

Abstract
DNA polymerase δ (Polδ) plays pivotal roles in eukaryotic DNA replication and repair. Polδ is conserved from yeast to humans, and mutations in human Polδ have been implicated in various cancers. Saccharomyces cerevisiae Polδ consists of catalytic Pol3 and the regulatory Pol31 and Pol32 subunits. Here, we present the near atomic resolution (3.2 Å) cryo-EM structure of yeast Polδ holoenzyme in the act of DNA synthesis. The structure reveals an unexpected arrangement in which the regulatory subunits (Pol31 and Pol32) lie next to the exonuclease domain of Pol3 but do not engage the DNA. The Pol3 C-terminal domain contains a 4Fe-4S cluster and emerges as the keystone of Polδ assembly. We also show that the catalytic and regulatory subunits rotate relative to each other and that this is an intrinsic feature of the Polδ architecture. Collectively, the structure provides a framework for understanding DNA transactions at the replication fork.
DNA polymerase δ (Polδ) is essential for DNA replication1–5. Saccharomyces cerevisiae Polδ consists of the catalytic Pol3 (125 kDa) subunit, and the regulatory Pol31 (55 kDa) and Pol32 (40 kDa) subunits (Fig. 1a). Pol3 is a member of the B-family of polymerases, and harbors an N-terminal catalytic module with both polymerase (pol) and 3′→5′ exonuclease (exo) proofreading activities2,6,7. Two cysteine-rich metal binding modules (CysAD and CysBD) lie at the C terminus of Pol3. CysBD serves to recruit Pol31, which in turn interacts with the N terminus of Pol32 (Pol32N)8–10,11. The C terminus of Pol32 (Pol32C) carries a canonical proliferating cell nuclear antigen (PCNA) interaction (PIP) motif that anchors the holoenzyme to PCNA for increased processivity12,13 (Fig. 1a). Studies in yeast show that the POL3 and POL31 genes are essential for cell viability, while POL32 deletion mutants are viable but defective in DNA replication, DNA repair and mutagenesis2,6–8,14. Thus, proper functioning of Polδ requires a complex of all three subunits, but despite repeated efforts the holoenzyme has failed to crystallize. As such, there is no high resolution information on the spatial organization of Pol3, Pol31 and Pol32, or how these subunits physically interact and affect Polδ function. Taking advantage of recent advances in cryo-EM, we present here a cryo-EM structure of the complete yeast Polδ holoenzyme in the act of DNA synthesis, at a nominal resolution of 3.2 Å (Fig. 1b). We also show through multi-body refinement that angular rotation between the catalytic and regulatory subunits is an intrinsic feature of Polδ architecture.
Fig. 1 |. Cryo-EM structure of DNA bound Polδ holoenzyme.
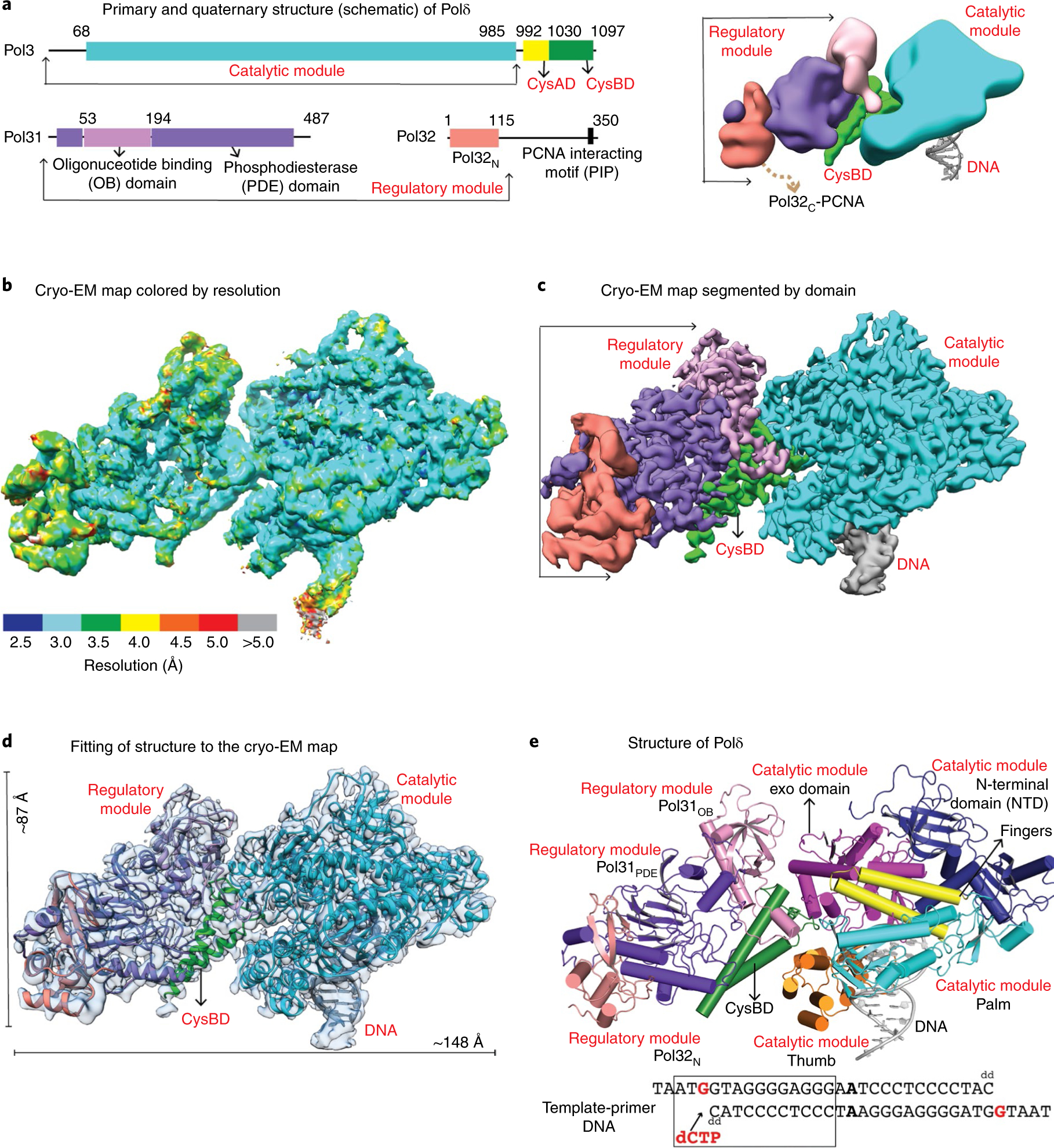
a, Schematic of the primary (left) and quaternary (right) structure of S. cerevisiae Polδ. b,c, Cryo-EM density map of Polδ colored by local resolution (b) and domains (c). d, Polδ structure fitted into the cryo-EM map. e, Structure of Polδ colored by domain. The region of the template-primer duplex enclosed in the box was built into the final model.
