

Abstract
Psoriasis is a common skin disorder characterized by hyperproliferation and aberrant differentiation of epidermal keratinocytes and inflammation. We previously demonstrated that phosphatidylglycerol (PG) can regulate keratinocyte function and suppress skin inflammation. Based on data suggesting that PG can inhibit toll-like receptor (TLR) activation induced by microorganisms and their components, we determined whether PG can inhibit TLR activation in response to anti-microbial peptides. These peptides, which are up-regulated in psoriasis, are known to function as danger-associated molecular patterns (DAMPs) to activate TLRs and the innate immune system. Since S100A9 is elevated in psoriatic skin and in animal models of psoriasis, we selected S100A9 as a representative anti-microbial peptide DAMP. In exciting results we show that in primary keratinocytes and a macrophage cell line PG suppressed inflammatory mediator production induced by recombinant S100A9 functioning through both TLR2 and TLR4. In addition, PG, but not phosphatidylcholine, inhibited downstream S100A9-elicited TLR2 and nuclear factor-κB activation. These results, to our knowledge previously unreported, demonstrate PG’s ability to inhibit DAMP-induced TLR activation, thereby reducing inflammatory signals. In addition, topical PG ameliorated skin lesions and inflammation in a mouse model of psoriasis. Together these results suggest the possibility of developing PG as a therapy for psoriasis.
INTRODUCTION
Keratinocytes comprise the major cell type of the epidermis to mediate its important barrier function. We have previously shown that the water and glycerol channel aquaporin-3 (AQP3) and the lipid-metabolizing enzyme phospholipase D2 (PLD2) physically and functionally associate in epidermal keratinocytes (Zheng and Bollag, 2003, Zheng et al., 2003). PLD2 can convert the glycerol transported by AQP3 into the phospholipid phosphatidylglycerol (PG) (Zheng et al., 2003), which is able to normalize keratinocyte function, by inhibiting or enhancing proliferation and/or differentiation depending on the proliferative status of the cells and the fatty acids comprising the PG (Bollag et al., 2007, Xie et al., 2014). Based on these data we proposed the use of PG to treat psoriasis, a skin disease characterized by hyperproliferation and abnormal differentiation of keratinocytes (Xie et al., 2014). However, another characteristic of psoriasis is (sterile) inflammation with extensive immune cell infiltration into the skin. Although we have previously shown that PG derived from soy is able to suppress these parameters in a contact irritant ear edema mouse model (Xie et al., 2018), the mechanism by which PG affects skin inflammation remains unknown.
Toll-like receptors (TLRs) are pattern recognition receptors that respond to microorganisms and their components, or pathogen-associated molecular patterns (PAMPs), to induce immune system activation. Nevertheless, many of these components also comprise non-pathogenic microbes, for which mounting an immune response is counter-productive. Matzinger and colleagues have proposed the “Danger Hypothesis,” the idea that signals associated with cell injury are required for induction of a complete immune response (Matzinger, 1994, Pradeu and Cooper, 2012, Seong and Matzinger, 2004), since such damage is likely to reflect pathogenic effects of the microorganism(s) present. Thus, endogenous proteins released by cell disruption, the so-called danger- or damage-associated molecular patterns (DAMPs), can also activate TLRs and induce an immune response, including inflammation (reviewed in (Erridge, 2010)).
Data in the lung have demonstrated an ability of PG found in pulmonary surfactant to inhibit the activation of TLR2 and TLR4 by PAMPs (Kandasamy et al., 2011, Kuronuma et al., 2009, Numata et al., 2010, Numata et al., 2012, Numata et al., 2013). In particular, PG effectively reduces TLR2-mediated arachidonic acid release from human and mouse macrophages treated with Mycoplasma pneumonia membranes (Kandasamy et al., 2011) and inhibits IL8 production in BEAS2B human bronchial epithelial cells stimulated with respiratory syncytial virus (Numata et al., 2013), as well as Type IIA secretory phospholipase A2 levels and activity in macrophages stimulated with endotoxin, a PAMP (Wu et al., 2003). Importantly, PG also limits the lung damage induced by microbial infection (Numata et al., 2010, Numata et al., 2012, Numata et al., 2013). Accessory proteins involved in the binding of microbial products to TLR2 and TLR4 (He et al., 2016, van Bergenhenegouwen et al., 2013), MD2 and CD14 are able to bind PG, suggesting that this may be the mechanism by which PG blocks inflammation (Kuronuma et al., 2009).
Psoriasis is a non-infectious immune-mediated disease (Davidovici et al., 2010). Indeed, despite a compromised epidermal barrier, the skin of psoriatic patients is not especially susceptible to infection (Niyonsaba et al., 2017), likely because several anti-microbial proteins, such as LL37 (cathelicidin) and β-defensins, are dramatically up-regulated in psoriasis (Niyonsaba et al., 2017). In fact, the levels of one such anti-microbial protein, S100A9, correlate with psoriasis severity, and successful treatment of the disease results in decreased serum concentrations of this protein (Nakajima et al., 2011, Racz et al., 2011, Waite and Skokos, 2012). Interestingly, this protein, as well as other anti-microbial proteins, are reported to act as DAMPs to activate TLR2 and TLR4 (Erridge, 2010, McInturff et al., 2005). We hypothesized that PG might inhibit DAMP-induced activation of TLRs, and we tested this idea using a representative DAMP, S100A9. Our results show that indeed S100A9 induced inflammatory mediator production through activation of TLR2 and 4 and that PG blocked this stimulation.
RESULTS
S100A9-induced inflammatory mediator expression in keratinocytes was inhibited by PG
Psoriasis, an immune-mediated inflammatory skin disorder (Davidovici et al., 2010), is characterized by extensive inflammation in the absence of infection. We recently demonstrated that PG inhibits skin inflammation induced by a contact irritant applied to the ears of mice in vivo (Xie et al., 2018). We reasoned that the ability of the contact irritant to elicit inflammation might be related in part to its capacity to induce anti-microbial peptides (Gebhardt et al., 2002, McNeill and Hogg, 2014), which are known to function as DAMPs to activate TLRs and stimulate the innate immune system to induce inflammation (Erridge, 2010). Therefore, we investigated the ability of PG to inhibit inflammatory mediator expression induced by S100A9, a recognized anti-microbial peptide DAMP (Chen et al., 2015, Kang et al., 2015, Moles et al., 2014, Schelbergen et al., 2012, Schiopu and Cotoi, 2013, Seong and Matzinger, 2004) known to be elevated in psoriasis (Nakajima et al., 2011, Racz et al., 2011, Waite and Skokos, 2012). Keratinocytes were stimulated with recombinant S100A9 in the presence and absence of 100μg/mL of the PG species, dioleoylphosphatidylglycerol (DOPG). As shown in Figure 1, DOPG inhibited the expression of interleukin-1β (IL1β), interleukin-6 (IL6) and tumor necrosis factor-α (TNFα in keratinocytes stimulated with recombinant S100A9, returning expression of the inflammatory mediators to essentially control levels. In additional experiments, we found that the effect of S100A9 on inflammatory mediator expression was dose-dependent and was likely mediated by the recombinant S100A9 itself rather than a bacterial contaminant (Supplemental Figure S1). The inhibitory effect of DOPG was also dose-dependent (data not shown).
Figure 1. DOPG inhibits expression of inflammatory mediators induced by the DAMP, recombinant S100A9, in keratinocytes and a macrophage cell line.
