

Abstract
Receptor‐interacting protein 3 (RIP3) plays an important role in the necroptosis signaling pathway. Our previous studies have shown that the RIP3/mixed lineage kinase domain‐like protein (MLKL)‐mediated necroptosis occurs in retinal ganglion cell line 5 (RGC‐5) following oxygen‐glucose deprivation (OGD). However, upstream regulatory pathways of RIP3 are yet to be uncovered. The purpose of the present study was to investigate the role of p90 ribosomal protein S6 kinase 3 (RSK3) in the phosphorylation of RIP3 in RGC‐5 cell necroptosis following OGD. Our results showed that expression of RSK3, RIP3, and MLKL was upregulated in necroptosis of RGC‐5 after OGD. A computer simulation based on our preliminary results indicated that RSK3 might interact with RIP3, which was subsequently confirmed by co‐immunoprecipitation. Further, we found that the application of a specific RSK inhibitor, LJH685, or rsk3 small interfering RNA (siRNA), downregulated the phosphorylation of RIP3. However, the overexpression of rip3 did not affect the expression of RSK3, thereby indicating that RSK3 could be a possible upstream regulator of RIP3 phosphorylation in OGD‐induced necroptosis of RGC‐5 cells. Moreover, our in vivo results showed that pretreatment with LJH685 before acute high intraocular pressure episodes could reduce the necroptosis of retinal neurons and improve recovery of impaired visual function. Taken together, our findings suggested that RSK3 might work as an upstream regulator of RIP3 phosphorylation during RGC‐5 necroptosis.
Keywords: necroptosis, oxygen and glucose deprivation, p90 ribosomal protein S6 kinase 3, phosphorylation, receptor‐interacting proteins 3, retinal ganglion cell‐5
Outline of RSK3 regulatory mechanism via p‐RIP3/p‐MLKL in OGD‐induced RGC‐5 necroptosis, which suggested that RSK3 might work as an upstream regulator of RIP3 phosphorylation during RGC‐5 necroptosis.
1. INTRODUCTION
In recent years, researchers have found that certain types of necrosis can be regulated by a series of signaling molecules (He et al., 2009; Zhang et al., 2009) that share similar morphological features with necrosis signaling molecules (Holler et al., 2000; Matsumura et al., 2000). This alternately regulated necrosis pathway is known as necroptosis. It is a sub‐type of regulated necrosis and depends on the activities of receptor‐interacting protein 3 (RIP3) and mixed lineage kinase domain‐like protein (MLKL; Degterev et al., 2005; Sun et al., 2012; Kim and Li, 2013; Li et al., 2017; Negroni et al., 2017; Zhang et al., 2017; Orozco et al., 2019). Earlier studies as well as our own previous work have shown that, other than in non‐neuronal cells, necroptosis also occurs in many types of neurons, such as retinal ganglion cells (RGCs) following acute retinal ischemia/reperfusion (I/R; Rosenbaum et al., 2010; Liao et al., 2017; Huang et al., 2013), oxygen‐glucose deprivation (OGD)/glutamate‐induced neuronal injury (Zhang et al., 2013a; 2013b; Wang et al., 2018a; 2018b; 2018c; 2018d; Mathew et al., 2019; 2019a; 2019b), oxidative stress, or methamphetamine‐induced cortical neuronal injury (Jiang et al., 2014; Ding et al., 2015; Xiong et al., 2016; Lu et al., 2019; Guo et al., 2020). Inhibition of necroptosis is extremely important for reducing neuronal death (Wu et al., 2015; Cruz et al., 2018; Wang et al., 2018a; 2018b; 2018c; 2018d). However, the regulatory mechanisms of necroptosis have not yet been fully elucidated.
RIP1 and RIP3 play important roles in the regulation of necroptosis (Moriwaki and Chan, 2016). During cell damage processes, when death signals are transmitted to the cell, RIP1 and RIP3 phosphorylate/activate each other via an RIP homotypic interaction motif or an amino‐terminal kinase domain to form the necrosome (Li et al., 2012; Orozco et al., 2019). Accumulation of activated RIP3 may lead to cell necroptosis via the phosphorylation and oligomerization of MLKL, which in turn leads to the disruption of cell membrane integrity (Sun et al., 2012; Nogusa et al., 2016; Li et al., 2017; Yang et al., 2018; Najafov et al., 2019). For the overall phosphorylation of the necrosome, RIP3 phosphorylation can promote the phosphorylation of RIP1 (Vandenabeele et al., 2010; Bozec et al., 2016). However, the regulatory mechanisms of RIP3 phosphorylation remain unknown. Previous studies have suggested that the extracellular signal‐regulated kinases 1/2 (ERK 1/2) may indirectly modulate necroptosis by regulating RIP3 expression (Gao et al., 2014). In contrast, a number of studies have reported that the heat shock protein 90 (HSP90) may regulate necroptosis via the formation of a necrosome (RIP1/RIP3) in HT‐29 cells and cortical neurons (Li et al., 2015; Jacobsen and Silke, 2016; Wang et al., 2018a; 2018b; 2018c; 2018d; ). These results showed that phosphorylation of RIP3 was reduced by regulatory molecules acting upstream of RIP3. However, necrotic neurons can only be partly rescued by targeted intervention therapy for two possible reasons: first, due to non‐regulated necrosis, it might not be possible to rescue some of the necrotic cells; second, some other molecules might also play a redundant role in the regulation of RIP3 phosphorylation. In fact, any type of cell death is cross‐regulated by multiple molecular mechanisms. These alternative aspects of the regulatory mechanisms of RIP3 phosphorylation need to be investigated.
In general, phosphorylation and dephosphorylation of substrate proteins are catalyzed by cytosolic kinases and phosphatases, respectively. Protein phosphorylation plays an important role in many biological processes and is regulated by a dynamic balance between kinase and phosphatase activities. Some serine/threonine (Ser/Thr) residues of RIP3 can be phosphorylated by protein Ser/Thr kinases (Meylan and Tschopp, 2005). Yuan and co‐workers analyzed the modification profiles of all proteins expressed using proteomics and RNA interference approaches, and found that three kinases were associated with the phosphorylation of substrate proteins involved in cell necroptosis (Hitomi et al., 2008). However, only the Ser/Thr residue of Rps6k (ribosomal protein S6 kinase) showed phosphorylating activity in their study. Rps6k is widely expressed in different types of neurons and is involved in cerebral hypoxia‐ischemia injury by phosphorylating its substrate proteins (Vazquez‐Higuera et al., 2011; Miyake et al., 2015). Also, Rps6k is likely involved in the process of phosphorylation of the key protein RIP3 in the necroptotic pathway. Currently, the molecular pathways underlying I/R injury of regulated necrosis in retinal neurons remain elusive.
Rps6k is classified into p90 ribosomal S6 kinase (p90RSK) and p70 ribosomal S6 kinase (p70RSK) subfamilies (Romeo et al., 2012). Eukaryotic ribosomes are composed of two subunits, namely, 40S and 60S. In 1985, Erikson and Maller reported a ribosomal S6 kinase (S6K) in Xenopus laevis eggs which phosphorylated the 40S ribosomal subunit S6 protein and promoted the translation of selected mRNAs (Erikson and Maller, 1985). S6 kinases of 85–90 kDa were identified by biochemical purification, which led to the cloning of cDNAs encoding highly homologous proteins later renamed p90RSK, or RSK (Jones et al., 1988; Romeo et al., 2012). The vertebrate RSK family is divided into RSK1, RSK2, RSK3, and RSK4 isoforms (Romeo et al., 2012). RSK3, which belongs to the Ser/Thr kinase family, plays a central role in the Ras‐MAPK and PI3K‐mTOR pathways by promoting protein phosphorylation (Carriere et al., 2008; Serra et al., 2013; Jagilinki et al., 2016; Pirbhoy et al., 2017; Shrestha et al., 2018; Wang et al., 2018a; 2018b; 2018c; 2018d; ). As an important first‐order effector, RSK3 regulates many cellular processes, including cell proliferation, cell cycle, cell survival, and differentiation (Romeo et al., 2012; Kwon et al., 2018). Moreover, our preliminary, unpublished micro‐array data also demonstrated that the mRNA levels of both rsk3 and rip3 in retinas associated with retinal acute high intraocular pressure (aHIOP), a classical retinal I/R model, were sixfold higher than in retinas of normal rats. Death of RGCs by ischemic injury is a predominant characteristic of aHIOP (Buchi, 1992; Mathew et al., 2019). Collectively, these reports raised the question whether RSK3 could directly interact with the phosphorylated form of RIP3, and further regulate the RIP3‐MLKL interaction during RGC necroptosis following I/R injury.
Our previous results showed that RIP3‐MLKL‐mediated neuronal necroptosis took place in the early stages of OGD injury in vitro (Wang et al., 2018a; 2018b; 2018c; 2018d; ). In the present study, we further evaluated the effect of RSK3 on the phosphorylation of RIP3/MLKL during retinal ganglion cell‐5 (RGC‐5) necroptosis following OGD injury. Additionally, we performed in vivo studies to verify the role of RSK3 in necroptosis in the rodent visual system elicited by acute high intraocular pressure (aHIOP). The outcome of our study will improve the understanding of the RIP3 regulatory mechanisms in neuronal necroptosis. It also provides a theoretical and experimental basis to validate the potential intervention targets for clinical treatment of ocular diseases.
2. MATERIALS AND METHODS
2.1. Animals
Forty adult Sprague‐Dawley rats weighing 200–250 g, available from the animal center of Central South University, were used in this study. All animals were housed in acrylic box cages with free access to food and water. Animals were maintained under conditions of constant temperature (25°C), humidity (50 ± 10%), and lighting cycle (12:12 hr). All experimental procedures in the present study were approved by the Ethics Committee of Xiangya School of Medicine, in accordance with the NIH guidelines for use and care of laboratory animals.
2.2. Cell cultures and in vitro OGD model preparation
The RGC‐5 cell line derived from mice was provided by the Department of Ophthalmology, Second Hospital of Jilin University. RGC‐5 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high‐glucose medium (HyClone Laboratories, Inc.) supplemented with 10% fetal bovine serum (FBS; Gibco). The RGC‐5 cells were used within five passages post thawing in the experiment. The cells were incubated at 37°C in a 5% CO2 incubator and the inoculation density was about 4–5 × 104 cells/ml. According to our previous studies, the homemade in vitro OGD device can simulate I/R injury in vivo (Chen et al., 2016a; 2016b; Wang et al., 2018a; 2018b; 2018c; 2018d; ). The density of RGC‐5 cells was around 80% in a T25 or T75 flask, 35‐mm dishes, or multi‐well plates before OGD. Cells were rinsed twice with phosphate‐buffered saline and the culture medium was replaced with glucose‐free medium. The OGD model was generated as described in our previous study. In brief, the culture medium in the cell bottle was completely replaced with DMEM sugar‐free medium. Cells were exposed to the atmosphere in a container with 5% CO2 and 95% N2 at a speed of 3 L/min for 5 min until low O2 levels were achieved in the container. After 4 hr of OGD injury, the glucose‐free medium was replaced with the previous medium and the RGC‐5 cells were cultured in conventional cell culture conditions for 0, 2, 4, 6, and 12 hr. Cell death was assessed by propidium iodide (PI) staining, lactate dehydrogenase (LDH) release, and flow cytometry analysis. Molecular changes were detected by western blotting, immunofluorescence, real‐time quantitative polymerase chain reaction (RT‐qPCR), and immunoprecipitation at the indicated time points after OGD.
2.3. Induction of aHIOP and PI treatment in vivo
The animal model was prepared following the procedure described by Tong et al. (2010). Briefly, animals were anesthetized with 10% chloral hydrate (4 ml kg–1). A droplet of 10% chloral hydrate was administered to the conjunctival sac. A 30‐gauge needle connected to the instillation instrument filled with normal saline was inserted into the anterior chamber. The intraocular pressure was elevated to 110 mmHg. This was maintained for 60 min and then gradually decreased to normal. The rats were allowed to survive for 4 hr before terminal use. Thirty minutes prior to animal perfusion, 3 μl PI (1.0 mg ml–1 in DW, Sigma‐Aldrich) was administered via intravitreal injection.
2.4. Animal tissue preparation
At the specified time point, animals were anesthetized with 10% chloral hydrate (4 ml/kg) and perfused transcardially with .9% sodium chloride, followed by 4% paraformaldehyde (PFA) in .1 M phosphate‐buffered saline (PBS). After perfusion, the eyes were enucleated, the anterior segments were removed, and the posterior eyecups were post‐fixed in 4% PFA overnight at 4°C. The eyes were then placed sequentially in 15% and 30% sucrose in .1 M PBS at 4°C. Next, the eyecups were embedded in Tissue‐Tek optimal cutting temperature medium and frozen in liquid nitrogen. Subsequently, frozen sections were cut to a thickness of 8 mm using a microtome (Thermo Scientific). The sections that included the optic nerve were stored at −20°C until use.
