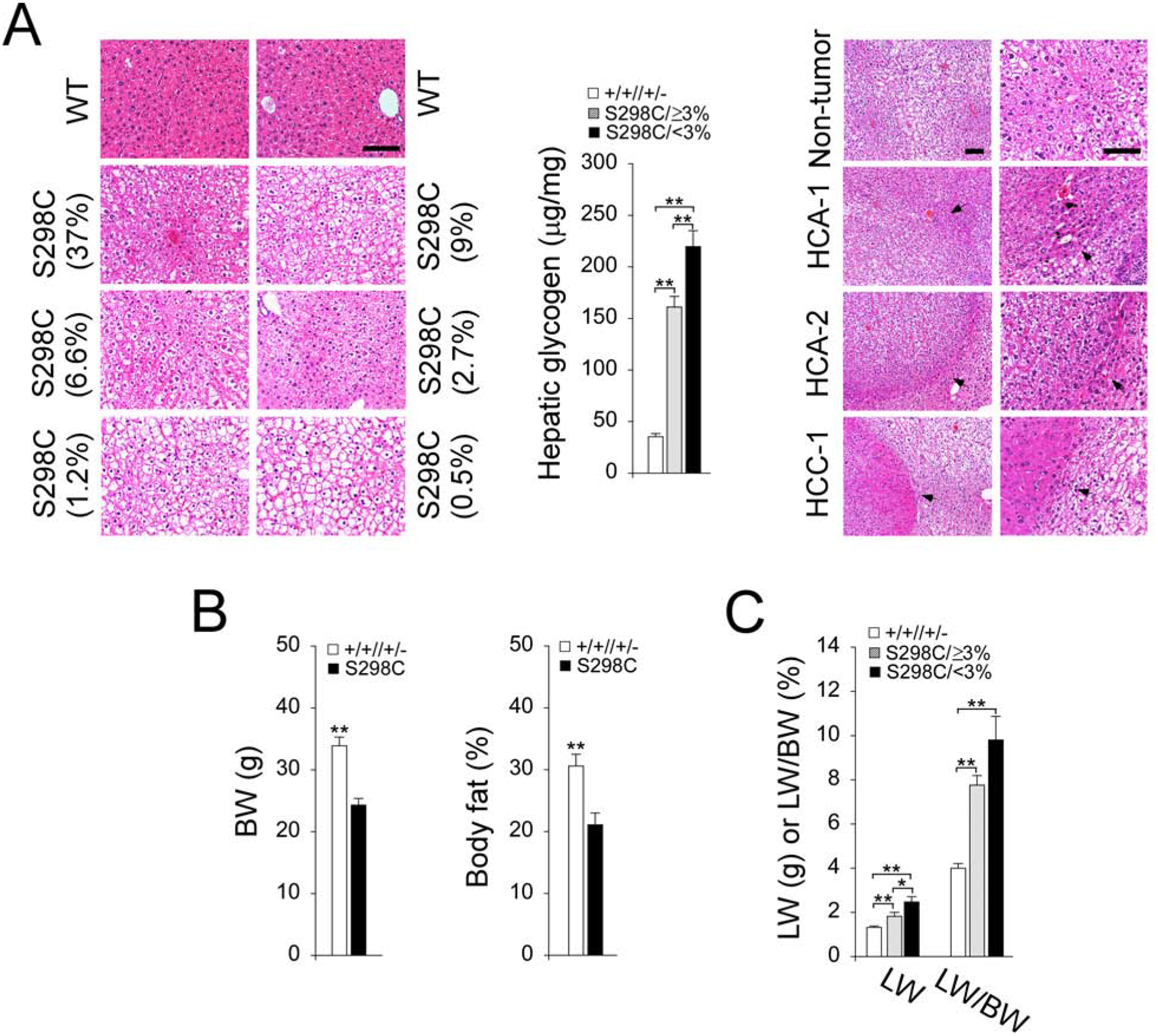
Phenotypic analysis of 62–66-week-old rAAV-G6PC-G6PC-S298C-treated G6pc−/− mice. (A) H&E stained liver sections and hepatic glycogen contents. Each plate represents an individual mouse. Numbers in parentheses represent % of hepatic G6Pase-α activity restored in the mice. The representative hematoxylin and eosin (H&E) stained non-tumor and tumor lesions in the tumor-bearing S298C/<3% mouse are shown. The arrow denotes HCA/HCC. Scale bar = 100μm. (B) Body weight (BW) and body fat values. Both values were similar between S298C/≥3% and S298C/<3% mice and were grouped as the S298C (n = 17) mice. (C) Liver weight (LW) and LW/BW values. Data represent the mean ± SEM. *p< 0.05, **p< 0.005.
