

Abstract
The construction of powerful cell factories requires intensive and extensive remodelling of microbial genomes. Considering the rapidly increasing number of these synthetic biology endeavors, there is an increasing need for DNA watermarking strategies that enable the discrimination between synthetic and native gene copies. While it is well documented that codon usage can affect translation, and most likely mRNA stability in eukaryotes, remarkably few quantitative studies explore the impact of watermarking on transcription, protein expression, and physiology in the popular model and industrial yeast Saccharomyces cerevisiae. The present study, using S. cerevisiae as eukaryotic paradigm, designed, implemented, and experimentally validated a systematic strategy to watermark DNA with minimal alteration of yeast physiology. The 13 genes encoding proteins involved in the major pathway for sugar utilization (i.e., glycolysis and alcoholic fermentation) were simultaneously watermarked in a yeast strain using the previously published pathway swapping strategy. Carefully swapping codons of these naturally codon optimized, highly expressed genes, did not affect yeast physiology and did not alter transcript abundance, protein abundance, and protein activity besides a mild effect on Gpm1. The markerQuant bioinformatics method could reliably discriminate native from watermarked genes and transcripts. Furthermore, presence of watermarks enabled selective CRISPR/Cas genome editing, specifically targeting the native gene copy while leaving the synthetic, watermarked variant intact. This study offers a validated strategy to simply watermark genes in S. cerevisiae.
Keywords: Saccharomyces cerevisiae, glycolysis, genome engineering, DNA and RNA watermarks, differential RNA expression analysis, pathway swapping
A DNA watermark is a unique synthetic nucleotide sequence that enables the identification and traceability of its carrier when applying PCR amplification and sequencing techniques. Application of the watermarks in living organisms started recently with a purpose to protect R&D investments, to create an information storage source, or to enable traceability of pathogenic or endangered species.1−3 The literature reports successful embedding and subsequent detection of the watermarks in DNA strands in vitro,4 as well as in vivo using several model microorganisms (i.e., Bacillus subtilis, Escherichia coli, Saccharomyces cerevisiae, Mycoplasma mycoides, and Mycoplasma capricolum), plants, and viruses.1,2,5−11 All these studies focused on a single locus for the watermark introduction, with a few notable exceptions. First, the Mycoplasma genome de novo synthesis in which four large watermarks (ca. 1 kb) were introduced to enable the differentiation between the natural and synthetic copies of the Mycoplasma genome.5 Second, the Synthetic Yeast 2.0 (Sc2.0) project, where approximately 28 bp regions of each open reading frame were recoded to distinguish synthetic from native genes by PCR.12 Lastly, the recoding of the E. coli genome, such that it uses 61 instead of 64 codons.10 The successes of these projects reveal the potential of the watermarks for future development in synthetic biology, particularly during large-scale genome remodeling projects, where tagging the synthetic gene copies can enable the discrimination between synthetic and native homologues. For instance, Kuijpers and co-workers recently reported the pathway swapping strategy that enables to redesign large, native essential pathways.13 Pathway swapping was demonstrated on the glycolytic and fermentation pathways of S. cerevisiae, involving 12 catalytic steps encoded by 26 genes. After a first genetic reduction leading to a minimal glycolysis set of 13 genes,14 a second, synthetic set of these 13 genes was integrated in a single locus on chromosome IX. Subsequently, the native copies of these 13 glycolytic genes were removed from their original chromosomal loci, leading to SwYG, a yeast strain with a single locus, minimal glycolytic pathway. However, the presence of two identical gene copies for all glycolytic genes during the strain construction process led to complications. First, in this intermediate strain carrying both native, scattered, and synthetic, colocalized glycolytic genes, removal of the native gene copies without harming the synthetic, identical copies integrated on chromosome IX was challenging. Second, expression of the native and synthetic genes could not be measured and compared. Both problems can easily be addressed by embedding watermarks in the synthetic genes. When judiciously placed in Protospacer Adjacent Motifs (PAM), watermarks can disable CRISPR/Cas editing in the synthetic genes.15 When designed in coding regions (CDS), watermarks can be used to identify native from watermarked mRNA molecules.
Whether inserted in coding or noncoding regions, the major downside of watermarks is the risk of unintended changes in the host physiology. Watermarking in coding regions is potentially less challenging as watermarks can be embedded in the CDS as silent mutation, taking advantage of the redundancy of the genetic code encompassing 61 codons for only 20 amino acids. However, while “silent” or synonymous mutations in CDS do not affect the amino acid sequence of the corresponding protein, they can alter cells at different levels. Codons can be classified as optimal and nonoptimal based on their frequency in the genome and the abundance of tRNAs with complementary anticodons.16−18 It is now well established that cells use codon optimality to tune protein expression. Highly expressed genes, such as genes encoding the highly abundant glycolytic proteins, are enriched for optimal codons.19,20 Furthermore, by tuning the translation rate, codon optimality regulates the cotranslational folding of polypeptides and plays a role in shaping proteins conformational states.21−24 More recently, it has been shown that codon optimality also modifies mRNA structure, splicing, and stability.25−28 Codon optimality preservation is therefore an important criterion to consider when introducing watermarks without causing undesirable changes in gene function. There is however little known about the impact of watermarking on cell physiology, and remarkably few studies are dedicated to Saccharomyces cerevisiae, a microbe intensively used in synthetic biology developments.29,30 Heider and Barnekow demonstrated that watermarking of VAM7 in S. cerevisiae did not affect the vacuolar function of the corresponding protein.6 Liss and co-workers expressed a watermarked GFP in S. cerevisiae and showed minimal impact on GFP protein by Western blotting.7 In the Sc2.0 project in every ORF larger than 500 bp at least two 19–28 bp PCRtags were introduced, which were recoded approximately 33–60%. Every strain with a native chromosome replaced by a synthetic version showed no or minor fitness defects, and transcript profiling showed only few genes changed in expression.9,12,31−36 Whether these transcript changes originated from the PCRtags was not always investigated, and it is unclear whether these PCRtags allow discrimination between native and synthetic mRNAs when both are present in the cell. Therefore, there remains a strong need for studies proposing a watermarking strategy with the ability to distinguish between native and synthetic DNA and mRNA, validated by a systematic, quantitative exploration of the impact of watermarking on transcription, translation, and general physiology.37
To fill this knowledge gap, using S. cerevisiae as eukaryotic paradigm, this study designed, implemented and experimentally validated a systematic approach to watermark DNA with minimal alteration of yeast physiology. The impact of simultaneously watermarking 13 genes encoding abundant proteins involved in the major pathway for sugar utilization (i.e., glycolysis and alcoholic fermentation) on metabolism, transcriptome, and enzyme activity was explored using batch cultures in tightly controlled bioreactors. Watermarked transcripts were segregated from native ones using the karyollelle specific expression detection method.38 Finally, the ability of watermarks to protect synthetic genes from CRISPR/Cas9 DNA editing was evaluated.
Results and Discussion
Design and In Silico Validation of the Watermarking Strategy
The presence of watermarks in the CDS of glycolytic genes shall enable discrimination of the watermarked versus native DNA and mRNA sequences with a minimal effect on transcript and protein levels, activity of enzymes in the glycolytic pathway and ultimately, yeast physiology. Finding the optimal trade-off between robust watermark detection by sequencing and minimal physiological impact was therefore the main design principle of the watermarking strategy. On the basis of current RNA sequencing resolution (Illumina platform with an error rate of <1%), at least five nucleotide substitutions were required to distinguish watermarked from native sequences using random single nucleotide polymorphisms (SNPs). Codon replacement was performed on the amino acids encoded by four to six alternative triplets (A, G, P, T, V, L, R, S), favoring triplets for which only the third base pair of the triplet was different from the original codon. The codon with the most similar percentage of abundance when referring to the codon usage table of S. cerevisiae (Table S1) was chosen, avoiding triplets leading to more than 20% variation in abundance when possible. The structure of the 5′ region of the mRNA is important for translation efficiency. Not only does the folding energy at the 5′ end affect translation initiation, but the presence of nonoptimal codons close to the initiation site can stall ribosomes, thereby hampering translation initiation.39,40 Furthermore, as translation initiation is considered as translation limiting step,41 and following the example of Annaluru and colleagues,9 the first 101 nt of the CDS were preserved. The optimal distribution of watermarks over the remaining CDS stretch was tested with two in silico approaches using ADH1. In the first approach, watermarks were colocalized in two 100 nt regions, one in the middle of the CDS, and the other located 10 nt upstream of the stop codon (Figure 1A). In the second approach, base pair substitutions were equally spread over the CDS sequence, every 85 nt (Figure 1B). In both approaches 11 watermarks were introduced, which resulted in an overall change in codon usage of 0.57 for the first and 0.47 for the second method (Figure 1, Figure S1. See Methods section for calculation of the change in codon usage). In order to evaluate the discriminatory potential of RNA sequencing with these two strategies, 100 bp paired-end sequence reads were simulated for both watermarked and native ADH1 copies (see Methods section). These data were processed using the k-mer method developed by Gehrmann et al.,38 only considering reads containing watermarks (see Methods section), to selectively quantify watermarked and native reads. On average 52.5% of the reads were captured when using the first approach with clustered watermarks, while 99.4% were detected using the second approach, with watermarks spread over the CDS (Figure 1). The Pearson correlation coefficient between generated and measured reads was above 0.99 for both methods, indicating that both methods are able to retrieve the variation in abundance across the samples, required for differential expression. The second approach resulted in a better sequence coverage and slightly lower codon usage change. However, the first approach is less labor intensive when manual design is performed, and is less likely to affect cotranslational folding,21 as a shorter part of the CDS undergoes codon usage change. The first method was therefore selected as watermarking strategy (detailed in Box 1 and Figure S2) and used to edit in silico the CDS of 13 genes of glycolysis and alcoholic fermentation (HXK2, PGI1, PFK1, PFK2, FBA1, TPI1, TDH3, PGK1, GPM1, ENO2, PYK1, PDC1, and ADH1, see example for watermarking for FBA1 and ENO2 in Figure S3). This resulted in a reasonably low change in codon usage of the watermarked genes of 0.5 on average (Table S2). Using simulated data, we compared the performance of the k-mer method with traditional alignment and found that the k-mer method was able to achieve a higher read retrieval rate than alignment indicating a more accurate transcription estimate (Table S3). The watermarked CDS were synthesized with flanks compatible with Golden Gate assembly (plasmids pGGKp137 to pGGKp150, Table S4).