Results
Overall arrangement.
The structure captures Polδ holoenzyme bound to a template-primer duplex DNA presenting G as the templating base and dCTP as the incoming nucleotide (Fig. 1, Table 1 and Supplementary Fig. 1). The cryo-EM map shows clear density for almost all secondary structures and 90% of the side chains, 11 of the 25 bases of the DNA duplex, templating base G, incoming dCTP, 44 solvent molecules, as well as the active side metal ions (see Methods). The resolution of Pol32N and portions of Pol31 improved majorly after multi-body refinement15, indicative of their flexibility relative to the catalytic module. Overall, the holoenzyme exhibits an extended architecture with approximate dimensions of 148 Å × 87 Å × 56 Å (Fig. 1c–e). For convenience and simplicity, we describe the structure as two discrete modules bridged by CysBD: a globular ‘catalytic module’ consisting of Pol3 amino acids 1–985 and a flat ‘regulatory module’ consisting of Pol31 and Pol32N. While the catalytic module is engaged with DNA, the regulatory module is positioned radially (at an angle of ~50°) with respect to the DNA duplex, proximal to the exonuclease active site (described below) of the catalytic module (Fig. 1e). This arrangement between the catalytic and regulatory modules has not been observed in any polymerase structure known so far.
Table 1 |.
Cryo-EM data collection, refinement and validation Statistics
| Polδ-DNA-dCTP complex (EMD-20235, PDB 6P1H) | |
|---|---|
| Data collection and processing | |
| Magnification | 130,000 |
| Voltage (kV) | 300 |
| Electron exposure (e−Å−2) | 64 |
| Defocus range (μm) | 1.0–2.8 |
| Pixel size (Å) | 0.8549 |
| Symmetry imposed | C1 |
| Initial particle images (no.) | 4,397,064 |
| Final particle images (no.) | 166,444 |
| Map resolution (Å) | 3.2 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 2.5–5.0 |
| Refinement | |
| Initial model used | 3IAY, 3E0J |
| Model resolution (Å) | 3.2 |
| FSC threshold | 0.143 |
| Model resolution range (Å) | 2.5–5.0 |
| Map sharpening B factor (Å2) | −121 |
| Model composition | |
| Non-hydrogen atoms | 13,006 |
| Protein residues | 1,539 |
| DNA/other | 27/7 |
| Water | 44 |
| B factors (Å2) | |
| Protein | 97.7 |
| DNA/other | 94.0/75.7 |
| Water | 70.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.728 |
| Validation | |
| MolProbity score | 2.02 |
| Clashscore | 10.92 |
| Poor rotamers (%) | 0 |
| Ramachandran plot | |
| Favored (%) | 92.6 |
| Allowed (%) | 6.81 |
| Disallowed (%) | 0.59 |
The catalytic module is primed for DNA synthesis.
The Polδ catalytic module is well resolved (~3 Å) and similar to the X-ray structure of the same module solved in the absence of CysBD and the regulatory module (PDB 3IAY)16. It is composed of N-terminal (NTD), exonuclease (exo), palm, fingers and thumb domains (Figs. 1e and 2a). The key differences are in the relative orientation of the exo and thumb domains and in regions involved in interactions with CysBD (described below). As with the isolated structure, the replicative end of the DNA template-primer duplex presents templating base G at the active site and establishes Watson-Crick base pairing with incoming dCTP (Fig. 2a). The palm domain carries the active site residues (Asp608 and Asp764) that cluster around two Ca2+ ions (metal A and B) for the polymerase catalytic reaction. The exo active site (defined by residues Asp320 and Glu322) is located ~45 Å away from the pol active site. Residues from the palm and thumb domain interact (in the minor groove) with up to seven base pairs of the DNA duplex immediately downstream of template G (Fig. 2a), while the remainder of the DNA is solvent exposed.
Fig. 2 |. Structure and cryo-EM density for selected regions of catalytic and regulatory modules and CysBD.
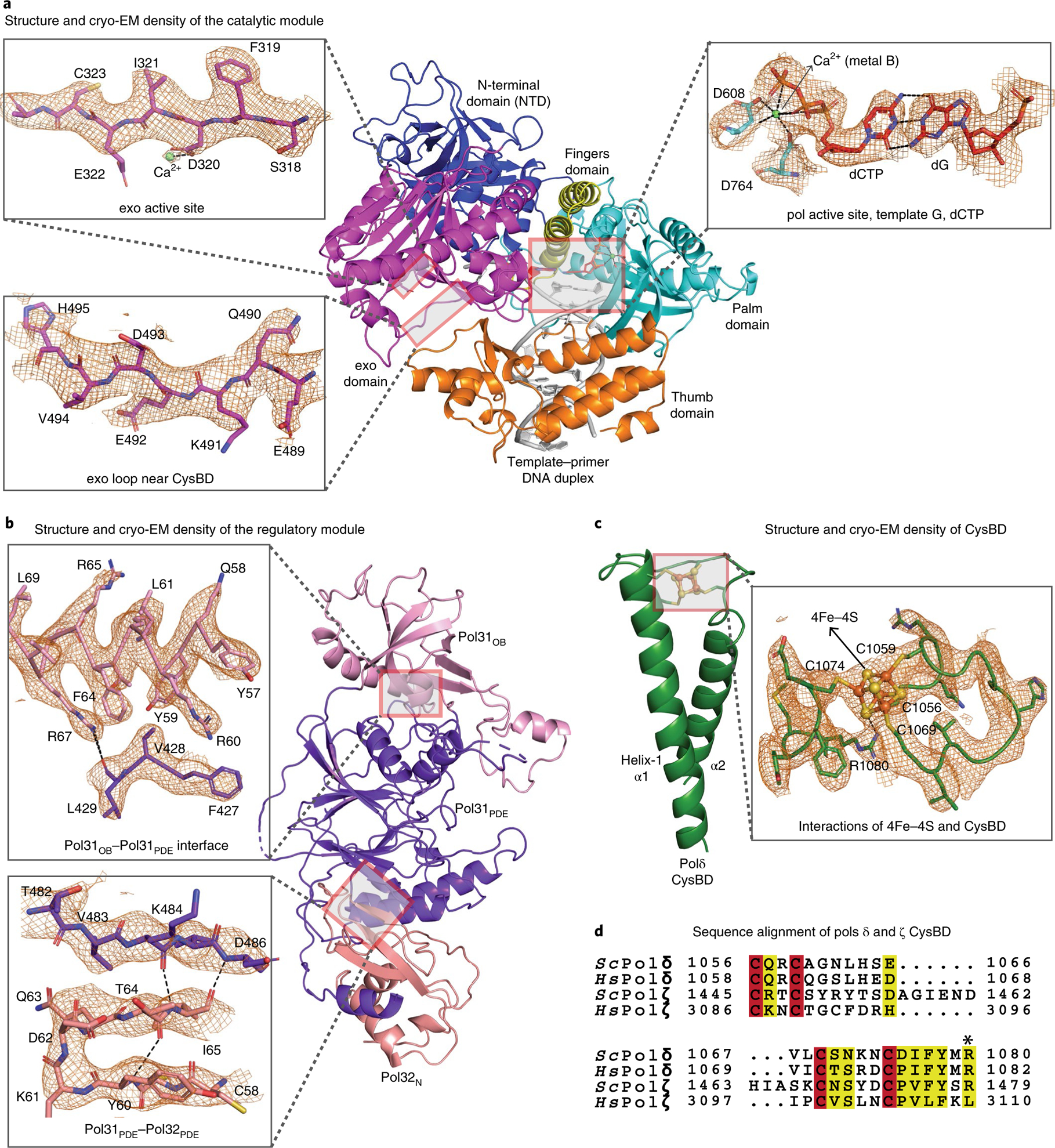
a, Well resolved density for the pol and exo active sites, including ligands, metal ions and water. Active sites in Polδ are configured as in the isolated catalytic module. (PDB 3IAY). b, Clear density for the regulatory module enabled modeling of regions that are not resolved in the human homolog (PDB 3E0J). c, Ab initio structure of 4Fe-4S cluster bound CysBD. Some side chains have been omitted for clarity. d, Sequence alignment of a section of CysBD spanning the conserved Arg1080.