Compared to 62–66-week-old control mice, fasting blood insulin levels were significantly lower in rAAV-G6PC-S298C-treated G6pc−/− mice (Fig. 3A), which were closer to the levels in 10–20-week-old young adult mice [17]. At age 17 weeks, following an intraperitoneal insulin injection, blood glucose levels in control and S298C (S298C/≥3% and S298C/<3%) mice decreased with time, although the decrease was more pronounced in the S298C mice (Fig. 3B). Blood glucose levels in the 63-week-old control mice failed to decrease following insulin injection (Fig. 3B), reflecting the known age-related decrease in insulin sensitivity [18]. In contrast, the S298C/≥3% mice continued exhibiting insulin sensitivity at age 63 weeks (Fig. 3B), showing that these mice were protected against an age-related insulin resistance, as had been noted previously in rAAV-G6PC-treated G6pc−/− mice [3]. Insulin tolerance was not examined in the 63-week-old S298C/<3% mice because of their low blood glucose levels.
Fig. 3.
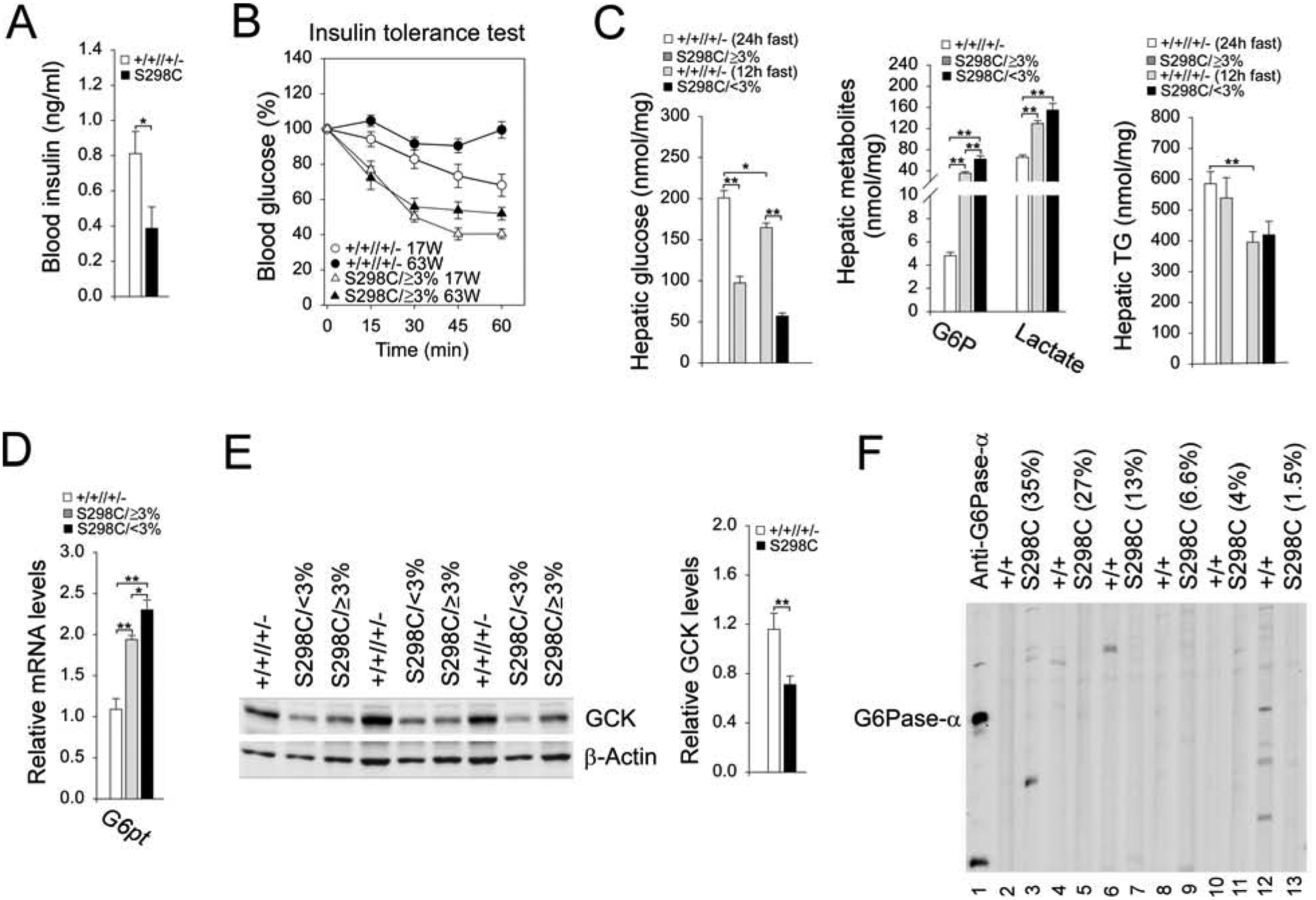
Analysis of 62–66-week-old rAAV-G6PC-S298C-treated G6pc−/− mice. The data were analyzed from wild-type (+/+, n = 15), S298C/≥3% (n = 9) and S298C/<3% (n = 8) mice. When values between S298C/≥3% and S298C/<3% mice were similar, they were grouped together as the S298C (n = 17) mice. (A) Blood insulin levels. (B) Insulin tolerance profiles. (C) Hepatic metabolites values. (D) Hepatic levels of G6pt transcript. (E) Western-blot and densitometry analyses of hepatic GCK and β-actin. (F) The serum antibodies against human G6Pase-α. Lanes 1: anti-human G6Pase-α antiserum; lanes 2, 4, 6, 8, 10, 12: serum samples (1: 50 dilution) from wild-type mice, or lanes 3, 5, 7, 9, 11, 13: serum samples (1: 50 dilution) from rAAV-G6PCS298C-treated G6pc−/− mice. Numbers in parenthesis represent % of normal hepatic G6Pase-α activity restored in the mice. Data represent the mean ± SEM. *p< 0.05, **p< 0.005.