2.5. Propidium iodide staining
Propidium iodide staining was initially performed to determine the necroptosis of RGC‐5 cells at different time points after restoration. Cell cultures on the coverslips with poly‐D‐lysine (PDL) coating were washed twice with PBS and incubated with PI (1.0 mg ml–1) at 37°C for 15 min in the dark. Subsequently, the coverslips were washed three times for 5 min gently each time with PBS and then fixed with 4% PFA at room temperature for 20 min. After fixation, the cells were washed thrice with PBS buffer for 5 min each time. Finally, the plate was sealed with anti‐fading mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI, Vector Laboratories) and the cell side of the slide with cells was inverted on a glass slide and photographed using a fluorescence microscope (Olympus). PI‐positive cells were counted and the result was expressed as the percentage of DAPI‐positive cells using image j software.
2.6. Immunofluorescence staining
The coverslips with fixed cells were blocked (1 × PBS, 5% bovine serum albumin, and .3% Triton X‐100) for 1 hr and subsequently incubated overnight at 4°C with the primary mouse anti‐RSK3 antibody (1:200, Abcam, ab201988) and rabbit anti‐p‐S232‐RIP3 antibody (1:200, Abcam, ab195117). The next day, the coverslips were removed and rewarmed at room temperature for 30 min. PBS was added and the coverslips were slowly washed three times at low speed on a shaker for 5 min each time. The fluorescent secondary antibody was diluted in .3% Triton X‐100 + 5% BSA and incubated with a Cy3‐Alexa‐conjugated anti‐mouse (1:200, Jackson ImmunoResearch, 715‐165‐151) and a 488‐Alexa‐conjugated anti‐rabbit (1:200, Jackson ImmunoResearch, 711‐545‐152) antibody at room temperature for 2 hr. After washing three times in PBS, the coverslips were covered with an anti‐fading mounting medium with DAPI before examination on a fluorescence microscope with an emission wavelength of 570 nm and an excitation wavelength of 550 nm.
2.7. The siRNA approach
The siRNA kit against rsk3 was obtained from RIBO‐Biology. The siRNA targeting rsk3 had the following sequence: #1 5′‐CCACCTACTTTGCTCTGAAdTdT‐3′, #2 5′‐GACTCACGTCTACCAGGTTdTdT‐3′, #3 5′‐GAGCCTGTGCTCTCATCTAdTdT‐3′.
When the density of RGC‐5 was around 30%–50%, the transfection was performed with Lipofectamine 2000 (Thermo Scientific, Invitrogen) and Opti‐MEM according to the manufacturer’s protocol. The same concentration of the nonspecific sequence was used as a negative control. Opti‐MEM (Gibco) was replaced with normal culture medium after transfection 6 hr later. Cells were incubated for 24–72 hr after transfection until they could be subjected to OGD for further detection and analyses.
2.8. Drug application
LJH685, a specific RSK inhibitor, inhibits RSK biochemical activities. It was dissolved in dimethylsulfoxide (DMSO, Sigma‐Aldrich) as a stock solution. The stock solution was further diluted in the culture medium to achieve DMSO concentrations below .1%. LJH685 (Selleck) was used at a concentration of 1, 3, and 10 μM. The drug solution was administered directly to RGC‐5 cultures or by intravitreal injection into rats, 4 hr before HIOP treatment (Aronchik et al., 2014).
Necrostatin‐1 (Nec‐1, Sigma‐Aldrich) is a potent, selective, and cell‐permeable necroptosis inhibitor that inhibits RIP1 kinase in the necroptosis pathway. It was dissolved in DMSO as a stock solution. The stock solution was further diluted in the culture medium to achieve DMSO concentrations below .1%. We pretreated RGC‐5 cells with 20 µM Nec‐1 for 2 hr before conducting the OGD model or TNF‐α/zVAD/SMAC‐mimetic (TSZ) model to inhibit necroptosis (Vieira et al., 2014).
TSZ is a typical necroptosis model (Cai et al., 2014; Wang et al., 2014) and was used as a positive control of OGD in vitro in our present study. Tumor necrosis factor‐α (TNF‐α; CST, #5178) was dissolved in water as a stock solution. zVAD (OMe)‐FMK (zVAD, CST, #60332) was dissolved in DMSO as a stock solution. SM‐164 is a bivalent mimetic of Smac and was dissolved in DMSO as a stock solution. The stock solution was further diluted in the culture medium to achieve DMSO concentrations below .1%. We pretreated RGC‐5 cells with 20 µM zVAD for 30 min, followed by treatment with 20 ng ml–1 TNF‐α and 100 nM SM‐164 for 6 hr to induce necroptosis (Weigert et al., 2017).
2.9. Western blot analysis
Cells were lysed in ice‐cold radioimmunoprecipitation assay buffer (CWBIO) supplemented with 1% protease and 1% phosphatase inhibitors. The extracts were maintained on ice for 30 min and centrifuged at 16,000 g for 20 min at 4°C and the supernatant was recovered. Protein concentration was determined using a BCA protein assay kit (CWBIO). Proteins were resolved by SDS‐PAGE (10% polyacrylamide), transferred to nitrocellulose membranes (GE Healthcare), and incubated with primary antibodies. The reactions were followed by incubation with peroxidase‐labeled secondary antibodies. Primary antibodies were: anti‐RSK3 (1:500, Abcam, ab201988), anti‐RIP3 (1:3,000, Sigma‐Aldrich, PRS2283), anti‐p‐S232‐RIP3 (1:1,500, Abcam, ab195117), anti‐RIP1 (1:1,000, CST, 3493), anti‐p‐S166‐RIP1 (1:1,000, CST, 31122), anti‐GAPDH (1:1,000, Beyotime, AF0006), anti‐MLKL (1:1,000, CST, #37705), and anti‐p‐S345‐MLKL (1:1,000, CST, #37333). The secondary antibodies were peroxidase‐conjugated affiniPure donkey anti‐rabbit IgG (1:6,000, Jackson Immuno Research, 711‐035‐152) and donkey anti‐mouse IgG (H+L, 1:6,000, Jackson Immuno Research, 715‐035‐150). The integrated density value (IDV) was quantified using image j (NIH).
2.10. Detection of phosphorylated proteins by Phos‐tag SDS‐PAGE
The preparation of protein samples, the operating procedure, and the reagents used for Phos‐tag SDS‐PAGE were almost identical to those for conventional SDS‐PAGE, except that the acrylamide‐pendant Phos‐tag, which binds to the phosphate groups, was copolymerized with the separation gel in Phos‐tag SDS‐PAGE. Proteins were resolved by Phos‐tag SDS‐PAGE (8% polyacrylamide with 50 μM Mn2+‐Phos‐tag), transferred to nitrocellulose membranes (GE Healthcare), and incubated with primary antibodies. The reactions were followed by incubation with peroxidase‐labeled secondary antibodies. The primary antibodies were anti‐RSK3 (1:500, Abcam, ab201988) and the IDV was quantified using image j (NIH).
2.11. RT‐qPCR assay
The total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific). For mRNA level detection, cDNA was synthesized using the Hifair® II 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus; YEASEN). RT‐qPCR was performed using the Hieff® qPCR SYBR Green Master Mix (Low Rox Plus; YEASEN) on an ABI 7500 Real‐Time PCR System (Thermo Fisher). The data were analyzed using the method with ABI7500 software (Rampazzo et al., 2019; Rehorova et al., 2019). GAPDH was used as an internal control.
The primer sequence in RT‐qPCR:
| Gene | Sequences | |
|---|---|---|
| rsk3 | F | CCTCATCCTCAAAGCCAAGC |
| R | GGTGACAAAGAACGGGTGAC | |
| rip3 | F | CGGGAAACAGTGTGTGACAG |
| R | GATGGCCTGTTTTCGGACTG | |
| rip1 | F | CCCCGATTTGAAGAGGCTTG |
| R | GTGCACAATGAGCCAGGAAA | |
| gapdh | F | TTGATGGCAACAATCTCCAC |
| R | CGTCCCGTAGACAAAATGGT |
2.12. pEGFP‐C1‐rip3 construction and transfection
According to the experimental requirements, the pEGFP‐C1 vector was selected to prepare the linearized vector by double digestion with EcoRI and BamHI. The experimental primer design is about RIP3 as follows:
Mouse‐rip3‐pEGFP‐C1‐Forward Primers: 5′‐GAGCTCAAGCTTCGAATTCTATGTCTTCTGTCAAGTTATG‐3′
Mouse‐rip3‐pEGFP‐C1‐Reverse Primers: 5′‐TATCTAGATCCGGTGGATCCCTACTTGTGGAAGGGCTGCC‐3′.
According to the manufacturer’s protocol, the target fragment was amplified via PCR (Q5® High‐Fidelity DNA Polymerase kit) and the purified PCR product was detected and recovered by agarose gel electrophoresis (EasyPure PCR Purification Kit). Subsequently, to determine the concentration of the insert and the linearized vector, an insert with overlapping sequences at both ends, a linearized vector, and a 2 × EasyGeno Assembly Mix were added to the reaction system, which was allowed to react for 15 min at 50°C to complete the recombination of the fragment and the vector. The experimental procedure was performed following the EasyGeno Assembly Cloning Kit manual. The recombinant product was transformed into competent Escherichia coli cells by rapid or conventional methods. Briefly, the tube was placed in a 42°C constant‐temperature water bath for a heat shock of 90 s. Immediately after removing the tube, the tube was placed in an ice bath for 2–3 min, taking care not to vibrate the tube during the period. Then 350 μl pre‐warmed Luria‐Bertani medium (without antibiotics) was added to the centrifuge tube. The bacteria were incubated at 5 g for 37–60 min at 37°C. After mixing the bacteria in the centrifuge tube by light or suction, we added 100–200 μl to the LB solid medium containing the corresponding antibiotic. The cells were gently spread evenly with a glass coating bar. After the surface of the plate was dried, the inverted plate was incubated at 37°C for 12–16 hr. After preliminary identification of the extracted plasmid, 2–4 positive clones of all newly constructed vectors were sent to Shanghai Biotech for sequencing identification. After sequence alignment, the fully qualified plasmid vector was transfected into the cells by Lipofectamine 2000 for subsequent overexpression‐related experimental detection.
2.13. Lactate dehydrogenase release
For in vitro experiments, an LDH cytotoxicity assay kit (Beyotime) was used to measure LDH release from necrotic cells into the extracellular space/supernatant upon the rupture of the plasma membrane (Li et al., 2016; Shang et al., 2017; Wang et al., 2018) after the different treatments. Cell‐free culture supernatants were collected from 96‐well microtiter plates and incubated with the appropriate reagent mixture according to the manufacturer’s instructions at room temperature for 30 min. For the in vivo experiment, the LDH cytotoxicity assay kit was purchased from Nanjing Jiancheng Bioengineering Institute and used according to the manufacturer’s instructions. Briefly, the retinae were homogenized by sonication in .86% NaCl and incubated with the appropriate reagent mixture at 37°C for 30 min. The intensity of the red color that formed in the assay, was measured at a wavelength of 490 or 450 nm, and was proportional to both the LDH activity and percentage of necrotic cells. The percentage of necrotic cells was calculated from the color intensity of treated cells minus control cells/LDH releasing reagent‐treated cells minus control cells, from four independent experiments.
2.14. Flow cytometry analysis
RGC‐5 cells were trypsinized by EDTA‐free trypsin, followed by three washes. Cells were then resuspended in 500 μl of binding buffer and 5 μl of Annexin V. A 10‐ μl aliquot of PI (YEASEN) was added. After incubation for 10 min at room temperature in the dark, the cells were washed and analyzed using a BD LSRFortessa™ Cell Analyzer (Becton, Dickinson and Company). The percentages of cells in each quadrant were analyzed using flowjo software (FlowJo LLC). Statistical analyses of flow cytometry results were conducted by calculating the number of PI‐positive cells (Yang et al., 2017).
2.15. Simulation of protein binding (RSK3‐RIP3) confirmation
The three‐dimensional structure of proteins was built based on available protein crystal diffraction structures and frozen electron microscopic structures by homologous modeling (Swiss Model; Schwede et al., 2003). The protein surface electrostatic potential energy was calculated using APBS (Figures were produced using pymol; Baker et al., 2001).
2.16. Co‐immunoprecipitation
We pre‐incubated 5 µg of primary antibody (rabbit anti‐RIP3 antibody, 1:200, Abcam, ab56164; rabbit anti‐RSK3 antibody, 1:200, Abcam, ab247271; rabbit anti‐RIP1 antibody, 1:1,000, CST, 3493; normal rabbit IgG, 1 µg µl–1, Beyotime, A7016) with 30 µl per sample in the resuspended Protein A+G Agarose for 8–12 hr at room temperature and washed it five times with GLB+ buffer. Cells were homogenized in cold immunoprecipitation (IP) lysis buffer (Thermo Fisher, 87788) containing 1% phenylmethylsulfonyl fluoride and 1% phosphatase inhibitor cocktail. Samples were incubated at 4°C for 30 min and centrifuged for 20 min at 16,000 g. The pellet was discarded. Protein (≥500 μg) from lysate was then combined with Protein A+G Agarose (Beyotime, P2055) coupled with primary antibody and incubated for 24 hr while gently mixing at 4°C. Next, the mixture was pre‐washed with cold GLB+ buffer 3–5 times and proteins were eluted with prepared 1 × loading buffer by boiling for 5 min and centrifuging at 16,000 g for 2 min to obtain the supernatant, which was then subjected to SDS‐PAGE.
