

Abstract
The cytokine transforming growth factor-β (TGF-β) regulates the development and homeostasis of several tissue-resident macrophage populations, including microglia. TGF-β is not critical for microglia survival but is required for the maintenance of the microglia-specific homeostatic gene signature1,2. Under defined host conditions, circulating monocytes can compete for the microglial niche and give rise to long-lived monocyte-derived macrophages residing in the central nervous system (CNS)3–5. Whether monocytes require TGF-β for colonization of the microglial niche and maintenance of CNS integrity is unknown. We found that abrogation of TGF-β signaling in CX3CR1+ monocyte-derived macrophages led to rapid onset of a progressive and fatal demyelinating motor disease characterized by myelin-laden giant macrophages throughout the spinal cord. Tgfbr2-deficient macrophages were characterized by high expression of genes encoding proteins involved in antigen presentation, inflammation and phagocytosis. TGF-β is thus crucial for the functional integration of monocytes into the CNS microenvironment.
Reporting Summary.
Further information on experimental design is available in the Nature Research Reporting Summary.
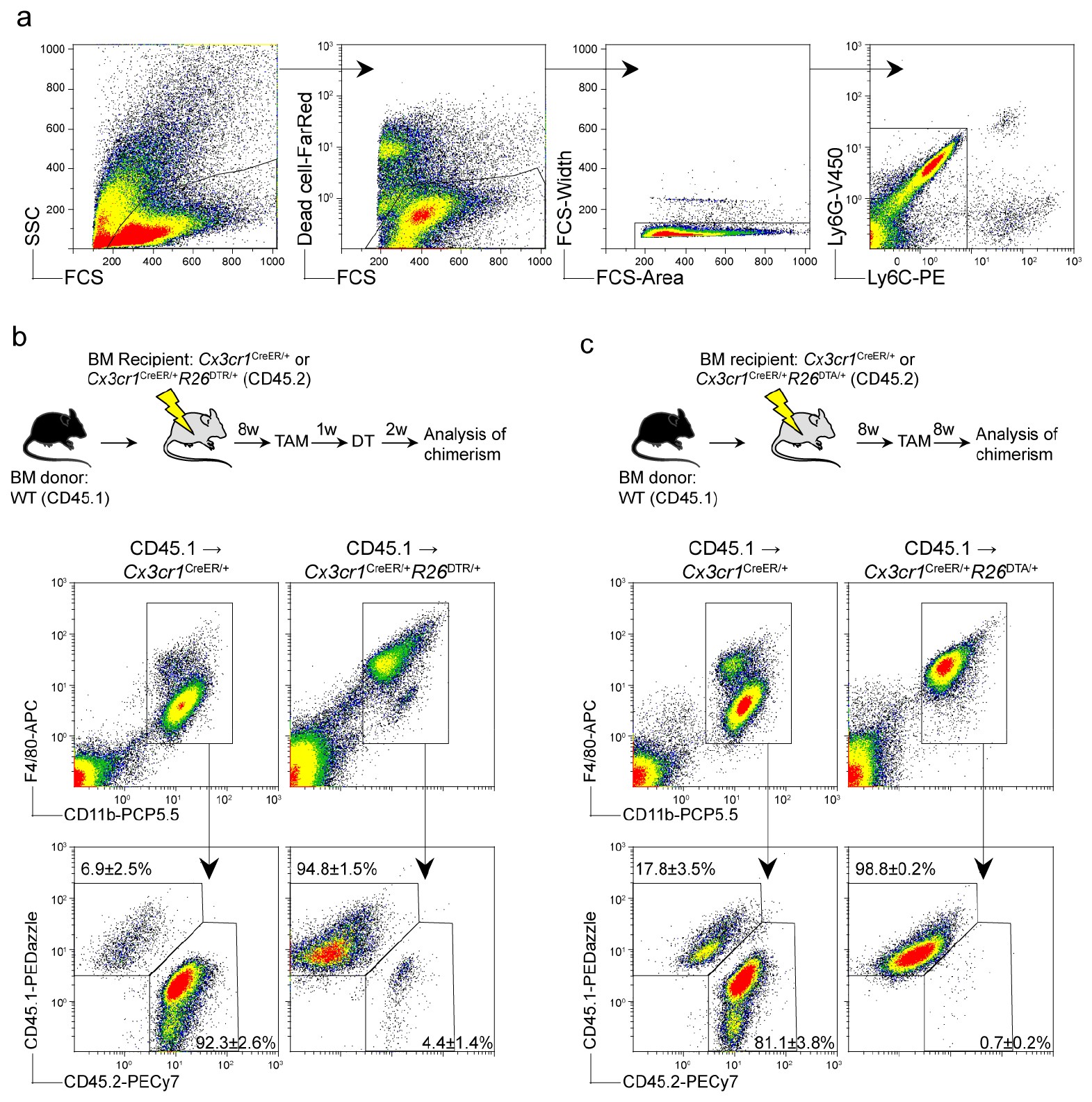
Circulating monocytes can, under settings such as organ growth, infection, tissue damage or macrophage depletion, compete for virtually any macrophage niche6. Monocytes can thus give rise to long-lived self-maintaining macrophage populations that can perform tissue-resident-macrophage functions; examples include Kupffer cells7, alveolar macrophages8 and microglia4,5. To investigate functions of peripherally derived macrophages in the CNS, we attempted to make the CNS myeloid niche available for monocyte engraftment. We thus bred microglia-inducible Cre-ERT2-EYFP (Cx3cr1CreER) mice with R26DTR mice9, which have previously been used to deplete microglia through tamoxifen (TAM) treatment and subsequent diphtheria toxin (DT) administration5,10. When we exposed Cx3cr1CreER/+R26DTR/+ mice to whole-body irradiation and reconstituted them with congenic bone marrow, DT-mediated depletion resulted in repopulation of the microglial niche (gating strategy in Supplementary Fig. 1a) by peripheral myeloid cells (Supplementary Fig. 1b), as previously reported2,5. In parallel, we generated Cx3cr1CreER/+R26DTA/+ mice in which TAM administration results in direct intracellular DT expression11. This experimental setup similarly resulted in efficient microglial depletion and peripheral repopulation (Supplementary Fig. 1c). As previously reported2,4,5, we did not observe obvious behavioral signs of CNS pathology as a consequence of replacing the microglial compartment with peripherally derived macrophages, thus indicating functional integration of monocyte-derived macrophages into the brain parenchyma. For simplicity, we elected to use Cx3cr1CreER/+R26DTA/+ mice (henceforth referred to as DTA) for subsequent investigations.