Hepatic free glucose levels in control mice after 12 and 24 hours of fast averaged 164.7 ± 5.5 and 200.9 ± 8.9 nmol/mg protein, respectively (Fig. 3C). The apparent increase in hepatic glucose in mice after 24 hours of fast may result from enhanced gluconeogenesis. Hepatic free glucose levels in S298C/≥3% and S298C/<3% mice after 24 or 12 hours of fast were 48.4% and 34.6% of their respective control glucose levels (Fig. 3C). Hepatic G6P and lactate levels were similar in control mice following 12 and 24 hours of fast at 4.8 ± 0.3 and 65.6 ± 4.5 nmol/mg protein, respectively (Fig. 3C). All rAAV-G6PC-S298C-treated G6pc−/− mice had markedly higher hepatic levels of G6P and lactate, compared to the controls. Hepatic triglyceride levels in control and rAAV-G6PC-S298C-treated G6pc−/− mice were statistically similar (Fig. 3C).
Glucose homeostasis is maintained by the coupled action of the G6Pase-α/G6P transporter (G6PT) complex [1], and the reduced G6Pase-α activity can be offset by an increase in G6PT expression [2]. In S298C/≥3% and S298C/<3% mice, hepatic G6pt mRNA levels were increased 1.8- to 2.1-fold, respectively over that of the controls (Fig. 3D). Studies have shown that hepatic levels of glucokinase (GCK), a glucose sensor, decreases when blood insulin levels are low [19]. Consistent with reduced blood insulin levels in rAAV-G6PC-S298C-treated mice, hepatic levels of GCK in the treated G6pc−/− mice were markedly lower than that in the control mice (Fig. 3E). To determine whether a humoral response directed against human G6Pase-α was generated in the infused mice, we performed Western-blot analysis of sera obtained from 62–66 week-old control and rAAV-G6PC-S298C-treated G6pc−/− mice, using a monoclonal antibody against human G6Pase-α [15] as a positive control. We detected no antibodies against human G6Pase-α in the sera of control or rAAV-G6PC-S298C-treated mice (Fig. 3F), demonstrating the G6PC-S298C variant was not more immunogenic than the wild-type G6PC protein.