The regulatory module does not engage the DNA.
Unlike the catalytic module, Pol31-Pol32N of the regulatory module do not engage the DNA (Figs. 1 and 2b). This belies the widely held assumption that the regulatory module tempers catalysis by interacting directly with the DNA duplex14,17, though it is conceivable that it may interact with the unpaired segment of the template strand far upstream of the nascent base pair. The Pol31-Pol32N substructure is reminiscent of human homologs p50-p66N (PDB 3E0J)17, albeit with differences in the conformation of loops engaged in interactions with CysBD (the p50-p66N structure does not contain regions equivalent to CysBD and the catalytic module), as well as in the relative orientation of Pol31 and Pol32N (Fig. 2b and Supplementary Fig. 2a). Pol31 contains an oligonucleotide binding domain (Pol31OB) and an inactive phosphodiesterase (Pol31PDE) domain that stack almost linearly with the winged helix-turn-helix domain of Pol32N. Although all three domains are potentially capable of binding single-stranded and/or double-stranded DNA, the relevant regions of Pol31 are partially occluded in the holoenzyme. Pol32C harboring the C-terminus PIP motif is disordered, but the spatial position of Pol32N is such that it would allow for the PIP motif of Pol32C to extend to the downstream portion of the DNA duplex for interactions with PCNA (Fig. 1a and Supplementary Fig. 3).
CysBD contains a Fe-S cluster.
Polδ CysAD is disordered, suggestive of a flexible region that may only become ordered when it interacts with other components of the replication fork, including PCNA18 (Supplementary Fig. 3). However, we see clear density for CysBD spanning amino acids 1031–1096 (Fig. 2c). CysBD is composed of two antiparallel α-helices (α1 and α2) linked by a loop that harbors the four cysteines (C1056, C1059, C1069 and C1074) required for binding to its metal cofactor (Fig. 2c). The four cysteines enclose a region of density that is most consistent with the presence of a 4Fe-4S cluster (Fig. 2c), providing a structural context for previous55Fe radiolabeling experiments that had indicated the presence of a Fe-S cofactor in CysBD18. Polδ CysBD is thus the smallest scaffold capable of binding a Fe-S cofactor, with the closest structural homolog being an artificial peptide (97 amino acid; PDB 6DLM) designed to form a coiled coil19. The equivalent domains in Polα14,20 and Polε21 are typically larger and observed to bind to a single divalent Zn2+ ion (Supplementary Fig. 2b). Intriguingly, the 4Fe-4S cluster in CysBD is coordinated with Arg1080, which is conserved in the sequences of all eukaryotic Polδ. An equivalent arginine is present in yeast but not in vertebrate Polζ, and is absent in other replicative polymerases (Fig. 2d). Arg1080 is engaged in a putative hydrogen bond with a sulfur atom of the 4Fe-4S cluster, with an NH1(Arg)-S distance of 2.8 Å and with the NH1 atom oriented more or less linearly (~162°) with the Fe-S bond (Fig. 2c). Although rare, Arg(NH)-S bonds have been proposed to stabilize the reduced state of the Fe-S cluster by raising its reduction potential22–25. This is likely to be important for Polδ function as an electrochemically oxidized version of the enzyme is significantly slower in processive DNA synthesis26, presumably as a mechanism to slow DNA synthesis in response to replication stress such as oxidation26. The slowing of Polδ in going from a reduced [4Fe-4S]2+ to an oxidized [4Fe-4S]3+ state may result from an increase in DNA binding affinity through an increase in electrostatic interactions with the DNA phosphate backbone, as in the case of the DNA repair enzyme endonuclease III (EndoIII)27. The oxidized [4Fe-4S]3+ cluster in yeast Polδ can be reversibly reduced to [4Fe-4S]2+ (ref.26), providing a mechanism for restoring Polδ to the more processive form after oxidative stress.
CysBD nucleates Polδ assembly.
CysBD emerges as the keystone of Polδ assembly (Fig. 3a). Synchronal interactions of CysBD with both the catalytic and regulatory modules nucleates the assembly of Polδ holoenzyme into its unique architecture (Figs. 1e and 3a). CysBD helix α1 and portions of α2 interact with the regulatory module, while the 4Fe-4S cluster and amino acids at its C terminus interact with the catalytic module, and amino acids from the linker between α1 and α2 interact with both (Fig. 3a). Both the Pol31OB and Pol31PDE domains interact with CysBD burying ~2,200 Å2 at the interface. Conserved amino acids from Pol31 such as 149-Glu-Asp-Glu-151 that confer temperature sensitivity in yeast lie at the interface with CysBD28. Similarly, many of the amino acids in CysBD (Glu1046, Gln1057, Arg1058 and His1064, for example) that have previously been implicated in genetic and biochemical studies as interacting with Pol31 also lie at this interface11 (Fig. 3a). Interactions of CysBD with the catalytic module occur with the exo and thumb domains (burying ~887 Å2), and help to draw these domains closer together than in the isolated structure of the catalytic module (Supplementary Fig. 4). The closer apposition of the exo and thumb domains creates a cavity between them that engages CysBD residues Lys1072, Asn1073 and Tyr1078 in van der Waals and polar interactions (Fig. 3a). Interactions between CysBD and the Polδ catalytic module are unexpected because previous genetic and biochemical experiments had mapped interactions between CysBD and the regulatory module, but not with the catalytic module11. Intriguingly, mutational studies had implicated Tyr1078 as interacting with Pol31, but the structure shows it interacting with the catalytic module instead11.