In adult mice, TGF-β has organ-specific effects on tissue-resident macrophage populations. Although it is dispensable for kidney and lamina propria macrophages12, TGF-β regulates the homeostatic properties of Langerhans cells13, alveolar macrophages12 and microglia1,2. Furthermore, TGF-β regulates the activation and subsequent effector functions of monocyte-derived cells and consequently can have beneficial or deleterious outcomes in various experimental settings including bacterial meningitis14, tumor growth15,16 and autoimmune encephalomyelitis17. To address the role of TGF-β receptor (TGF-βR) signaling in CNS-repopulating myeloid cells, we produced bone marrow chimeras by adoptively transferring LysMCre/+Tgfbr2fl/fl donor cells into DTA recipients in which Tgfbr2 was deleted in lysozyme+ (encoded by LysM or Lyz2) phagocytes, including monocytes and monocyte-derived macrophages16,17. However, we achieved only a 30% decrease in Tgfbr2 expression in bone marrow–derived monocytes (data not shown). We therefore bred LysMCre/+Tgfbr2fl/flR26YFP/+ mice with the aim of tracking Tgfbr2 targeting by using yellow fluorescent protein (YFP) expression as a proxy. In bone marrow–derived monocytes of LysMCre/+Tgfbr2fl/flR26YFP/+→DTA chimeras, we observed ~30% YFP expression (Fig. 1a, b). We confirmed almost-complete loss of Tgfbr2 in YFP+Ly6Chi monocytes without any deletion in YFP–Ly6Chi monocytes in the bone marrow (Fig. 1c). We observed similar or higher YFP frequencies in blood Ly6Chi monocytes (Fig. 1a, b) and equally efficient targeting of Tgfbr2 in the YFP+ compartment (Fig. 1c), thus indicating that TGF-β signaling was dispensable for monocyte exit from the bone marrow. However, after repopulation, there was a selective loss of YFP+ cells in the CNS in LysMCre/+Tgfbr2fl/flR26YFP/+→DTA chimeras compared with control LysMCre/+R26YFP/+→DTA chimeras (Fig. 1a, b). Furthermore, sorting of donor-derived YFP– and YFP+ CNS macrophages revealed that both populations exhibited normal expression of Tgfbr2 (Fig. 1c), thus indicating that TGF-βR2-deficient monocytes could not compete for the microglial niche or were impaired from entering the CNS parenchyma.
Fig. 1 |. Tgfbr2 expression in circulating monocytes is required for microglial-niche colonization.

DTA mice were irradiated and reconstituted with LysMCre/+Tgfbr2fl/flR26YFP/+ or LysMCre/+R26YFP/+ bone marrow. After reconstitution, microglia were depleted through TAM administration and analyzed 5 weeks later. a, Representative dot plots showing YFP frequencies in CD115+Ly6Chi monocytes (bone marrow (BM) and blood) and CD11b+CD45+Ly6C– Ly6G–F4/80hi macrophages (CNS). b, YFP frequencies of individual mice. The data are representative of three pooled experiments; n = 9 and 13 mice. c, Analysis of Tgfbr2 deletion in sorted YFP– and YFP+ fractions of the indicated bone marrow, blood and CNS populations, as assessed by qRT–PCR analysis of the floxed exon 2 normalized to the unfloxed exon 1. Bar graphs display mean ± s.e.m. of n = 2, 4, 4, 4, 4, 8, 8, 2 and 2 mice per group. WT, wild type.
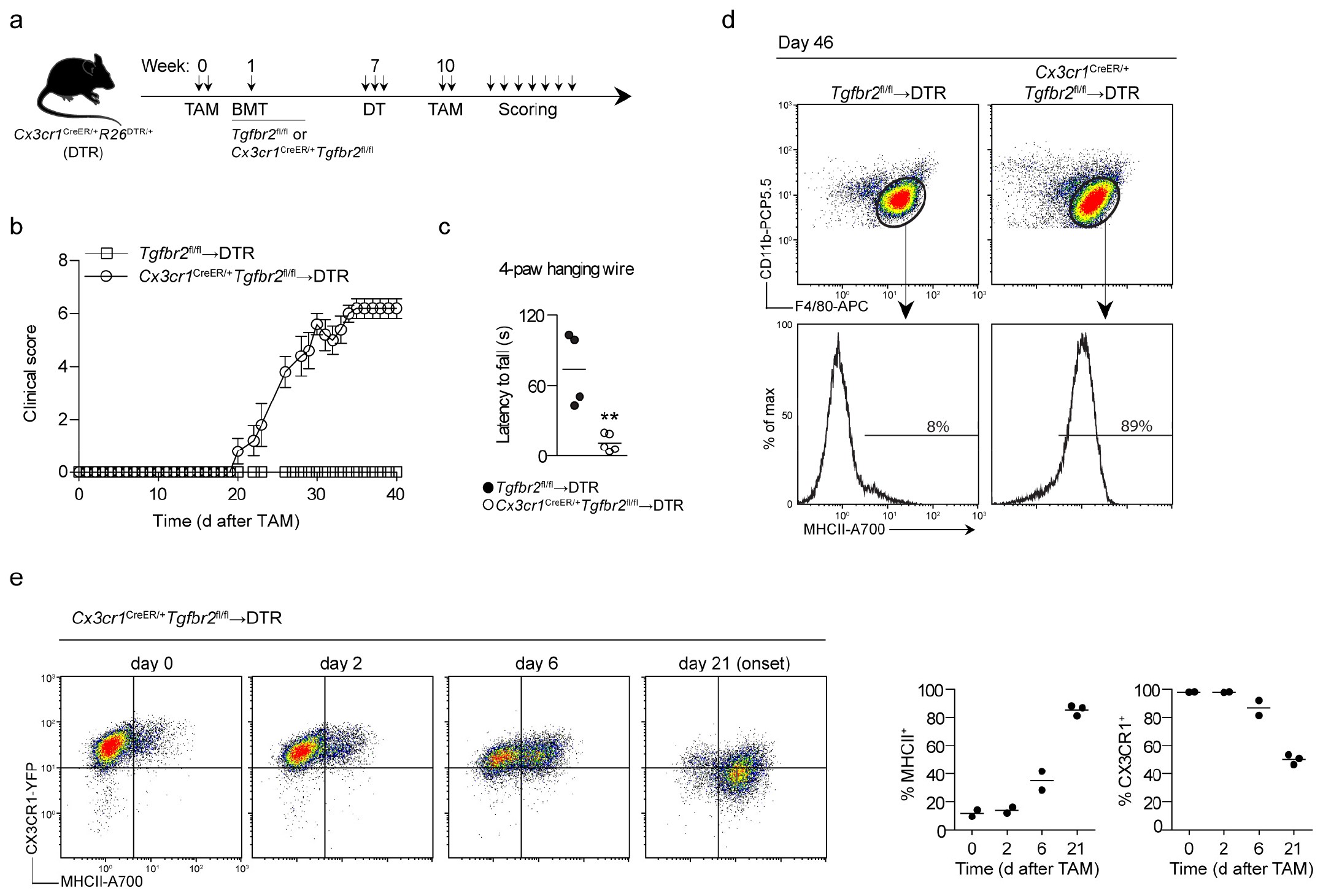