Figure 1.

Comparison of two watermarking strategies. (A) First strategy with clustered watermarks. (B) Second strategy with watermarks distributed over the whole coding region. The tables in panel A and B represent the % of sequencing reads that can be captured by the two watermarking strategies, calculated from in silico simulated 100 bp paired-end sequencing reads.
Strain Construction Strategy and Confirmation
In the SwYG strain,13 the set of genes involved in glycolysis and fermentation was reduced from 26 to 13 and relocalized to a single locus (Single Locus Glycolysis, SinLoG) on chromosome IX. The SwYG strain is a perfect platform to rapidly remodel glycolysis and alcoholic fermentation and test multiple (heterologous) variants. SwYG was therefore used as starting strain to express the watermarked genes. Using simultaneous Cas9-mediated genome editing and in vivo assembly, the entire glycolytic and fermentation pathways composed of 13 watermarked genes were integrated in one step in the CAN1 locus on chromosome V. The watermarked genes were framed by the native, standardized corresponding promoters and terminators (800 bp and 300 bp respectively). Three helper elements, two Autonomously Replicating Sequences (ARS) and a selection marker were included in the SinLoGs design (Figure 2). Two active ARSs (ARS418 and ARS1211) were added on both ends of the ca. 35 Kb long SinLoGs to minimize the risk of perturbing DNA replication of this long DNA stretch. A selection marker was used to facilitate screening for correct integration and removal of the SinLoGs. The native SinLoG, present in the SGA1 locus on chromosome IX, was then removed using the Cas9 endonuclease, resulting in strain IMX1770 (Figure 2 and Figure 3). To obtain an isogenic control strain, the same procedure was followed to construct a strain with native SinLoG, framed by the same promoters and terminators as the watermarked genes, and integrated in the same CAN1 locus on chromosome V (Figure 2 and 3). This control strain was named IMX1771. The genome of both strains was sequenced, confirming the presence of a single, correctly assembled glycolytic pathway at the targeted chromosomal location. Sequencing revealed the absence of mutations in the coding regions of the glycolytic and marker genes but identified a few mutations in the promoter and terminator regions of the glycolytic and selection marker expression cassettes (Table S5). In IMX1770, a single Single Nucleotide Variation (SNV) was found in the promoter of PFK1 and HIS3 and in the terminator of PGK1 and ENO2. In IMX1771, a single SNV was identified in the HIS3 terminator, and a short TA stretch was missing in the promoter of GPM1.
Figure 2.

Construction of SinLoG (Single Locus Glycolysis) strains IMX1770 and IMX1771 using the glycolysis swapping strategy.13 (A) A newly designed glycolysis is integrated in the CAN1 locus by simultaneous CRISPR/Cas9-aided editing of CAN1 and in vivo assembly of glycolytic expression cassettes and helper fragments (ARS418, ARS1211 and the selection marker HIS3). The > and < signs next to the gene names indicate the directionality of transcription and letters indicate the synthetic homologous recombination (SHR) sequence which was used for assembly. (B) Subsequently, the Single Locus Glycolysis present in the SGA1 locus was excised by double editing using CRISPR/Cas9 and replaced by the URA3 selection marker. The set of genes integrated in CAN1 is then the sole set of glycolytic genes present in the newly constructed strain and is essential for growth on glucose.
Figure 3.
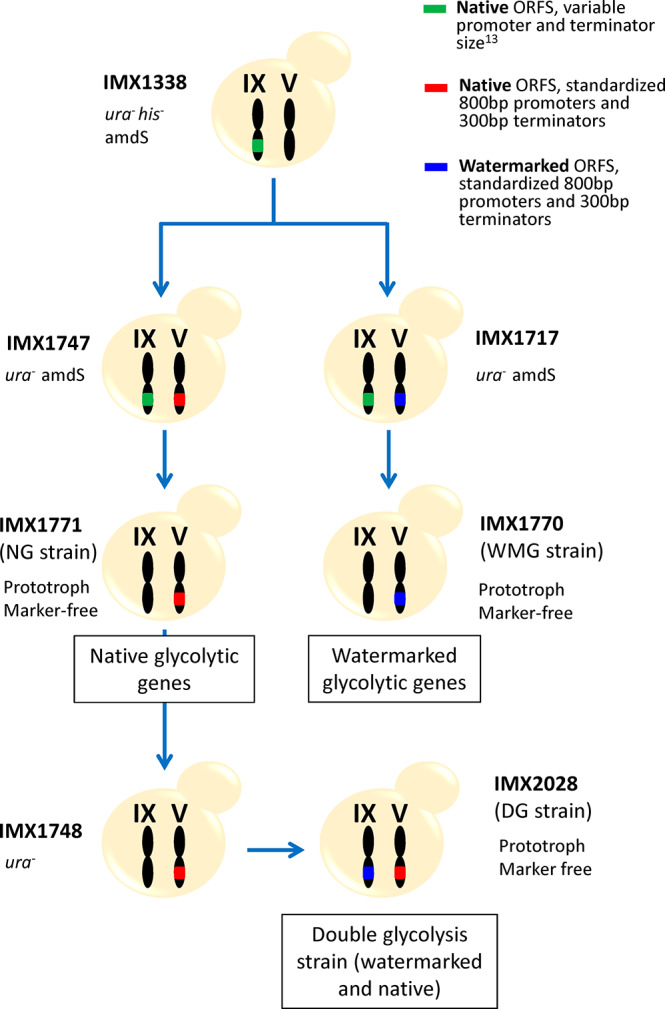
Strain construction workflow. The Switchable Yeast Glycolysis (SwYG) strain, IMX1338, served as parental strain to introduce in chromosome V a SinLoG (Single Locus Glycolysis) with native ORFs and standardized promoters/terminators (IMX1747) as well as with watermarked ORFs and standardized promoters/terminators (IMX1717). From both strains the native SinLoG in chromosome IX with variable promoters and terminators was removed (resulting in strain IMX1771 and IMX1770, respectively). After removal of URA3 from strain IMX1771 (native ORFs) the SinLoG with watermarked ORFs was introduced in chromosome IX, resulting in a strain with double glycolysis (IMX2028).
A third strain, IMX2028 was constructed. IMX2028 harbored a double SinLoG, one located on chromosome IX carrying the watermarked genes and another on one carrying the native yeast genes on chromosome V (IMX2028, Figure 1 and Table 1). Unfortunately, genome sequencing revealed the deletion of a large region of the mitochondrial DNA (Figure S4). The strain IMX2028 was constructed to evaluate the performance of the k-mer method to discriminate between watermarked and native genes when present in the same strain. Despite IMX2028 respiration deficiency, the watermarked and native SinLoG that this strain carried were essentially faithful to the in silico design (Table S5), which made this strain still valuable for differential quantification of watermarked and native genes.
Table 1. List of Strains Used in This Study.
| strain name | genotype | source and brief description |
|---|---|---|
| Strains Characterized in This Study | ||
| IMX1770 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-I-SceI-tCYC1)can1::(ARS418CAN1 AHFBA1_*AH HTPI1_*HPPGK1_*PQADH1_*QNPYK1_*NOTDH3_*OAENO2_*ABHXK2_*BCPGI1_*CDPFK1_*DJPFK2_*J BPHIS3BP LGPM1_*LMPDC1_*M ARARS1211AR CAN1) sga1::KlURA3 | This study; Prototrophic strain with watermarked single locus glycolysis (WMG strain); Derived from IMX1717 |
| IMX1771 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-I-SceI-tCYC1)can1::(ARS418CAN1 AHFBA1AH HTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2J BPHIS3BP LGPM1LMPDC1M ARARS1211AR CAN1)sga1::KlURA3 | This study; Prototrophic strain with native single locus glycolysis (NG strain); Derived from IMX1747 |
| IMX2028 | MATa ura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4glk1::Sphis5Δ::(pGAL1-I SceI-tCYC1)can1::(ARS418CAN1 AHFBA1AH HTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2J BPHIS3BP LGPM1LMPDC1M ARARS1211AR CAN1)sga1::(ARS418sga1 AHFBA1_*AH HTPI1_*HPPGK1_*PQADH1_*QNPYK1_*NOTDH3_*OAENO2_*ABHXK2_*BCPGI1_*CDPFK1_*DJPFK2_*J BPHIS3BP LGPM1_*LMPDC1_*M ARARS1211AR sga1) | This study; Prototrophic strain with native and watermarked single locus glycolysis (DG strain); Derived from IMX1748 |
| Strains Used As Starting Point or Intermediate in the Construction of the above Strains | ||
| CEN.PK113–7D | MATaMAL2–8cSUC2 | Control strain;46,47 Starting strain for all construction work;13 Contains a SinLoG in Chr. IX, with variable promoter and terminator length; Uracil auxotroph |
| IMX589 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2glk1::Sphis5hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4sga1::(FBA1GHTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2JKAmdSYMKLGPM1LMPDC1-SYNMF)pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2 | |
| IMX1338 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4sga1::(FBA1GHTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2JKAmdSYMKLGPM1LMPDC1-SYNMF)pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-ISceI-tCYC1) | This study; Derived from IMX589; Contains a SinLoG in Chr. IX; Uracil and histidine auxotroph |
| IMX1717 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4sga1::(FBA1GHTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2JKAmdSYMKLGPM1LMPDC1-SYNMF)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-I SceI-tCYC1)can1::(ARS418AHFBA1_*AH.HTPI1_*HPPGK1_*PQADH1_*QNPYK1_*NOTDH3_*OAENO2_*ABHXK2_*BCPGI1_*CDPFK1_*DJPFK2_*J.BPHIS3BP.LGPM1_*LMPDC1_*M.ARARS1211AR) | This study; Derived from IMX1338; Strain with native (variable prom and term length) and watermarked SinLoG in Chr. IX and V respectively; Histidine auxotroph |
| IMX1747 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4 sga1::(FBA1GHTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2JKAmdSYMKLGPM1LMPDC1-SYNMF)pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-I SceI-tCYC1)can1::(pGAL1-I-SceI-tCYC1)can1::(ARS418CAN1 AHFBA1AH HTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2J BPHIS3BP LGPM1LMPDC1M ARARS1211AR CAN1) | This study; Derived from IMX1338; Strain with native SinLoG with variable prom and term length in Chr. IX and native SinLoG with standardized prom and term length in Chr. V; Histidine auxotroph |
| IMX1748 | MATaura3–52his3–1leu2–3,112MAL2–8cSUC2hxk1::KlLEU2tdh1tdh2gpm2gpm3eno1pyk2pdc5pdc6adh2adh5adh4pyk1pgi1tpi1tdh3pfk2::(pTEF-cas9-tCYC1 natNT1)pgk1gpm1fba1hxk2pfk1adh1pdc1eno2glk1::Sphis5Δ::(pGAL1-I-SceI-tCYC1)can1::(ARS418CAN1 AHFBA1AH HTPI1HPPGK1PQADH1QNPYK1NOTDH3OAENO2ABHXK2BCPGI1CDPFK1DJPFK2J BPHIS3BP LGPM1LMPDC1M ARARS1211AR CAN1) | This study; Derived from IMX1771; Strain with watermarked SinLoG in Chr. IX and native SinLoG with standardized prom and term length in Chr. V; Uracil auxotroph |
Watermarks Do Not Affect Yeast Physiology
To evaluate the impact of DNA watermarking on yeast physiology, the watermarked strain IMX1770 and its isogenic control IMX1771 were grown in aerobic batch bioreactors and their growth kinetics were compared. Both strains were prototrophic, meaning that they fully relied on glucose, the sole carbon and energy source catabolized via glycolysis, to produce the required cellular building blocks and therefore to grow. The two strains displayed identical growth rates (0.33 ± 0.004 h–1 and 0.32 h–1 ± 0.002 h–1 for IMX1770 and IMX1771, respectively) as well as glucose and O2 uptake rates, ethanol and CO2 production rates and yields (Figure 4 and Table 2). Both strains passed the diauxic shift and grew equally well using the ethanol, which was produced during fermentative growth on glucose, as carbon and energy source (Figure S5). Watermarking of glycolytic and alcoholic fermentation genes therefore did not alter metabolic fluxes and the overall physiological responses during fast respiro-fermentative on glucose and full respiratory growth on ethanol.