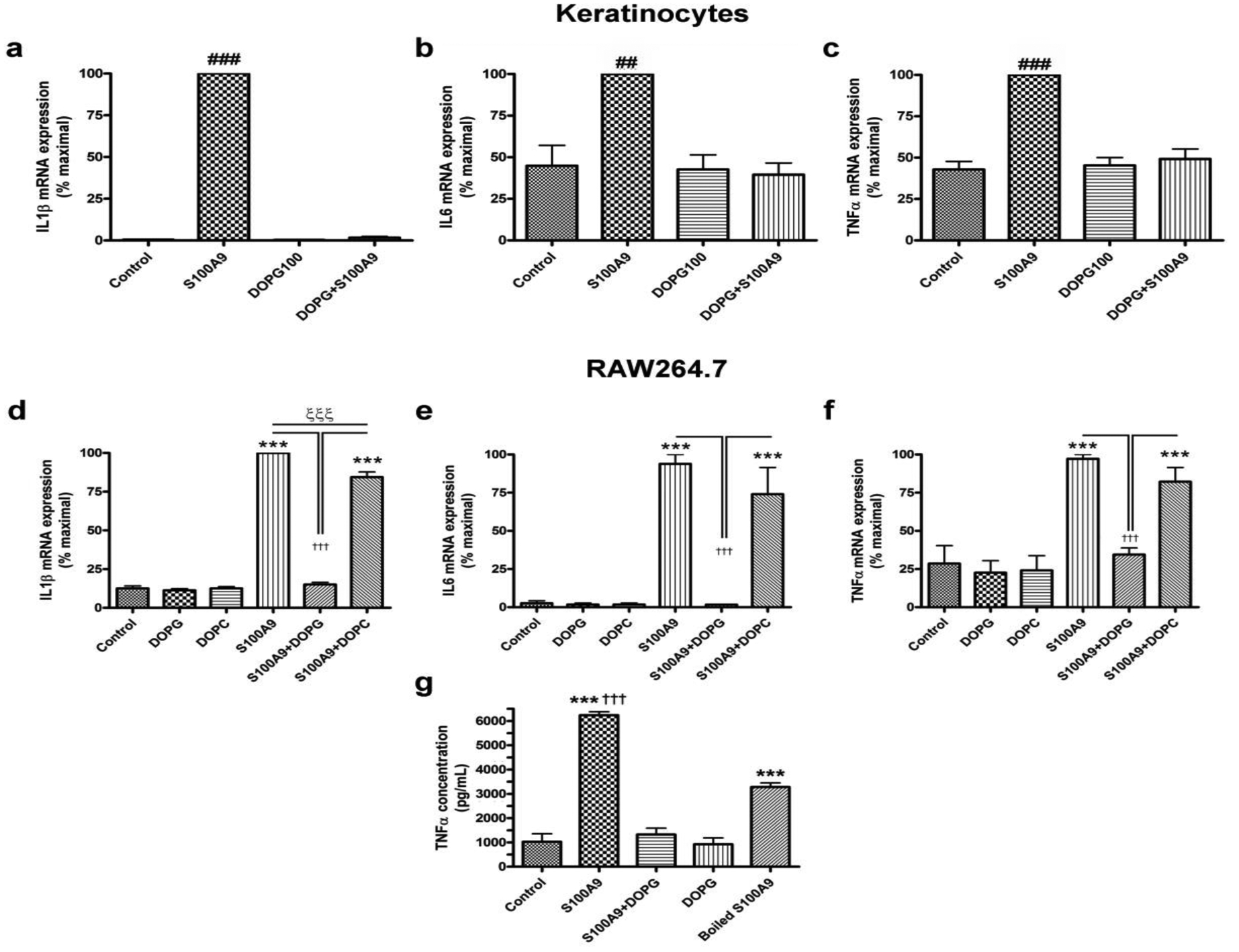
(a-c) Keratinocytes or (d-g) RAW264.7 cells were treated with 2.5μg/mL S100A9 in the presence and absence of 100μg/mL DOPG or DOPC for 2 hours. RNA was then isolated and the expression of the inflammatory mediators, (a and d) IL1β, (b and e) IL6 and (c and f) TNFα monitored by quantitative RT-PCR with GAPDH used as the housekeeping gene. Results represent the means ± SEM of 3 separate experiments; ##p<0.01, ###p<0.001 versus all other conditions; *p<0.05, ***p<0.001 versus control; †††p<0.001 as indicated. (g) Media collected from RAW264.7 cells treated with or without 2μg/mL recombinant S100A9 (or S100A9 that had been boiled) in the presence and absence of 100μg/mL DOPG were analyzed by ELISA for TNFα levels. Results represent the means ± SEM of 3 separate experiments; ***p<0.001 versus the control; †††p<0.001 versus boiled S100A9.
DOPG, but not dioleoylphosphatidylcholine (DOPC), inhibited S100A9-induced inflammatory mediator expression in a macrophage cell line
Although keratinocytes and keratinocyte-produced inflammatory mediators likely play an important role in psoriasis, the disease is considered an immune-mediated disease (Brotas et al., 2012, Lowes et al., 2013, Sabat and Wolk, 2011). We, therefore, wished to investigate the action of PG on inflammatory mediator expression in an immune cell and selected the macrophage cell line, RAW264.7. This choice was based on (1) data in the literature suggesting the importance of macrophages in psoriasis(Clark and Kupper, 2006, Stratis et al., 2006, Wang et al., 2006) and (2) the fact that PG is thought to act via CD14, a PG-binding protein(Kandasamy et al., 2016, Kuronuma et al., 2009) and TLR2 co-receptor(van Bergenhenegouwen et al., 2013), which is abundantly expressed in macrophages (Jersmann, 2005). RAW264.7 cells were stimulated with recombinant S100A9 and monitored for the expression of inflammatory mediators (in particular, IL1β, IL6 and TNFα) in the presence and absence of DOPG. We found that in these cells S100A9 increased inflammatory mediator expression, and that DOPG inhibited these increases (Figure 1d–f). In the experiment shown we included dioleoylphosphatidylcholine (DOPC) as a control and showed that there was little or no effect of this phospholipid on S100A9-induced inflammatory mediator expression, indicating the specificity of PG’s effect.
To determine if the changes in mRNA levels were translated into alterations in protein, we treated RAW264.7 cells with S100A9 in the presence and absence of DOPG and measured TNFα concentrations in the medium using an ELISA. We initially focused on TNFα because of the clear role of this cytokine in psoriasis, such that the disease is often successfully treated with anti-TNFα agents (Brotas et al., 2012). We observed that S100A9 increased the amount of TNFα secreted into the medium by approximately 6-fold, whereas co-stimulation with DOPG resulted in a complete abrogation of the increased TNFα concentrations (Figure 1g). In addition, and in contrast to the relative lack of effect with regard to TNFα mRNA levels, boiling the recombinant protein significantly inhibited its ability to induce TNFα secretion (Figure 1g). Note that boiling S100A9 also reduced its ability to induce mRNA expression of IL1β and IL6 (Supplemental Figure S2). These results suggest that the S100A9 protein, rather than a lipid-based contaminant, was acting to induce this inflammatory mediator response.
In additional experiments we used a multiplex inflammation array to determine the effects of a 2-hour exposure to S100A9 in the presence and absence of DOPG on thirteen inflammatory markers in RAW264.7 cells. Although the majority of tested cytokines were below the limit of detection after this short stimulation, results were obtained for TNFα, IL6 and interferon-β (IFNβ). S100A9 significantly elevated levels of each cytokine and DOPG inhibited this increase (Supplemental Figure S3).