To target monocyte-derived macrophages after their passage into the CNS parenchyma, we took advantage of their upregulation of the chemokine receptor CX3CR1 during differentiation into brain macrophages (data not shown). We thus produced bone marrow chimeras by using Cx3cr1CreER/+Tgfbr2fl/fl donor cells transferred into DTA recipients such that TAM administration simultaneously depleted microglia and deleted Tgfbr2 in CX3CR1+ repopulating macrophages. We observed that Cx3cr1CreER/+Tgfbr2fl/fl→DTA chimeras invariably developed motor impairments (incidence 100%) with onset at day 12 ± 2 (Fig. 2a, b and Supplementary Videos 1 and 2). The symptoms initially presented as tail and hind-leg weakness and impaired escape extension during tail suspension. Muscle weakness was apparent as a complete failure in performing the four-paw hanging-wire test at day 12, which assesses combined front-and hind-paw strength (Fig. 2c and Supplementary Videos 3 and 4). The mice progressed to develop incontinence and hind-leg paralysis, and most of the mice reached the ethical endpoint by day 40 (Fig. 2d). At that stage, the mice had developed both severe fore-and hind-limb paralysis and either were in a side-resting position or were unable to move around the cage (Supplementary Video 5). We analyzed the CNS during ongoing disease to investigate any changes in immune-cell composition that might explain the observed phenotype. The predominant finding in both the brains and spinal cords of Cx3cr1CreER/+Tgfbr2fl/fl→DTA mice was the presence of ‘activated’ macrophages, as evidenced by their high expression of major histocompatibility complex (MHC) II (Fig. 2e). qRT–PCR analysis of sorted CD11b+F4/80hiLy6C– macrophages demonstrated concomitant loss of Tgfbr2 expression (Fig. 2f). We observed no significant differences in the numbers of T cells retrieved from the CNS between Cx3cr1CreER/+Tgfbr2fl/fl→DTA and control Tgfbr2fl/fl→ DTA chimeras (Supplementary Fig. 2a), nor did we detect differences in peripheral T cell activation, as measured by proliferation, cytokine production or induction of regulatory T cells in the spleen (Supplementary Fig. 2b). Together, our data suggested that TGF-β signaling in monocyte-derived CNS macrophages is essential to prevent the onset of fatal motor disease.
Fig. 2 |. Tgfbr2 deficiency in monocyte-derived macrophages results in fatal motor disease.

DTA mice were irradiated and reconstituted with bone marrow from Tgfbr2fl/fl or Cx3cr1CreER/+Tgfbr2fl/fl mice and given TAM after reconstitution. a, Incidence of motor symptoms after TAM administration. Data are from three pooled independent experiments; n = 17 and 37 mice. b, Clinical scores assessing motor symptoms. Data are presented as mean ± s.e.m. from three pooled independent experiments; n = 17 and 26 mice. c, Four-paw hanging-wire test to assess grip strength, performed at the onset of motor symptoms (day 12). Lines represent mean values for n = 4 and 9 mice. Data are representative of two independent experiments. d, Survival of Cx3cr1CreER/+Tgfbr2fl/fl→DTA chimeric mice until day 40 after TAM administration (n = 19). Mice were euthanized when they reached an immobile or moribund state. Neither of the Tgfbr2fl/fl→ DTA control chimeras displayed any symptoms throughout the 40-d period (n = 4). e, Flow cytometry analysis of the brain and spinal cord on day 20 after TAM administration. Top, gated on live singlet cells; bottom, gated on CD11b+CD45+Ly6C–Ly6G–. Values in plots are mean ± s.d. for n = 3 and 4 mice. Data are representative of two independent experiments. f, Expression of the floxed exon 2 of Tgfbr2 in sorted CD11b+F4/80hiLy6C– macrophages at day 20. Lines represent mean values of n = 3 samples per group, and each sample was sorted from a pool of 2–4 mice.