Figure 4.

Physiological characterization of strains with native (IMX1771) and watermarked (IMX1770) glycolytic genes during aerobic batch cultures in bioreactors. (A) Left panel, biomass concentration (gram dry biomass per liter), central panel, glucose concentration (mM); right panel, ethanol concentration (mM). Three independent culture replicates are represented for each strain. Shades of blue with square symbols, IMX1770, shades of red with round symbols, IMX1771. (B) Specific enzyme activities of the 12 reactions encoded by the 13 glycolytic enzymes (Pfk1 and Pfk2 form an enzyme complex) of the strains with native (IMX1771, red bars) and watermarked glycolysis (IMX1770, blue bars). Samples were taken in midexponential phase. Bars represent the average and standard deviation of measurements from three independent batch cultures for each strain. Stars indicate enzyme activities that are significantly different between the two strains (Student t test, p-value threshold 0.05, two-tailed test, homoscedastic).
Table 2. Physiological Characterization in Bioreactor of Yeast Strains with Native and Watermarked Glycolysisa.
| IMX1771 | IMX1770 | |
|---|---|---|
| Yields | ||
| Ysx (gdry weight/gglucose) | 0.12 ± 0.01 | 0.13 ± 0.01 |
| Ys,glycerol (mol/mol) | 0.07 ± 0.01 | 0.05 ± 0.01 |
| Ys,ethanol(mol/mol) | 1.47 ± 0.05 | 1.46 ± 0.02 |
| Ys,acetate(mol/mol) | 0.05 ± 0.00 | 0.06 ± 0.00 |
| Specific rates | ||
| μmax (h–1) | 0.32 ± 0.00 | 0.33 ± 0.01 |
| qglucose (mmol/g–1·h–1) | –14.6 ± 0.7 | –14.7 ± 0.9 |
| qglycerol (mmol/g–1·h–1) | 0.96 ± 0.03 | 0.76 ± 0.17 |
| qethanol(mmol/g–1·h–1) | 21.4 ± 0.3 | 21.4 ± 1.7 |
| qacetate(mmol/g–1·h–1) | 0.79 ± 0.05 | 0.88 ± 0.06 |
| Carbon balances (%) | 105 ± 2 | 103 ± 3 |
Physiological characterization of strains with native (IMX1771) and watermarked (IMX1770) glycolytic genes during aerobic batch cultures in bioreactors. Data represent the average and standard deviation of measurements from three independent batch cultures for each strain. Statistical analysis (Student t test, p-value threshold 0.05, two-tailed test, homoscedastic) revealed no significant differences between the two strains.
Watermarking might affect protein folding and consequently function. However, as yeast glycolysis is characterized by an overcapacity of its enzymes, mild variations of glycolytic enzymes activities might not be detectable by growth kinetics. The 12 specific activity assays of the 13 enzymes (Pfk1 and Pfk2 are subunits of a hetero-octameric phosphofructokinase42) encoded by the watermarked genes were therefore assayed in vitro. The specific activity of these 13 enzymes was, with the exception of Gpm1, remarkably similar between watermarked and native strains (p-values above 0.05; Student t test, two-tailed, homoscedastic). For all enzymes, specific activities were remarkably similar to protein abundance, including a 1.6-fold decrease in specific activity and protein abundance for Gpm1 (Figure 4, Figure S8 and S9). Watermarking therefore did not affect or marginally affect protein expression and functionality (Figure 4).
To further explore the potential impact of watermarking on yeast physiology, the transcriptome of IMX1771 and IMX1770 grown in aerobic batch reactors was compared. The transcriptional response of these two strains was remarkably similar (Figure 5). The native and watermarked glycolytic genes were the only differentially expressed genes between the two strains. This differential expression reflects the absence of the native genes and therefore their lack of expression in IMX1770, and the absence and lack of expression of the watermarked genes in IMX1771. However, expression levels of the native and watermarked genes in IMX1771 and IMX1770, respectively, were highly similar (Figure 6A).
Figure 5.

Genome-wide transcriptome analysis of IMX1770 and IMX1771. The x-axis represents the log fold change in expression, and the y-axis represents the −log p-value. Each point represents a transcript. A negative log fold change reflects higher expression in the native strain than in the watermarked strain, and vice versa. The horizontal, dashed line represents the FDR corrected p-value threshold of 0.05, and the vertical dashed lines represent a log fold change threshold of 1. Red points indicate significantly differentially expressed transcripts (FDR-corrected p-value above 0.05 and Log fold change higher than 1).
Figure 6.
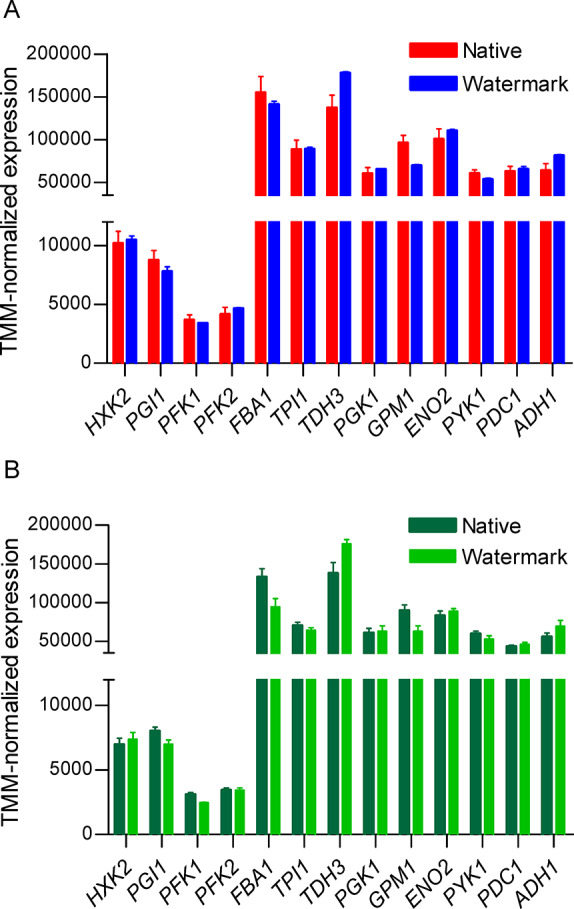
Glycolysis and fermentation transcript levels of S. cerevisiae grown in aerobic batch cultures in bioreactors. (A) Watermarked transcript levels of IMX1770 (blue) and native transcript levels of IMX1771 (red). (B) Watermarked and native transcript levels of IMX2028. Bars represent the average and standard deviation of three independent cultures replicates. Samples were taken in midexponential phase (Table S9). No significant change in expression was found between watermarked and native genes (Student t test, p-value threshold 0.05, two-tailed test, homoscedastic) between IMX1770 and IMX1771 and within IMX2028.
Physiological characterization of IMX2028 confirmed the respiration deficiency suggested by the absence of mitochondrial DNA. The k-mer method was able to selectively quantify expression of the native and watermarked genes. While expression of glycolytic and respiration genes might differ in IMX2028 as compared to IMX1770 and IMX1771 due to the mutations in mitochondrial DNA and associated respiration deficiency, the relative expression of glycolytic and fermentation genes, expressed from the same promoters in the native and watermarked SinLoG, was not expected to differ between the native and watermarked genes in this strain. Accordingly, and in agreement with the similarity of the expression levels between IMX1770 and IMX1771, transcript levels of native and watermarked genes in IMX2028 were identical (Figure 6B).