Fig. 3 |. CysBD is the keystone of Polδ assembly.
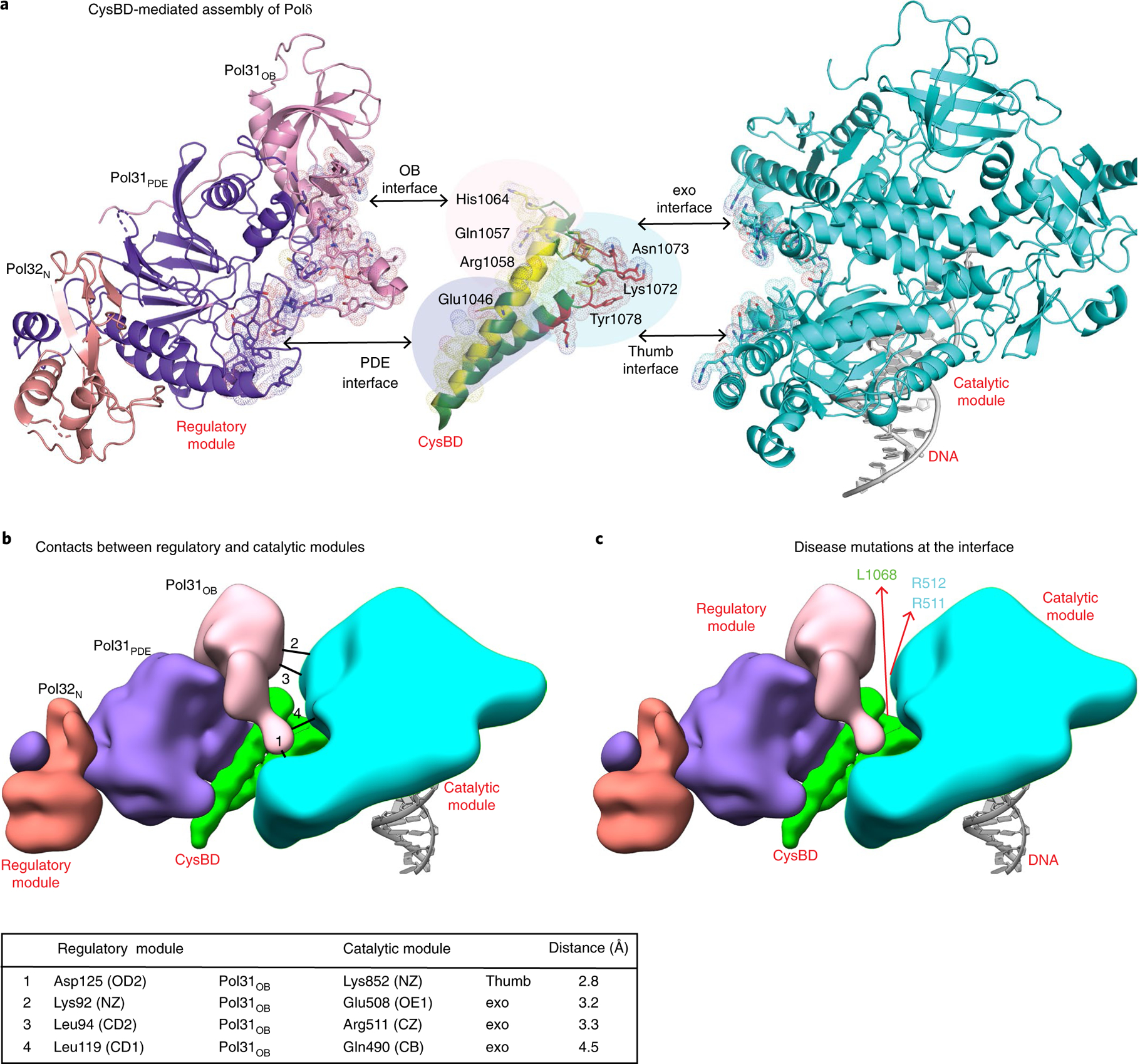
a, Simultaneous interactions of CysBD with the regulatory and catalytic modules (in yellow and red respectively and dotted surface) nucleates the assembly of Polδ. CysBD amino acids tested previously in genetic experiments for interactions and those identified from our structure are labeled. b, Potential contacts between the regulatory and catalytic modules are mapped on the schematic and listed in the legend below. c, Human Polδ disease mutations R506H, R507C and I1070N corresponding to R511, R512 and L1068 mapped on a schematic of the structure of yeast Polδ. These mutations localize at the interface between the catalytic and regulatory modules.
Overall, the association of CysBD with both the thumb and exo domains lends to a compaction of the catalytic module, wherein the axial dimension of the catalytic module diminishes by >2.5 Å when compared to the isolated catalytic module (Supplementary Fig. 4a). This general tightening of the catalytic module may underlie the observed increase in activity of the holoenzyme when compared to Pol3 alone29. Association with CysBD also changes the shape of the exo active site. In particular, a loop (amino acids 490–497) bordering the exo site becomes ordered in the holoenzyme and Tyr496 is within van der Waals distance from a mismatched primer modeled at the exo active site (Supplementary Fig. 4b). Notably, the transfer of a mismatched primer from the pol to the exo active site in B family polymerases is critical for ensuring replication fidelity and repair of DNA lesions30, and typically involves large movements of the thumb and exo domains31,32. The fact that CysBD interacts with both of these domains suggests an accompanying role for CysBD in the intermolecular switch of a primer between the pol and exo active sites. Consistent with this idea, subtle changes to the 4Fe-4S binding pocket of human Polδ have shown to affect the exonuclease activity of the holoenzyme both in vivo and in vitro33,34.
Flexibility between the catalytic and regulatory modules.