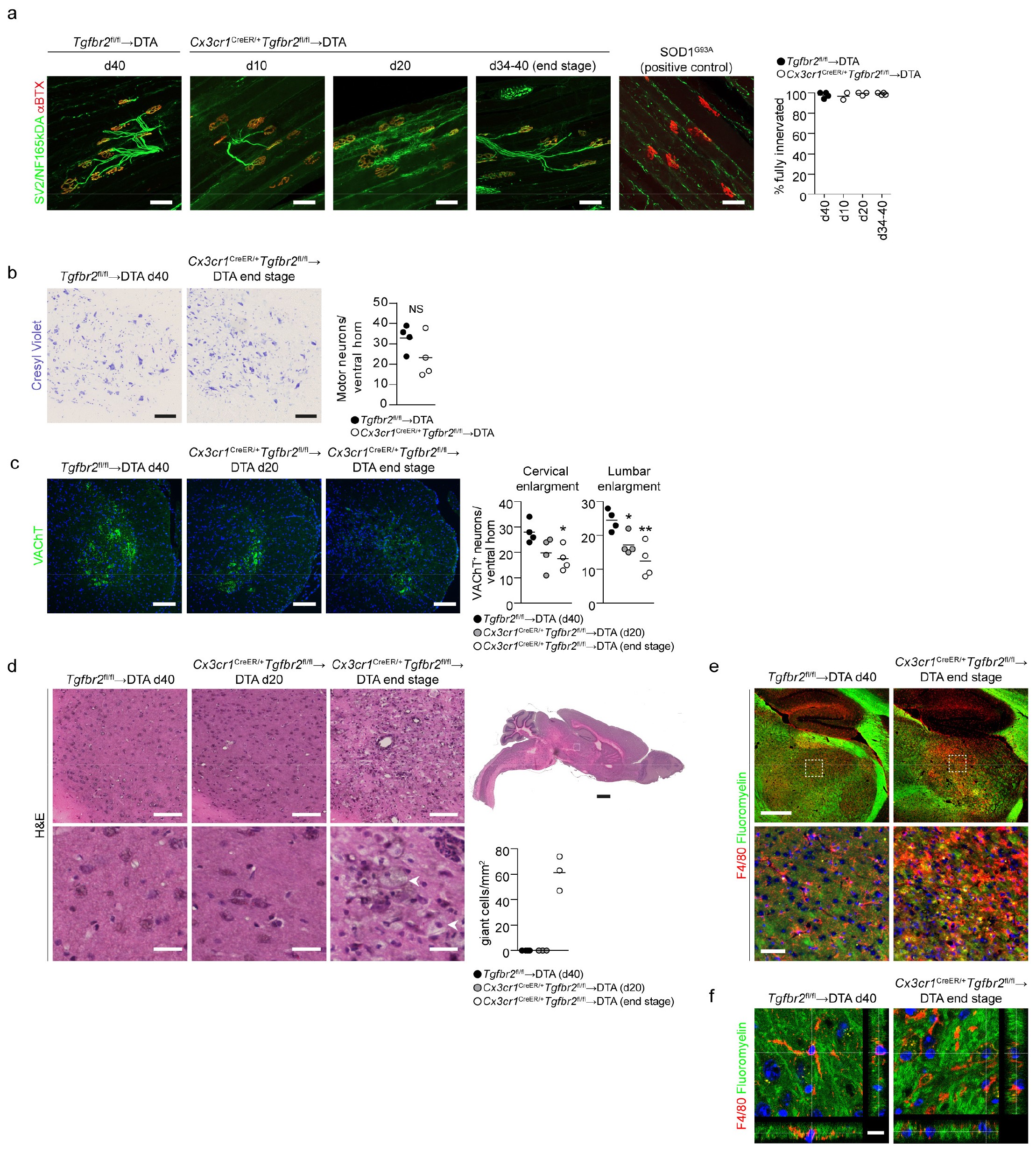
Because the phenotype was predominantly motor related, we analyzed the CNS and peripheral muscle innervation by using immunohistochemistry. Analysis of neurofilament innervation of α-bungarotoxin+ motor endplates in the tibialis anterior, which are early targets of pathology in mouse models of amyotrophic lateral sclerosis (Supplementary Fig. 3a and ref. 18), did not demonstrate any signs of denervation at any time point in Cx3cr1CreER/+Tgfbr2fl/fl→ DTA mice, even though the mice were completely paralyzed in the hind legs at the end stage of disease (Supplementary Fig. 3a). Nissl staining of the spinal cord did not reveal a substantial decrease in motor neurons in the ventral horns of Cx3cr1CreER/+Tgfbr2fl/fl→DTA mice (Supplementary Fig. 3b). However, there was a progressive loss of the vesicular acetylcholine transporter, a neurotransmitter transporter in motor neurons in both cervical and lumbar enlargements (Supplementary Fig. 3c). This observation indicated altered motor neuron connectivity similar to that observed in amyotrophic lateral sclerosis and its mouse model19,20 and suggested ongoing motor neuron pathology occurring through a mechanism initiating in the spinal cord and not in the muscle. Importantly, however, we observed progressive accumulation of lesions along the entire spinal cord that were predominantly located in the dorsal and lateral columns of the cervical spinal cord (Fig. 3a,b). These lesions were filled with giant cells containing what appeared to be ingested material (Fig. 3a). Immunofluorescence staining of spinal cord sections for F4/80 revealed that these cells were giant macrophages laden with intracellular myelin (Fig. 3c,d). Quantification of myelin staining in the dorsal column revealed progressive demyelination (Fig. 3e) that was accompanied by loss of neuronal/axonal staining (Fig. 3f). At end-stage disease, large portions of the dorsal and lateral columns were degenerated, thus probably affecting ascending/descending neurotransmission. Furthermore, analysis of the brain revealed lesions that were specifically located to the thalamus and were pres-ent only in mice at the end stage of disease (Supplementary Fig. 3d). These lesions similarly contained giant F4/80+ macrophages (Supplementary Fig. 3e,f).
Fig. 3 |. Motor disease is characterized by myelin-ingesting macrophages in the spinal cord.