Inflammatory mediator expression appeared to be mediated by both TLR2 and TLR4 in keratinocytes and RAW264.7 cells
S100A9 is reported to activate both TLR2 and TLR4 (Erridge, 2010). To determine which TLR seemed to be mediating the effects of this DAMP in our study, we examined the effect of a TLR2 and a TLR4 antagonist on inflammatory mediator expression induced by recombinant S100A9 in keratinocytes and in RAW264.7 cells. In keratinocytes the TLR1/2 antagonist CU-CPT22 inhibited the ability of S100A9 to increase the expression of IL1β and IL6 (Figure 2a and b) but had no significant effect on the S100A9-induced expression of TNFα (Figure 2c), suggesting that TLR2 can be activated by the S100A9 DAMP. This interpretation was largely supported by studies in keratinocytes isolated from TLR2 knockout relative to wild-type mice: TLR2 knockout keratinocytes exhibited a reduced S100A9-mediated induction of IL6 expression, with no significant difference in the S100A9-elicited TNFα mRNA levels observed (Supplemental Figure S4).
Figure 2. A TLR1/2 and/or TLR4 antagonist inhibits S100A9-induced inflammatory mediator expression in keratinocytes and the macrophage cell line.
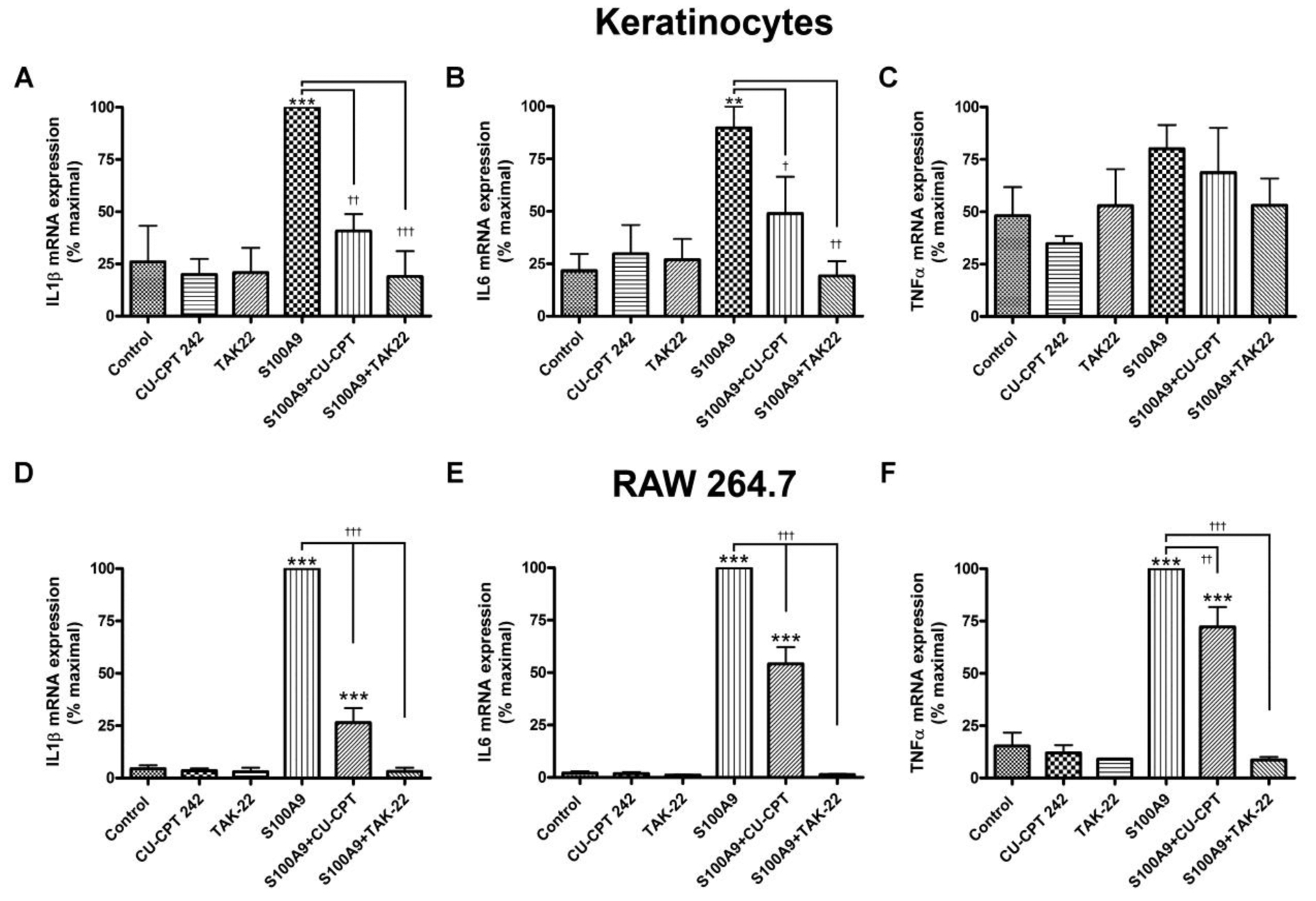
(a-c) Keratinocytes or (d-f) RAW264.7 cells were treated with or without 2μg/mL recombinant S100A9 in the presence and absence of 25μM CU-CPT22, a TLR1/2 antagonist, or 10μM TAK242, a TLR4 antagonist, for 2 hours. RNA was then isolated and the expression of the inflammatory mediators, (a and d) IL-1β, (b and e) IL6 and (c and f) TNFα was monitored by quantitative RT-PCR with GAPDH used as the housekeeping gene. Results represent the means ± SEM of 4–5 separate experiments; **p<0.01, ***p<0.001 versus the control; † p<0.05, †† p<0.01, and †††p<0.001 as indicated.
In contrast, the TLR4 antagonist TAK242 inhibited the effect of S100A9 on the keratinocyte expression of all three cytokines, IL1β, IL6 and TNFα (Figure 2a–c), indicating that TLR4 can also mediate the inflammatory effects of S100A9 and may, in fact, be more important in the response. Again, this result was supported by data obtained with a TLR4-neutralizing antibody, which inhibited S100A9-induced IL1β and IL6 expression, although it had a minimal effect of TNFα mRNA levels in keratinocytes (Supplemental Figure S5). In the macrophage cell line both the TLR1/2 and the TLR4 antagonist significantly decreased the mRNA levels of IL1β, IL6 and TNFα (Figure 2d–f), suggesting the likelihood that S100A9 can induce the expression of inflammatory mediators through either TLR2 or TLR4 in these cells and that PG can also inhibit the S100A9-induced activation of either TLR.
DOPG inhibited p65-NFκB activation by S100A9
Nuclear factor-κB (NFκB) is downstream of TLR activation. To examine the ability of DOPG to inhibit TLR activation we examined its ability to reduce p65-NFκB phosphorylation at serine536, a marker of its activation. We found that S100A9 triggered p65-NFκB phosphorylation/activation and DOPG blocked this response (Figure 3a and b), although DOPC had no effect. Consistent with western analysis, DOPG, but not DOPC, also inhibited the translocation of p65-NFκB into the nucleus induced by S100A9 (Figure 3c and Supplemental Figure S6); translocation of NFκB into the nucleus is considered to be a marker of stimulated NFκB activity (Wan and Lenardo, 2010). Thus, p65-NFκB localized predominantly to the cytoplasm in control cells and in cells treated with DOPG or DOPC alone. Exposure to S100A9 induced the translocation of p65-NFκB to the nucleus, and this response was unaffected by DOPC. However, DOPG inhibited the nuclear redistribution, again supporting the idea that DOPG inhibited TLR activation in response to S100A9. These results were confirmed by quantitation of the percentage of cells with p65-NFκB-positive nuclei (Figure 3d).
Figure 3. DOPG, but not DOPC, inhibits p65-NFκB activation by recombinant S100A9 protein.