3.3. The S298C/<3% mice display impaired hepatic autophagy
Recent studies showed that G6Pase-α deficiency leads to impaired hepatic autophagy [15, 16] and that autophagy impairment can lead to hepatocarcinogenesis [9]. We therefore examined the expression of genes in components of the autophagy network [8] to evaluate whether the increased risk of tumor development in the S298C/<3% mice correlated to a deficiency in autophagy. Compared to control mice, hepatic levels of LC3B-II (microtubule-associated protein 1 light chain 3B-II) and Beclin-1 were decreased in both S298C/≥3% and S298C/<3% mice (Fig. 4A). However, hepatic levels of LC3B-I, ATG101, and BNIP3 (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3) were decreased primarily in the S298C/<3% mice (Fig. 4A). Significantly, hepatic levels of LC3B-I, LC3B-II, ATG101, and BNIP3 were significantly lower in the S298C/<3% mice, compared to the S298C/≥3% mice (Fig. 4A), indicating that the S298C/<3% mice displayed impaired autophagy.
Fig. 4.
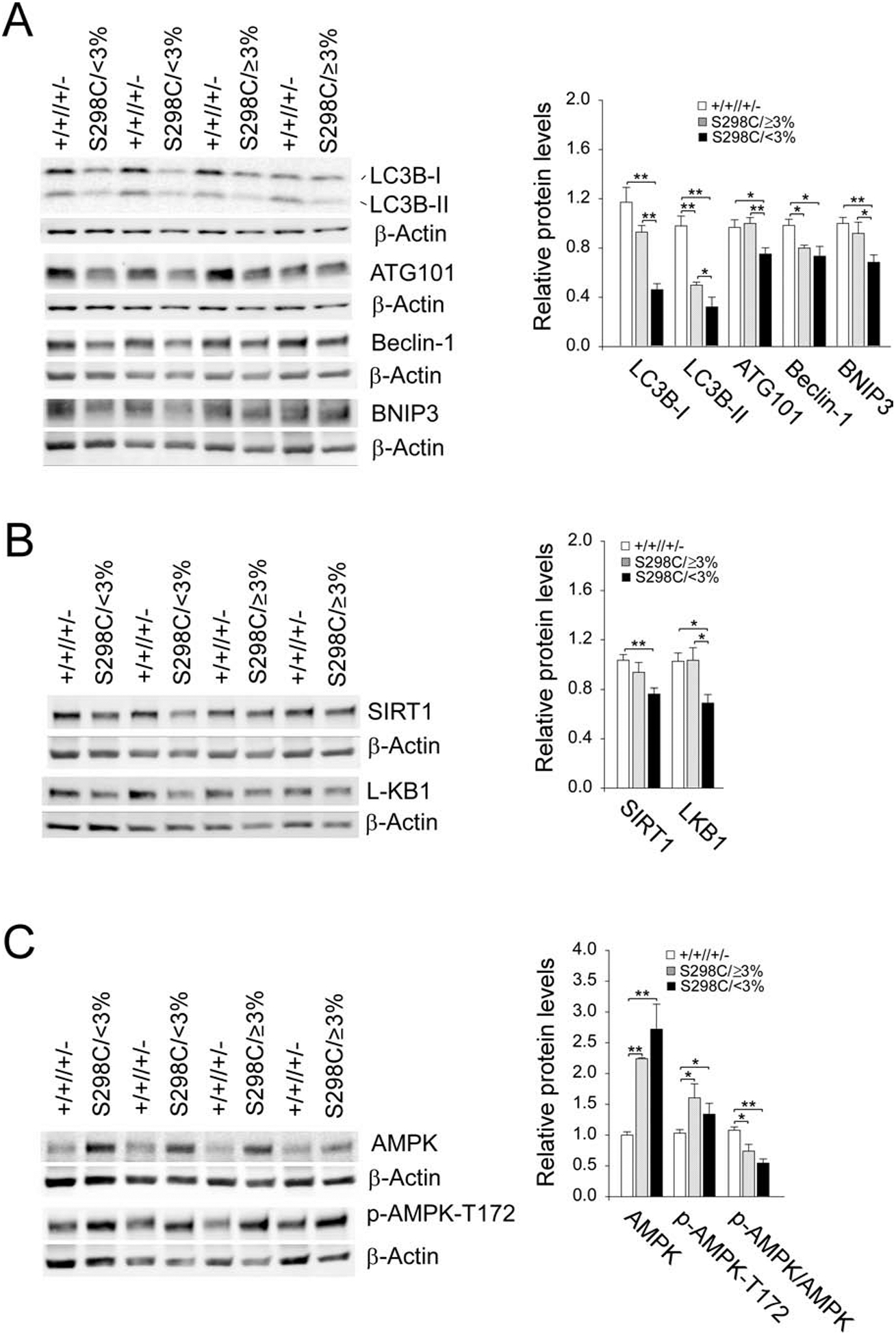
Impaired hepatic autophagy in the S298C/<3% mice. The data were analyzed from wild-type (+/+//+/− , n = 15), S298C/≥3% (n = 9) and S298C/<3% (n = 8) mice. (A) Western-blot and densitometry analyses of hepatic LC3B, ATG101, Beclin-1, BNIP3, and β-actin. (B) Western-blot and densitometry analyses of hepatic SIRT1, LKB1, and β-actin. (C) Western-blot and densitometry analyses of hepatic AMPK, p-AMPK-T172, and β-actin. Data represent the mean ± SEM. *P < 0.05, **P < 0.005.
SIRT1 [10, 11] and AMPK [12, 13] are positive regulators of autophagy. We have shown that SIRT1 signaling downregulation underlies hepatic autophagy impairment in L-G6pc−/− [15] and G6pc−/− [16] mice. Compared to the controls, hepatic levels of SIRT1 were unaltered in the S298C/≥3% mice (Fig. 4B). In contrast, hepatic levels of SIRT1 were significantly decreased in the S298C/<3% mice (Fig. 4B), suggesting that SIRT1 downregulation can contribute to autophagy deficiency seen in the S298C/<3% mice. In the liver, LKB1 is the upstream kinase that phosphorylates T-172 on the activation loop of AMPK-α and subsequently activates AMPK [14]. Compared to control mice, hepatic LKB1 levels were decreased only