Watermarking of 13 highly expressed genes of central carbon metabolism, essential for glucose utilization, had therefore no impact on yeast transcriptome and physiology.
Watermarking Enables Selective CRISPR/Cas9 Genome Editing
DNA binding and editing by CRISPR/Cas9 requires the presence at the targeted site of a specific PAM recognition sequence.15 A single nucleotide variation in this sequence can abolish Cas9 ability to introduce a double strand DNA break.15 This feature is particularly interesting when considering selective editing of identical or highly similar sequences. If strategically designed, watermarks can enable targeted editing of a watermarked gene, leaving the native copy intact or conversely, prevent editing of the watermarked gene while cutting the native copy. Guide RNAs (gRNAs) selectively targeting the native copies of PYK1 and TPI1 for CRISPR/Cas9 editing were designed (Table S6), inserted into expression vectors and transformed to IMX1717, a double SinLoG strain and direct ancestor of IMX1770 (Figure 3). Double-stranded DNA fragments of 120 nt were supplied during transformation to repair via homologous recombination the break induced by CRISPR/Cas9. As the sequence of the native and watermarked genes is identical with the exception of watermarks, a single primer set designed just outside the open reading frame can be used to amplify both copies of PYK1 or TPI1 in a single PCR reaction using IMX1717 genomic DNA as template. Ran on a gel, the PCR products of this reaction would lead to a single band corresponding to both the native and watermarked copies of PYK1 or TPI1. Selective editing would lead to the appearance of a second, smaller band on gel, corresponding to the edited copy of PYK1 or TPI1. Out of 15 colonies of IMX1717 transformed with the gRNA targeting PYK1, three displayed two bands demonstrating editing of a single PYK1 copy (Figure 7). Five out of 15 colonies of IMX1717 transformed with the gRNA targeting TPI1 showed selective editing (Figure 7). For two transformants per gene (TPI1 and PYK1) showing two bands on the gel, sequencing the largest band confirmed the presence of the watermarked sequence only, confirming selective editing of the native TPI1 and PYK1 (Figure S6). It has recently been shown that cells can use chromosomal DNA with high homology to repair a CRISPR/Cas mediated DNA break, leading to loss of heterozygosity.43 In the present case, it means that cells could repair the induced DNA break in the targeted, native gene copy with its watermarked homologue, resulting in two copies of the watermarked gene, but a single PCR product and therefore a single band on gel. Sequencing of the unique PCR product of four colonies in which editing of PYK1 and TPI1 was considered unsuccessful revealed that, for all tested colonies, the PYK1 and TPI1 genes were cut by CRISPR/Cas but repaired by (part of) the watermarked allele. Editing of the targeted, native genes by CRISPR/Cas9 was therefore highly efficient (100% of the tested colonies), however the DNA break was repaired either by the supplied repair DNA fragment or by the watermarked homologue.
Figure 7.

Diagnostic PCR for selective editing of native glycolytic genes. Separation of PCR products resulting from outside–outside amplification to identify edited (nonwatermarked) and nonedited (watermarked) loci for PYK1 (A) and TPI1 (B) from transformants of IMX1717 (double SinLoG). (A) Lanes 1–15 show the PCR results of amplification of the PYK1 locus of randomly picked colonies. Successful editing of the locus results in a DNA fragment with a length of 670 bp. No editing of the locus results in a DNA fragment with a length of 2177 bp. Primers 11915 and 4667 were used. Lanes 1, 5, and 15 display bands of both sizes revealing selective editing. (B) Lane 1–15 show the PCR results of amplification of the TPI1 locus of randomly picked colonies. Successful editing of the locus results in a DNA fragment with a length of 378 bp. No editing of the locus results in a DNA fragment with a length of 1125 bp. Primers 3514 and 6406 were used. Lanes 9–11, 13, and 15 display bands of both sizes revealing selective editing. A negative control is indicated with “C-“ (IMX1338, SinLog). In the lanes indicated with “L”, GeneRuler DNA ladder mix was loaded. 1% (w/v) agarose in TAE.
Conclusion
The present study offers an innocuous watermarking strategy for coding regions that enables the discrimination of DNA and mRNA by sequencing through a k-mer approach and facilitates selective editing of watermarked and nonwatermarked sequences. While the design of watermarked genes was performed manually in the present study, it can easily be automated when a larger number of genes is concerned, with software similar to for example GeneDesign.44
The set of genes chosen to test the watermarking strategy encodes highly abundant proteins that are generally considered to operate at overcapacity, which means that the capacity of the enzymes is considerably larger than the actual in vivo flux. While this overcapacity might obscure physiological responses, a closer inspection of transcript levels and enzyme activities confirmed the watermarks' harmlessness for S. cerevisiae. Only one of the 13 tested genes showed an activity of the enzyme encoded by the watermarked allele significantly decreased (ca. 1.6-fold) as compared to the activity of the enzyme encoded by the native allele (phosphoglucomutase encoded by GPM1), which could be explained by a similar decrease in Gpm1 protein abundance. As native and watermarked transcript levels were identical for GPM1, the lower enzyme abundance in the watermarked strain might result from a slightly lower translation efficiency. Neither the watermarking specifics (type of codon substitution, change in codon usage, etc.) of this particular protein, nor information from literature hinted toward the mechanism underlying this decreased protein abundance. A recent study, combining measurements of protein synthesis rate with ribosome footprinting data confirmed that Gpm1, like most glycolytic proteins, has a fast synthesis rate.45 Applying the same approach to watermarked strains could help characterizing the impact of nucleotide substitution on translational efficiency.
Another particularity of glycolytic genes is their high codon optimality (on average ca. 90% of optimal codons according to Hanson and Coller24). While one could argue that this set of genes might not be representative of the yeast genome, to the best of our knowledge there is no evidence that such genes are more or less robust toward changes in codon frequency than genes with lower levels of codon optimality. For future studies it would be interesting to explore if codon optimality affects genes sensitivity to watermarking.
Methods
Strains and Cultivation Conditions
The S. cerevisiae strains used in the study belong to CEN.PK family46−48 and are listed in Table 1. Liquid cultures were grown in 500 mL shake flasks filled with 100 mL of medium at 30 °C with 200 rpm agitation. Complex media (further referred to as YPD) contained 10 g·L–1 yeast extract, 20 g·L–1 peptone and 20 g·L–1 glucose. Synthetic minimal medium (further referred as SMG) consisted of 3 g g·L–1 KH2PO4, 0.5 g·L–1 MgSO4·7H2O, 5 g·L–1 (NH4)2SO4, 1 mL·L–1 of a trace element solution, and 1 mL·L–1 of a vitamin solution as previously described13 and supplemented with 20 g·L–1 glucose. For solid medium, 20 g·L–1 agar was added prior autoclaving. When selection in SMG was required, (NH4)2SO4 was replaced with 3 g·L–1 K2SO4 and 2.3 g·L–1 filter-sterilized urea, and the medium was supplemented with 200 mg·L–1 of G418, hygromycin B or 10 mM acetamide.49,50 For the counterselection purpose, 1 mg·mL–1 5-FOA (Zymo Research, Irvine, US) was added to SMG supplemented with uracil (150 mg·L–1).49 For plasmid propagation, E. coli XL1-Blue cells (Agilent Technologies, Santa Clara, CA, USA) were grown in Lysogeny broth (LB) medium supplied with 100 mg Lampicillin or 25 mg·L–1 chloramphenicol at 37 °C with 180 rpm agitation. Yeast and bacterial frozen stocks were prepared by addition of 30% (v/v) glycerol to exponentially growing cultures. Strain aliquots were stored at −80 °C.
Molecular Biology Techniques
PCR reactions for diagnostic purposes were performed using DreamTaq DNA polymerase Master Mix (Thermo Fisher Scientific, Walthman, MA, USA) according to the manufacturer’s instructions. For high fidelity PCR reactions, Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific) was used following the supplier’s manual. Oligonucleotides of desalted or PAGE quality, depending on the purpose, were purchased from Sigma-Aldrich (St Louis, MO, USA). DNA fragments were resolved in agarose gels and purified using PCR cleanup kit from the reaction mixture (Sigma-Aldrich, St Louis, MO, USA) or excised from the agarose gel and purified using Zymoclean gel purification kit (Zymo Research, Irvine, CA, USA) when required. Circular templates were removed by applying DpnI enzyme restriction according to the producer’s manual (Thermo Fisher Scientific).
Plasmids were isolated from E. coli cultures using Sigma GenElute Plasmid kit (Sigma-Aldrich, St Louis, MO, USA). E. coli transformations were performed using chemical competent XL-1 Blue cells (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer instructions. Golden Gate Assembly was performed as previously described51 using equimolar concentrations of 20 fmol for each fragment. For a 10 μL reaction mixture 1 μL T4 DNA ligase buffer (Thermo Fisher Scientific), 0.5 μL T7 DNA ligase (NEB New England Biolabs, Ipswich, MA), and 0.5 μL of either FastDigest Eco31I (BsaI) or BsmBI (NEB) were added.
Gibson Assembly was performed using Gibson Assembly Master Mix (New England Biolabs, Ipswich, MA) according to the manufacturer’s protocol.
All plasmids are reported in Table S4 and primers in Table S7.
In Silico Design of the Watermarks
Watermarks were introduced in the genes of interest according to the guidelines described in Box 1 using the Clone Manager software.
Watermarked CDS were ordered as a synthetic gene from GenArt (Thermo Fisher, Regensburg). The list of synthesized plasmids encoding watermarked CDS (pGGKp137 to pGGKp150) can be found in Table S4.
The change in codon usage in a gene caused by watermarking was calculated as
where i represents each codon substitution in a gene.

Construction of Libraries Encoding Transcriptional Units of Watermarked Glycolysis
The sequences of the watermarked genes, promoters (800 bp) and terminators (300 bp) were ordered from GeneArt (Thermo Fisher, Regensburg, Germany). For compatibility with Golden Gate Cloning, the sequences were ordered flanked with BsaI and BsmBI restriction sites. The promoters and terminators were delivered by GeneArt subcloned in the entry vector pUD565 and for the watermarked genes the subcloning into pUD565 was done in house using BsmBI Golden Gate cloning. An exception was made for pTDH3, pPGK1, tPGK1, tENO2, and tADH1 which were amplified from genomic DNA of CEN.PK113–7D using primers with flanks containing BsaI restriction sites listed in Table S7.