The interface between the catalytic and regulatory modules is relatively sparse and best described as a space lined by Pol31 and the exo domain from opposite sides, linked by CysBD at the base (Fig. 3b). The closest approach places poorly ordered residues Lys92, Leu94 and Leu119 from Pol31 within 3.5 Å of Glu508, Arg511 and Gln490of the exo domain (Fig. 3b), which could potentially make hydrogen bond or van der Waals interactions via a small rotation between the catalytic and regulatory modules. The only observed hydrogen bond is between Asp125 from Pol31 with Lys852 from the thumb domain of the catalytic module.
The paucity of interactions between the catalytic and regulatory modules behooves a relatively flexible complex able to accommodate changes in DNA direction. In a previous low resolution SAXS analysis of the Polδ holoenzyme in the absence of DNA, we observed significant flexibility between the catalytic and regulatory modules35. From the current cryo-EM analysis, flexibility between the modules appears to be a feature even when Polδ is complexed to DNA. Indeed, when we performed multi-body refinement15 with the catalytic module and regulatory module as discrete bodies (see Methods), it resulted in major improvement in resolution for regions of Pol32N and Pol31 that are most distant from the catalytic module (Fig. 4a and Supplementary Fig. 5a), indicative of flexibility between the catalytic and regulatory modules. From principal component analysis of the multi-body refinement derived positional variance, ~35% of the flexibility is accounted by motion along the first two eigenvectors (Supplementary Fig. 5b) with monomodal distribution of amplitudes, indicative of continuous (rather than ratcheted) motion between the catalytic and regulatory modules (Supplementary Fig. 5c). Motion represented by the first eigenvector corresponds to a ~20° rocking motion of the regulatory module parallel to the catalytic module (Fig. 4b, Supplementary Fig. 6 and Supplementary Video 1). The second eigenvector represents a ~25° rocking motion of the regulatory module in the direction of the catalytic module (Fig. 4c, Supplementary Fig. 6 and Supplementary Video 2). The observed conformational dynamics is in agreement with the predicted dynamics of Polδ using normal mode analysis36 (Supplementary Fig. 6). A small angular freedom between the catalytic and regulatory modules appears thus to be an inherent feature of the Polδ architecture.
Fig. 4 |. Flexibility between catalytic and regulatory modules.
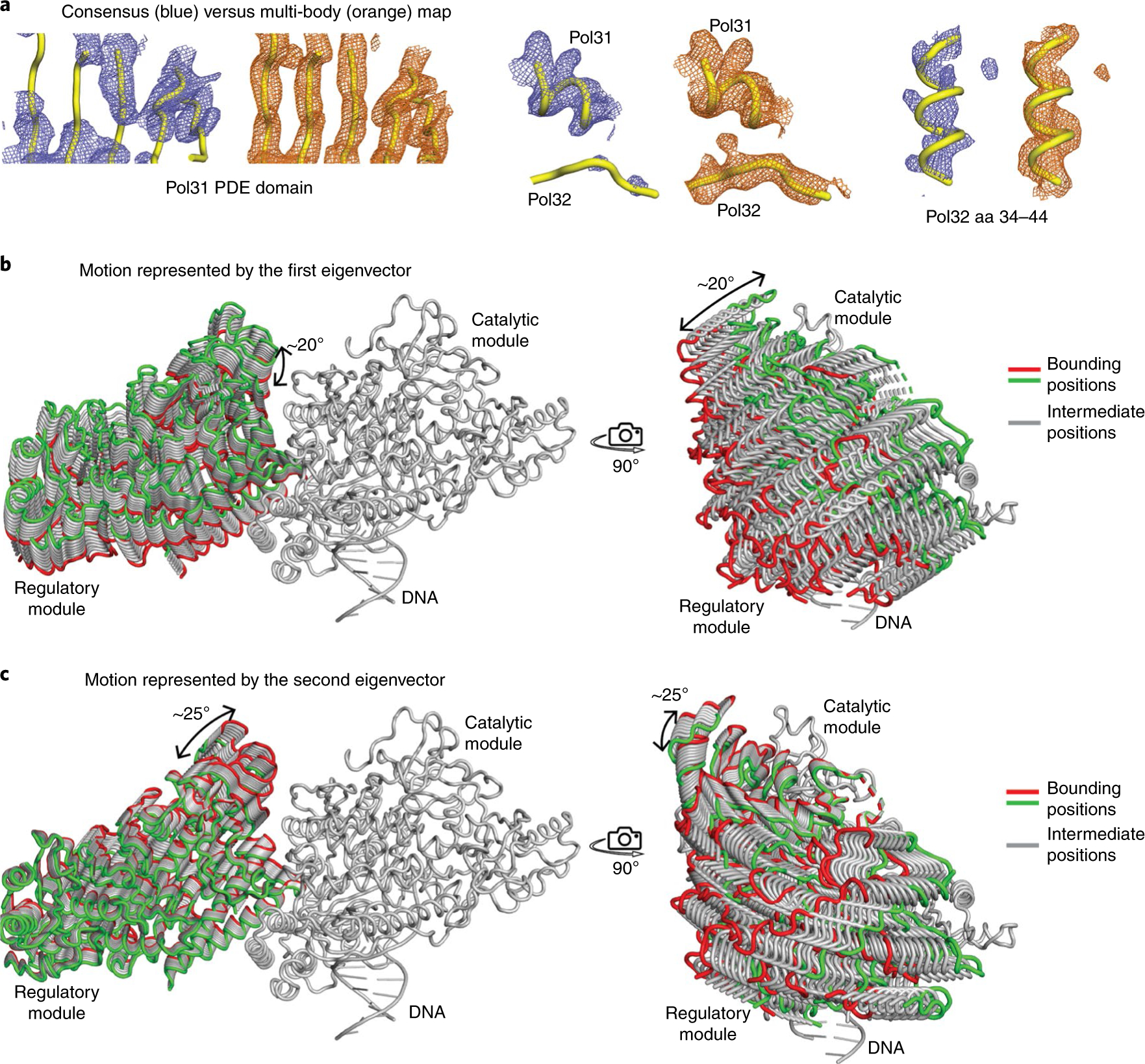
a, Comparison of consensus and multi-body maps for selected regions of Pol31 and Pol32. Multi-body refinement results in significant improvement in the resolution of Pol32N and regions of Pol31 that are far from the catalytic module. b,c, Motion represented by the first and second eigenvectors from multi-body analysis. The first vector represents a rocking motion parallel to the exo domain of the catalytic module, while the second vector represents a rocking motion towards the exo domain.
Discussion
Although Polδ has been studied for decades, major questions have remained about its overall architecture and dynamics. Here, we report the near atomic resolution structure of the Polδ holoenzyme.
Polδ has a unique architecture.