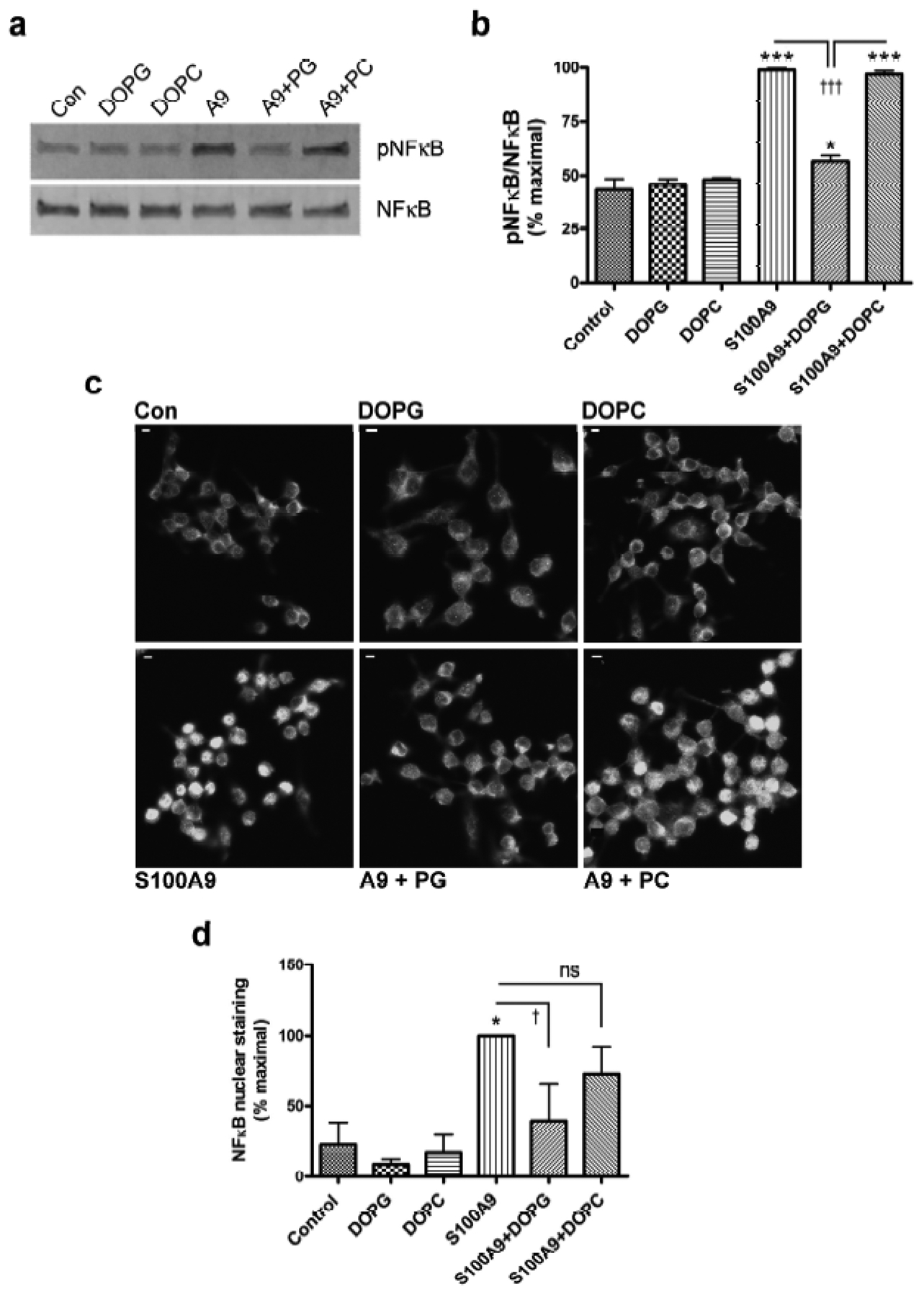
RAW264.7 cells were treated with or without 2μg/mL recombinant S100A9 (A9) in the presence and absence of 100μg/mL DOPG or 100μg/mL DOPC for 30 minutes. Cells were harvested and the phosphorylation (activation) status of p65-NFκB determined as described in Methods. (a) A representative Western blot is shown. (b) The cumulative results from 3 separate experiments are presented as means ± SEM. (c) RAW264.7 cells plated on coverslips were treated with or without 2μg/mL recombinant S100A9 (A9) in the presence and absence of 100μg/mL DOPG or DOPC, as indicated, for 60 minutes. Immunocytochemistry for p65-NFκB was performed as described in Methods and shown in grayscale (scale bar = 5μm). The figures are representative of 3 separate experiments, all showing similar results. (d) Quantification of nuclear staining by two observers blinded to the experiment and counted in at least two random micrographs; results from each observer were averaged and expressed as the percent maximal value. Values represent the means ± SEM of 3 independent experiments and were analyzed by ANOVA with Student-Newman-Keuls post-hoc tests; *p<0.05, ***p<0.001 versus the control; †p<0.05, ††††p<0.001 as indicated.
DOPG inhibited S100A9-induced TLR activation in a TLR2 reporter cell line
Further support for the importance of S100A9-induced TLR2 and NFκB activation is provided by results using a TLR2 reporter cell line, HEK-Blue-TLR2 cells. These cells stably express the genes for human TLR2, CD14, MD-2 and a reporter construct in which an interferon-β minimal promoter fused to five NFκB and activating protein-1 (AP-1) binding sites drives transcription of secreted embryonic alkaline phosphatase (SEAP). Stimulation of these cells with agents that activate TLR2 and NFκB/AP-1 results in secretion of SEAP into the medium, which can be measured using an appropriate chromagen as determined by absorbance at 620 nm. Using these cells we initially verified that they responded in a dose-dependent manner to a synthetic triacylated lipopeptide TLR2 agonist, Pam3CSK4 (data not shown). We then demonstrated that reporter activity could be induced by S100A9 and that this increase was inhibited by DOPG, but not DOPC (Figure 4a). In additional studies we demonstrated that β-defensin-2, another anti-microbial DAMP up-regulated in psoriasis (van Bergenhenegouwen et al., 2013), also stimulated TLR2 and DOPG prevented this activation (Figure 4b). Thus, the effect of PG was not specific to S100A9. Thus, these data represent to our knowledge previously unreported findings to show that PG can inhibit inflammatory mediator production induced by various DAMPs.
Figure 4. DOPG inhibits anti-microbial peptide DAMP-induced TLR activation in a TLR2 reporter cell line.