a, Analysis of lesion size in hematoxylin and eosin (H&E)-stained spinal cords at the cervical enlargement. Bottom, magnifications of the dorsal column (location shown in Tgfbr2fl/fl→DTA). Scale bars, 500 μm (top) and 50 μm (bottom). Lines in the graph display mean values for n = 3 mice per group and 4 sections per mouse. *P = 0.0171; **P = 0.0012 by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test. b, H&E staining showing the length of the dorsal lesions in the cervical spinal cord in sagittal brain sections (location of magnification shown in low-power image). Images are representative of n = 3 mice per group. Scale bars, 250 μm (magnifications) and 1 mm. c, F4/80 and Fluoromyelin staining in the dorsal and lateral columns of the spinal cord. Images are representative of n = 4, 3 and 4 mice. Scale bars, 50 μm. d, Confocal images of F4/80 and Fluoromyelin staining in dorsal column lesions. Scale bars, 10 μm. In graph, lines represent mean values of intrac ellular myelin (integrated density) quantified in n = 18 and 35 individual cells, on the basis of an average of 3 z-stack images per cell from n = 2 mice per group and 2 sections per mouse at day 40 after TAM. e, Demyelination assessed by quantification of Fluoromyelin staining in the dorsal column. Scale bars, 200 μm. In graph, lines represent mean values of n = 4, 3 and 4 mice and at least 4 sections per mouse. ***P < 0.0001 by one-way ANOVA with Dunnett’s multiple comparison test. f, Loss of nerve fibers, as assessed with a pan-neuronal-antibody cocktail (Mab2300). Quantification was performed for the dorsal column. Scale bars, 200 μm. In graph, lines represent mean values of n = 4 mice per group and at least 3 sections per mouse. **P = 0.0066 by Student’s two-tailed unpaired t test.
The rapid activation of brain and spinal cord macrophages after loss of TGF-β signaling and the absence of adaptive immune cells indicated that Tgfbr2−/− macrophages directly induced demyelination and neuronal damage. To confirm that the motor phenotype arose as a direct consequence of Tgfbr2 deletion in CX3CR1+ CNS macrophages, we produced Cx3cr1CreER/+Tgfbr2fl/fl→ Cx3cr1CreER/+R26DTR/+ (DTR) chimeras, in which TAM administration before bone marrow transplantation made the CNS-resident microglia susceptible to ablation by DT (Supplementary Fig. 4a). Three weeks after microglia depletion, Tgfbr2 was recombined in repopulating macrophages through TAM administration. This procedure resulted in development of motor impairments with the same high incidence and disease course as those observed in the Cx3cr1CreER/+Tgfbr2fl/fl→DTA chimeras (Supplementary Fig. 4b), as well as a similar impairment in the four-paw hanging-wire test (Supplementary Fig. 4c). However, the onset of these symptoms was somewhat delayed (day 23 ± 3) relative to the onset in the Cx3cr1CreER/+Tgfbr2fl/fl→DTA chimeras (day 12 ± 2), thus indicating that integration into the microglial niche affected the vulnerability of macrophages to loss of TGF-β signaling. End-stage motor symptoms in the Cx3cr1CreER/+Tgfbr2fl/fl→DTR mice were accompanied by the presence of MHCIIhi macrophages in the CNS (Supplementary Fig. 4d). Kinetic analyses after TAM administration further demonstrated that CX3CR1+ macrophages progressively gained MHCII expression and lost CX3CR1 expression until the onset of motor-impairment symptoms (Supplementary Fig. 4e). Together, these observations indicated that loss of Tgfbr2 results in reprogramming of CNS macrophages, thus leading to spontaneous damage to neurons and subsequent development of motor disease.
As previously reported, mice lacking Tgfb1 expression specifically in the CNS develop motor impairments 4–6 months after birth. Although that phenotype has been attributed to loss of TGF-β signaling in microglia, the mice are characterized by a heterogeneous CNS myeloid compartment1. To address whether motor impairments similarly developed after loss of Tgfbr2 in CNS-resident microglia, we administered TAM directly to Cx3cr1CreER/+Tgfbr2fl/fl mice. This treatment resulted in efficient deletion of Tgfbr2 in microglia (Supplementary Fig. 5a), which was accompanied by microglial activation, as evidenced by changes in cell-surface-marker expression (Supplementary Fig. 5b–d) and morphology (Supplementary Fig. 5e), as previously reported2. The peripheral contribution to the microglial compartment due to microglial loss of Tgfbr2 was negligible (Supplementary Fig. 5f). In four-paw hanging-wire tests, we observed progressive deterioration of muscle strength starting in the fourth week after TAM administration, an effect apparent in all mice by the eighth week (Supplementary Fig. 5g). However, visible motor impairments were not evident until day 42 ± 5. Symptoms progressed to include tail weakness, gait abnormalities, hind-leg weakness/paralysis and incontinence (Supplementary Fig. 5h). This progression of motor changes correlated with a gradual increase in MHCII expression in microglia (Supplementary Fig. 5i). These results demonstrated that Tgfbr2 deficiency in CNS-resident microglia is sufficient to drive the motor disease phenotype, although the disease progressed at a markedly slower pace.
The devastating motor disease that developed in TGF-β R2-deficient chimeras indicated that in the absence of TGF-β signaling, the CNS environment licenses monocyte-derived macrophages for tissue damage. We therefore next addressed how the gene expression signature of CNS macrophages is controlled by TGF-β signaling. We thus sorted TGF-βR2-deficient monocyte-derived macrophages from Cx3cr1CreER/+Tgfbr2fl/fl→DTA animals with ongoing motor disease and from their Tgfbr2fl/fl→DTA littermate counterparts. For comparison, we sorted wild-type and TGF-βR2-deficient microglia and performed RNA sequencing for all four groups (Fig. 4a). Unbiased hierarchical clustering of global gene expression profiles revealed that the samples organized into their respective groups and that most of the variance was attributable to differences between wild-type microglia and the remaining groups (Fig. 4b). Almost 90% of the variance within the samples could be explained by a two-dimensional principal-component-analysis plot demonstrating a clear segregation of sample groups (Fig. 4c). This analysis demonstrated that Tgfbr2 loss in microglia and monocyte-derived macrophages resulted not only in partly functionally diverse outcomes but also in transcriptionally distinct cell populations.
Fig. 4 |. Tgfbr2-deficient macrophages are characterized by expression of antigen presentation, inflammation and phagocytosis genes.

RNA-seq analysis of wild-type and Tgfbr2-deficient microglia (MG) and monocyte-derived macrophages (Mac), sorted from the CNS on days 18–22 after TAM administration. a, n = 3 samples per group, sorted from a pool of 1 (WT MG), 2 (Tgfbr2−/− MG), 2 or 3 (WT Mac) or 2–4 (Tgfbr2−/− Mac) mice per sample. b,c, Unbiased hierarchical clustering (b) and principal component (PC) analysis (visualizing 89% of variance; c) of DESeq2-normalized data from n = 3 samples per group. d, Volcano plots of DESeq2-normalized RNA-seq data visualizing the log2 ratio versus –log10 adjusted P value (Tgfbr2−/− Mac/WT Mac) of n = 3 samples per group. e, Expression (log2 ratio) of selected genes differentially expressed between Tgfbr2–/– macrophages and wild-type macrophages, clustered according to their pathways.