We show that Polδ has a unique architecture that has not been observed previously in any other polymerase complex. In our structure of Polδ, the catalytic module engages the template-primer DNA duplex and is well poised for catalysis. Unexpectedly, the regulatory module does not engage the DNA duplex, contrary to the belief that the regulatory module affects catalysis by associating with the DNA. The regulatory module is positioned proximal to the exo and thumb domains of the catalytic module, but does not make any significant contacts with either domain (Fig. 1e). Given the lack of interactions between the catalytic and regulatory modules, one question is what drives the assembly of Polδ. From our work, CysBD emerges as the keystone of this unique architecture and assembly. CysBD engages with the catalytic module (via the exo and thumb domains) as well as with the Pol31 subunit from the regulatory module. The coordinated interactions of CysBD with both the catalytic and regulatory modules are central to Polδ assembly. Many of the mutations that have previously been implicated in Polδ function11,28 lie at the interface between CysBD and the catalytic and regulatory modules. Interestingly, the interaction of CysBD with the catalytic module pulls the exo and thumb domains closer by >2.5 Å relative to their position in the isolated catalytic core (Supplementary Fig. 4a). This general tightening of the catalytic module may underlie the observed increase in DNA polymerase activity of the Polδ holoenzyme relative to the Pol3 catalytic core by itself29.
A redox sensor in Polδ.
The Fe-S cluster bound to CysBD and its coordination chemistry provide a mechanism for the reversible inactivation or activation of Polδ in response to oxidative stress. Consistent with previous biochemical studies18, the 4Fe-4S cluster of CysBD is coordinated by four cysteines. Intriguingly, the 4Fe-4S cluster is also coordinated with Arg1080, which is conserved in the sequences of all eukaryotic Polδ. A positively charged group, such as the guanidinium moiety of arginine, has the capacity to stabilize the reduced state of the 4Fe-4S cluster by raising its reduction potential22–25. The reduced state of the Fe-S cluster is likely to be important for Polδ activity as an electrochemically oxidized version of the enzyme is significantly slower in processive DNA synthesis26, presumably as a mechanism to slow DNA synthesis in response to replication stress such as oxidation26.
Impact of flexibility.
From the structure, the interface between the catalytic and regulatory modules is relatively sparse and suggestive of a relatively flexible complex able to accommodate changes in DNA direction. Indeed, multi-body analysis of our cryo-EM data suggests that ~35% of the flexibility between the catalytic and regulatory modules is due to motion of the regulatory domain either parallel to or in the direction of the catalytic module (Supplementary Figs. 5 and 6). Flexibility between the regulatory and catalytic modules, coupled with the proximity of the regulatory module to the exo and thumb domains, appears to be important for the 3′→5′ exonuclease activity of Polδ. The transfer of a mismatched primer from the pol to the exo active site for proofreading by B family polymerases typically involves a large movement of the thumb domain31,32. A loose association between the regulatory and catalytic modules may better allow for the thumb movement without incurring a steric penalty. Also, in our structure, the regulatory module and associated CysBD order a loop in the exo domain for putative contacts with a primer modeled at the exo active site (Supplementary Fig. 4b). There may be additional stabilizing interactions stemming from the close juxtaposition of the regulatory module and exo domain, with the observed flexibility as part of the conformational transition in accommodating a primer at the exo active site. In addition to DNA synthesis and proofreading, Polδ is required for Okazaki fragment maturation30,37 and gap filling synthesis in DNA mismatch repair. Local flexibility between the catalytic and regulatory modules may also facilitate the binding of various DNA substrates in these diverse pathways.
Disease mutations.
A number of germline and somatic mutations have been mapped to the catalytic module of human Polδ in cancers associated with ‘hypermutated’ or ‘ultramutated’ tumors38,39. Supplementary Fig. 7 shows the location of driver mutations mapped on the structure of yeast Polδ holoenzyme. Interestingly, the recurrent oncogenic R506H40,41 mutation (corresponding to Arg511 in yeast Pol3) maps at the interface between the regulatory and catalytic modules and is within van der Waals distance from the regulatory module (Fig. 3b). Mutations such as R507C and I1070N in the catalytic subunit of human Polδ are also associated with multi-symptom mandibular hypoplasia, deafness, progeroid features and lipodystrophy syndrome (MDPL) syndrome42. Both R507C and I1070N (corresponding to R512 and L1068 in CysBD of yeast Pol3) again map to the interface between the regulatory and catalytic modules (Fig. 3b). These disease mutations at the interface may modulate the inherent motion between the catalytic and regulatory modules. The correspondence between ‘observed’ motion via multi-body refinement and ‘predicted’ motion via normal mode analysis in our structure is further evidence that this motion is functionally important. In other words, the structure (or the amino acid sequence) of the Polδ holoenzyme has evolved to execute this motion as part of its function.
Methods
Protein and DNA preparation.
S. cerevisiae Polδ holoenzyme was overexpressed and purified from protease-deficient yeast strain, encoding the full length FLAG tagged Pol3, GST tagged Pol31, and Pol3243. Cells containing the overexpressed protein were resuspended in two volumes of lysis buffer containing 50 mM Tris, pH 7.4, 10% sucrose, 150 mM NaCl, 175 mM (NH4)2SO4, 1 mM EDTA, 10 mM β-mercaptoethanol and protease inhibitors benzamidine, leupeptin, aprotinin, chymostatin and pepstatin A. Cells were lysed with a mechanical bead beater using 0.5-mm zirconia beads pre-cooled with an ice/ethanol/dry ice mixture. Clarified cell lysate was first subjected to 55% ammonium sulfate precipitation. Pellet containing the Polδ holoenzyme was resuspended in 50 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol and 2 mM DTT and purified by passing through a column containing glutathione-sepharose beads. Bound protein was eluted by cleaving the GST tag from GST-Pol31 with PreScission protease. Protein was purified further to apparent homogeneity by heparin chromatography. Duplex template-primer DNA was assembled by annealing a 30-nucleotide palindromic DNA (5′ TAATGGTAGGGGAGGAAATTCCTCCCCTACdd 3′) to itself, yielding G as the templating base.
Sample and grid preparation.
For cryo-EM studies, purified protein was mixed with a two-fold molar excess of the template-primer duplex and run over a SD200 gel filtration column. Incoming nucleotide dCTP and CaCl2 were added to capture Polδ in a state just before adding dNTP to the primer 3′ end. A 5 μl volume of the Polδ-DNA complex was applied to lacey gold-coated homemade grids that had been treated with H2/O2 plasma (Gatan Solarus) for 30 s. Grids were blotted and plunge frozen in liquid ethane with a manual cryoplunger.
Data collection.