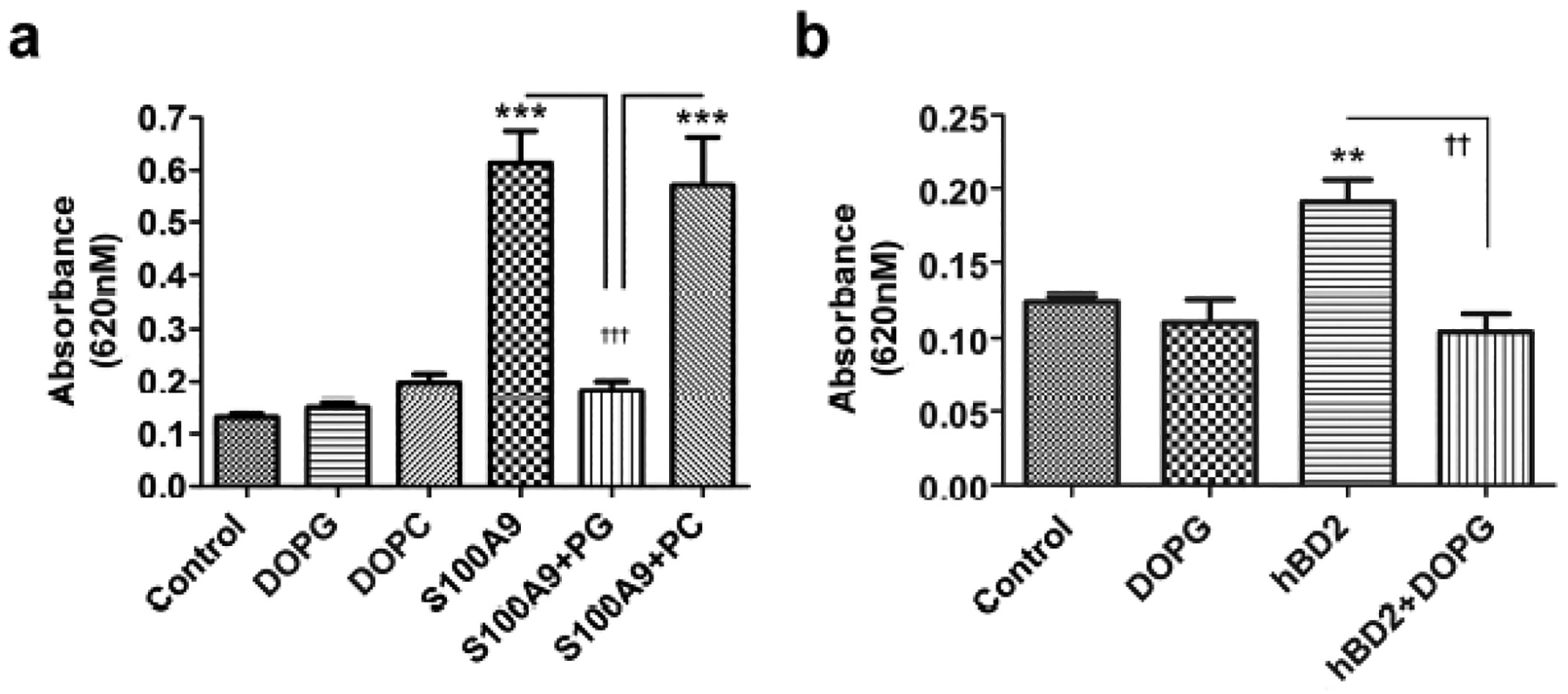
(a) HEK-Blue-hTLR2 cells were incubated with or without S100A9 (10μg/mL) in the presence and absence of 100μg/mL DOPG or DOPC for 24 h or (b) with and without human β-defensin-2 (hBD2, 25μg/mL) in the presence and absence of 100μg/mL DOPG in the HEK-Blue detection medium for 18 h. SEAP activity was measured as absorbance at 620 nm. Values represent the means ± SEM from 3 separate experiments; ***p<0.001 versus the control; ††p<0.01, †††p<0.001 as indicated.
DOPG inhibited inflammation and skin lesion development in the imiquimod-induced mouse model of psoriasis
Based on our in vitro data, we predicted that PG would inhibit inflammation and skin lesion development in mice treated with imiquimod (IMQ) to induce a psoriasis phenotype. Indeed, we found that the IMQ-induced psoriasiform phenotype (Figure 5a) was inhibited by DOPG applied topically in the form of liposomes. In addition, at days 3 and 4 of treatment, DOPG significantly reduced the Psoriasis Area and Severity Index (PASI) value, a measure of the appearance and area of the skin lesions, and tended to maintain a reduced PASI score at day 5 (Figure 5b). DOPG significantly inhibited the increase in ear thickness observed with IMQ treatment and returned the IMQ-induced increase in ear biopsy weight to a value that was not significantly different from the control (Figure 5c and d). DOPG also reduced the epidermal hyperplasia (Figure 5e and f) and increase in epidermal TNFα immunoreactivity (Figure 5g and h) elicited by IMQ. Aldara® is known to function through TLR7 and TLR8 (Flutter and Nestle, 2013, Schon and Schon, 2008); in vitro DOPG only minimally inhibited, to a value not statistically different from control DOPC, keratinocyte inflammatory mediator expression induced by a TLR7/8 agonist (Supplemental Figure S7). This result suggests that the ability of DOPG to improve psoriasiform lesions likely did not result from inhibition of the generation of the model through TLR7 and TLR8.
Figure 5.
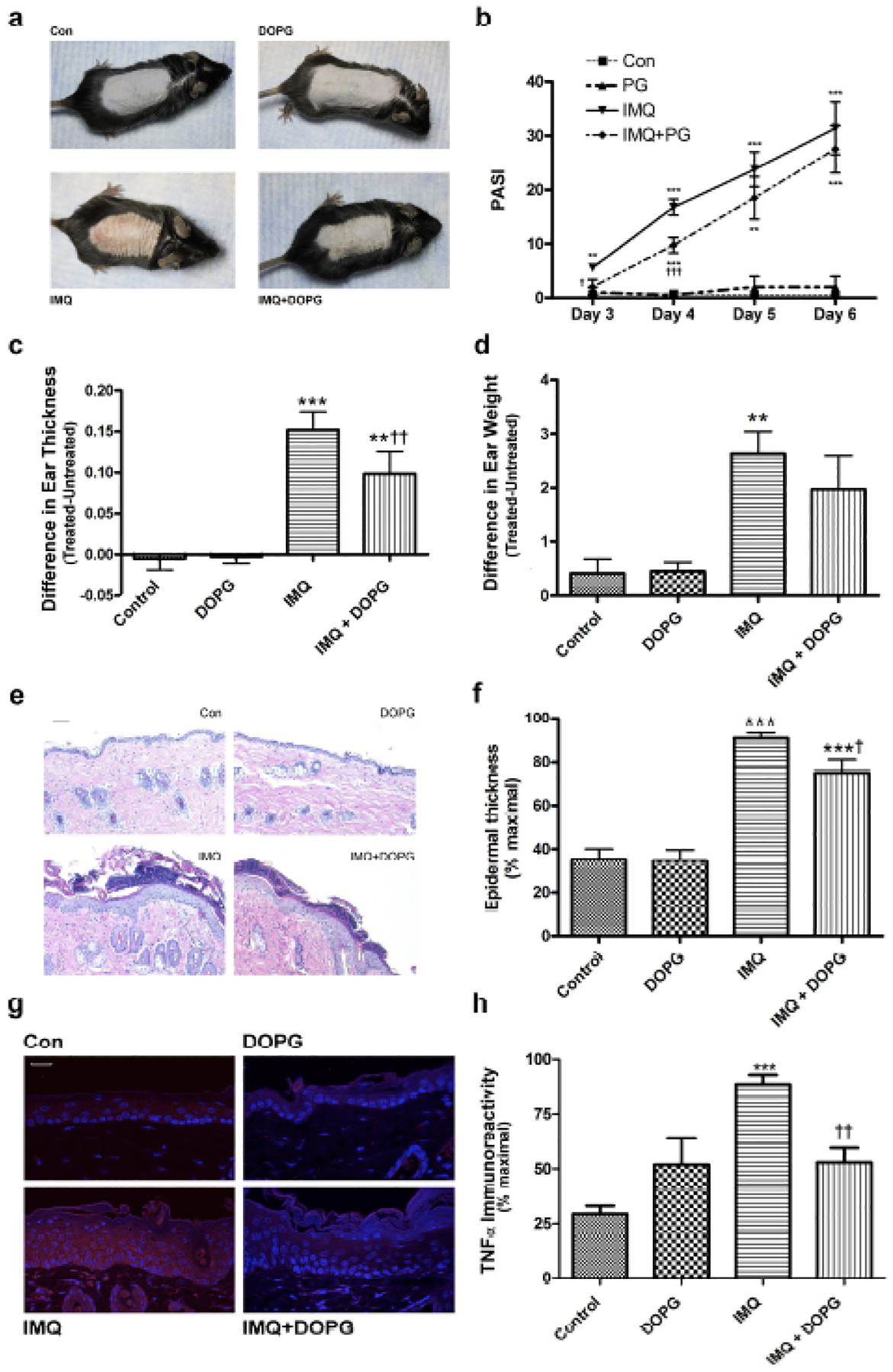
DOPG improves psoriasiform lesions in the imiquimod-induced mouse model of psoriasis. Mice (n=4–5) were treated with vehicle (vaseline) or imiquimod daily for 5 days in the morning. In the afternoon water or DOPG in water (prepared by sonicating a DOPG lipid film in deionized water) was applied to the treated skin. (a) Ear thickness and (b) weight were measured and are shown as means ± SEM. (c) Psoriasis area severity index (PASI) scores were quantified in a blinded fashion (means ± SEM). Sections of formalin-fixed, paraffin-embedded back skin were stained with H&E. (d) Representative images are shown and (e) epidermal thickness quantified in multiple sections from each mouse as described in Methods. Additional sections were incubated with an antibody recognizing TNFα and visualized with a Cy3-conjugated secondary antibody. (f) Representative images and (g) TNFα immunoreactivity quantified in multiple sections from each mouse as described in Methods. One-way analysis of variance with Student-Newman-Keuls multiple comparison post-hoc testing was used to determine significant differences; **p<0.01, ***p<0.001 versus control; †p<0.05, †††p<0.001 versus IMQ alone.