2.17. Flash electroretinogram
The RM6240 system (Chengdu Instrument Factory) was used for flash electroretinogram (fERG) recording. After drug treatment, rats were dark‐adapted for 6 hr. Under dim red illumination, the rats were anesthetized using an intraperitoneal injection with a 1:1 mixed solution (5 ml kg–1) of 10% chloral hydrate and 25% urethane. A recording electrode was then inserted into the anterior chamber. The reference and ground electrodes were placed on the subcutaneous layer of the forehead and tail base, respectively. A bandpass filter of 10 Hz was used and the lash luminance was 1.6 cd s−1 m−2. Each eye was exposed to flashes three times at 5‐min intervals. When one eye was recorded, the contralateral eye was covered. All procedures were repeated at least four times. The amplitude of the b‐wave was calculated from the trough of a wave to the peak of the b‐wave.
2.18. Statistical analysis
Figure panels were assembled using photoshop CS6 (Adobe Systems Incorporated). The measurement data are presented as the mean ± standard deviation (SD). One‐way analysis of variance and independent sample t tests were used to analyze the data with GraphPad prism 5 software (GraphPad Software Inc.). Statistical significance was set at p < .05.
3. RESULTS
3.1. OGD injury induces necrosis of RGC‐5
We investigated whether necrosis occurred in RGC‐5 following OGD injury. Our results showed a change in OGD‐induced membrane permeability by PI‐positive staining and LDH release at 0, 2, 4, 6, and 12 hr after 4 hr of OGD injury. The PI and DAPI double‐staining results showed that PI‐positive cells were rarely found in the normal group (Figure 1a,b). Further, the number of PI‐positive cells gradually increased in a time‐dependent manner following OGD injury. Quantitative analysis showed a marked increase in necrotic RGC‐5 levels within 2, 4, and 6 hr of recovery from OGD injury. LDH release assay results (Figure 1c) also showed that the percentage of necrotic cells was significantly higher in the OGD group at 2, 4, 6, and 12 hr recovery time points compared with normal or 0 hr groups (p < .01). Collectively, these results showed that the necrosis of RGC‐5 occurred 2–6 hr after OGD injury.
Figure 1.
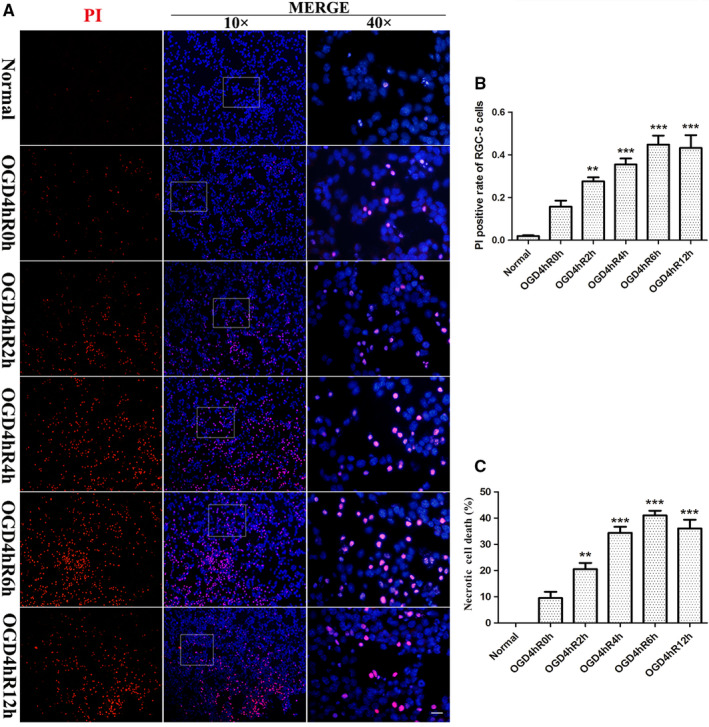
Necrotic RGC‐5 was determined by PI staining and LDH release assay after OGD injury. (a) PI (red) staining of RGC‐5 after OGD injury. Nuclei were counterstained with DAPI (blue). The Scale bars: 40 µm (all images on the left and middle bar); 10 µm (all panels on the right bar). (b) The statistical analysis of PI/DAPI staining of necrotic cells. **p < .01 vs. normal group; ***p < .001 vs. normal group. (c) The percentage of RGC‐5 necrosis was determined by LDH release assay. **p < .01 vs. normal or OGD 4 hr/R 0 hr; ***p < .001 vs. normal or OGD 4 hr/R 0 hr
3.2. OGD upregulates RSK3 expression and phosphorylation of RIP3 in RGC‐5
To analyze whether RSK3 interacted with RIP3 in RGC‐5, we manually modeled hypothetical complexes of the two molecules according to the potential energies and structures of RSK3 and RIP3. We found a possible interaction site between positive and negative charges of the two proteins by simulating the stereoscopic conformation of RSK3 and RIP3, which was calculated using APBS software, followed by energy minimization using NAMD v2.12 (Figure 2a). Based on these results, the RSK3 kinase domains are likely to associate with RIP3 through electrostatic interactions. Next, we performed co‐immunoprecipitation (Co‐IP; Figures 2b,c and S1A) assay, which showed that the RSK3 and RIP1/RIP3 were bound to each other in the OGD groups of RGC‐5. These results suggested that the binding of these molecules could be involved in OGD‐induced necrosis in RGC‐5. Results of the western blot (WB) results showed that the phosphorylation of RSK3, RIP3, and MLKL gradually increased in all injury groups (2, 4, 6, and 12 hr after OGD 4 hr) compared with the normal group (Figure 2d). Quantification and statistical analysis of protein expression (Figure 2f) showed a significant difference between OGD 4 hr/R 4 hr and the normal group (p < .05). The ratio of p‐RIP3/RIP3 (Figure 2e) was higher than that of the normal group (p < .01). These results suggested that the change in RSK3 phosphorylation might be involved in RGC‐5 necrosis after OGD injury. Immunofluorescence (IF) staining (Figure 3) showed that both RSK3 and p‐RIP3 were mainly localized to the cytoplasm and nucleus. Meanwhile, green fluorescence intensities of RSK3 and p‐RIP3 in RGC‐5 were enhanced in the injury groups (2, 4, 6, and 12 hr after OGD 4 hr) compared with the normal group, indicating that the expression levels of both RSK3 and p‐RIP3 were increased. In addition, compared with the normal group, the expression of RSK3 and p‐RIP3 was slightly increased at 2 hr. However, the fluorescence intensity of p‐RIP3 further increased until 4 hr after OGD, indicating that RSK3 expression changed earlier than p‐RIP3 levels. Taken together, these results suggested that RSK3 and RIP3 might be activated after OGD injury.
Figure 2.
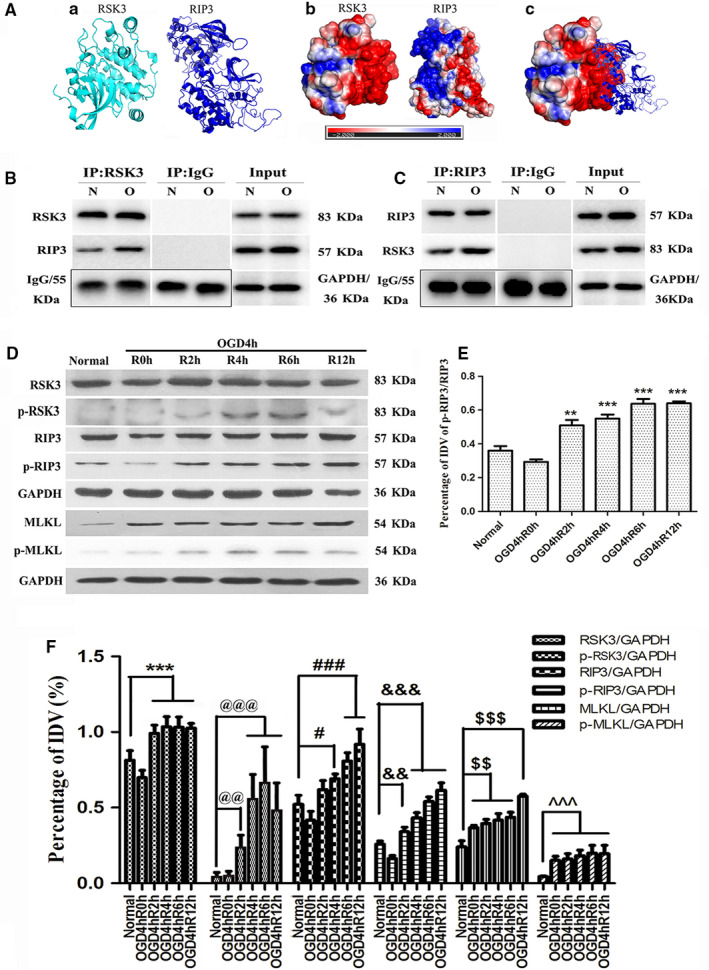
The changes of RIP3/MLKL pathway following OGD, and the interaction of RSK3 with RIP3. (a) A software simulation diagram of the relationship between RSK3 and RIP3. (a) The protein spatial structure of RSK3 and RIP3. (b) The protein surface electrostatic potential energy diagram of RSK3 and RIP3. (c) The initial binding mode of RSK3 and RIP3. (b,c) IP assay of RSK3 and RIP3 in RGC‐5. IgG was used as a negative control and input was used as a positive control. N represents normal RGC‐5, O represents RGC‐5 undergoing OGD 4 hr/R 4 hr. (d) The expression level of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL in RGC‐5 following OGD determined by Western Blot. (E) The statistical analysis of p‐RIP3/RIP3 in RGC‐5 after OGD treatment. **vs. normal, p < .01; ***vs. normal, p < .001. (f) The statistical analysis of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL expression level in RGC‐5 following OGD. ***vs. normal, p < .001; @@vs. normal, p < .01; @@@vs. normal, p < .001; #vs. normal, p < .05; ###vs. normal, p < .001; &&vs. normal, p < .01; &&&vs. normal, p < .001; $$vs. normal, p < .01; $$$vs. normal, p < .001; ^^^vs. normal, p < .001. RSK3 phosphorylation was analyzed by a Phos‐tag assay
Figure 3.
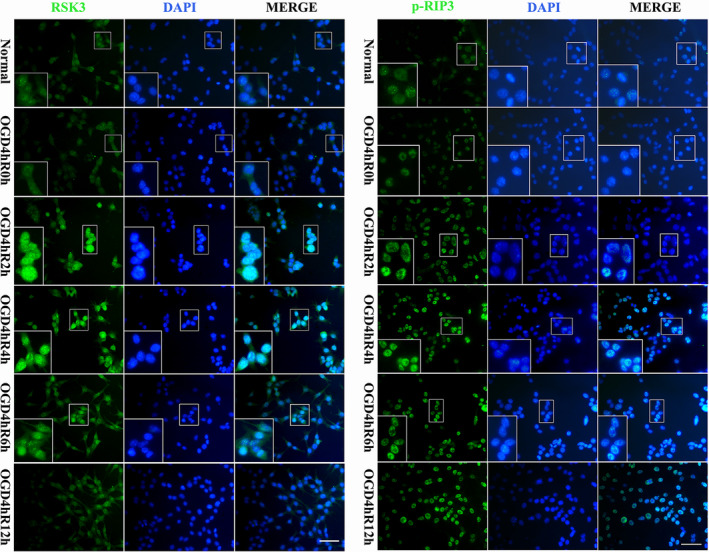
Immunofluorescence staining of RSK3 and p‐RIP3 in RGC‐5 following OGD at different survival time points. Green fluorescence was positive for immunofluorescence staining of RSK3 and p‐RIP3. Blue fluorescence was the DAPI‐labeled nuclei. Scale bars: 30 μm (applies to all images except the high magnification images); 60 μm (applies to all high magnification images)
3.3. LJH685 decreases RIP3 phosphorylation in RGC‐5 following OGD
To investigate the effect of RSK3 on RIP3/MLKL phosphorylation, the RSK3‐specific inhibitor LJH685 was added to the RGC‐5 culture 4 hr before OGD. The WB results showed that the expression of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL increased significantly in the OGD 4 hr/R 4 hr group compared with the normal group (Figure 4a). Notably, the levels of p‐RSK3, p‐RIP3, and p‐MLKL decreased in the groups pretreated with 3 or 10 μM LJH685 before OGD injury (Figure 4a,b). Statistical analysis showed a statistically significant difference between the OGD injury group pretreated with 3 or 10 μM LJH685 and the OGD 4 hr/R 4 hr group (p < .05; Figure 4c). However, no significant differences were found between the RSK3, RIP3, and MLKL immunoreactive bands in the groups pretreated with 3 or 10 μM LJH685 compared with the OGD 4 hr/R 4 hr group (Figure 4A,C). These results indicated that inhibition of RSK3 phosphorylation by LJH685 application might block the phosphorylation of RIP3 and MLKL.
Figure 4.