in the S298C/<3% mice (Fig. 4B). Hepatic levels of AMPK and the active p-AMPK-T172 were elevated in both S298C/≥3% and S298C/<3% mice and the ratios of p-AMPK-T172 to total AMPK were lower in both mouse groups (Fig. 4C). However, the decrease in the p-AMPK-T172/AMPK values was more pronounced in the S298C/<3% mice (Fig. 4C). This suggests that downregulation of LKB1/AMPK may also play a role in autophagy impairment in the S298C/<3% mice.
4. Discussion
To improve the efficacy of rAAV-mediated gene transfer and minimize the potential problems associated with codon optimization [6], we developed the rAAV-G6PC-S298C vector [7]. In short-term gene transfer studies, we had previously shown that rAAV-G6PC-S298C is ~3-fold more efficacious in directing hepatic G6Pase-α expression than the rAAV-G6PC vector [7]. We have also shown previously that rAAV-G6PC-treated G6pc−/− mice restoring ≥3% of normal hepatic G6Pase-α activity do not develop HCA/HCC but 18% of the treated mice expressing <3% of normal hepatic G6Pase-α activity are at risk of tumor development [2–5]. To examine whether the rAAV-G6PC-S298C vector is a valuable alternative vector for clinical translation in human GSD-Ia, we undertook the current long-term safety and efficacy study in G6pc−/− mice. We show that both the S298C/≥3% and S298C/<3% mice maintain glucose homeostasis and do not develop detectable antibodies against human G6Pase-α. While the S298C/≥3% mice do not develop HCA/HCC, one of the eight S298C/<3% mice developed HCA/HCC nodules. Taken together, there was no observable difference in pathophysiology between rAAV-G6PC or rAAVG6PC-S298C transduced G6pc−/− mice expressing equivalent G6Pase-α enzymatic activities and that 3% normal hepatic G6Pase-α activity is the threshold for tumor prevention as had been noted previously [2–5]. Studies have shown that the efficiency and persistence of rAAV-mediated hepatic gene transfer are lower during early development because the fast rate of liver growth which can dilute out the number of cells effectively infected with rAAV [20]. Importantly, the increased efficacy of rAAV-G6PC-S298C has enabled 2-week-old G6pc−/− mice treated with a low dose (1012 vp/kg) of this vector to survive long-term.