DISCUSSION
Psoriasis represents a sterile inflammatory disease in which anti-microbial DAMPs are markedly up-regulated, leading to TLR activation and inflammation. We show here in to our knowledge previously unreported findings that PG inhibits activation of TLRs by these anti-microbial peptide DAMPs. This result suggests that PG may be an effective therapeutic to treat sterile inflammation, i.e., inflammation resulting from non-infectious causes, such as occurs in skin diseases, but also in corneal disorders such as chemical burns and dry eye syndrome and in stroke, neurodegenerative diseases, myocardial infarction and atherosclerosis (Oh et al., 2012). In this regard, we suggest that inhibition of DAMP-induced inflammation may explain the previously observed ability of PG to decrease skin inflammation in response to the contact irritant, 12-O-tetradecanoylphorbol 13-acetate (TPA) (Xie et al., 2018). TPA is known to up-regulate anti-microbial peptides like S100A8 and S100A9 (Gebhardt et al., 2002, McNeill and Hogg, 2014); therefore, we hypothesize that PG inhibits TPA-induced inflammation in this model in part by blocking inflammatory mediator production elicited by TPA-increased anti-microbial peptides.
We elected to use S100A9 as the representative anti-microbial peptide DAMP because of the possible involvement of S100A9 in psoriasis. Thus, S100A9 is consistently up-regulated in psoriatic lesions and various mouse models of psoriasis (Gudjonsson and Elder, 2006, Schonthaler et al., 2013), and the expression of S100A9 and other S100A proteins is increased by cytokines that are associated with psoriasis (Nograles et al., 2010, Schonthaler et al., 2013, Waite and Skokos, 2012). In addition, recent studies have reported epigenetic alterations (hypomethylation) that result in enhanced S100A9 in psoriatic individuals (Roberson et al., 2012, Zhou et al., 2016). Other anti-microbial proteins elevated in psoriasis (Niyonsaba et al., 2017) are also thought to serve as DAMPs, including other S100A proteins as well as β-defensins and cathelicidin (van Bergenhenegouwen et al., 2013). Therefore, therapies designed to target only one DAMP may not be efficacious in treating psoriasis, whereas PG should inhibit TLR activation by multiple DAMPs and thus should suppress inflammation induced by various anti-microbial peptides and possibly other DAMPs that are increased in psoriasis. Indeed, we also demonstrated an ability of β-defensin-2 to activate TLR2 and this effect was blocked by DOPG. Nevertheless, it is possible that DAMPs such as S100A9 are functioning through pattern recognition receptors other than or in addition to TLR2 and TLR4, some of which may not be inhibited by PG. Furthermore, keratinocytes and macrophages are not the only cells that express TLRs, and other immune cells might respond to DAMPs in a similar or disparate manner compared to these two cell types. Clearly, further investigation is needed. Nevertheless, our in vivo results suggest the usefulness of PG in inhibiting the inflammation and skin lesions induced by imiquimod.
Our results demonstrate that PG inhibits the mRNA expression of several cytokines in both primary keratinocytes and RAW264.7 macrophages, as well as the secretion of TNFα from the macrophage cell line. Most of the TNFα in psoriasis is thought to originate from immune cells (Brotas et al., 2012) rather than keratinocytes. Nevertheless, there is increasing awareness of the ability of keratinocytes to produce various cytokines. Indeed, injury, infection and UV irradiation increase keratinocyte release of cytokines including TNFα (Balato et al., 2012), possibly accounting for the Koebner phenomenon (Weiss et al., 2002), a well-known response in which individuals prone to psoriasis develop lesions at sites of skin injury. Furthermore, the ability of PG to inhibit TNFα secretion from RAW264.7 cells seems to reflect similar actions on other cytokines, such IL-6 and IFNβ. Of note, IL6 and type I interferons like IFNβ have also been reported to play important roles in psoriasis (Grossman et al., 1989, Nestle et al., 2009).
The mechanism of action of PG to inhibit TLR activation likely involves the TLR accessory molecule CD14 (Kandasamy et al., 2016), which binds PG (Kuronuma et al., 2009). CD14 serves as a co-receptor for TLR2 (van Bergenhenegouwen et al., 2013) and TLR4 (He et al., 2016), and PG’s ability to inhibit TLR activation may reside in its ability to disrupt CD14’s ability to present PAMPs to TLRs (Kuronuma et al., 2009). Thus, PG inhibits the interaction of LPS with CD14, and antibodies specific for the LPS binding site on CD14 inhibit its ability to bind PG (Kuronuma et al., 2009). CD14 is primarily expressed in macrophages, with some expression in neutrophils (and dendritic cells), as well as basophils and B-lymphocytes and in non-myeloid cells such as keratinocytes (Jersmann, 2005). Thus, PG may inhibit DAMP-induced inflammatory responses by a variety of cell types. Nevertheless, TLR activation contributes to the innate immune response, so it is possible that inhibition of this process might increase susceptibility to infection. However, PG was actually found to inhibit lung infection by respiratory syncytial virus and influenza virus (Numata et al., 2010, Numata et al., 2012, Numata et al., 2013). These results indicate that PG is not globally immunosuppressive, supporting its use as an anti-inflammatory agent in inflammation.
Our data to date indicate that PG is produced in keratinocytes by the combined action of aquaporin-3 (AQP3), which transports glycerol into the cell, and phospholipase D2 (PLD2), which converts the glycerol to PG (Bollag et al., 2007, Zheng and Bollag, 2003, Zheng et al., 2003). The observed interaction between PLD2 and AQP3 suggests that PG represents a physiologically relevant lipid signal that regulates TLR activation and inflammation and may be deficient in conditions of sterile inflammation. Indeed, in the lung PG is a component of surfactant, and the known anti-inflammatory hormone cortisol increases PG production (Warburton, 1983), as do other glucocorticoids (Rooney, 1984). These agents are anti-inflammatory in skin, and epidermal-specific genetic ablation of the glucocorticoid receptor (GR) leads to skin inflammation resembling inflammatory skin diseases (Bollag and Isales, 2013). These observations raise the possibility that PG may be produced under normal conditions to dampen and/or resolve inflammation induced by exposure of the skin to microorganisms and anti-microbial peptide DAMPs, and that this process may be disrupted in pathological conditions. Indeed, abnormalities in AQP3 and/or PLD2 levels have been observed in various skin conditions (Voss et al., 2011)(Patel et al., 2017).