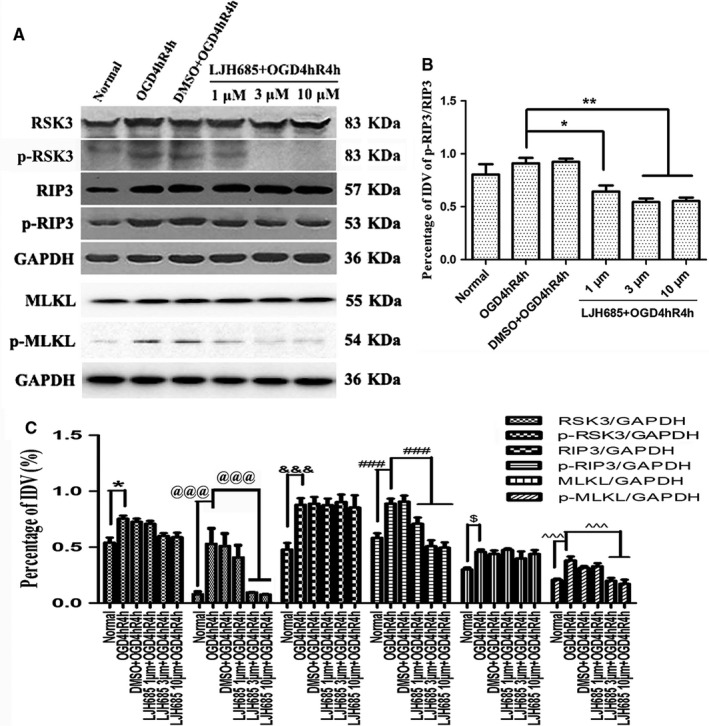
Changes in the phosphorylation levels of RIP3/MLKL in RGC‐5 after LJH685 pretreatment following OGD. (a) WB analysis of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL following LJH685 + OGD 4 hr/R 4 hr. (b) The statistical analysis of the changes in protein level of p‐RIP3/RIP3. *vs. OGD 4 hr/R 4 hr, p < .05; **vs. OGD 4 hr/R 4 hr, p < .01. (c) The statistical analysis of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL expression level in RGC‐5 following OGD and LJH685 + OGD. *vs. normal, p < .05; @@@vs. normal or OGD 4 hr/R 4 hr, p < .001; &&&vs. normal, p < .001; ###vs. normal or OGD 4 hr/R 4 hr, p < .001; $vs. normal, p < .05; ^^^vs. normal or OGD 4 hr/R 4 hr, p < .001. RSK3 phosphorylation was analyzed by a Phos‐tag assay
3.4. Protein levels of p‐RIP3 are decreased upon rsk3 knockdown in RGC‐5 following OGD
Three siRNA sequences were used to knock down rsk3 gene expression. The WB results showed that RSK3 protein levels decreased in the rsk3‐siRNA #1/#3 interference groups in comparison with the normal group (Figure 5a,b), indicating successful knockdown of the RSK3 expression. Thus, the rsk3‐siRNA #1/#3 sequence was selected for use in subsequent experiments. The immunoreactive bands positive for p‐RSK3, p‐RIP3, and p‐MLKL were thinner and smaller for the rsk3‐siRNA #1/#3‐treated groups compared with the OGD 4 hr/R 4 hr groups (Figure 5c,f). Statistical analysis of the ratio of p‐RIP3/RIP3 showed that the phosphorylation of RIP3 was also reduced in the rsk3‐siRNA #1/#3 groups (Figure 5d). The IDVs differed significantly among normal RGC‐5 cells, OGD, OGD+MOCK, negative control siRNA (NC)‐treated OGD, and rsk3‐siRNA‐treated OGD groups (p < .05). RT‐qPCR results showed that the expression of rsk3 mRNA was significantly reduced in the groups treated with rsk3‐siRNA #1/#3 than with the OGD 4 hr/R 4 hr groups (Figure 5e), whereas the expression of rip3 mRNA in the rsk3‐siRNA #1/#3 group did not change significantly compared with that of the OGD group (Figure 5E). These results implied that the inhibition of RSK3 expression in RGC‐5 after OGD injury could decrease phosphorylation of RIP3/MLKL.
Figure 5.
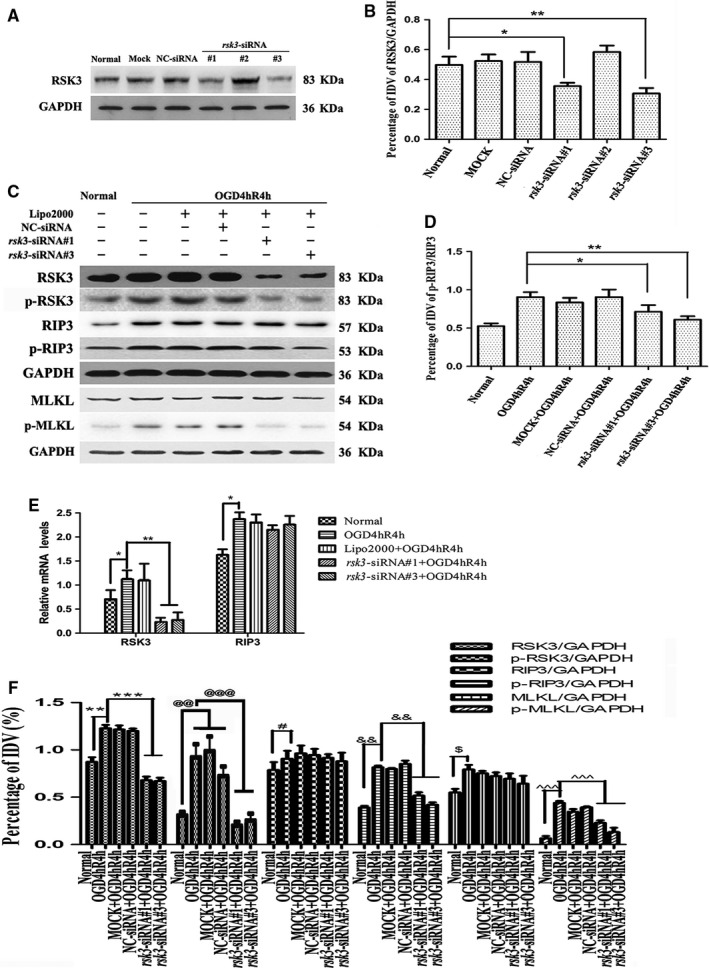
The protein levels of RSK3, p‐RIP3, RIP3, p‐RIP3, MLKL, and p‐MLKL in RGC‐5 transfected with rsk3‐siRNA following OGD. (a) Changes in RSK3 expression level after rsk3‐siRNA intervention. Mock: transfection control group; NC‐siRNA: negative control siRNA; rsk3‐siRNA: rsk3‐knockdown group. (b) Statistical analysis of the changes in protein level of RSK3. *vs. normal, p < .05; **vs. normal, p < .01. (c) WB analysis of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL following rsk3‐siRNA + OGD 4 hr/R 4 hr. (d) The statistical analysis of the changes in protein level of p‐RIP3/RIP3. *vs. OGD 4 hr/R 4 hr, p < .05; **vs. OGD 4 hr/R 4 hr, p < .01. (e) RSK3 and RIP3 mRNA levels in RGC‐5 cells following OGD and rsk3 siRNA+OGD treatment. *p < .05 vs. CTL group, **p < .01 vs. OGD 4 hr/R 4 hr group. (f) The statistical analysis of RSK3, p‐RSK3, RIP3, p‐RIP3, MLKL, and p‐MLKL expression levels in RGC‐5 following rsk3‐siRNA+OGD. **vs. normal, p < .01; ***vs. OGD 4 hr/R 4 hr, p < .001; @@vs. normal, p < .01; @@@vs. OGD 4 hr/R 4 hr, p < .001; #vs. normal or OGD 4 hr/R 4 hr, p < .05; &&vs. normal or OGD 4 hr/R 4 hr, p < .01; $vs. normal, p < .05; ^^^vs. normal or OGD 4 hr/R 4 hr, p < .001. RSK3 phosphorylation was analyzed by a Phos‐tag assay
3.5. Immunofluorescence staining (IF) of RSK3 and p‐RIP3 in RGC‐5 following OGD
IF was used to further evaluate the regulatory role of RSK3 in the phosphorylation of RIP3 during RGC‐5 necroptosis (Figure 6). IF intensities of RSK3 and p‐RIP3 were found to be increased in the cytoplasm and nucleus after OGD 4 hr/R 4 hr (Figure 6). Fluorescence intensities of RSK3 and p‐RIP3 in the DMSO treatment group, NC siRNA treatment group, and OGD 4 hr/R 4 hr group were gradually enhanced. However, phosphorylation of RIP3 was inhibited by LJH685 pretreatment after OGD 4 hr/R 4 hr as demonstrated by a decrease in fluorescence intensity (Figure 6). On the other hand, we observed that the fluorescence intensities of RSK3 and p‐RIP3 decreased when RGC‐5 cells were treated with rsk3‐siRNA (Figure 6). These results indicated that RSK3 might be involved in the phosphorylation of RIP3 in RGC‐5 after OGD injury.
Figure 6.
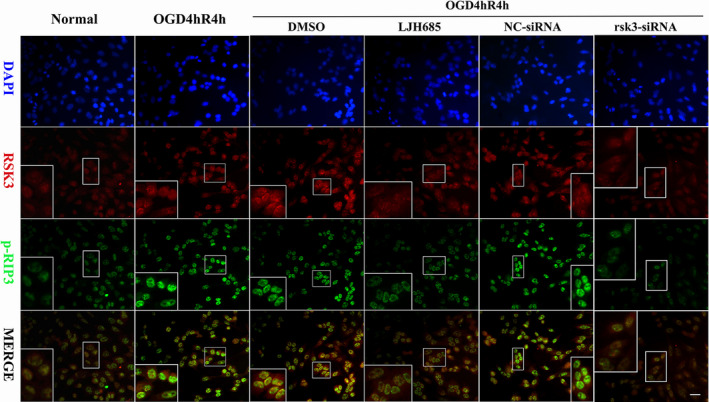
Immunofluorescence staining of RSK3 and p‐RIP3 in RGC‐5 via LJH685 and rsk3‐siRNA pretreatment following OGD. DMSO: vehicle control group; NC‐siRNA: the scrambled base control group. rsk3‐SiRNA: the siRNA treatment group. RSK3 (red)/p‐RIP3 (green)/DAPI (blue) triple‐staining in RGC‐5. Scale bars: 10 µm (all panels except the high magnification images); 20 µm (all high magnification images)
3.6. Interference with RSK3 activity reduces necroptosis in RGC‐5 after OGD injury
In our previous study, we found that RIP3 and MLKL knockdown could reduce the number of necrotic cells during OGD injury. Here, we evaluated the effect of RSK3 via LJH685 or rsk3‐siRNA pretreatment on RGC‐5 necroptosis using flow cytometry and LDH release assays. The flow cytometry results (Figure 7a) showed a large number of living cells in the lower left quadrant, whereas necrotic cells were found in the upper left and/or right quadrants of all groups. The number of necrotic cells in the OGD 4 hr/R 4 hr, DMSO, and NC siRNA pretreatment groups was significantly higher than in the normal group (Figure 7a). However, the number of necrotic cells was significantly decreased in the LJH685 or rsk3‐siRNA groups compared with the OGD group (Figure 7a). Statistical analysis showed that LJH685 or rsk3‐siRNA intervention significantly inhibited OGD‐induced RGC‐5 necrosis (p < .01; Figure 7b). Consistent with the flow cytometry results, the LDH release assay also demonstrated that the ratio of RGC‐5 necrosis increased in the OGD 4 hr/R 4 hr group (Figure 7c,d). In addition, the release of LDH by LJH685‐ or rsk3‐siRNA‐treated groups was significantly reduced (p < .01). Combined with the previous RIP3 phosphorylation data, these results indicated that the RSK3 could reduce necroptosis by regulating the phosphorylation of RIP3 in RGC‐5 following OGD.
Figure 7.
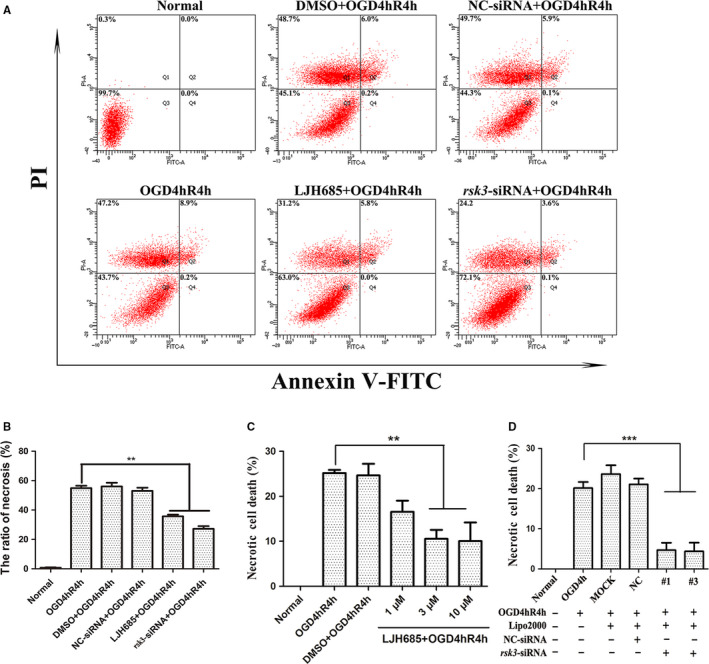
The role of RSK3 in regulating RGC‐5 necroptosis. (a) Annexin V/PI staining of RGC‐5 detected by flow cytometry. (b) The statistical analysis of flow cytometry results. **vs. OGD 4 hr/R 4 hr, p < .01. (c) The percentage of RGC‐5 necrosis was determined by LDH release assay following LJH685+OGD. **vs. OGD 4 hr/R 4 hr, p < .01. (d) The percentage of RGC‐5 necrosis was determined by LDH release following rsk3‐siRNA+OGD. ***vs. OGD 4 hr/R 4 hr, p < .001
3.7. Overexpression of the rip3 gene in RGC‐5 affects neither RSK3 expression nor phosphorylation after OGD injury
We confirmed that the inhibition of RSK3 could effectively modulate the phosphorylation of RIP3 in RGC‐5 after OGD. However, whether RIP3 could regulate RSK3 remained unclear. To further investigate whether RIP3 could regulate RSK3 activity, we constructed the pEGFP‐C1‐rip3 plasmid, transfected it into RGC‐5 cells to overexpress the rip3 gene, and analyzed transfected cells by PCR (Figure 8b). The different groups (1, 2, or 3 group) were reflected in the PCR results by the addition of different template concentrations. The PCR results showed that the rip3 coding sequence was successfully amplified in the sample 3 group.