To determine the consequence of loss of Tgfbr2 in macrophages, we compared their transcriptome with that of wild-type macrophages. The results revealed 313 differentially expressed genes (more than fourfold change in both directions, adjusted P < 0.001; Fig. 4d,e). Gene Ontology–enrichment analysis revealed that transcripts for antigen processing and presentation were enriched in TGF-βR2-deficient macrophages, including several MHCII genes (H2-Aa, H2-Ab1 and H2-Eb1) and genes encoding MHCII-associated molecules (Cd74 and Ciita). These results corroborated our finding of high surface expression of MHCII observed in TGF-β R2-deficient macrophages and the correlation between MHCII expression and disease onset. In TGF-βR2-deficient macrophages, we also observed enrichment of genes associated with inflammatory responses (Tnfsf18, Tlr5 and Pparg) as well as chemotaxis (Ccl6, Ccl7, Ccl8 and Ccl9) and the complement system (C4b). C4 promotes synaptic pruning in the developing mouse brain and is linked to schizophrenia21. There was also a general enrichment in genes associated with the cell cycle. Furthermore, we observed upregulation of metallopeptidases that break down the extracellular matrix, as well as upregulation of scavenger receptors (Cd163 and Scarb1). Numerous C-type lectins were upregulated in macrophages lacking Tgfbr2; these lectins recognize ligands from ‘nonself ‘ or ‘damaged self ‘22. In addition, several CD300 molecules were highly expressed in TGF-βR2-deficient macrophages; these receptors recognize lipids exposed on dead and activated cells and have been linked to several pathological conditions23. This gene signature was consistent with the ability of brain and spinal cord TGF-βR2-deficient macrophages to promote inflammation, deposit complement and phagocytose neuronal cells and myelin. We observed downregulation of genes associated with microglial identity (Cx3cr1, Siglech, Tmem119 and Olfml3), a result consistent with the importance of TGF-β in the induction of these genes in monocyte-derived macrophages. We also observed enrichment of genes negatively regulating TGF-β signaling (Smad6 and Smad7), thus indicating release of negative feedback as a result of loss of Tgfbr2. Analysis of upstream regulators through ingenuity pathway analysis predicted that the gene set was the result of inhibited TGF-β signaling as well as activation by lipopolysaccharide, interferon-γ and the proinflammatory cytokine GM-CSF (Supplementary Table 1), all of which have been implicated in perpetuating microglia/macrophage-driven CNS inflammation.
Together, our results highlight a critical function of TGF-β in preventing microglia/macrophage-mediated CNS pathology. This conclusion is supported by a recent study reporting that suppressed TGF-β signaling is a common feature of microglia isolated from neurodegenerative disease models24. Similar neurological disorders to those observed in our study develop spontaneously in mice deficient in NRROS, a myeloid-expressed protein that regulates production of reactive oxygen species, although these mice have not been reported to exhibit histological signs of CNS pathology25. Nrros−/− mice lack conventional microglia cells and are instead populated by myeloid cells exhibiting a perivascular macrophage-like phenotype. Motor symptoms in CNS-conditional Tgfb1−/− mice are similarly accompanied by loss of typical microglia phenotypes and the appearance of peripherally derived monocytes in the CNS1.
In conclusion, responding to TGF-β is crucial for the functional integration of monocytes into the CNS microenvironment. For a number of neuropathological conditions in which microglia and macrophages have an active role in disease, the failure to respond to TGF-β may explain the onset of neurological disease and may aid in understanding of how to design therapies.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at https://doi.org/10.1038/s41590–018-0091–5.
Methods
Mice.
All mice were bred and maintained under specific-pathogen-free conditions at the Karolinska Institutet, in accordance with national animal-care guidelines. All animal experiments were approved by the appropriate ethical review board (Stockholms djurförsöksetiska nämnd). Cx3cr1CreER, R26DTR, R26DTA, R26YFP and LysMCre mice were obtained from The Jackson Laboratory. Tgfbr2fl/fl mice were obtained from M. Li (Sloan Kettering Institute), and CD45.1 mice were obtained from F. Wermeling (Karolinska Institutet). Experiments were generally initiated when mice were 6–12 weeks old.
In vivo treatments.
For tamoxifen administration, tamoxifen (TAM; Sigma) was suspended in corn oil at 75 °C, and 5 mg (200 μl), unless otherwise specified, was administered subcutaneously on three consecutive days. For diphtheria toxin administration, mice were injected with diphtheria toxin (25 ng/g body weight) intraperitoneally on three consecutive days.
Generation of bone marrow chimeras.
Mice were irradiated with 9.5 gray with an X-RAD 320 irradiation source (0.95 gray/min) with an irradiation field of 20 × 20 cm. Head protection was accomplished by keeping mice under isoflurane anesthesia and placing the head (from the neck up) outside the field of irradiation. Mice were monitored throughout the irradiation period to ensure that their heads stayed outside the field (the irradiation source was equipped with a lamp to visualize the irradiation field). Mice were reconstituted on the same day with 2 × 106 to 5 × 106 bone marrow cells through tail-vein injection. Mice were considered reconstituted and used for experiments 6–8 weeks later. Whole-body irradiation invariably resulted in complete chimerism (>99%), and head-protected mice generally showed 50–80% chimerism.
Clinical scoring.
Paralysis and motor symptoms were scored by assignment of one point for the presence of each of the following symptoms: loss of tail tonus, impaired tail lifting, gait disturbance, dragging of one hind leg, dragging of two hind legs, incontinence and moribundity/death.
Four-paw hanging-wire test.
Mice were placed on a wire bar that was then inverted over a table covered with soft bedding or alternatively over a plastic mouse cage with soft bedding. The time until each mouse released its grip or fell was recorded. Three trials were performed for each mouse, and the longest time was recorded. The maximum time was set to 180 s, after which the trial was terminated.