The grids were imaged in three sessions on two 300 keV Titan Krios (FEI) microscopes equipped with Gatan K2 summit cameras and controlled by the Leginon software44 at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center. For all sessions, frame alignment was performed with Motioncor245 and CTF determination was performed with CTFFind446 using Appion47 for realtime pre-processing. The first session was collected on ‘Krios2’, equipped with a Cs corrector and an energy filter. Images were collected over 10 s with 200 ms frame time in counting mode with a calibrated pixel size of 0.8549 Å and a slit width of 20 eV. The total exposure was 64 e− Å−2. A total of 2,155 images were collected, of which 1,206 were selected for processing based on Thon ring extent. Images for the second and third sessions were collected on ‘Krios1’ over 10 s with a 200 ms frame time in super resolution mode with a super resolution pixel size of 0.416 Å. The total exposure was 63.0 and 63.7 e− Å−2 for the two sessions. A total of 2,824 and 2,856 images were collected in these sessions, with 2,228 and 1,837 micrographs used for further processing, respectively. Aligned movies were rescaled to a final pixel size of 0.8549 Å using MotionCorr2.
Image processing.
Particles were initially picked for the first dataset using DoGPicker48. Rough two-dimensional (2D) classes were created from this initial picking using XMIPP249–51 and, from these, templates were made. These templates were used to pick particles from all three sessions using FindEM52, with parameters set to pick virtually everything that might be a particle. Further processing was performed using RELION 2.053 and RELION 3.0 beta54. Particles from the first session were subjected to 2D classification and 12,500 particles were chosen to create a 3D model. This model was subjected to 3D refinement and used for 3D classification of particles from the first dataset. The cleanest 3D classes and corresponding particles were refined and used as a reference for the second and third datasets, which were processed using both 2D and 3D classification to produce clean particle stacks. Initial maps showed clear density for the catalytic module and CysBD, but the density for Pol31-Pol32 was weak.
To select particles that contain all three subunits, we performed focused 3D classification with signal subtraction. The region corresponding to Pol31-Pol32 was masked, and the density outside the mask was subtracted from all particles. Focused 3D classification was performed without alignment, and classes that showed clear density for all the subunits were selected for all three datasets. The three datasets were combined at this stage and processed further as a group. 3D classification was performed on the combined dataset and one out of the four classes that lacked the Pol31-Pol32 subunit was discarded. Particles belonging to classes showing clear density for Pol31-Pol32 were selected for subsequent refinement with cryoSPARC55, resulting in a consensus map with an overall resolution of 3.2 Å using the Fourier shell correlation (FSC) value of 0.143 between independently refined half sets56,57. Local resolution estimates were calculated with ResMap58.
Multi-body refinement.
Inspection of the consensus map from cryoSPARC revealed weak density for parts of Pol31 that were located away from the catalytic module and for Pol32. Based on the weakening of density with distance from the catalytic module, we reasoned that Polδ exhibits structural flexibility, with regions further from the catalytic module sweeping larger angular arcs. To improve the density and characterize the conformational dynamics, we performed multi-body refinement with RELION 3. Polδ was divided into two discrete bodies composed of the Pol3 catalytic module as one rigid body and the regulatory module plus the CysBD as the second discrete body. Masks for multi-body refinement were made in Chimera59 from the consensus map after applying a Gaussian blur. The maps for the two discrete bodies after multi-body refinement were post-processed individually and combined using PHENIX60. Substantial improvement was observed in the density for Pol32 and regions of Pol31 that were farthest from the catalytic module. Importantly, regions of Pol32 that were not visible in the consensus maps were resolved in the multi-body maps and enabled interpretation of the density for Pol32N, including side chains of amino acids in regions that are involved in interactions with Pol31. PyMOL61 was used to generate movies (Supplementary Videos 1 and 2) to aid visual inspection of motions represented by the first two eigenvectors.
Atomic model building and refinement.
First, the X-ray structure of the catalytic module of Pol3 (amino acids 68–985, PDB 3IAY) bound to DNA and the incoming nucleotide was fitted manually into the cryo-EM map16. The Pol3 catalytic module and Pol31-Pol32N homology models were adjusted manually with COOT62. The density for the Pol3 catalytic module, bound DNA and incoming nucleotide was very well resolved and allowed building of side chains including regions that were previously unresolved in the crystal structure16, 44 water molecules, metal ions and the incoming nucleotide. Similarly, well resolved density for the second metal binding domain (CysBD) of Pol3 allowed for ab initio tracing of amino acids 1031–1096 and the bound 4Fe-4S cluster. Regions of Pol31 that were close to the interface with CysBD had well defined density and allowed reliable interpretation of the secondary structure and side chains, including those that were not visible in the human homolog17. Modeling of the peripheral regions (including Pol32N) relied on a combination of consensus and multi-body maps. The model was refined to a final resolution of 3.2 Å using real space refinement in PHENIX and was validated using MolProbity63 and EMRinger64. Figures were prepared using Chimera and PyMOL61.
Normal mode analysis.
Polδ dynamics was determined by the normal mode analysis approach using the DynaMut web server36. The C-alpha force field based on fitting the Amber94 all-atom potential was selected for normal mode analysis calculations. Results were displayed in PyMOL for visualization and making figures.
Reporting Summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank B. Carragher, C. Potter and E. Eng for helpful advice and discussions throughout the project. Some of the work was supported by grant GM129689 from the NIH (S.P.). Initial EM screening was performed at the Icahn School of Medicine microscope facility supported by a shared instrumentation grant from the NIH (1S10RR026473). Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR and the NIH National Institute of General Medical Sciences (GM103310), with additional support from Agouron Institute (F00316), NIH (OD019994) and NIH (RR029300). Computing resources needed for this work were provided in part by the High Performance Computing facility of the Icahn School of Medicine at Mount Sinai. Molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41594-019-0305-z.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41594-019-0305-z.
Peer review information Beth Moorefield was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Data availability
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank under accession code EMD-20235. Atomic coordinates have been deposited in the Protein Data Bank (https://www.rcsb.org) with accession code 6P1H. All reagents and relevant data are available from the authors upon reasonable request.