Figure 8.
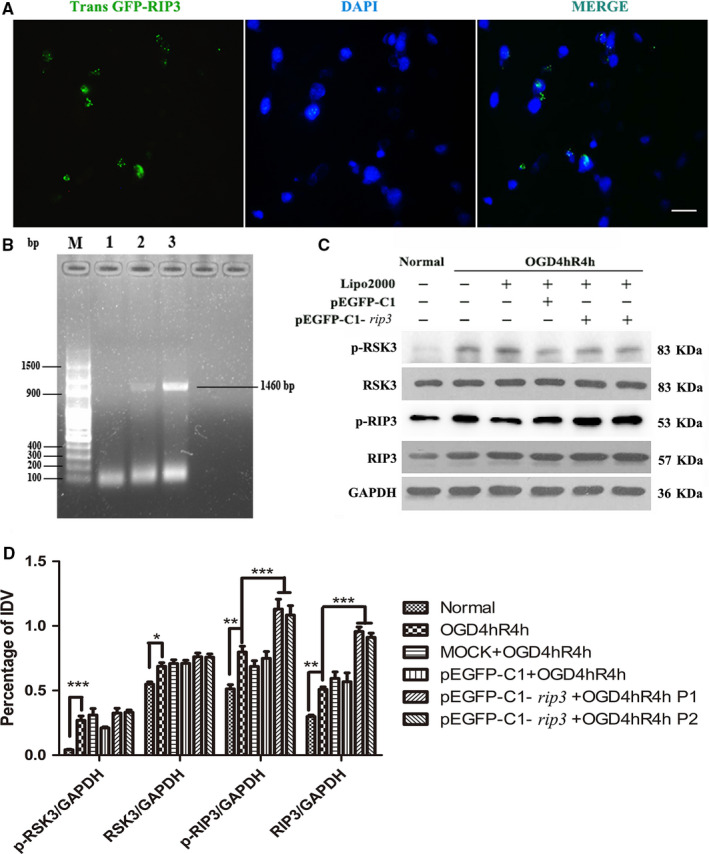
The effects of rip3 gene over expression on RSK3 in RGC‐5. (a) Over‐expression of rip3 gene and transfecting RGC‐5. The green fluorescence was positively expressed by the GFP fluorescence, which was carried by the pEGFP‐C1‐rip3 vector itself. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (b) The target gene of rip3 was amplified by PCR. M represents a 100‐bp DNA Ladder. 1, 2, and 3 respectively represent PCR results of adding different concentrations of templates. (c) The expression levels of RSK3, p‐RSK3, RIP3, and p‐RIP3 were detected by WB pretreatment over‐expressing rip3 following OGD in RGC‐5. Mock: transfection control group; pEGFP‐C1: empty plasmid vector group; pEGFP‐C1‐rip3: rip3‐over‐expressing group. There were two pEGFP‐C1‐rip3 duplicate samples. (d) The statistical analysis of RSK3, p‐RSK3, RIP3, and p‐RIP3 expression. P1 and P2 were duplicate samples. *vs. normal, p < .05; **vs. normal, p < .01; ***vs. normal or OGD 4 hr/R 4 hr, p < .01. RSK3 phosphorylation was analyzed by a Phos‐tag assay
Further, we transfected recombinant pEGFP‐C1‐rip3 plasmid with Lipo2000 into RGC‐5 to achieve an overexpression of rip3 (Figure 8a,c). Transfection efficiency of the recombinant plasmid improved, as demonstrated by the increased expression of GFP green fluorescent protein by the plasmid vector itself. The IF staining showed that the fraction of GFP‐positive RGC‐5 reached 50% (Figure 8a). The WB results demonstrated that RSK3, p‐RSK3, RIP3, and p‐RIP3 protein levels were upregulated in OGD injury compared with the normal group (Figure 8c). Statistical analysis showed that RIP3 expression was significantly upregulated (Figure 8d) in the pEGFP‐C1‐rip3‐transfected group after OGD injury, whereas RSK3 and p‐RSK3 expression levels remained unchanged (Figure 8d). These results showed that the overexpression of the rip3 gene did not affect the expression and phosphorylation of RSK3, thereby indicating that RSK3 could be an upstream molecule that regulates the phosphorylation of RIP3 during OGD‐induced RGC‐5 necroptosis.
3.8. RSK3 regulates cell necroptosis in the retinal GCL and INL after HIOP
While establishing an in vivo model for uncovering the role of RSK3 in necroptosis, we first analyzed visual function and necrosis of retinal neurons in rats after aHIOP. PI dye liquor was injected intravitreally to examine the dispersal of necrotic cells in the rat retina. The LDH release assays were performed on retinal tissue to measure the necrosis of rat retinal neurons. The fERG was used to detect the changes in the visual function of the rats. The results of fluorescence double staining with PI (red) and DAPI (blue) did not show PI‐positive cells in the normal (Figure 9a,b) or sham groups (Figure 9c,d). On the other hand, PI‐positive necrotic cells numbers were increased in the retinal GCL and INL in the aHIOP group (Figure 9e,f). Quantitative analysis of PI‐stained cells in the retinal GCL and INL is shown in Figure 9(m). After pretreatment with LJH685 before aHIOP, the number of necrotic cells was reduced in the retinal GCL and INL (Figure 9g,h). In addition, statistical analysis of the LDH release assay (Figure 9n) showed that the release of LDH in the LJH685 pretreatment group was significantly lower than in the normal and sham groups (p < .05). These results indicated that the inhibition of RSK3 activity could protect neurons in retinal GCL and INL against aHIOP‐induced necrosis. Additionally, the fERG results showed that the amplitude of b‐waves in the aHIOP group was almost zero. However, following pretreatment with the RSK3 inhibitor LJH685, the amplitude of b‐waves recovered (Figure 9i–l,n). Collectively, these results further demonstrated that RSK3 played a key role in the regulation of necroptosis in rat retinal neurons.
Figure 9.
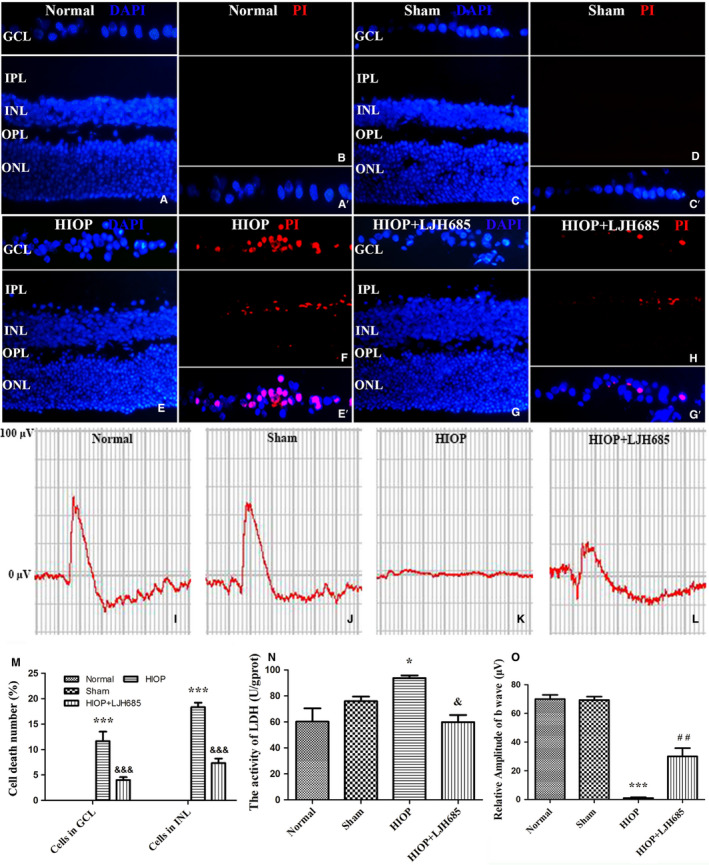
RSK3 regulated necroptosis in retinal GCL and INL following HIOP in vivo. (a–h) PI (red, necrotic neurons)/DAPI (blue) double staining in rat retina. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; Sham, a sham operation group. Scale bar: 20 μm (all panels). LJH685 inhibitor and PI reagent were injected through the vitreous cavity in rat eye. (a′,c′,e′,g′) The framed areas show merged nuclei and PI staining in the GCL as indicated. (i–l) Representative fERG results. (m) Quantitative analysis of PI‐stained cells in GCL and INL of retina. ***vs. normal group, p < .001; &&&vs. HIOP group, p < .001. (n) The percentage of necrotic cells was determined by LDH release assay. *vs. normal group, p < .05; &vs. HIOP group, p < .05. (o) Quantitative analysis of the amplitudes of b‐wave. ***vs. normal group, p < .001; ##vs. HIOP group, p < .01
4. DISCUSSION
In the present study, we investigated the role of RSK3 in RIP3/MLKL‐modulated retinal necroptosis in vivo and in vitro in an acute I/R injury model. First, we examined necroptosis 4–6 hr after OGD injury in RGC‐5 cells, followed by I/R injury in rat retina by in vivo PI staining and LDH release assay. The levels of phosphorylated RIP3 and MLKL were found to increase significantly following OGD injury in RGC‐5. We further demonstrated that RSK3 played a partial role in p‐RIP3/p‐MLKL‐modulated necroptosis via LJH685 or rsk3‐siRNA intervention. After injury, the number of necrotic neurons was reduced by the suppression of both RSK3 activity and expression. We transfected the pEGFP‐C1‐rip3 plasmid into RGC‐5 cells to verify that RSK3 is an upstream regulator of RIP3. Finally, using an animal model, we also confirmed the neuroprotective role of inhibiting RSK3 activity in aHIOP‐induced necroptosis in the rat retina. In summary, these results indicate that RSK3 is implicated in the p‐RIP3/p‐MLKL pathway as an upstream regulator of necroptosis.
Retinal ganglion cell loss and axon degeneration are key features of glaucoma (Xie and Jiang, 2018). As two major risk factors, increased intraocular pressure (IOP) and insufficient blood supply to the optic nerve alter the retinal micro‐environment of RGCs (Buchi, 1992; Feng et al., 2013). Rosenbaum et al. (2010) reported that necroptosis occurred in the retinal GCL after 6 hr in a rat aHIOP model. Many studies have confirmed that the phosphorylation of RIP3 is also required in the regulation of necroptosis (Moriwaki and Chan, 2016; Honda et al., 2017). The RIPs trigger inflammatory responses by participating in the formation of the necrosome, in which RIP1 and RIP3, in particular, relate to necroptosis. The RIP homotypic interaction motif (RHIM) can mediate an interaction between RIP1 and RIP3 to form an amyloid β‐protein structure, which recruits intracellular free RIP3 to promote the formation of an active necrosome (Li et al., 2012; Daskalov et al., 2016). Subsequently, the death signals transmitted through the TNF and Toll‐like receptor signaling pathways are able to recruit and phosphorylate MLKL, thereby triggering cell disintegration and necroptosis or inflammation (Lin et al., 2016; Nogusa et al., 2016; Cabal‐Hierro and O’Dwyer, 2017). Our experiment was performed in an in vitro OGD model of retinal ischemia to simulate the aHIOP model in vivo (Mathew et al., 2019). Our results showed that RSK3 and RIP3/MLKL were increased significantly in RGC‐5 following OGD injury, which was accompanied by an increase in p‐RSK3, p‐RIP3, and p‐MLKL levels. However, the mechanisms of retinal neuronal necrosis remain poorly identified in glaucoma. In recent years, several types of regulated cell necrosis have been found and defined (Roca and Ramakrishnan, 2013; Chen et al., 2016a; 2016b; Kong et al., 2017; Cheng et al., 2018), such as necroptosis, ferroptosis, pyroptosis, parthanatos, and MPT‐mediated regulated necrosis, among others. Therefore, it is important to investigate the necrosis mechanism of RGCs under I/R injury. Our previous experiments focused on RIP3/MLKL‐mediated necroptosis pathways in RGCs under conditions of elevated hydrostatic pressure (EHP; Liao et al., 2017). In the current study, the phosphorylation of RSK3 and RIP3/MLKL was increased following reperfusion injury, with a necrosis rate that peaked 6 hr after reperfusion. In addition, p‐RIP3 was blocked by LJH685 or rsk3‐siRNA after OGD injury. Based on these results, we speculated that the RSK3 may be involved in OGD‐induced necroptosis in RGC‐5. Moreover, results from the software simulation and confirmatory Co‐IP showed that RSK3 and RIP3 interacted with each other. Thus, we conclude that RSK3 may have a regulatory function in the phosphorylation of RIP3 during necroptosis.