Subsequently, the assembly of the promoter, gene and terminator was done in the preassembled vector pGGKd012 using Golden Gate cloning as described in the previous section. pGGKd012 was assembled from the Yeast toolkit51 plasmids pYTK-002, 047, 072, 078, 081, and 083 (Table S4).
Correct plasmid assembly was verified by enzyme digestion with either BsaI, BsmBI (New England Biolabs) or FastDigest enzymes (Thermo Fisher Scientific) following the manufacturer’s instructions.
Construction of gRNA Plasmids Used in the Study
The guide RNA (gRNA) plasmids pUDR413 and pUDR529 for the yeast strain construction were designed and constructed according to Mans et al. (2015).52 gRNA targets were selected using the Yeastriction tool52 in case of pUDR413, or designed manually for the K. lactis URA3 target in plasmid pUDR529. For pUDR413, the 2 μm fragment was constructed in two parts using the primer 6131 and 5975 and primer 6296 together with 5941 using pROS12 as a template. For pUDR529, the 2 μm fragment was obtained by PCR using primer 14549 and pROS12 as a template. The backbone for pUDR413 was amplified with primers 6005 and 6006 using pROS13 as a template, while for pUDR529 same primer pair was used to amplify the backbone from pROS12. For both plasmids, 100 ng of each purified fragment was used in the Gibson Assembly and correct plasmid assembly was verified with the primers 3841, 5941, and 6070 in case of pUDR413 and 4034 and 5941 for pUDR529.
The guide RNA plasmids for selective native copy gene removal of TPI1 and PYK1, named pUDR531 and pUDR532 respectively (Table S4), were constructed as described in Mans et al. (2015)52 with the modifications regarding the design of the gRNA. gRNAs were designed manually to target the native CDS containing a PAM which was removed in the watermarked copy of the CDS. Each gRNA was ordered as a primer (Table S7, primers 14515, 14517, 14519, 14521). The 2 μm fragment for four gRNA plasmids was obtained by PCR using corresponding gRNA primer (Table S7, primers 14515, 14517, 14519, 14521) and pROS13 as a template. The backbone for the four plasmids was obtained by amplification with primers 6005 and 6006 using pROS12 as a template. For the assembly, 100 ng of purified backbone and gRNA fragments were used in the Gibson Assembly and correct plasmid assemblies were verified with the primers 3841 and 5941 in combination with gRNA specific primers listed in Table S7.
Construction of SwYG Strains with Native and Watermarked Glycolysis and Double Glycolysis Strain
A schematic overview of the strain construction approach is shown in Figure 3. All yeast transformations were performed according to Gietz and Woods (2002).53 For highly efficient targeted integration CRISPR/Cas9 mediated editing was applied. To this end, 350 ng of a plasmid carrying a corresponding guide RNA (further gRNA) was transformed into the yeast strain together with a purified PCR fragment (150 fmol) containing 60 bp homology to the integration site and acting as donor DNA4 (see primers listed in Table S7). gRNA plasmids and the donor DNA were specific for each strain construction step and will be specified below. When donor DNA was consisting of multiple fragments, 60 bp sequences for homologous recombination (SHR) were flanking each of the fragments to enable in vivo assembly by homologous recombination. PCR fragments for the native SinLoG genes and for ARS418, ARS1211 and HIS3 were obtained using CEN.PK113–7D genomic DNA as a template, while fragments encoding the watermarked SinLoG were amplified from plasmids encoding the corresponding transcriptional units (Table S4, Table S7).
To obtain a double auxotrophic host strain named IMX1338, the Schizosaccharomyces pombe HIS5 gene previously inserted in the glk1 locus of IMX589 containing the SinLoG in chromosome IX13 was replaced by the I-SceI expression cassette (pGAL1 – I-SceI – tADH1), which was amplified from the plasmid pUDC073 (primers 10708 and 10709). The replacement was mediated by a Cas9 gRNA plasmid assembled in vivo from two PCR fragments amplified from the pMEL10 plasmid using primers 6005 and 6006 in combination with 10904 (gRNA primer). Transformants were selected on SMG, and the gRNA plasmid with URA3 marker was removed by two sequential restreaks on SMG with 5-FOA. The correct genotype was confirmed by diagnostic PCR using primers 6190 + 1525 and 1553 + 6189 and later by whole genome sequencing. To construct IMX1717 and IMX1747, IMX1338 was transformed with the p426-SNR52p-gRNA.CAN1.Y-SUP4t plasmid targeting the CAN1 locus,54 and PCR fragments of the 13 native or watermarked SinLoG glycolytic genes together with ARS418, ARS1211, and the HIS3 marker gene (Figure 2). Transformants were selected on SMG media and after strain confirmation by PCR (Table S7, Figure S7) the gRNA plasmid encoding the KlURA3 marker was removed. As the next step, the SinLoG with variable length of promoters and terminators was removed from the SGA1 locus in the strains IMX1717 and IMX1747. To this end, both strains were transformed with plasmid pUDR413 and 1 μg of KlURA3 repair fragment amplified with primers 13273 and 13274 introducing homology flanks to the SGA1 site (Figure 2). Transformants were selected on SMG supplemented with G418 and after strain confirmation by PCR using primers 11898 + 7479, 11898 + 2363, and 170 + 7479, the plasmid was removed. For the construction of the strain IMX2028 containing the native SinLoG in Ch V and watermarked SinLoG in Ch IX, first, the intermediate strain IMX1748 was constructed by removing the KlURA3 gene from IMX1771. This was done by transformation with plasmid pUDR529 encoding a gRNA for the KlURA3 gene and a repair fragment amplified with the primers 4223 and 4224 and containing homology to the SGA1 locus. Colonies were selected on YPD media supplemented with Hygromycin B and correct strain construction was confirmed by PCR using primers 4223 and 4224. After KlURA3 marker removal, IMX1748 was transformed with plasmid pUDR314 and the mixture of fragments for the watermarked SinLoG, ARS418, ARS1211, and KlURA3 marker gene resulting in strain IMX2028. Correct assembly of the fragments was confirmed by PCR.
Selective CRISPR/Cas9 Genome Editing
For selective CRISR/Cas9 genome editing, IMX1717 (Table 1) was transformed with 1 μg of a 120 bp repair fragment with homology to the beginning and end of the gene and with 1 μg of plasmids pUDR531 or pUDR532 containing a gRNA for TPI1 and PYK1 respectively as described in the section Construction of gRNA plasmids and Table S4. Cells were plated on YPD with Hygromycin B. Repair fragments (120 nt-long) and diagnostic primers are listed in Table S7-G.
Whole Genome Sequencing and Data Analysis
Yeast genomic DNA was isolated using the Qiagen Genomic DNA Buffer Set and Genomic-tip 100/G tips (Qiagen, Hilden, Germany) according to the manufacturer’s manual. The incubation step with zymolyase was performed for 11 h and the incubation step for digestion with proteinase K was performed overnight. The concentration of the genomic DNA mixture was measured with the BR ds DNA kit (Invitrogen, Carlsbad, CA, USA) using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and the purity was verified with a Nanodrop 2000 UV–vis Spectrophotometer (Thermo Fisher Scientific).
IMX1770, IMX1771, and IMX2028 genomes were sequenced on an Illumina MiSeq Sequencer (Illumina, San Diego, CA, USA) using the MiSeq Reagent Kit v3 with 2 × 300 bp read length. Extracted DNA was mechanically sheared to an aimed average size of 550 bp with the M220 ultrasonicator (Covaris, Wolburn, MA, USA). DNA libraries were prepared using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina) according to the manufacturer’s manual. Quantification of the libraries was done by qPCR using the KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems, Wilmington, MA, USA) on a Rotor-Gene Q PCR cycler (Qiagen). The genome of CEN.PK113–7D, the in silico constructed watermarked and reference (native) SinLoG sequences and the KlURA3 repair fragment were used as a reference to map sequence reads of genomic DNA onto using the Burrows-Wheeler Alignment tool (BWA).55 The sequence alignment was further processed using SAMtools.56 Coverage of the sequence reads was also calculated using the Magnolya algorithm.57
All Illumina sequences are available at NCBI (http://www.ncbi.nlm.nih.gov/) under the bioproject accession number PRJNA554743.
Batch Cultivations of IMX1770, IMX1771, and IMX2028
Batch cultivations were performed in biologically independent triplicates in 2-Liter fermenters (Applikon, Delft, The Netherlands) with a working volume of 1.4 L. Cells from exponentially growing SMG shake flask cultures were inoculated into the fermenters containing SMG supplied with 0.2 g·L–1 antifoam Emulsion C (Sigma-Aldrich, St Louis, MO) at an OD660 of 0.4. The fermenters were sparged with dried compressed air at a rate of 700 mL/min (Linde, Gas Benelux, The Netherlands). The broth was stirred constantly at 800 rpm, kept at a constant temperature of 30 °C and at a pH of 5 by automatic addition of 2 M KOH by an Applikon ADI 1030 Bio Controller.
Optical density was measured every hour at 660 nm with a Jenway 7200 spectrophotometer (Staffordshire, United Kingdom). For extracellular metabolite analysis 1 mL of the broth was centrifuged for at least 10 min at 13 000 rpm and the supernatant was analyzed with high-performance liquid chromatography (HPLC) using an Agilent 1100 (Agilent Technologies, Santa Clara, CA, USA) with an Aminex HPX-87H ion-exchange column (BioRad, Veenendaal, The Netherlands) operated with 5 mM sulfuric acid as mobile phase at a flow rate of 0.6 mL/min. The carbon dioxide and oxygen concentration in the gas outflow were analyzed by a Rosemount NGA 2000 analyzer (Baar, Switzerland), after cooling of the gas by a condenser (2 °C) and drying using a PermaPure Dryer (model MD 110-8P-4; Inacom Instruments, Veenendaal, The Netherlands). Biomass dry weight was measured 5–6 times by filtering (pore size 0.45 μm, Gelman Laboratory, Ann Arbor, MI, USA), as described previously.58 Sampling for RNA was done directly from the reactor in liquid nitrogen as described by Piper et al.(59) The cells were stored at −80 °C for maximally 2 weeks until further processing and RNA was extracted as previously described.23 An equivalent of 48 mg dry weight per sample was used.