We have shown that hepatic G6Pase-α deficiency in L-G6pc−/− [15] and G6pc−/− [16] mice lead to autophagy deficiency that can contribute to hepatocarcinogenesis [9]. In this study, we show that the S298C/≥3% mice do not exhibit impaired autophagy but the S298C/<3% mice display autophagy impairment, evident by reduced expression of many components of the autophagy network, including LC3B-I, LC3B-II, ATG101, Beclin-1, and BNIP3. During vesicle elongation of autophagy, LC3B-I is converted to LC3B-II, a marker of autophagosome formation and its decreased expression is consistent with impaired autophagosome formation [21]. Our study shows that hepatic autophagy deficiency may be one factor contributing to the risk of tumor development in mice expressing <3% of normal hepatic G6Pase-α activity. SIRT1 is a positive autophagy regulator [10, 11]. We have shown that SIRT1signaling downregulation underlies hepatic autophagy impairment in GSD-Ia mice [15, 16]. In this study, we show that SIRT1 expression was reduced only in the S298C/<3% mice that are at risk of tumor development, suggesting SIRT1 downregulation may underlie autophagy impairment in these mice.
The interesting observation seen in the preclinical GSD-Ia gene therapy studies is metabolic correction and tumor prevention require only a 3% restoration of normal hepatic G6Pase-α enzymatic activity. Blood glucose homeostasis is maintained by the G6Pase-α/G6PT complex [1]. We have shown that in rAAV-G6PC-treated G6pc−/− mice, the decreased G6Pase-α expression is accompanied by increased G6PT and reduced GCK expression [2]. As expected, in rAAV-G6PC-S298C-treated G6pc−/− mice, the reduced G6Pase-α activity is characterized by elevated G6pt expression. Moreover, the rAAV-G6PC-S298C-treated G6pc−/− mice also displayed reduced levels of blood insulin and hepatic glucose, leading to inhibition of GCK expression. Collectively, our results suggest that elevated G6pt expression along with inhibition of GCK has enabled the rAAV-treated G6pc−/− mice expressing ≥3% of normal hepatic G6Pase-α activity to maintain glucose homeostasis.
In summary, we show that wild-type G6PC and the G6PC-S298C variant behave in a similar manner and the increased efficacy of the rAAV-G6PC-S298C vector has enabled 2-week-old G6pc−/− mice treated with low dosage (1012 vp/kg) of this vector to survive long-term. The rAAV-G6PC-S298C-treated G6pc−/− mice expressing ≥3% of normal hepatic G6Pase-α activity maintain glucose homeostasis, display no autophagy defect, and lack HCA/HCC. However, the treated mice expressing <3% of normal hepatic G6Pase-α activity display impaired autophagy along with reduced SIRT1 expression that can play a role in HCA/HCC development in the S298C/<3% mice.
Highlights.
The rAAV vector expressing a novel G6PC-S298C protein is safe and efficacious.
The rAAV-G6PC-S298C vector avoids the sequence changes in codon optimization.
Autophagy impairment may contribute to tumor development in GSD-Ia.
Acknowledgements
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health and the Children’s Fund for Glycogen Storage Disease Research.