In summary, then, these results, to our knowledge previously unreported, show that PG can inhibit TLR activation and inflammation induced by a representative DAMP, recombinant S100A9. Our data also indicate that further studies into the mechanism by which PG inhibits TLR activation are warranted. Finally, since several DAMPs are up-regulated in inflammatory skin diseases such as psoriasis, our results suggest the possibility of developing PG as a topical therapy to treat these diseases.
MATERIALS AND METHODS
Keratinocyte Preparation and Cell Culture
Primary cultures of mouse epidermal keratinocytes were prepared from ICR strain CD-1 outbred neonatal mice 1–3 days of age or from tail skin as described in (Bailey et al., 2014). RAW264.7 cells (from American Type Culture Collection) were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin and streptomycin.
Quantitative RT-PCR (qRT-PCR)
Keratinocytes or RAW264.7 cells were treated with or without recombinant S100A9 for 2 hours in the presence and absence of 100μg/mL DOPG or DOPC or 25μM TLR1/2 (CU-CPT22) or 10μM TLR4 antagonist (TAK242). Cells were harvested, RNA isolated and expression quantified as described in (Choudhary et al., 2015).
Enzyme-linked Immunosorbent Assay (ELISA)
TNFα in the culture supernatants was measured using ELISA kits as described previously (Helwa et al., 2015). Multiplex flow-based cytometric bead cytokine arrays were performed as indicated in Supplemental Methods.
Western blotting
After the indicated treatments, cells were harvested and proteins separated by SDS-PAGE and transferred to Immobilon-P membranes and visualized using antibodies recognizing p65-NFκB phosphorylated at serine 536 (pNFκB) or total p65-NFκB as described previously (Choudhary et al., 2015).
Immunocytochemistry
Cells plated on coverslips were treated, fixed with 4% paraformaldehyde, stained for p65-NFκB immunoreactivity and imaged using a Zeiss confocal microscope as described in (Choudhary et al., 2015). The percentage of cells with nuclear p65-NFκB immunoreactivity (relative to total number of cells) was determined in micrographs of at least two random fields by two independent observers blinded to treatments.
Determination of TLR2 activation in TLR2 reporter cell lines
HEK-Blue-hTLR2 reporter cells were cultured in growth medium and TLR2 activation determined using HEK-Blue Detection medium according to the supplier’s instructions and as described in Supplemental Methods.
Generation and analysis of the IMQ mouse model of psoriasis
Two to three days after removal of back hair by shaving and depilation, 8- to 10-week-old male C57BL/6 mice were treated with IMQ to induce psoriasiform lesions as in (van der Fits et al., 2009) and described in Supplemental Materials. Briefly, the mice were treated for five days with vehicle or IMQ (Aldara™) on their ears and back skin. PG was applied topically in water, in comparison with water alone. The PASI score was estimated in a blinded manner from day 3. On the sixth day ear thickness and ear punch biopsy weight were determined. Epidermal thickness and TNFα immunoreactivity were also determined. All procedures were approved by the Institutional Animal Care and Use Committee.
Statistical Analyses
Statistical analyses were performed using ANOVA followed by Newman-Keuls post-hoc tests, with GraphPad Prism (La Jolla, CA) and significance at p≤0.05.
Supplementary Material
Acknowledgements
The authors thank Purnima Merai for excellent technical assistance. HP and WB were supported by the Medical College of Georgia Medical Scholars Program (MSP) and BH by the Augusta University Graduate School Student Training and Research (STAR) Program. EC was supported by an Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship Award. CMI and QZ were supported by National Institutes of Health/National Institute of Aging #P01 AG036675. WBB was supported in part by a Research Career Scientist Award. The contents of this article do not represent the official views of the Department of Veterans Affairs, the United States Government or the National Institutes of Health.
Abbreviations:
- AQP3
aquaporin 3
- DAMP
danger- or damage-associated molecular pattern
- DOPG
dioleoylphosphatidylglycerol
- IL
interleukin
- PAMP
pathogen-associated molecular pattern
- PG
phosphatidylglycerol
- PLD2
phospholipase D2
- TLR
toll-like receptor
- TNFα
tumor necrosis factor-α
Footnotes
Conflict of Interest
Dr. Bollag is an inventor on a patent awarded to Augusta University for the use of phosphatidylglycerol to modulate skin function.
REFERENCES
- Bailey LJ, Choudhary V, Merai P, Bollag WB. Preparation of primary cultures of mouse epidermal keratinocytes and the measurement of phospholipase D activity. Meth Mol Biol 2014;1195:111–31. [DOI] [PubMed] [Google Scholar]
- Balato A, Balato N, Megna M, Schiattarella M, Lembo S, Ayala F. Pathogenesis of Psoriasis: The Role of Pro-Inflammatory Cytokines Produced by Keratinocytes,. In: Soung J, editor. Psoriasis; http://cdn.intechweb.org/pdfs/28300.pdf InTech; 2012. [Google Scholar]
- Bollag WB, Isales CM. GRowing an epidermal tumor. J Invest Dermatol 2013;133(12):2659–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag WB, Xie D, Zhong X, Zheng X. A potential role for the phospholipase D2-aquaporin-3 signaling module in early keratinocyte differentiation: Production of a novel phosphatidylglycerol lipid signal. J Invest Dermatol 2007;127:2823–31. [DOI] [PubMed] [Google Scholar]
- Brotas AM, Cunha JM, Lago EH, Machado CC, Carneiro SC. Tumor necrosis factor-alpha and the cytokine network in psoriasis. An Bras Dermatol 2012;87(5):673–81; quiz 82–3. [DOI] [PubMed] [Google Scholar]
- Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, et al. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One 2015;10(2):e0115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary V, Olala LO, Qin H, Helwa I, Pan ZQ, Tsai YY, et al. Aquaporin-3 re-expression induces differentiation in a phospholipase D2-dependent manner in aquaporin-3-knockout mouse keratinocytes. J Invest Dermatol 2015;135:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Kupper TS. Misbehaving macrophages in the pathogenesis of psoriasis. J Clin Invest 2006;116(8):2084–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovici BB, Sattar N, Prinz J, Puig L, Emery P, Barker JN, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol 2010;130(7):1785–96. [DOI] [PubMed] [Google Scholar]
- Erridge C Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol 2010;87(6):989–99. [DOI] [PubMed] [Google Scholar]
- Flutter B, Nestle FO. TLRs to cytokines: Mechanistic insights from the imiquimod mouse model of psoriasis. Eur J Immunol 2013;43(12):3138–46. [DOI] [PubMed] [Google Scholar]
- Gebhardt C, Breitenbach U, Tuckermann JP, Dittrich BT, Richter KH, Angel P. Calgranulins S100A8 and S100A9 are negatively regulated by glucocorticoids in a c-Fos-dependent manner and overexpressed throughout skin carcinogenesis. Oncogene 2002;21(27):4266–76. [DOI] [PubMed] [Google Scholar]
- Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA 1989;86(16):6367–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson JE, Elder JT. Mouse models: psoriasis: an epidermal disease after all? Eur J Hum Genet 2006;14(1):2–4. [DOI] [PubMed] [Google Scholar]
- He Z, Riva M, Bjork P, Sward K, Morgelin M, Leanderson T, et al. CD14 Is a Co-Receptor for TLR4 in the S100A9-Induced Pro-Inflammatory Response in Monocytes. PLoS One 2016;11(5):e0156377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwa I, Patel R, Karempelis P, Kaddour-Djebbar I, Choudhary V, Bollag WB. The antipsoriatic agent monomethylfumarate has antiproliferative, prodifferentiative, and anti-inflammatory effects on keratinocytes. J Pharmacol Exp Ther 2015;352(1):90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jersmann HP. Time to abandon dogma: CD14 is expressed by non-myeloid lineage cells. Immunol Cell Biol 2005;83(5):462–7. [DOI] [PubMed] [Google Scholar]