At the same time points as the samples that were taken for RNA isolation, approximately 62.5 mg dry weight was sampled for the enzyme assays, stored at −20 °C in 4 mL aliquots and further process as previously described.58 Optical densities of the cultures at the moment of sampling for RNA analysis and enzyme assays can be found in Table S9.
Determination of In Vitro Enzyme Activities
On the day of the enzyme assays, frozen samples were thawed and prepared for assays as described by Postma et al.(58) Assays were performed using a U-3010 spectrophotometer (Hitachi, Tokio, Japan) at 30 °C and 340 nm as described by Jansen et al. (2005),60 with the exception of Pfk, which was performed according to Cruz et al. (2012).61 Reported activities are based on at least two technical replicates, measured with different cell extract concentrations. When necessary, cell extracts were diluted in 100 mM monopotassium phosphate buffer and with 2 mM magnesium chloride (pH 7.5), or in demineralized water when triose phosphate isomerase activity was measured. The protein concentration of the cell extracts was determined as described by Lowry et al.(62) using bovine serum albumin as a standard.
RNA Sequencing Simulation
To evaluate the watermarking methods, and to compare the markerQuant tool with traditional alignment, we generated artificial RNA-Seq reads from the native and watermarked sequences. Using the polyester R package,63 we simulated two conditions, in which the second condition has a 4-fold expression of each transcript compared to the first condition. We used the standard error_rate parameter of 0.005. Generated reads were paired-end, each end 100 bp in length.
RNA Sequencing and Data Analysis
RNA libraries and sequencing were performed by Novogene Bioinformatics Technology Co., Ltd. (Yueng Long, Hong Kong). Sequencing was performed using HiSeq 150 bp paired-end reads system using 250–300 bp insert strand-specific library. As described by Novogene, library preparation involved mRNA enrichment using oligo (dT) beads, followed by random fragmentation of the mRNA. cDNA was synthesized from mRNA using random hexamer primers and a second strand synthesis was done applying a custom second strand synthesis buffer (Illumina), dNTPs, RNase H, and DNA polymerase I. After adaptor ligation, double-stranded cDNA library was finalized by size selection and PCR enrichment and samples were sequenced.
Obtained data had an average of 23.08 M reads per sample (Table S8). To quantify the abundance of glycolytic genes with and without watermarks, a similar scheme as the k-mer algorithm of Gehrmann and co-workers38 was applied. Briefly, for each transcript, we identify sequence markers of 21bp that are unique in the transcript relative to the entire transcriptome and genome. With an exact matching algorithm, these markers can uniquely identify the transcript of origin of a read in RNA-Seq data. In contrast to the previous work,38 we did not remove overlapping markers (we did not remove redundant markers) but merged them into larger sequences in which any 21 bp k-mer would uniquely identify the transcript of origin. This allowed us to recover a higher percentage of reads per transcript. Gaps in these merged sequences that are not unique relative to the genome and transcriptome were ignored in the marker quantification step. As in previous work, we used an Aho-Corasick exact string-matching algorithm to quantify transcripts. Differential expression was performed using DE-Seq2.64 All RNA-seq sequences are available at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under GEO accession number: GSE135470.
RNA Sequencing Data Analysis Implementation and Code Availability
The marker discovery and quantification tools were developed in scala, and the entire pipeline is implemented in python using Snakemake.65 In addition to the k-mer method, a traditional alignment pipeline is also implemented in the markerQuant utility. All code, including an example data set, is available at https://github.com/thiesgehrmann/markerQuant.
Label Free Quantification (LFQ) by Shot-Gun Proteomics
Cultivation and Sampling
For proteomics analysis, the yeast strains pregrown to exponential phase in SMG in shake flask were used to inoculate fresh SMG flasks. Five ml of these cultures in midexponential phase were centrifuged for 10 min at 5000g at 4 °C and the cell pellet was directly stored at −80 °C. Cultures were performed in biological triplicates for strains with watermarked and native glycolysis. To verify if the difference in Gpm1 activity observed in bioreactor between the strains with watermarked and native glycolysis was also present in shake flask culture, Gpm1 activity was assayed in cell samples from the shake flasks. This additional analysis confirmed the lower specific activity of Gpm1 in the watermarked strain (Figure S9).
Protein Extraction and Trypsin Proteolytic Digestion
Cell pellets were resuspended in lysis buffer composed of 100 mM TEAB containing 1% SDS and phosphatase/protease inhibitors. Yeast cells were lysed by glass bead milling and thus shaken 10 times for 1 min with a bead beater alternated with 1 min rest on ice. Proteins were reduced by addition of 5 mM DTT and incubated for 1 h at 37 °C. Subsequently, the proteins were alkylated for 60 min at room temperature in the dark by addition of 50 mM acrylamide. Protein precipitation was performed by addition of four volumes of ice-cold acetone (−20 °C) and proceeded for 1 h at −20 °C. The proteins were solubilized using 100 mM ammonium bicarbonate. Proteolytic digestion was performed by Trypsin (Promega, Madison, WI), 1:100 enzyme to protein ratio, and incubated at 37 °C overnight. Solid phase extraction was performed with an Oasis HLB 96-well μElution plate (Waters, Milford, USA) to desalt the mixture. Eluates were dried using a SpeedVac vacuum concentrator at 45 °C. Dried peptides were resuspended in 3% ACN/0.01% TFA prior to MS-analysis to give an approximate concentration of 250 ng per μL.
Large-Scale Shotgun Proteomics
An aliquot corresponding to approximately 250 ng protein digest was analyzed using an one-dimensional shot-gun proteomics approach.66 Briefly, the samples were analyzed using a nanoliquid-chromatography system consisting of an EASY nano LC 1200, equipped with an Acclaim PepMap RSLC RP C18 separation column (50 μm × 150 mm, 2 μm), and an QE plus Orbitrap mass spectrometer (Thermo). The flow rate was maintained at 350 nL/min over a linear gradient from 6% to 26% solvent B over 45 min, followed by back equilibration to starting conditions. Data were acquired from 5 to 60 min. Solvent A was H2O containing 0.1% formic acid, and solvent B consisted of 80% acetonitrile in H2O and 0.1% formic acid. The Orbitrap was operated in data depended acquisition mode acquiring peptide signals from 385 to 1250 m/z at 70 K resolution. The top 10 signals were isolated at a window of 2.0 m/z and fragmented using a NCE of 28. Fragments were acquired at 17 K resolution.
Database Search, Label Free Quantification, and Visualization
Data were analyzed against the proteome database from Saccharomyces cerevisiae (Uniprot, strain ATCC 204508/S288C, Tax ID: 559292, July 2018) using PEAKS Studio X (Bioinformatics Solutions Inc.)2 allowing for 20 ppm parent ion and 0.02 m/z fragment ion mass error, 2 missed cleavages, acrylamide as fixed and methionine oxidation and N/Q deamidation as variable modifications. Peptide spectrum matches were filtered against 1% false discovery rates (FDR) and identifications with ≥2 unique peptides. Changes in protein abundances between both strains IMX1770 and IMX1771 using the label free quantification (LFQ) option provided by the PEAKS Q software tool (Bioinformatics Solutions Inc.).67 Protein areas were normalized to the total ion count (TIC) of the respective analysis run before performing pairwise comparison between the above-mentioned strains. LFQ was performed using protein identifications containing at least 2 unique peptides, which peptide identifications were filtered against 1% FDR. The significance method for evaluating the observed abundance changes was set to ANOVA. The abundances of the glycolytic enzymes were further visualized as bar graphs using Matlab2018b. The area of the biological triplicates were averaged and standard deviations were represented as error bars.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD016914.
Acknowledgments
We thank Marijke Luttik for assaying the glycolytic enzymes activity, Jordi Geelhoed for the confirmation of selective CRISPR/Cas editing by PCR and sequencing, and Pilar de la Torre for RNA samples processing. This work was supported by a consolidator grant from the European Research Council (ERC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00045.
Table S1: Codons used for codon optimization and their abundance; Table S2: Watermarking of the glycolytic and fermentative genes; Table S3: Comparing Alignment and markerQuant for differential quantification of watermarked and native glycolytic transcripts; Table S4: List of plasmids used in this study; Table S5: List of mutations in glycolytic expression cassettes and helper elements in the constructed strains; Table S6: gRNA sequence for selective gene editing; Table S7: List of primers used in this study; Table S8: RNA sequencing depth; Table S9: Sampling OD in batch cultures for RNA seq analysis; Figure S1: In silico comparison of two watermarking approaches for ADH1; Figure S2: Step-by-step watermarking of CDS workflow; Figure S3: Watermarking of FBA1 and ENO2; Figure S4: Genetic characterization of IMX2028 by whole genome sequencing; Figure S5: CO2 profiles of batch cultures with IMX1770 and IMX1771; Figure S6: Confirmation by Sanger sequencing of selective DNA editing; Figure S7: PCR confirmation IMX1770 and IMX1771; Figure S8: Label-free quantification (LFQ) of glycolytic protein abundance; Figure S9: Specific activity of Gpm1 (phosphoglucomutase) in shake flask cultures of IMX1770 and IMX1771 (PDF)
Author Contributions
# F.J.B., S.D., and P.D.-L. equally contributed to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Jiao S., and Goutte R. (2008) Code for encryption hiding data into genomic DNA of living organisms. In International Conference on Signal Processing Proceedings, ICSP, pp 2166–2169, IEEE.