Abbreviations
- AAV
adeno-associated virus
- AMPK
AMP-activated protein kinase
- BNIP3
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- G6P
glucose-6-phosphate
- G6Pase-α
glucose-6-phosphatase-α
- G6PT
glucose-6-phosphate transporter
- GCK
glucokinase
- GSD-Ia
glycogen storage disease type Ia
- HCA
hepatocellular adenoma
- HCC
hepatocellular carcinoma
- LC3B
microtubule-associated protein 1 light chain 3B
- LKB1
liver kinase B1
- Sirtuin 1
SIRT1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None declared
References
- [1].Chou JY, Jun HS, Mansfield BC, Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat. Rev. Endocrinol 6 (2010) 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee YM, Jun HS, Pan CJ, Lin SR, Wilson LH, Mansfield BC, Chou JY, Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology 56 (2012) 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim GY, Lee YM, Cho JH, Pan CJ, Jun HS, Springer DA, Mansfield BC, Chou JY, Mice expressing reduced levels of hepatic glucose-6-phosphatase-α activity do not develop age-related insulin resistance and obesity. Hum. Mol. Genet 24 (2015) 5115–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee YM, Kim GY, Pan CJ, Mansfield BC, Chou JY, Minimal hepatic glucose-6-phosphatase-alpha activity required to sustain survival and prevent hepatocellular adenoma formation in murine glycogen storage disease type Ia. Mol. Genet. Metab. Rep 3 (2015) 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim GY, Lee YM, Kwon JH, Cho JH, Pan CJ, Starost MF, Mansfield BC, Chou JY, Glycogen storage disease type Ia mice with less than 2% of normal hepatic glucose-6-phosphatase-α activity restored are at risk of developing hepatic tumors. Mol. Genet. Metab 120 (2017) 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mauro VP, Chappell SA, A critical analysis of codon optimization in human therapeutics. Trends Mol. Med 20 (2014) 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang L, Cho JH, Arnaoutova I, Mansfield BC, Chou JY, An evolutionary approach to optimizing glucose-6-phosphatase-α enzymatic activity for gene therapy of glycogen storage disease type Ia. J. Inherit. Metab. Dis 42 (2019) 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mizushima N, Komatsu M, Autophagy: renovation of cells and tissues. Cell 147 (2011) 728–741. [DOI] [PubMed] [Google Scholar]
- [9].Madrigal-Matute J, Cuervo AM, Regulation of Liver Metabolism by Autophagy. Gastroenterology 150 (2016) 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ng F, Tang BL, Sirtuins’ modulation of autophagy. J. Cell. Physiol 228 (2013) 2262–2270. [DOI] [PubMed] [Google Scholar]
- [11].Galluzzi L, Pietrocola F, Levine B, Kroemer G, Metabolic control of autophagy. Cell 159 (2014) 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR, Treatment of nonalcoholic fatty liver disease: role of AMPK. Am. J. Physiol. Endocrinol. Metab 311 (2016) E730–E740. 10.1152/ajpendo.00225.2016. [DOI] [PubMed] [Google Scholar]
- [13].Jeon SM, Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med 48 (2016) e245 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shaw RJ, Lamia KA, Vasquez D, et al. , The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310 (2005) 642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cho JH, Kim GY, Pan CJ, Anduaga J, Choi EJ, Mansfield BC, Chou JY, Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLOS Genet. 13 (2017) e1006819 10.1371/journal.pgen.1006819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gautam S, Zhang L, Arnaoutova I, Mansfield BC, Chou JY, The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum. Mol. Genet 29 (2020) 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Flatt PR, Bailey CJ, Development of glucose intolerance and impaired plasma insulin response to glucose in obese hyperglycemic (ob/ob) mice. Horm. Metab. Res 13 (1981) 556–560. [DOI] [PubMed] [Google Scholar]
- [18].Barzilai N, Huffman DM, Muzumdar RH, Bartke A, The critical role of metabolic pathways in aging. Diabetes 61 (2012) 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Massa ML, Gagliardino JJ, Francini F, Liver glucokinase: An overview on the regulatory mechanisms of its activity. IUBMB Life 63 (2011) 1–6. [DOI] [PubMed] [Google Scholar]
- [20].Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE, Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol. Ther 16 (2008) 1081–1088. [DOI] [PubMed] [Google Scholar]
- [21].Fullgrabe J, Klionsky DJ, Joseph B, The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell. Biol 15 (2014) 65–74. [DOI] [PubMed] [Google Scholar]