Expression of RSK3 in ventricular myocytes of neonatal rats was upregulated and activated by α‐adrenergic stimulation in vitro (Zhang et al., 2013a; 2013b; Martinez et al., 2015). Passariello et al. (2013) and Passariello et al. (2016) also demonstrated that RSK3 knockout could improve cardiac function as well as prevent excessive pressure and catecholamine infusion‐associated cardiac hypertrophy in TM180 mice (Li et al., 2013; Ryba et al., 2017). Moreover, RSK3/4 has been found to mediate resistance to PI3K pathway inhibitors in breast cancer (Serra et al., 2013). However, whether RSK3 is involved in RGC‐5 necroptosis following OGD is still not known. Considering the different roles and changes of RSK3 in cell survival, we focused on whether the activation of RSK3 and RIP3 mediates neuronal necroptosis after OGD injury. Several highly selective and potent RSK inhibitors, such as BI‐D1870, LJH685, and LJI308, are currently known (Sapkota et al., 2007; Aronchik et al., 2014; Davies et al., 2015). Aronchik et al. (2014) found that the inhibition efficiency of LJH685 and LJI308 on RSK3 was significantly higher than that of BI‐D1870 (Roffe et al., 2015). The crystal structure analysis suggests that LJH685 has three aromatic rings in a non‐planar conformation that form a spiral‐shaped arrangement, allowing it to bind to RSK3. As a result, RSK3 was functionally inhibited and the downstream molecules could not activate phosphorylation. Compared with LJI308, LJH685 has a superior suppression effect. In the present study, we used a specific small‐molecule inhibitor of RSK3, LJH685, and rsk3‐siRNA to determine the roles of RSK3 in RIP3/MLKL‐mediated necroptosis induced by OGD injury in RGC‐5 cells or aHIOP injury in the rat retina. Inhibition of RSK3 by the inhibitor or siRNA decreased the phosphorylation of RIP3, which in turn reduced neuronal necroptosis. Although accumulation of p‐RIP3 was inhibited by LJH685 interfering with RSK3 activity, the expression of RIP3 itself did not change significantly. This suggests that LJH685 exerts its effects exclusively by preventing the phosphorylation of RSK3 substrates.
RIP1 and RIP3 are key molecules of necroptosis. In many cases, RIP1 and RIP3 combine with each other to form a cytosolic complex that mediates necroptosis, wherein RIP1 plays the upstream role to phosphorylate RIP3. As such, we explored the potential role of RIP1 alone in necroptosis. The WB results showed no significant differences between the expression of RSK3 and p‐RSK3 in the OGD+Nec‐1 group compared with the OGD group (Figure S1B). This suggested that Nec‐1 had no effect on RSK3. Second, we did not observe any changes in RIP1 expression when RSK3 activity or expression was inhibited in RGC‐5 after OGD injury, compared with the OGD group (Figure S1C,E,G). Collectively, these results indicated that RSK3 did not modulate the expression of RIP1 in the OGD model. Unfortunately, we could not detect a band for p‐RIP1 in the WB results of the OGD model; however, it was detected in the TSZ treatment experiment using the same antibody (Figure S3A). RIP1 is phosphorylated at several sites, including Ser14, Ser15, Ser161, and Ser166, within the kinase domain (Ofengeim and Yuan, 2013; Zhang et al., 2017). However, we were unable to obtain any alternative RIP1 antibodies (no commercial products) and were therefore unable to determine whether phosphorylation of RIP1 was regulated by RSK3 in the OGD model (Figure 10).
Figure 10.
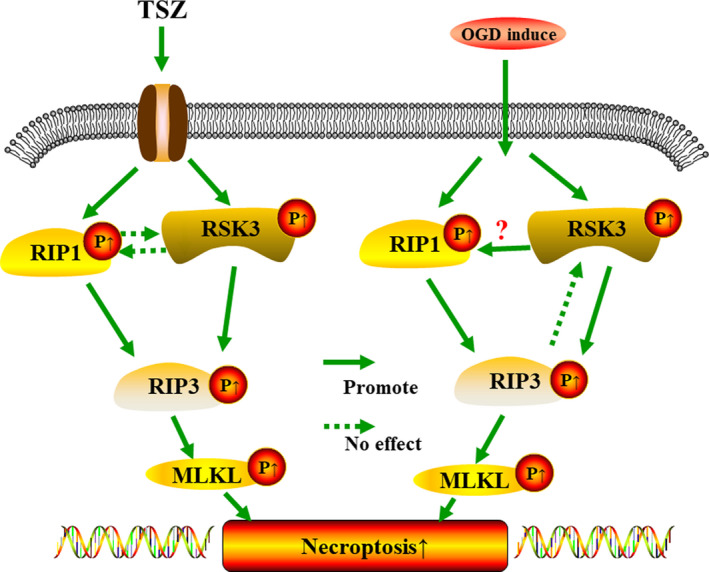
Outline of RSK3 regulatory mechanism via p‐RIP3/p‐MLKL in OGD/TSZ‐induced RGC‐5 necroptosis. The question marks indicate the need for further investigation
The induction of necroptosis by TNF‐α is widely accepted (Newton et al., 2019; Zhang et al., 2019). Compared with the OGD model, we used TSZ to induce necroptosis in RGC‐5 and found similar results as with OGD injury. The annexin V/PI staining and LDH assay results suggested that LJH685 and rsk3‐siRNA could rescue RGC‐5 from necrosis induced by TSZ (Figure S2). Additionally, the WB results suggested that Nec‐1 affected the phosphorylation of RIP1 and RIP3 of the TSZ group (Figure S3A,B), which was consistent with the literature, and indicates that the RIP1/RIP3/MLKL regulatory axis played an important role in TSZ‐induced necroptosis of RGC‐5 (Figure 10). However, the phosphorylation and expression of RSK3 in the Nec‐1+TSZ group (Figure S3A,B) did not change significantly compared with the TSZ group, which was consistent with the OGD model.
In our study, LJH685 + TSZ or rsk3‐siRNA + TSZ inhibited RIP3 phosphorylation without changing the expression of RIP1 or RIP3 significantly compared with the TSZ group (Figure S3A,B). Further analysis by RT‐qPCR showed that the expression of rsk3 was significantly reduced in rsk3‐siRNA#1/#3 + TSZ compared with the TSZ group (Figure S3C), but the expression of rip3 and rip1 in the LJH685 + TSZ or rsk3‐siRNA#1/#3 + TSZ groups did not change significantly compared with the TSZ group (Figure S3C). Collectively, these results suggested that RSK3 could regulate RIP3 phosphorylation without affecting the expression of RIP1/RIP3 of RGC‐5 in the TSZ model, which confirmed our hypothesized molecular mechanism in the OGD model (Figure 10). It should be noted that although the Co‐IP results (Figure S1A) showed that RSK3 may bind to the cytosolic complex containing the kinases RIP1/RIP3, the band for p‐RIP1 in the LJH685 + TSZ or rsk3‐siRNA#1/#3 + TSZ groups did not change significantly compared with the TSZ group. Collectively, these findings suggest that RSK may regulate the necroptosis of RGC‐5 by enhancing the phosphorylation of RIP3, but not the phosphorylation of RIP1, in the TSZ model (Figure 10).
Previous studies have shown that RSK3 is mainly located in the cytoplasm under physiological conditions. However, when cells are subjected to a pathological stimulus or when the activation of intracellular‐related factors is triggered by extracellular signals, RSK can directly activate substrate proteins in the cytoplasm or switch to membrane‐associated proteins. RSK could partially translocate to the nucleus to trigger the phosphorylation of various downstream nuclear proteins, such as c‐Fos, cAMP response element‐binding protein (CREB), nuclear factor‐κB (NF‐κB), and estrogen receptor α (ERα), among others (Zhao et al., 1995; MacKenzie et al., 2000; Yamnik and Holz, 2010; Park et al., 2018). Our IF results also showed that RSK3 proteins were present in both the cytoplasm and nucleus under normal conditions. After OGD injury, the expression of RSK3 increased compared with the normal control, and the distribution of p‐RIP3 protein was visibly increased in the cellular nucleus. The green fluorescence intensity of RSK3 increased significantly after 2 hr of reperfusion, but p‐RIP3 signals changed only slightly. Immunofluorescence intensities of p‐RIP3‐derived signals increased significantly after 4 hr of reperfusion, indicating that RSK3 expression changed earlier than p‐RIP3 levels (Figure 3). To the best of our knowledge, this is the first report to show that RSK3 acts as an upstream regulator of the RIP3/MLKL pathway to promote RIP3 phosphorylation. We transfected the recombinant plasmid pEGFP‐C1‐rip3 into RGC‐5 cells but the overexpression of the rip3 gene did not affect the expression of RSK3 in RGC‐5 after OGD. These results confirmed that RIP3 could not act as an upstream regulator of RSK3 in the process of necroptosis in RGC‐5 cells after OGD injury (Figure 10).
Phosphorylation is an important modification that serves to regulate the biological functions of proteins (Wu et al., 2019). Some studies have shown that the N‐terminal phosphorylation sites at positions Ser199 and Ser227 of RIP3 are autophosphorylated during necroptosis (Sun et al., 2012). When caspase‐8 is inhibited, Toll‐like receptor 4 (TLR4) interacts directly with RIP3 and MLKL via the RIP homotypic interaction motif (RHIM) of its adaptor Toll/interleukin (IL)‐1 receptor domain‐containing adaptor interferon (IFN)‐β (TRIF; Najjar et al., 2016). Subsequently, they phosphorylate amino acids T357 and S358 of MLKL, thereby resulting in the release of intercellular materials after cell necroptosis (Kang et al., 2015). In the present study, we showed that an inhibition or silencing of RSK3 reduced the phosphorylation of RIP3 and MLKL. Further, the phosphorylated RIP3 could induce the phosphorylation of the pseudokinase domain of MLKL, which is critical for cell necroptosis (Gunther et al., 2016). We also found that RSK3 interacts with RIP3 and acts as a kinase‐promoting protein to phosphorylate RIP3, eventually regulating RGC‐5 necroptosis after OGD injury. Other research has shown that ERK activates RSK by binding to inactive RSK at the carboxyl‐terminal kinase domain (CTKD; Doehn et al., 2009; Martinez et al., 2015; Kidger and Cook, 2018). Phosphoinositide‐dependent protein kinase‐1 (PDK1) was shown to be recruited to RSK following CTKD autophosphorylation of the RSK hydrophobic motif (Frodin et al., 2000; Zaru et al., 2015). Using antibodies against the N‐terminus of RSK3, Zhao et al. (1995) showed that RSK3 was translocated to the nucleus of HeLa cells, where it subsequently phosphorylated nuclear proteins, such as c‐Fos and histones (Pereira et al., 2014). However, the specific domains of RIP3 to which RSK3 binds, and which are regulated by RSK3, remain unknown and will require further study.
Other studies have suggested that the necroptosis of RGCs is an early event responsible for the loss of vision (Zhou et al., 2018). The compound LJH685 may serve as a useful tool in disrupting the potential signaling pathways by inhibiting RSK3 activity (Poomakkoth et al., 2016). In our study, we demonstrated that RSK3 plays a neuroprotective role in an in vivo model of aHIOP‐induced necroptosis in the rat retina. Inhibition of RSK3 activity can be used to rescue some of the retinal neurons from necroptosis, thereby maintaining some visual function in rats. Furthermore, these results demonstrated that RSK3 may play a role in regulating retinal neuronal necroptosis in vivo. As such, the findings presented in this study provide a novel theoretical basis for targeting the necroptosis pathway in RGCs.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
KX designed the experiment. MW and HW performed the experiment. LSL and YXH analyzed the data. MW and HW drafted the manuscript. KX, LS, and JFH participated in modification of the paper. SCW, LMG, and FXL revised the manuscript for the English. DJ, XBX, and BJ edited the manuscript. All authors participated in critical revision of the manuscript, and read and approved the final manuscript.
Supporting information
Figure S1
Figure S2
Figure S3
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation of China (Nos 81571939, 81772134, 81971891, 81671225, and 81860781), the Key Research and Development Program of Hunan Province (Nos 2018SK2090, 2018SK2091, and 2018SK2094), Wu Jie‐Ping Medical Foundation of the Minister of Health of China (No. 320.6750.14118), Teacher Research Foundation of Central South University (No. 2014JSJJ026). We are grateful to Dr. Hongkang Zhou (Department of Anatomy and Neurobiology, School of Basic Medical Science, Central South University) for editing the article.
Wang M, Wan H, Wang S, et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. J. Anat. 2020;237:29–47. 10.1111/joa.13185
Mi Wang and Hao Wan contributed equally to this study.
Contributor Information
Dan Chen, Email: chendan0101@csu.edu.cn.
Kun Xiong, Email: xiongkun2001@163.com.