- Arita M.; Ohashi Y. (2004) Secret signatures inside genomic DNA. Biotechnol. Prog. 20 (5), 1605–7. 10.1021/bp049917i. [DOI] [PubMed] [Google Scholar]
- Wong P. C.; Wong K.; Foote H. (2003) Organic data memory using the DNA approach. Commun. ACM 46 (1), 95–98. 10.1145/602421.602426. [DOI] [Google Scholar]
- Clelland C. T.; Risca V.; Bancroft C. (1999) Hiding messages in DNA microdots. Nature 399 (6736), 533–4. 10.1038/21092. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Glass J. I.; Lartigue C.; Noskov V. N.; Chuang R. Y.; Algire M. A.; Benders G. A.; Montague M. G.; Ma L.; Moodie M. M.; Merryman C.; Vashee S.; Krishnakumar R.; Assad-Garcia N.; Andrews-Pfannkoch C.; Denisova E. A.; Young L.; Qi Z. Q.; Segall-Shapiro T. H.; Calvey C. H.; Parmar P. P.; Hutchison C. A. 3rd; Smith H. O.; Venter J. C. (2010) Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329 (5987), 52–6. 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Heider D.; Barnekow A. (2008) DNA watermarks: a proof of concept. BMC Mol. Biol. 9, 40. 10.1186/1471-2199-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss M.; Daubert D.; Brunner K.; Kliche K.; Hammes U.; Leiherer A.; Wagner R. (2012) Embedding permanent watermarks in synthetic genes. PLoS One 7 (8), e42465 10.1371/journal.pone.0042465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N.; Kajiura H.; Takeno S.; Suzuki N.; Nakazawa Y. (2014) A watermarking system for labeling genomic DNA. Plant Biotechnol. 31, 241–248. 10.5511/plantbiotechnology.14.0609b. [DOI] [Google Scholar]
- Annaluru N.; Muller H.; Mitchell L. A.; Ramalingam S.; Stracquadanio G.; Richardson S. M.; Dymond J. S.; Kuang Z.; Scheifele L. Z.; Cooper E. M.; Cai Y.; Zeller K.; Agmon N.; Han J. S.; Hadjithomas M.; Tullman J.; Caravelli K.; Cirelli K.; Guo Z.; London V.; Yeluru A.; Murugan S.; Kandavelou K.; Agier N.; Fischer G.; Yang K.; Martin J. A.; Bilgel M.; Bohutski P.; Boulier K. M.; Capaldo B. J.; Chang J.; Charoen K.; Choi W. J.; Deng P.; DiCarlo J. E.; Doong J.; Dunn J.; Feinberg J. I.; Fernandez C.; Floria C. E.; Gladowski D.; Hadidi P.; Ishizuka I.; Jabbari J.; Lau C. Y.; Lee P. A.; Li S.; Lin D.; Linder M. E.; Ling J.; Liu J.; Liu J.; London M.; Ma H.; Mao J.; McDade J. E.; McMillan A.; Moore A. M.; Oh W. C.; Ouyang Y.; Patel R.; Paul M.; Paulsen L. C.; Qiu J.; Rhee A.; Rubashkin M. G.; Soh I. Y.; Sotuyo N. E.; Srinivas V.; Suarez A.; Wong A.; Wong R.; Xie W. R.; Xu Y.; Yu A. T.; Koszul R.; Bader J. S.; Boeke J. D.; Chandrasegaran S. (2014) Total synthesis of a functional designer eukaryotic chromosome. Science 344 (6179), 55–58. 10.1126/science.1249252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredens J.; Wang K.; de la Torre D.; Funke L. F. H.; Robertson W. E.; Christova Y.; Chia T.; Schmied W. H.; Dunkelmann D. L.; Beranek V.; Uttamapinant C.; Llamazares A. G.; Elliott T. S.; Chin J. W. (2019) Total synthesis of Escherichia coli with a recoded genome. Nature 569 (7757), 514–518. 10.1038/s41586-019-1192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius I. S.; Boeke J. D. (2018) Yeast 2.0-connecting the dots in the construction of the world’s first functional synthetic eukaryotic genome. FEMS Yeast Res. 10.1093/femsyr/foy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymond J. S.; Richardson S. M.; Coombes C. E.; Babatz T.; Muller H.; Annaluru N.; Blake W. J.; Schwerzmann J. W.; Dai J.; Lindstrom D. L.; Boeke A. C.; Gottschling D. E.; Chandrasegaran S.; Bader J. S.; Boeke J. D. (2011) Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 477 (7365), 471–476. 10.1038/nature10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers N. G.; Solis-Escalante D.; Luttik M. A.; Bisschops M. M.; Boonekamp F. J.; van den Broek M.; Pronk J. T.; Daran J. M.; Daran-Lapujade P. (2016) Pathway swapping: Toward modular engineering of essential cellular processes. Proc. Natl. Acad. Sci. U. S. A. 113 (52), 15060–15065. 10.1073/pnas.1606701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis-Escalante D.; Kuijpers N. G.; Barrajon-Simancas N.; van den Broek M.; Pronk J. T.; Daran J. M.; Daran-Lapujade P. (2015) A minimal set of glycolytic genes reveals strong redundancies in Saccharomyces cerevisiae central metabolism. Eukaryotic Cell 14 (8), 804–816. 10.1128/EC.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337 (6096), 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A. C. (2012) Decoding properties of tRNA leave a detectable signal in codon usage bias. Bioinformatics 28 (18), i340–i348. 10.1093/bioinformatics/bts403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya S.; Yamada Y.; Kudo Y.; Ikemura T. (1999) Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238 (1), 143–55. 10.1016/S0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- Novoa E. M.; Ribas de Pouplana L. (2012) Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 28 (11), 574–81. 10.1016/j.tig.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Sharp P. M.; Li W. H. (1986) An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24 (1–2), 28–38. 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- Presnyak V.; Alhusaini N.; Chen Y. H.; Martin S.; Morris N.; Kline N.; Olson S.; Weinberg D.; Baker K. E.; Graveley B. R.; Coller J. (2015) Codon optimality is a major determinant of mRNA stability. Cell 160 (6), 1111–24. 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S.; Frydman J. (2013) Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 20 (2), 237–43. 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazzo P.; Cervenansky C.; Marin M.; Reiss C.; Ehrlich R.; Deana A. (2002) Silent mutations affect in vivo protein folding in Escherichia coli. Biochem. Biophys. Res. Commun. 293 (1), 537–41. 10.1016/S0006-291X(02)00226-7. [DOI] [PubMed] [Google Scholar]
- Cannarozzi G.; Schraudolph N. N.; Faty M.; von Rohr P.; Friberg M. T.; Roth A. C.; Gonnet P.; Gonnet G.; Barral Y. (2010) A role for codon order in translation dynamics. Cell 141 (2), 355–67. 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- Hanson G.; Coller J. (2018) Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19 (1), 20–30. 10.1038/nrm.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G.; Murray A. W.; Tollervey D.; Plotkin J. B. (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324 (5924), 255–8. 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary J. V.; Hurst L. D. (2005) Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 6 (9), R75. 10.1186/gb-2005-6-9-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary J. V.; Hurst L. D. (2005) Biased codon usage near intron-exon junctions: selection on splicing enhancers, splice-site recognition or something else?. Trends Genet. 21 (5), 256–9. 10.1016/j.tig.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Cartegni L.; Chew S. L.; Krainer A. R. (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3 (4), 285–98. 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Nandy S. K.; Srivastava R. K. (2018) A review on sustainable yeast biotechnological processes and applications. Microbiol. Res. 207, 83–90. 10.1016/j.micres.2017.11.013. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Zhang Y.; Nielsen J. (2019) Synthetic Biology of Yeast. Biochemistry 58 (11), 1511–1520. 10.1021/acs.biochem.8b01236. [DOI] [PubMed] [Google Scholar]
- Richardson S. M.; Mitchell L. A.; Stracquadanio G.; Yang K.; Dymond J. S.; DiCarlo J. E.; Lee D.; Huang C. L. V.; Chandrasegaran S.; Cai Y.; Boeke J. D.; Bader J. S. (2017) Design of a synthetic yeast genome. Science 355 (6329), 1040–1044. 10.1126/science.aaf4557. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Wang Y.; Chen T.; Gao F.; Gong J.; Abramczyk D.; Walker R.; Zhao H.; Chen S.; Liu W.; Luo Y.; Muller C. A.; Paul-Dubois-Taine A.; Alver B.; Stracquadanio G.; Mitchell L. A.; Luo Z.; Fan Y.; Zhou B.; Wen B.; Tan F.; Wang Y.; Zi J.; Xie Z.; Li B.; Yang K.; Richardson S. M.; Jiang H.; French C. E.; Nieduszynski C. A.; Koszul R.; Marston A. L.; Yuan Y.; Wang J.; Bader J. S.; Dai J.; Boeke J. D.; Xu X.; Cai Y.; Yang H. (2017) Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science 355 (6329), eaaf4791. 10.1126/science.aaf4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z. X.; Li B. Z.; Mitchell L. A.; Wu Y.; Qi X.; Jin Z.; Jia B.; Wang X.; Zeng B. X.; Liu H. M.; Wu X. L.; Feng Q.; Zhang W. Z.; Liu W.; Ding M. Z.; Li X.; Zhao G. R.; Qiao J. J.; Cheng J. S.; Zhao M.; Kuang Z.; Wang X.; Martin J. A.; Stracquadanio G.; Yang K.; Bai X.; Zhao J.; Hu M. L.; Lin Q. H.; Zhang W. Q.; Shen M. H.; Chen S.; Su W.; Wang E. X.; Guo R.; Zhai F.; Guo X. J.; Du H. X.; Zhu J. Q.; Song T. Q.; Dai J. J.; Li F. F.; Jiang G. Z.; Han S. L.; Liu S. Y.; Yu Z. C.; Yang X. N.; Chen K.; Hu C.; Li D. S.; Jia N.; Liu Y.; Wang L. T.; Wang S.; Wei X. T.; Fu M. Q.; Qu L. M.; Xin S. Y.; Liu T.; Tian K. R.; Li X. N.; Zhang J. H.; Song L. X.; Liu J. G.; Lv J. F.; Xu H.; Tao R.; Wang Y.; Zhang T. T.; Deng Y. X.; Wang Y. R.; Li T.; Ye G. X.; Xu X. R.; Xia Z. B.; Zhang W.; Yang S. L.; Liu Y. L.; Ding W. Q.; Liu Z. N.; Zhu J. Q.; Liu N. Z.; Walker R.; Luo Y.; Wang Y.; Shen Y.; Yang H.; Cai Y.; Ma P. S.; Zhang C. T.; Bader J. S.; Boeke J. D.; Yuan Y. J. (2017) “Perfect” designer chromosome V and behavior of a ring derivative. Science 355 (6329), eaaf4704. 10.1126/science.aaf4704. [DOI] [PubMed] [Google Scholar]