Preparation of single-cell suspensions.
Mice were euthanized through intraperitoneal injection of 100 μl pentobarbital. When applicable, blood was collected from the right ventricle before perfusion. Mice were perfused with ice-cold PBS, and organs were dissected. CNS cells were prepared by enzymatic digestion with collagenase (1 mg/ml, Roche) and DNase (0.2 mg/ml, Roche) or a Neural Tissue Dissociation Kit (T, Miltenyi Biotec). Myelin was removed with 38% Percoll.
Blood samples of 100–200 μl were collected into tubes containing EDTA, lysed in ACK buffer and centrifuged. The pellet was resuspended in PBS and used for staining. Spleen cell suspensions were prepared through mechanical dissociation in PBS and straining through 40-μm strainers. Bone marrow cells were prepared through flushing of femurs with PBS. Spleen and bone marrow preparations were treated with ACK buffer to lyse red blood cells.
Flow cytometry.
Single-cell suspensions were plated in 96-well V-bottom plates and stained at 4 °C. Dead cells were removed with a LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen). Antibodies to the following proteins were used: CD3 (145– 2C11, BioLegend), CD4 (RM4–5, BioLegend), CD11b (M1/70, BioLegend), CD45 (30F11, BioLegend), CD45.1 (A20, BioLegend), CD45.2 (104, BioLegend), CD115 (AFS98, BioLegend), F4/80 (BM8, BioLegend), Foxp3 (FJK-16s, eBioscience), IFN-γ (XMG1.2, BD Biosciences), IL-17 (TC11–18H10, BD Biosciences), Ki67 (B56, BD Biosciences), Ly6C (HK1.4, BioLegend), Ly-6G (1A8, BD Biosciences), MHCII (M5/114.15.2, BioLegend) and TNF (MP6-XT22, BioLegend). Intracellular cytokine staining was performed with an Intracellular Staining Kit (eBioscience) after 4 h treatment with PMA (50 ng/ml, Sigma Aldrich), ionomycin (1 μg/ml, Sigma Aldrich) and GolgiPlug (1 μl/ml, BD Biosciences) in complete DMEM (Sigma). Cells were sorted with a Gallios flow cytometer (Beckman Coulter), and the data were analyzed in Kaluza software (Beckman Coulter).
Cell sorting.
Cells were sorted to >95% purity with a BD influx cell sorter and the following sorting strategies; microglia in Tgfbr2fl/fl mice, Dead-Ly6C-CD11b+F4/80lo; microglia in Cx3cr1CreER/+Tgfbr2fl/fl mice, Dead-Ly6C-CD11b+F4/80hi; monocyte-derived macrophages in Tgfbr2fl/fl→DTA chimeras, Dead-Ly6C-CD11b+F4/80hi; Cx3cr1CreER/+Tgfbr2fl/fl→DTA chimeras, Dead-Ly6C-CD11b+CX3CR1-YFP+F4/80hi; and blood Ly6Chi monocytes, CD115+Ly6ChiLysM-YFP– or CD115+Ly6ChiLysM-YFP+. BM Ly6Chi monocytes were presorted with a monocyte isolation kit (BM, Miltenyi Biotec) to ~95% purity and then sorted as LysM-YFP– or LysM-YFP+.
Next-generation sequencing.
For RNA preparation, cells were sorted into a solution of RNA later (Thermo Fisher Scientific). RNA was prepared with an RNeasy Micro Kit (Qiagen). RNA quality and integrity were assessed with a Bioanalyzer 2100 (Agilent). All samples included had high quality (RIN = 7.1–10). NA (>100 pg) was amplified with a SMARTer Stranded Total RNA-Seq Kit–Pico Input Mammalian (Clontech). Next-generation sequencing and bioinformatics analysis was performed by the National Genomics Infrastructure (NGI) at the Science for Life Laboratory on a HiSeq 2500 System with a HiSeq Rapid SBS Kit v2 (Illumina), thus generating >13.5 M reads/sample. Data normalization and analysis of differential gene expression were done in the DESeq2 R package26 with a negative binomial test. The false-discovery-rate-adjusted P value was estimated with Benjamini–Hochberg correction27.
Gene Ontology and pathway analysis.
Enrichment analysis for Gene Ontology terms was performed in DAVID28. Ingenuity pathway analysis (Qiagen) was used to analyze upstream regulators.
qRT–PCR.
cDNA was synthesized with an iScript cDNA Synthesis Kit (Bio-Rad). qRT–PCR was performed with SYBR green reactions (Bio-Rad).
Immunohistochemistry and image analysis.
Perfused brain and spinal cord tissues were immersion-fixed in 4% PFA for 24 h, then sucrose protected (20%) for at least 24 h. Brains and spinal cords were then embedded in OCT cryomount (Histolab), frozen in isopentane and sectioned at 14 μm. Sections were stained with antibodies to the following proteins: Iba-1 (Wako), F4/80-biotin (Cl:A3–1, AbD Serotec), P2ry12 (generated at Harvard Medical School), and vesicular acetylcholine transporter (Synaptic Systems); pan-neuronal-marker cocktail (Millipore, Mab2300) was also used. The secondary antibodies were donkey anti-mouse Cy3 (715–165-150, Jackson ImmunoResearch), goat anti-rabbit Alexa Fluor 488 (A-11008, Thermo Fisher), goat anti-rabbit Alexa Fluor 594 (R37117) or streptavidin-conjugated Alexa Fluor 488 (S11223, Thermo Fisher). H&E staining was performed with Mayer’s hematoxylin (Histolab) and 0.2% eosin (Histolab). Nissl staining was performed with cresyl violet (Sigma). Myelin was stained with Fluoromyelin according to the manufacturer’s instructions (Thermo Fisher Scientific). Confocal images were acquired with a DMI6000 microscope (Leica Biosystems). Whole tissue sections were scanned with a Pannoramic 250 Flash digital scanner (3D histech).