REFERENCES
- Aronchik, I. , Appleton, B.A. , Basham, S.E. , Crawford, K. , Del Rosario, M. , Doyle, L.V. , et al. (2014) Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK‐driven cancers. Molecular Cancer Research, 12, 803–812. [DOI] [PubMed] [Google Scholar]
- Baker, N.A. , Sept, D. , Joseph, S. , Holst, M.J. and McCammon, J.A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozec, D. , Iuga, A.C. , Roda, G. , Dahan, S. and Yeretssian, G. (2016) Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget, 7, 46384–46400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchi, E.R. (1992) Cell death in the rat retina after a pressure‐induced ischaemia‐reperfusion insult: an electron microscopic study. I. Ganglion cell layer and inner nuclear layer. Experimental Eye Research, 55, 605–613. [DOI] [PubMed] [Google Scholar]
- Cabal‐Hierro, L. and O’Dwyer, P.J. (2017) TNF signaling through RIP1 kinase enhances SN38‐induced death in colon adenocarcinoma. Molecular Cancer Research, 15, 395–404. [DOI] [PubMed] [Google Scholar]
- Cai, Z. , Jitkaew, S. , Zhao, J. , Chiang, H.‐C. , Choksi, S. , Liu, J. , et al. (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nature Cell Biology, 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere, A. , Ray, H. , Blenis, J. and Roux, P.P. (2008) The RSK factors of activating the Ras/MAPK signaling cascade. Frontiers in Bioscience, 13, 4258–4275. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Yan, J. , Deng, H.X. , Long, L.‐L. , Hu, Y.‐J. , Wang, M. , et al. (2016a) Inhibition of calpain on oxygen glucose deprivation‐induced RGC‐5 necroptosis. Journal of Huazhong University of Science and Technology [Medical Sciences], 36, 639–645. [DOI] [PubMed] [Google Scholar]
- Chen, X. , He, W.T. , Hu, L. , Li, J. , Fang, Y. , Wang, X. , et al. (2016b) Pyroptosis is driven by non‐selective gasdermin‐D pore and its morphology is different from MLKL channel‐mediated necroptosis. Cell Research, 26, 1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, S.Y. , Wang, S.C. , Lei, M. , Wang, Z. and Xiong, K. (2018) Regulatory role of calpain in neuronal death. Neural Regeneration Research, 13, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, S.A. , Qin, Z. , Stewart, A. , and Chen, H.‐H. (2018) Dabrafenib, an inhibitor of RIP3 kinase‐dependent necroptosis, reduces ischemic brain injury. Neural Regeneration Research, 13, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalov, A. , Habenstein, B. , Sabate, R. , Berbon, M. , Martinez, D. , Chaignepain, S. , et al. (2016) Identification of a novel cell death‐inducing domain reveals that fungal amyloid‐controlled programmed cell death is related to necroptosis. Proceedings of the National Academy of Sciences of the United States of America, 113, 2720–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, A.H. , Reipas, K. , Hu, K. , Berns, R. , Firmino, N. , Stratford, A.L. , et al. (2015) Inhibition of RSK with the novel small‐molecule inhibitor LJI308 overcomes chemoresistance by eliminating cancer stem cells. Oncotarget, 6, 20570–20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev, A. , Huang, Z. , Boyce, M. , Li, Y. , Jagtap, P. , Mizushima, N. , et al. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature Chemical Biology, 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Ding, W. , Shang, L. , Huang, J.F. , Li, N.A. , Chen, D. , Xue, L.‐X. , et al. (2015) Receptor interacting protein 3‐induced RGC‐5 cell necroptosis following oxygen glucose deprivation. BMC Neuroscience, 16, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehn, U. , Hauge, C. , Frank, S.R. , Jensen, C.J. , Duda, K. , Nielsen, J.V. , et al. (2009) RSK is a principal effector of the RAS‐ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Molecular Cell, 35, 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson, E. and Maller, J.L. (1985) A protein kinase from Xenopus eggs specific for ribosomal protein S6. Proceedings of the National Academy of Sciences of the United States of America, 82, 742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, L. , Zhao, Y. , Yoshida, M. , Chen, H. , Yang, J.F. , Kim, T.S. , et al. (2013) Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location. Investigative Ophthalmology & Visual Science, 54, 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin, M. , Jensen, C.J. , Merienne, K. and Gammeltoft, S. (2000) A phosphoserine‐regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO Journal, 19, 2924–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. , Andreeva, K. and Cooper, N.G. (2014) Ischemia‐reperfusion injury of the retina is linked to necroptosis via the ERK1/2‐RIP3 pathway. Molecular Vision, 20, 1374–1387. [PMC free article] [PubMed] [Google Scholar]
- Gunther, C. , He, G.W. , Kremer, A.E. , Murphy, J.M. , Petrie, E.J. , Amann, K. , et al. (2016) The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. Journal of Clinical Investigation, 126, 4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L.M. , Wang, Z. , Li, S.P. , Wang, M. , Yan, W.T. , Liu, F.X. , et al. (2020) RIP3/MLKL‐mediated neuronal necroptosis induced by methamphetamine at 39 degrees C. Neural Regeneration Research, 15, 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, S. , Wang, L. , Miao, L. , Wang, T. , Du, F. , Zhao, L. , et al. (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell, 137, 1100–1111. [DOI] [PubMed] [Google Scholar]
- Hitomi, J. , Christofferson, D.E. , Ng, A. , Yao, J. , Degterev, A. , Xavier, R.J. , et al. (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell, 135, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler, N. , Zaru, R. , Micheau, O. , Thome, M. , Attinger, A. , Valitutti, S. , et al. (2000) Fas triggers an alternative, caspase‐8‐independent cell death pathway using the kinase RIP as effector molecule. Nature Immunology, 1, 489–495. [DOI] [PubMed] [Google Scholar]
- Honda, T. , Yamamoto, O. , Sawada, Y. , Egawa, G. , Kitoh, A. , Otsuka, A. , et al. (2017) Receptor‐interacting protein kinase 3 controls keratinocyte activation in a necroptosis‐independent manner and promotes psoriatic dermatitis in mice. The Journal of Allergy and Clinical Immunology, 140, 619–622.e6. [DOI] [PubMed] [Google Scholar]
- Huang, J.F. , Shang, L. , Zhang, M.Q. , Wang, H. , Chen, D. , Tong, J.‐B. , et al. (2013) Differential neuronal expression of receptor interacting protein 3 in rat retina: involvement in ischemic stress response. BMC Neuroscience, 14, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen, A.V. and Silke, J. (2016) The importance of being chaperoned: HSP90 and necroptosis. Cell Chemical Biology, 23, 205–207. [DOI] [PubMed] [Google Scholar]
- Jagilinki, B.P. , Choudhary, R.K. , Thapa, P.S. , Gadewal, N. , Hosur, M.V. , Kumar, S. , et al. (2016) Functional basis and biophysical approaches to characterize the C‐terminal domain of human‐ribosomal S6 kinases‐3. Cell Biochemistry and Biophysics, 74, 317–325. [DOI] [PubMed] [Google Scholar]
- Jiang, S.H. , Shang, L. , Xue, L.X. , Ding, W. , Chen, S. , Ma, R.‐F. , et al. (2014) The effect and underlying mechanism of Timosaponin B‐II on RGC‐5 necroptosis induced by hydrogen peroxide. BMC Complementary and Alternative Medicine, 14, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S.W. , Erikson, E. , Blenis, J. , Maller, J.l. and Erikson, R.l. (1988) A Xenopus ribosomal protein S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Proceedings of the National Academy of Sciences of the United States of America, 85, 3377–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S. , Fernandes‐Alnemri, T. , Rogers, C. , Mayes, L. , Wang, Y. , Dillon, C. , et al. (2015) Caspase‐8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nature Communications, 6, 7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidger, A.M. and Cook, S.J. (2018) De‐RSKing ERK—regulation of ERK1/2‐RSK dissociation by phosphorylation within a disordered motif. FEBS Journal, 285, 42–45. [DOI] [PubMed] [Google Scholar]
- Kim, S.J. and Li, J. (2013) Caspase blockade induces RIP3‐mediated programmed necrosis in Toll‐like receptor‐activated microglia. Cell Death & Disease, 4, e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, D. , Zhu, J. , Liu, Q. , Jiang, Y. , Xu, L. , Luo, N. , et al. (2017) Mesenchymal stem cells protect neurons against hypoxic‐ischemic injury via inhibiting parthanatos, necroptosis, and apoptosis, but not autophagy. Cellular and Molecular Neurobiology, 37, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, N.H. , Lee, M.R. , Kong, J. , Park, S.K. , Hwang, B.J. , Kim, B.G. , et al. (2018) Transfer‐RNA‐mediated enhancement of ribosomal proteins S6 kinases signaling for cell proliferation. RNA Biology, 15, 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Kritzer, M.D. , Michel, J.J. , Le, A. , Thakur, H. , Gayanilo, M. , et al. (2013) Anchored p90 ribosomal S6 kinase 3 is required for cardiac myocyte hypertrophy. Circulation Research, 112, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , McQuade, T. , Siemer, A.B. , Napetschnig, J. , Moriwaki, K. , Hsiao, Y.‐S. , et al. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell, 150, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Meng, L. , Xu, T. , Su, Y. , Liu, X., Zhang, Z. , et al. (2017) RIPK1‐RIPK3‐MLKL‐dependent necrosis promotes the aging of mouse male reproductive system. Elife, 6, e27692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Shang, L. , Wang, S.C. , Liao, L.‐S. , Chen, D. , Huang, J.‐F. , et al. (2016) The toxic effect of ALLN on primary rat retinal neurons. Neurotoxicity Research, 30, 392–406. [DOI] [PubMed] [Google Scholar]
- Li, D. , Xu, T. , Cao, Y. , Wang, H. , Li, L. , Chen, S. , et al. (2015) A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proceedings of the National Academy of Sciences of the United States of America, 112, 5017–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, L. , Shang, L. , Li, N. , Wang, S. , Wang, M.I. , Huang, Y. , et al. (2017) Mixed lineage kinase domain‐like protein induces RGC‐5 necroptosis following elevated hydrostatic pressure. Acta Biochimica et Biophysica Sinica (Shanghai), 49, 879–889. [DOI] [PubMed] [Google Scholar]
- Lin, X. , Chen, Q. , Huang, C. and Xu, X. (2016) CYLD promotes TNF‐alpha‐induced cell necrosis mediated by RIP‐1 in human lung cancer cells. Mediators of Inflammation, 2016, 1542786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S. , Liao, L. , Zhang, B. , Yan, W. , Chen, L. , Yan, H.e. , et al. (2019) Antioxidant cascades confer neuroprotection in ethanol, morphine, and methamphetamine preconditioning. Neurochemistry International, 131, 104540. [DOI] [PubMed] [Google Scholar]
- MacKenzie, S.J. , Baillie, G.S. , McPhee, I. , Bolger, G.B. and Houslay, M.D. (2000) ERK2 mitogen‐activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP‐specific phosphodiesterases. The involvement of COOH‐terminal docking sites and NH2‐terminal UCR regions. Journal of Biological Chemistry, 275, 16609–16617. [DOI] [PubMed] [Google Scholar]
- Martinez, E.C. , Passariello, C.L. , Li, J. , Matheson, C.J. , Dodge‐Kafka, K. , Reigan, P. , et al. (2015) RSK3: a regulator of pathological cardiac remodeling. IUBMB Life, 67, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, B. , Ravindran, S. , Liu, X. , Torres, L. , Chennakesavalu, M. , Huang, C.‐C. , et al. (2019) Mesenchymal stem cell‐derived extracellular vesicles and retinal ischemia‐reperfusion. Biomaterials, 197, 146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, H. , Shimizu, Y. , Ohsawa, Y. , Kawahara, A. , Uchiyama, Y. and Nagata, S. (2000) Necrotic death pathway in Fas receptor signaling. Journal of Cell Biology, 151, 1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan, E. and Tschopp, J. (2005) The RIP kinases: crucial integrators of cellular stress. Trends in Biochemical Sciences, 30, 151–159. [DOI] [PubMed] [Google Scholar]
- Miyake, S. , Wakita, H. , Bernstock, J.D. , Castri, P. , Ruetzler, C. , Miyake, J. , et al. (2015) Hypophosphorylation of ribosomal protein S6 is a molecular mechanism underlying ischemic tolerance induced by either hibernation or preconditioning. Journal of Neurochemistry, 135, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki, K. and Chan, F.K. (2016) Necroptosis‐independent signaling by the RIP kinases in inflammation. Cellular and Molecular Life Sciences, 73, 2325–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafov, A. , Mookhtiar, A.K. , Luu, H.S. , Luu, H.S. , Ordureau, A. , Pan, H. , et al. (2019) TAM kinases promote necroptosis by regulating oligomerization of MLKL. Molecular Cell, 75, 457–468.e4. [DOI] [PubMed] [Google Scholar]
- Najjar, M. , Saleh, D. , Zelic, M. , Nogusa, S. , Shah, S. , Tai, A. , et al. (2016) RIPK1 and RIPK3 kinases promote cell‐death‐independent inflammation by toll‐like receptor 4. Immunity, 45, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negroni, A. , Colantoni, E. , Pierdomenico, M. , Palone, F. , Costanzo, M. , Oliva, S. , et al. (2017) RIP3 and pMLKL promote necroptosis‐induced inflammation and alter membrane permeability in intestinal epithelial cells. Digestive and Liver Disease, 49, 1201–1210. [DOI] [PubMed] [Google Scholar]