- Mitchell L. A.; Wang A.; Stracquadanio G.; Kuang Z.; Wang X.; Yang K.; Richardson S.; Martin J. A.; Zhao Y.; Walker R.; Luo Y.; Dai H.; Dong K.; Tang Z.; Yang Y.; Cai Y.; Heguy A.; Ueberheide B.; Fenyo D.; Dai J.; Bader J. S.; Boeke J. D. (2017) Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355 (6329), eaaf4831. 10.1126/science.aaf4831. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Li B. Z.; Zhao M.; Mitchell L. A.; Xie Z. X.; Lin Q. H.; Wang X.; Xiao W. H.; Wang Y.; Zhou X.; Liu H.; Li X.; Ding M. Z.; Liu D.; Zhang L.; Liu B. L.; Wu X. L.; Li F. F.; Dong X. T.; Jia B.; Zhang W. Z.; Jiang G. Z.; Liu Y.; Bai X.; Song T. Q.; Chen Y.; Zhou S. J.; Zhu R. Y.; Gao F.; Kuang Z.; Wang X.; Shen M.; Yang K.; Stracquadanio G.; Richardson S. M.; Lin Y.; Wang L.; Walker R.; Luo Y.; Ma P. S.; Yang H.; Cai Y.; Dai J.; Bader J. S.; Boeke J. D.; Yuan Y. J. (2017) Bug mapping and fitness testing of chemically synthesized chromosome X. Science 355 (6329), eaaf4706. 10.1126/science.aaf4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Zhao G.; Luo Z.; Lin Y.; Wang L.; Guo Y.; Wang A.; Jiang S.; Jiang Q.; Gong J.; Wang Y.; Hou S.; Huang J.; Li T.; Qin Y.; Dong J.; Qin Q.; Zhang J.; Zou X.; He X.; Zhao L.; Xiao Y.; Xu M.; Cheng E.; Huang N.; Zhou T.; Shen Y.; Walker R.; Luo Y.; Kuang Z.; Mitchell L. A.; Yang K.; Richardson S. M.; Wu Y.; Li B. Z.; Yuan Y. J.; Yang H.; Lin J.; Chen G. Q.; Wu Q.; Bader J. S.; Cai Y.; Boeke J. D.; Dai J. (2017) Engineering the ribosomal DNA in a megabase synthetic chromosome. Science 355 (6329), eaaf3981. 10.1126/science.aaf3981. [DOI] [PubMed] [Google Scholar]
- Martinez M. A.; Jordan-Paiz A.; Franco S.; Nevot M. (2019) Synonymous genome recoding: a tool to explore microbial biology and new therapeutic strategies. Nucleic Acids Res. 47 (20), 10506–10519. 10.1093/nar/gkz831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann T.; Pelkmans J. F.; Ohm R. A.; Vos A. M.; Sonnenberg A. S. M.; Baars J. J. P.; Wosten H. A. B.; Reinders M. J. T.; Abeel T. (2018) Nucleus-specific expression in the multinuclear mushroom-forming fungus Agaricus bisporus reveals different nuclear regulatory programs. Proc. Natl. Acad. Sci. U. S. A. 115 (17), 4429–4434. 10.1073/pnas.1721381115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.; Kazana E.; Bellanger N.; Singh T.; Tuite M. F.; von der Haar T. (2014) Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 33 (1), 21–34. 10.1002/embj.201385651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T.; Waldman Y. Y.; Kupiec M.; Ruppin E. (2010) Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. U. S. A. 107 (8), 3645–50. 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P.; Ding Y.; Niemczyk M.; Kudla G.; Plotkin J. B. (2013) Rate-limiting steps in yeast protein translation. Cell 153 (7), 1589–601. 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopperschlager G.; Bar J.; Nissler K.; Hofmann E. (1977) Physicochemical parameters and subunit composition of yeast phosphofructokinase. Eur. J. Biochem. 81 (2), 317–25. 10.1111/j.1432-1033.1977.tb11954.x. [DOI] [PubMed] [Google Scholar]
- Gorter de Vries A. R.; van Couwenberg L. G. F.; Van den Broek M.; De la Torre Cortes P.; ter Horst J.; Pronk J. T.; Daran J. G. (2019) Allele-specific genome editing using CRISPR–Cas9 is associated with loss of heterozygosity in diploid yeast. Nucleic Acids Res. 47 (3), 1362–1372. 10.1093/nar/gky1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson S. M.; Wheelan S. J.; Yarrington R. M.; Boeke J. D. (2006) GeneDesign: rapid, automated design of multikilobase synthetic genes. Genome Res. 16 (4), 550–6. 10.1101/gr.4431306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riba A.; Di Nanni N.; Mittal N.; Arhne E.; Schmidt A.; Zavolan M. (2019) Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc. Natl. Acad. Sci. U. S. A. 116 (30), 15023–15032. 10.1073/pnas.1817299116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entian K. D., and Kötter P. (2007) Yeast genetic strain and plasmid collections. In Yeast Gene Analysis, 2nd ed. (Stansfield I., and Stark M. J. R., Eds.), Vol. 36, pp 629–666, Academic Press, Elsevier, Amsterdam. [Google Scholar]
- Salazar A. N.; Gorter de Vries A. R.; van den Broek M.; Wijsman M.; de la Torre Cortes P.; Brickwedde A.; Brouwers N.; Daran J. G.; Abeel T. (2017) Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113–7D. FEMS Yeast Res. 10.1093/femsyr/fox074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp J. F.; van den Broek M.; Datema E.; de K. S.; Bosman L.; Luttik M. A.; Daran-Lapujade P.; Vongsangnak W.; Nielsen J.; Heijne W. H.; Klaassen P.; Paddon C. J.; Platt D.; Kotter P.; van Ham R. C.; Reinders M. J.; Pronk J. T.; de R. D.; Daran J. M. (2012) De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113–7D, a model for modern industrial biotechnology. Microb. Cell Fact. 11, 36. 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk J. T. (2002) Auxotrophic yeast strains in fundamental and applied research. Appl. Environ. Microbiol. 68 (5), 2095–2100. 10.1128/AEM.68.5.2095-2100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis-Escalante D.; Kuijpers N. G.; Bongaerts N.; Bolat I.; Bosman L.; Pronk J. T.; Daran J. M.; Daran-Lapujade P. (2013) amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res. 13 (1), 126–139. 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. E.; DeLoache W. C.; Cervantes B.; Dueber J. E. (2015) A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 4 (9), 975–986. 10.1021/sb500366v. [DOI] [PubMed] [Google Scholar]
- Mans R.; van Rossum H. M.; Wijsman M.; Backx A.; Kuijpers N. G.; van den Broek M.; Daran-Lapujade P.; Pronk J. T.; van Maris A. J.; Daran J. M. (2015) CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 10.1093/femsyr/fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D.; Woods R. A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96. 10.1016/S0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- DiCarlo J. E.; Norville J. E.; Mali P.; Rios X.; Aach J.; Church G. M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336–4343. 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 (16), 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp J. F.; van den Broek M. A.; Geertman J. M.; Reinders M. J.; Daran J. M.; de R. D. (2012) De novo detection of copy number variation by co-assembly. Bioinformatics 28 (24), 3195–3202. 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
- Postma E.; Verduyn C.; Scheffers W. A.; van Dijken J. P. (1989) Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 55 (2), 468–477. 10.1128/AEM.55.2.468-477.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M. D.; Daran-Lapujade P.; Bro C.; Regenberg B.; Knudsen S.; Nielsen J.; Pronk J. T. (2002) Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J. Biol. Chem. 277 (40), 37001–8. 10.1074/jbc.M204490200. [DOI] [PubMed] [Google Scholar]
- Jansen M. L. A.; Diderich J. A.; Mashego M. R.; Hassane A.; de Winde J. H.; Daran-Lapujade P.; Pronk J. T. (2005) Prolonged selection in aerobic, glucose-limited chemostat cultures of Saccharomyces cerevisiae causes a partial loss of glycolytic capacity. Microbiology 151 (5), 1657–1669. 10.1099/mic.0.27577-0. [DOI] [PubMed] [Google Scholar]
- Cruz A. L.; Verbon A. J.; Geurink L. J.; Verheijen P. J.; Heijnen J. J.; van Gulik W. M. (2012) Use of sequential-batch fermentations to characterize the impact of mild hypothermic temperatures on the anaerobic stoichiometry and kinetics of Saccharomyces cerevisiae. Biotechnol. Bioeng. 109 (7), 1735–1744. 10.1002/bit.24454. [DOI] [PubMed] [Google Scholar]
- Lowry O. H.; Rosebrough H. J.; Farr A. L.; Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- Frazee A. C.; Jaffe A. E.; Langmead B.; Leek J. T. (2015) Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics 31 (17), 2778–84. 10.1093/bioinformatics/btv272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I.; Huber W.; Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J.; Rahmann S. (2012) Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28 (19), 2520–2. 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- Kocher T.; Pichler P.; Swart R.; Mechtler K. (2012) Analysis of protein mixtures from whole-cell extracts by single-run nanoLC-MS/MS using ultralong gradients. Nat. Protoc. 7 (5), 882–90. 10.1038/nprot.2012.036. [DOI] [PubMed] [Google Scholar]
- Valikangas T.; Suomi T.; Elo L. L. (2017) A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Briefings Bioinf. 19 (6), 1344–1355. 10.1093/bib/bbx054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD016914.