- Kandasamy P, Numata M, Berry KZ, Fickes R, Leslie CC, Murphy RC, et al. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J Lipid Res 2016;57(6):993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy P, Zarini S, Chan ED, Leslie CC, Murphy RC, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits Mycoplasma pneumoniae-stimulated eicosanoid production from human and mouse macrophages. J Biol Chem 2011;286(10):7841–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Hwang SM, Chung IY. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-kappaB pathways. Immunology 2015;144(1):79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuronuma K, Mitsuzawa H, Takeda K, Nishitani C, Chan ED, Kuroki Y, et al. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J Biol Chem 2009;284(38):25488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol 2013;34(4):174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolerance Matzinger P., danger, and the extended family. Ann Rev Immunol 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- McInturff JE, Modlin RL, Kim J. The role of toll-like receptors in the pathogenesis and treatment of dermatological disease. J Invest Dermatol 2005;125(1):1–8. [DOI] [PubMed] [Google Scholar]
- McNeill E, Hogg N. S100A9 has a protective role in inflammation-induced skin carcinogenesis. Int J Cancer 2014;135(4):798–808. [DOI] [PubMed] [Google Scholar]
- Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol 2014;60(4):782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Kanda T, Takaishi M, Shiga T, Miyoshi K, Nakajima H, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol 2011;186(7):4481–9. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009;361(5):496–509. [DOI] [PubMed] [Google Scholar]
- Niyonsaba F, Kiatsurayanon C, Chieosilapatham P, Ogawa H. Friends or Foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp Dermatol 2017; 26(11):289–98. [DOI] [PubMed] [Google Scholar]
- Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg 2010;29(1):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M, Chu HW, Dakhama A, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc Natl Acad Sci USA 2010;107(1):320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M, Kandasamy P, Nagashima Y, Posey J, Hartshorn K, Woodland D, et al. Phosphatidylglycerol suppresses influenza A virus infection. Am J Respir Cell Mol Biol 2012;46(4):479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M, Nagashima Y, Moore ML, Berry KZ, Chan M, Kandasamy P, et al. Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection. J Lipid Res 2013;54(8):2133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JY, Choi H, Lee RH, Roddy GW, Ylostalo JH, Wawrousek E, et al. Identification of the HSPB4/TLR2/NF-kappaB axis in macrophage as a therapeutic target for sterile inflammation of the cornea. EMBO Mol Med 2012;4(5):435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Kevin Heard L, Chen X, Bollag WB. Aquaporins in the Skin. Adv Exp Med Biol 2017;969:173–91. [DOI] [PubMed] [Google Scholar]
- Pradeu T, Cooper EL. The danger theory: 20 years later. Frontiers Immunol 2012;3:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz E, Prens EP, Kurek D, Kant M, de Ridder D, Mourits S, et al. Effective treatment of psoriasis with narrow-band UVB phototherapy is linked to suppression of the IFN and Th17 pathways. J Invest Dermatol 2011;131(7):1547–58. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Liu Y, Ryan C, Joyce CE, Duan S, Cao L, et al. A subset of methylated CpG sites differentiate psoriatic from normal skin. J Invest Dermatol 2012;132(3 Pt 1):583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney SA. Lung surfactant. Environ Health Perspect 1984;55:205–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat R, Wolk K. Research in practice: IL-22 and IL-20: significance for epithelial homeostasis and psoriasis pathogenesis. J Dtsch Dermatol Ges 2011;9(7):518–23. [DOI] [PubMed] [Google Scholar]
- Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheumat 2012;64(5): 1477–87. [DOI] [PubMed] [Google Scholar]
- Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm 2013;2013: 828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008;27(2):190–9. [DOI] [PubMed] [Google Scholar]
- Schonthaler HB, Guinea-Viniegra J, Wculek SK, Ruppen I, Ximenez-Embun P, Guio-Carrion A, et al. S100A8–S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity 2013;39(6):1171–81. [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004;4(6):469–78. [DOI] [PubMed] [Google Scholar]
- Stratis A, Pasparakis M, Rupec RA, Markur D, Hartmann K, Scharffetter-Kochanek K, et al. Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J Clin Invest 2006;116(8):2094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergenhenegouwen J, Plantinga TS, Joosten LA, Netea MG, Folkerts G, Kraneveld AD, et al. TLR2 & Co: a critical analysis of the complex interactions between TLR2 and coreceptors. J Leukoc Biol 2013;94(5):885–902. [DOI] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009;182(9):5836–45. [DOI] [PubMed] [Google Scholar]
- Voss KE, Bollag RJ, Fussell N, By C, Sheehan DJ, Bollag WB. Abnormal aquaporin-3 protein expression in hyperproliferative skin disorders. Arch Dermatol Res 2011;303:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite JC, Skokos D. Th17 response and inflammatory autoimmune diseases. Int J Inflam 2012;2012:819467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan F, Lenardo MJ. The nuclear signaling of NF-kappaB: current knowledge, new insights, and future perspectives. Cell Res 2010;20(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N, et al. Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest 2006;116(8):2105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton D Chronic hyperglycemia with secondary hyperinsulinemia inhibits the maturational response of fetal lamb lungs to cortisol. J Clin Invest 1983;72(2):433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Shemer A, Trau H. The Koebner phenomenon: review of the literature. J Eur Acad Dermatol Venereol 2002;16(3):241–8. [DOI] [PubMed] [Google Scholar]
- Wu YZ, Medjane S, Chabot S, Kubrusly FS, Raw I, Chignard M, et al. Surfactant protein-A and phosphatidylglycerol suppress type IIA phospholipase A2 synthesis via nuclear factor-kappaB. Am J Respir Crit Care Med 2003;168(6):692–9. [DOI] [PubMed] [Google Scholar]
- Xie D, Choudhary V, Seremwe M, Edwards JG, Wang A, Emmons AC, et al. Soy Phosphatidylglycerol Reduces Inflammation in a Contact Irritant Ear Edema Mouse Model in vivo. J Pharmacol Exp Ther 2018;366(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D, Seremwe M, Edwards JG, Podolsky R, Bollag WB. Distinct effects of different phosphatidylglycerol species on mouse keratinocyte proliferation. PLoS One 2014;9(9):e107119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Bollag WB. Aquaporin 3 colocates with phospholipase D2 in caveolin-rich membrane microdomains and is regulated by keratinocyte differentiation. J Invest Dermatol 2003;121:1487–95. [DOI] [PubMed] [Google Scholar]
- Zheng X, Ray S, Bollag WB. Modulation of phospholipase D-mediated phosphatidylglycerol formation by differentiating agents in primary mouse epidermal keratinocytes. Biochim Biophys Acta 2003;1643:25–36. [DOI] [PubMed] [Google Scholar]
- Zhou F, Wang W, Shen C, Li H, Zuo X, Zheng X, et al. Epigenome-Wide Association Analysis Identified Nine Skin DNA Methylation Loci for Psoriasis. J Invest Dermatol 2016;136(4): 779–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.