- Newton, K. , Wickliffe, K.E. , Dugger, D.L. , Maltzman, A. , Roose‐Girma, M. , Dohse, M. , et al. (2019) Cleavage of RIPK1 by caspase‐8 is crucial for limiting apoptosis and necroptosis. Nature, 574, 428–431. [DOI] [PubMed] [Google Scholar]
- Nogusa, S. , Thapa, R.J. , Dillon, C.P. , Liedmann, S. , Oguin, T. , Ingram, J. , et al. (2016) RIPK3 activates parallel pathways of MLKL‐driven necroptosis and FADD‐mediated apoptosis to protect against influenza A virus. Cell Host & Microbe, 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim, D. and Yuan, J. (2013) Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nature Reviews Molecular Cell Biology, 14, 727–736. [DOI] [PubMed] [Google Scholar]
- Orozco, S.L. , Daniels, B.P. , Yatim, N. , Messmer, M.N. , Quarato, G. , Chen‐Harris, H. , et al. (2019) RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Reports, 28, 2275–2287.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J.H. , Moon, S.H. , Kang, D.H. , Um, H.J. , Kang, S.‐S. , Kim, J.Y. , et al. (2018) Diquafosol sodium inhibits apoptosis and inflammation of corneal epithelial cells via activation of Erk1/2 and RSK. In vitro and in vivo dry eye model. Investigative Ophthalmology & Visual Science, 59, 5108–5115. [DOI] [PubMed] [Google Scholar]
- Passariello, C.L. , Gayanilo, M. , Kritzer, M.D. , Thakur, H. , Cozacov, Z. , Rusconi, F. , et al. (2013) p90 ribosomal S6 kinase 3 contributes to cardiac insufficiency in alpha‐tropomyosin Glu180Gly transgenic mice. American Journal of Physiology. Heart and Circulatory Physiology, 305, H1010–H1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passariello, C.L. , Martinez, E.C. , Thakur, H. , Cesareo, M. , Li, J. and Kapiloff, M.S. (2016) RSK3 is required for concentric myocyte hypertrophy in an activated Raf1 model for Noonan syndrome. Journal of Molecular and Cellular Cardiology, 93, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, A. , Zhang, B. , Malcolm, P. , Sugiharto‐Winarno, A. and Sundram, S. (2014) Quetiapine and aripiprazole signal differently to ERK, p90RSK and c‐Fos in mouse frontal cortex and striatum: role of the EGF receptor. BMC Neuroscience, 15, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirbhoy, P.S. , Farris, S. and Steward, O. (2017) Synaptically driven phosphorylation of ribosomal protein S6 is differentially regulated at active synapses vs. dendrites and cell bodies by MAPK and PI3K/mTOR signaling pathways. Learning & Memory, 24, 341–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poomakkoth, N. , Issa, A. , Abdulrahman, N. , Abdelaziz, S.G. and Mraiche, F. (2016) p90 ribosomal S6 kinase: a potential therapeutic target in lung cancer. Journal of Translational Medicine, 14, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampazzo, R. , Graziani, A.C. , Leite, K.K. , Surdi, J.A. , Biondo, C.A. , Costa, M.L.N. , et al. (2019) Proof of concept for a portable platform for molecular diagnosis of tropical diseases: on‐chip ready‐to‐use real‐time quantitative PCR for detection of Trypanosoma cruzi or Plasmodium spp. The Journal of Molecular Diagnostics, 21, 839–851. [DOI] [PubMed] [Google Scholar]
- Rehorova, M. , Vargova, I. , Forostyak, S. , Vacková, I. , Turnovcová, K. , Kupcová Skalníková, H. , et al. (2019) A combination of intrathecal and intramuscular application of human mesenchymal stem cells partly reduces the activation of necroptosis in the spinal cord of SOD1(G93A) rats. Stem Cells Translational Medicine, 8, 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca, F.J. and Ramakrishnan, L. (2013) TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell, 153, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffe, M. , Lupinacci, F.C. , Soares, L.C. , Hajj, G.N. and Martins, V.R. (2015) Two widely used RSK inhibitors, BI‐D1870 and SL0101, alter mTORC1 signaling in a RSK‐independent manner. Cellular Signalling, 27, 1630–1642. [DOI] [PubMed] [Google Scholar]
- Romeo, Y. , Zhang, X. and Roux, P.P. (2012) Regulation and function of the RSK family of protein kinases. The Biochemical Journal, 441, 553–569. [DOI] [PubMed] [Google Scholar]
- Rosenbaum, D.M. , Degterev, A. , David, J. , Rosenbaum, P.S. , Roth, S. , Grotta, J.C. , et al. (2010) Necroptosis, a novel form of caspase‐independent cell death, contributes to neuronal damage in a retinal ischemia‐reperfusion injury model. Journal of Neuroscience Research, 88, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba, D.M. , Li, J. , Cowan, C.L. , Russell, B. , Wolska, B.M. and Solaro, R.J. (2017) Long‐term biased beta‐arrestin signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation, 135, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota, G.P. , Cummings, L. , Newell, F.S. , Armstrong, C. , Bain, J. , Frodin, M. , et al. (2007) BI‐D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. The Biochemical Journal, 401, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede, T. , Kopp, J. , Guex, N. and Peitsch, M.C. (2003) SWISS‐MODEL: an automated protein homology‐modeling server. Nucleic Acids Research, 31, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra, V. , Eichhorn, P.J. , Garcia‐Garcia, C. , Ibrahim, Y.H. , Prudkin, L. , Sánchez, G. , et al. (2013) RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. Journal of Clinical Investigation, 123, 2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, L. , Ding, W. , Li, N. , Liao, L. , Chen, D. , Huang, J. , et al. (2017) The effects and regulatory mechanism of RIP3 on RGC‐5 necroptosis following elevated hydrostatic pressure. Acta Biochimica et Biophysica Sinica (Shanghai), 49, 128–137. [DOI] [PubMed] [Google Scholar]
- Shrestha, D. , Choi, D. and Song, K. (2018) Actin dysfunction induces cell cycle delay at G2/M with sustained ERK and RSK activation in IMR‐90 normal human fibroblasts. Molecules and Cells, 41, 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L. , Wang, H. , Wang, Z. , He, S. , Chen, S. , Liao, D. , et al. (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell, 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Tong, J.B. , Chen, D. , Zeng, L.P. , Mo, X.‐y. , Wang, H. , Huang, J. , et al. (2010) Differential changes of local blood supply in rat retinae are involved in the selective loss of retinal ganglion cells following the acute high intraocular pressure. Current Eye Research, 35, 425–434. [DOI] [PubMed] [Google Scholar]
- Vandenabeele, P. , Galluzzi, L. , Vanden, B.T. and Kroemer, G. (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Reviews Molecular Cell Biology, 11, 700–714. [DOI] [PubMed] [Google Scholar]
- Vazquez‐Higuera, J.L. , Mateo, I. , Sanchez‐Juan, P. , Rodríguez‐Rodríguez, E. , Pozueta, A. , Calero, M. , et al. (2011) Genetic variation in the tau protein phosphatase‐2A pathway is not associated with Alzheimer’s disease risk. BMC Research Notes, 4, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, M. , Fernandes, J. , Carreto, L. , Anuncibay‐Soto, B. , Santos, M. , Han, J. , et al. (2014) Ischemic insults induce necroptotic cell death in hippocampal neurons through the up‐regulation of endogenous RIP3. Neurobiology of Diseases, 68, 26–36. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Sun, L. , Su, L. , Rizo, J. , Liu, L. , Wang, L.‐F. , et al. (2014) Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Molecular Cell, 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Liao, L. , Wang, M. , Zhou, H. , Huang, Y. , Wang, Z. , et al. (2018) Pin1 promotes regulated necrosis induced by glutamate in rat retinal neurons via CAST/Calpain2 pathway. Frontiers in Cellular Neuroscience, 11, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Liu, J. , Zhou, J.S. , Huang, H.‐Q. , Li, Z.‐Y. , Xu, X.‐C. , et al. (2018a) MTOR suppresses cigarette smoke‐induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. The Journal of Immunology, 200, 2571–2580. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Guo, L.M. , Wang, S.C. , Chen, D. , Yan, J. , Liu, F.‐X. , et al. (2018b) Progress in studies of necroptosis and its relationship to disease processes. Pathology, Research and Practice, 214, 1749–1757. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Guo, L.M. , Wang, Y. , Zhou, H.K. , Wang, S.C. , Chen, D. , et al. (2018c) Inhibition of HSP90alpha protects cultured neurons from oxygen‐glucose deprivation induced necroptosis by decreasing RIP3 expression. Journal of Cellular Physiology, 233, 4864–4884. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Guo, L.M. , Zhou, H.K. , Qu, H.K. , Wang, S.C. , Liu, F.X. , et al. (2018d) Using drugs to target necroptosis: dual roles in disease therapy. Histology and Histopathology, 33, 773–789. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Huang, Y. , Yan, Y. , Zhou, H. , Wang, M.i. , Liao, L. , et al. (2019a) Calpain2 but not calpain1 mediated by calpastatin following glutamate‐induced regulated necrosis in rat retinal neurons. Annals of Anatomy, 221, 57–67. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Liao, L. , Huang, Y. , Wang, M.i. , Zhou, H. , Chen, D. , et al. (2019b) Pin1 is regulated by CaMKII activation in glutamate‐induced retinal neuronal regulated necrosis. Frontiers in Cellular Neuroscience, 13, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert, M. , Binks, A. , Dowson, S. , Leung, E.Y.L. , Athineos, D. , Yu, X. , et al. (2017) RIPK3 promotes adenovirus type 5 activity. Cell Death & Disease, 8, 3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J.R. , Wang, J. , Zhou, S.K. , Yang, L. , Yin, J.‐l. , Cao, J.‐P. , et al. (2015) Necrostatin‐1 protection of dopaminergic neurons. Neural Regen Res, 10, 1120–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Xing, X. , Dowlut, D. , Zeng, Y. , Liu, J. and Liu, X. (2019) Integrating phosphoproteomics into kinase‐targeted cancer therapies in precision medicine. Journal of Proteomics, 191, 68–79. [DOI] [PubMed] [Google Scholar]
- Xie, L.L. and Jiang, B. (2018) The relationship between necroptosis and blinding eye diseases. Zhonghua Yan Ke Za Zhi, 54, 234–240. [DOI] [PubMed] [Google Scholar]
- Xiong, K. , Liao, H. , Long, L. , Ding, Y. , Huang, J. and Yan, J. (2016) Necroptosis contributes to methamphetamine‐induced cytotoxicity in rat cortical neurons. Toxicology in Vitro, 35, 163–168. [DOI] [PubMed] [Google Scholar]
- Yamnik, R.L. and Holz, M.K. (2010) mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Letters, 584, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Jiang, B. , Wang, Y. , Ni, H. , Zhang, J. , Xia, J. , et al. (2017) 2‐HG inhibits necroptosis by stimulating DNMT1‐dependent hypermethylation of the RIP3 promoter. Cell Reports, 19, 1846–1857. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Li, C. , Wang, Y. , Yang, J. , Yin, Y. , Liu, M. , et al. (2018) Melatonin attenuates chronic pain related myocardial ischemic susceptibility through inhibiting RIP3‐MLKL/CaMKII dependent necroptosis. Journal of Molecular and Cellular Cardiology, 125, 185–194. [DOI] [PubMed] [Google Scholar]
- Zaru, R. , Matthews, S.P. , Edgar, A.J. , Prescott, A.R. , Gomez‐Nicola, D. , Hanauer, A. , et al. (2015) The PDK1‐Rsk signaling pathway controls Langerhans cell proliferation and patterning. The Journal of Immunology, 195, 4264–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.W. , Shao, J. , Lin, J. , Zhang, N. , Lu, B.‐J. , Lin, S.‐C. , et al. (2009) RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science, 325, 332–336. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Li, J. , Geng, R. , Ge, W. , Zhou, Y. , Zhang, C. , et al. (2013a) The inhibition of ERK activation mediates the protection of necrostatin‐1 on glutamate toxicity in HT‐22 cells. Neurotoxicity Research, 24, 64–70. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Liu, R. , Townsend, P.A. , and Proud, C.G. (2013b) p90(RSK)s mediate the activation of ribosomal RNA synthesis by the hypertrophic agonist phenylephrine in adult cardiomyocytes. Journal of Molecular and Cellular Cardiology, 59, 139–147. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Su, S.S. , Zhao, S. , Yang, Z. , Zhong, C.‐Q. , Chen, X. , et al. (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nature Communications, 8, 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, H. , Xu, C. , Li, X. , Li, M. , Wu, X. , et al. (2019) Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nature Communications, 10, 4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Bjorbaek, C. , Weremowicz, S. , Morton, C.C. and Moller, D.E. (1995) RSK3 encodes a novel pp90rsk isoform with a unique N‐terminal sequence: growth factor‐stimulated kinase function and nuclear translocation. Molecular and Cellular Biology, 15, 4353–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Chen, W. , Lin, D. , Hu, W. and Tang, Z. (2018) Neuronal apoptosis, axon damage and synapse loss occur synchronously in acute ocular hypertension. Experimental Eye Research, 180, 77–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Supplementary Material