H&E lesions were quantified in the cervical enlargement of the spinal cord (four sections per mouse) or in the thalamus (two areas with two sections each per mouse). Fluoromyelin and neuronal (Mab2300) staining were quantified in dorsal-column lesion areas in Image J. VAChT+ or Nissl-stained (>200 μm2) motor neurons were counted in a blinded manner in the ventral horns in at least four sections per mouse. Fluoromyelin uptake in F4/80+ macrophages was quantified in confocal z-stack images as the product of the area and mean pixel intensity (integrated density) in ImageJ.
For analysis of neuromuscular-junction innervation, legs from PBS-perfused mice were immersion-fixed in 4% PFA for 40 min and then placed in PBS. Tibialis anterior muscles were dissected and stored in 30% sucrose for 24 h before sectioning at 40-μm thickness. Neurofilament was stained with anti–synaptic vesicle protein (SV2, Developmental Studies Hybridoma Bank), anti–neurofilament 165 kDa (2H3, Developmental Studies Hybridoma Bank) and donkey anti-mouse Alexa Fluor 488 (R37114, Thermo Fisher). Endplates were then visualized by staining with TRITC-conjugated α-bungarotoxin (Thermo Fisher) for 15 min. Images were acquired with a Zeiss LSM800 confocal microscope. Neuromuscular-junction innervation was quantified by assessing the overlay between neurofilament (SV2/2H3) and α-bungarotoxin-labeled endplates. Endplates were quantified as occupied when two-thirds of the endplate was covered with neurofilament staining, partial when colocalization was less than two-thirds of the endplate and vacant when no neurofilament staining was overlaying the endplate. A minimum of 40 endplates were analyzed per mouse. The analysis was performed in a blinded manner.
Statistical analysis.
General statistical analyses were performed in GraphPad Prism software. Two group comparisons were done with Student’s two-tailed unpaired t test. Comparison of two groups with a control group were done with one-way ANOVA and Dunnett’s multiple comparison test. P < 0.05 was considered statistically significant. Analysis of RNA-seq data was done in the DESeq2 package, which uses Wald testing with Benjamini–Hochberg adjustment for multiple testing. The overlap P value of the upstream regulator analysis was calculated with Fisher’s exact test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank A. van Vollenhoven for flow cytometry. We thank the staff at AKM, particularly E. Qvist, for animal caretaking and M. Gustafsson for performing tail-vein injections. We thank A. Svensson, D. Sunnemark and A. Dahlstrand for help with slide scanning, performed at Offspring Biosciences, Södertälje. We thank M. Li (Sloan Kettering Institute) for Tgfbr2fl/fl mice and F. Wermeling (Karolinska Institutet) for CD45.1 mice. We acknowledge support from the Science for Life Laboratory, the National Genomics Infrastructure (NGI) and Uppmax for providing assistance in next-generation sequencing and computational infrastructure.
This work was supported by grants from the Swedish Alzheimer Foundation (Alzheimerfonden, AF-74004, R.A.H.), Swedish Research Council (Vetenskapsrådet, 2014–02087, R.A.H. and K2015–61X-20776–08-3, M.J.), Swedish Childhood Cancer Foundation (Barncancerfonden, PR2014–0154 and NCP2015–0064, X.-M.Z.), Åke Wibergs stiftelse (M14–0263, X.-M.Z.), Foundation of Swedish MS Research (MS Forskningsfonden, L.K.), Swedish Heart-Lung Foundation (Hjärt-Lungfonden, M.J.F. and D.F.J.K.), the Novo Nordisk Foundation (NNF15CC0018346, M.J.F. and D.F.J.K.), NIH-NINDS (1R01NS088137, O.B.), NIH-NIA (R01AG051812 and R01AG054672, O.B.), National Multiple Sclerosis Society (5092A1, O.B.) and Amyotrophic Lateral Sclerosis Association (ALSA2087, O.B.), and by a Nancy Davis Foundation Faculty Award (O.B.). X.-M.Z. was supported by a fellowship from the Swedish Childhood Cancer Foundation (Barncancerfonden, NC2014–0046, NBCNS). L.K. was supported by a fellowship from the Margaretha af Ugglas Foundation.
Footnotes
Data availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA-seq raw and processed data have been deposited in the Gene Expression Omnibus data repository under accession number GSE111385.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41590–018-0091–5.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Butovsky O. et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buttgereit A. et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol 17, 1397–1406 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Mildner A. et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci 10, 1544–1553 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Varvel NH et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc. Natl. Acad. Sci. USA 109, 18150–18155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruttger J. et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Guilliams M. & Scott CL Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol 17, 451–460 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Scott CL et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun 7, 10321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van de Laar L. et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Buch T. et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods 2, 419–426 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Parkhurst CN et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu S, Wu Y. & Capecchi MR Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development 133, 581–590 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Yu X. et al. The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity 47, 903–912.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Bobr A. et al. Autocrine/paracrine TGF-β1 inhibits Langerhans cell migration. Proc. Natl. Acad. Sci. USA 109, 10492–10497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malipiero U. et al. TGFbeta receptor II gene deletion in leucocytes prevents cerebral vasculitis in bacterial meningitis. Brain 129, 2404–2415 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Novitskiy SV et al. Deletion of TGF-β signaling in myeloid cells enhances their anti-tumorigenic properties. J. Leukoc. Biol 92, 641–651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J. et al. Myeloid TGF-β signaling contributes to colitis-associated tumorigenesis in mice. Carcinogenesis 34, 2099–2108 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Parsa R. et al. TGFβ regulates persistent neuroinflammation by controlling Th1 polarization and ROS production via monocyte-derived dendritic cells. Glia 64, 1925–1937 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nijssen J, Comley LH & Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagao M, Misawa H, Kato S. & Hirai S. Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 57, 329–333 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Wootz H. et al. Alterations in the motor neuron–renshaw cell circuit in the Sod1G93A mouse model. J. Comp. Neurol 521, 1449–1469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekar A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dambuza IM & Brown GD C-type lectins in immunity: recent developments. Curr. Opin. Immunol 32, 21–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borrego F. The CD300 molecules: an emerging family of regulators of the immune system. Blood 121, 1951–1960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krasemann S. et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong K. et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat. Immunol 18, 633–641 (2017). [DOI] [PubMed] [Google Scholar]
References
- 26.Love MI, Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y. & Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995). [Google Scholar]
- 28.Huang W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.