References
- 1.Johnson RE, Klassen R, Prakash L & Prakash S A major role of DNA polymerase delta in replication of both the leading and lagging DNA strands. Mol. Cell 59, 163–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartwell LH Sequential function of gene products relative to DNA synthesis in the yeast cell cycle. J. Mol. Biol 104, 803–817 (1976). [DOI] [PubMed] [Google Scholar]
- 3.Stillman B Reconsidering DNA polymerases at the replication fork in eukaryotes. Mol. Cell 59, 139–141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jain R, Aggarwal AK & Rechkoblit O Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol 53, 77–87 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Burgers PMJ & Kunkel TA Eukaryotic DNA replication fork. Annu. Rev. Biochem 86, 417–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulet A, Simon M, Faye G, Bauer GA & Burgers PM Structure and function of the Saccharomyces cerevisiae CDC2 gene encoding the large subunit of DNA polymerase III. EMBO J 8, 1849–1854 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon M, Giot L & Faye G The 3′ to 5′ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 10, 2165–2170 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerik KJ, Li X, Pautz A & Burgers PM Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem 273, 19747–19755 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Johansson E, Garg P & Burgers PM The Pol32 subunit of DNA polymerase delta contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J. Biol. Chem 279, 1907–1915 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Johansson E, Majka J & Burgers PM Structure of DNA polymerase delta from Saccharomyces cerevisiae. J. Biol. Chem 276, 43824–43828 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Sanchez Garcia J, Ciufo LF, Yang X, Kearsey SE & MacNeill SA The C-terminal zinc finger of the catalytic subunit of DNA polymerase delta is responsible for direct interaction with the B-subunit. Nucleic Acids Res. 32, 3005–3016 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mondol T, Stodola JL, Galletto R & Burgers PM PCNA accelerates the nucleotide incorporation rate by DNA polymerase delta. Nucleic Acids Res. 47, 1977–1986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acharya N, Klassen R, Johnson RE, Prakash L & Prakash S PCNA binding domains in all three subunits of yeast DNA polymerase delta modulate its function in DNA replication. Proc. Natl Acad. Sci. USA 108, 17927–17932 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinge S, Nunez-Ramirez R, Llorca O & Pellegrini L 3D architecture of DNA Pol alpha reveals the functional core of multi-subunit replicative polymerases. EMBO J. 28, 1978–1987 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakane T, Kimanius D, Lindahl E & Scheres SH Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. Elife 7, e36861(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swan MK, Johnson RE, Prakash L, Prakash S & Aggarwal AK Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol 16, 979–986 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranovskiy AG et al. X-ray structure of the complex of regulatory subunits of human DNA polymerase delta. Cell Cycle 7, 3026–3036 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Netz DJ et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol 8, 125–132 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z et al. Programmable design of orthogonal protein heterodimers. Nature 565, 106–111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suwa Y et al. Crystal structure of the human Pol alpha B subunit in complex with the C-terminal domain of the catalytic subunit. J. Biol. Chem 290, 14328–14337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baranovskiy AG et al. Crystal structure of the human Pol B-subunit in complex with the C-terminal domain of the catalytic subunit. J. Biol. Chem 292, 15717–15730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adman E, Watenpaugh KD & Jensen LH NH…S hydrogen bonds in Peptococcus aerogenes ferredoxin, Clostridium pasteurianum rubredoxin and Chromatium high potential iron protein. Proc. Natl Acad. Sci. USA 72, 4854–4858 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters MA, Roche CL, Rheingold AL & Kassel SW N-H…S hydrogen bonds in a ferredoxin model. Inorg. Chem 44, 3777–3779 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Carter CW Jr, Kraut J, Freer ST & Alden RA Comparison of oxidation-reduction site geometries in oxidized and reduced Chromatium high potential iron protein and oxidized Peptococcus aerogenes ferredoxin. J. Biol. Chem 249, 6339–6346 (1974). [PubMed] [Google Scholar]
- 25.Chen K et al. Crystal structures of ferredoxin variants exhibiting large changes in [Fe-S] reduction potential. Nat. Struct. Biol 9, 188–192 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Bartels PL, Stodola JL, Burgers PMJ & Barton JK A redox role for the [4Fe4S] cluster of yeast DNA polymerase delta. J. Am. Chem. Soc 139, 18339–18348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tse ECM, Zwang TJ & Barton JK The oxidation state of [4Fe4S] clusters modulates the DNA-binding affinity of DNA repair proteins. J. Am. Chem. Soc 139, 12784–12792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez Garcia J et al. Functional mapping of the fission yeast DNA polymerase delta B-subunit Cdc1 by site-directed and random pentapeptide insertion mutagenesis. BMC Mol. Biol 10, 82(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson RE, Prakash L & Prakash S Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc. Natl Acad. Sci. USA 109, 12455–12460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin YH et al. The multiple biological roles of the 3′→5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell Biol 25, 461–471 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marquez LA & Reha-Krantz LJ Using 2-aminopurine fluorescence and mutational analysis to demonstrate an active role of bacteriophage T4 DNA polymerase in strand separation required for 3′→5′-exonuclease activity. J. Biol. Chem 271, 28903–28911 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Shamoo Y & Steitz TA Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 99, 155–166 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Giot L, Chanet R, Simon M, Facca C & Faye G Involvement of the yeast DNA polymerase delta in DNA repair in vivo. Genetics 146, 1239–1251 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jozwiakowski SK, Kummer S & Gari K Human DNA polymerase delta requires an iron-sulfur cluster for high-fidelity DNA synthesis. Life Sci. Alliance 2, e201900321(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain R et al. Structural insights into yeast DNA polymerase delta by small angle X-ray scattering. J. Mol. Biol 394, 377–382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrigues CH, Pires DE & Ascher DB DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin YH, Ayyagari R, Resnick MA, Gordenin DA & Burgers PM Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′−5′-exonuclease activities of Pol delta in the creation of a ligatable nick. J. Biol. Chem 278, 1626–1633 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Campbell BB et al. Comprehensive analysis of hypermutation in human cancer. Cell 171, 1042–1056 e1010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rayner E et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 16, 71–81 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Flohr T et al. Detection of mutations in the DNA polymerase delta gene of human sporadic colorectal cancers and colon cancer cell lines. Int. J. Cancer 80, 919–929 (1999). [DOI] [PubMed] [Google Scholar]
- 41.da Costa LT et al. Polymerase delta variants in RER colorectal tumours. Nat. Genet 9, 10–11 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Elouej S et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features and lipodystrophy syndrome (MDPL). Metabolism 71, 213–225 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Zhuang Z et al. Regulation of polymerase exchange between Poleta and Poldelta by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc. Natl Acad. Sci. USA 105, 5361–5366 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suloway C et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohou A & Grigorieff N CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voss NR, Yoshioka CK, Radermacher M, Potter CS & Carragher B DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheres SH et al. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol 348, 139–149 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Sorzano CO et al. XMIPP: a new generation of an open-source image processing package for electron microscopy. J. Struct. Biol 148, 194–204 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Sorzano CO et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J. Struct. Biol 171, 197–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roseman AM FindEM-a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol 145, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-. Elife 7, e42166(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Scheres SH & Chen S Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.The PyMOL Molecular Graphics System, Version 2.0 (Schrödinger; ). [Google Scholar]
- 62.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barad BA et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.