

Abstract
The prevalence of type 2 diabetes (T2D) and obesity has recently increased dramatically. These common diseases are likely to arise from the interaction of multiple genetic, socio-demographic and environmental risk factors. While previous research has found genetic risk and education to be strong predictors of these diseases, few studies to date have examined their joint effects. This study investigates whether education modifies the association between genetic background and risk for type 2 diabetes (T2D) and obesity. Using data from non-Hispanic Whites in the Health and Retirement Study (HRS, n = 8398), we tested whether education modifies genetic risk for obesity and T2D, offsetting genetic effects; whether this effect is larger for individuals who have high risk for other (unobserved) reasons, i.e., at higher quantiles of HbA1c and BMI; and whether effects differ by gender. We measured T2D risk using Hemoglobin A1c (HbA1c) level, and obesity risk using body-mass index (BMI). We constructed separate genetic risk scores (GRS) for obesity and diabetes respectively based on the most current available information on the single nucleotide polymorphism (SNPs) confirmed as genome-wide significant predictors for BMI (29 SNPs) and diabetes risk (39 SNPs). Linear regression models with years of schooling indicate that the effect of genetic risk on HbA1c is smaller among people with more years of schooling and larger among those with less than a high school (HS) degree compared to HS degree-holders. Quantile regression models show that the GRS × education effect systematically increased along the HbA1c outcome distribution; for example the GRS × years of education interaction coefficient was −0.01 (95% CI = −0.03, 0.00) at the 10th percentile compared to −0.03 (95% CI = −0.07, 0.00) at the 90th percentile. These results suggest that education may be an important socioeconomic source of heterogeneity in responses to genetic vulnerability to T2D.
Keywords: Genetic risk, Education, Diabetes, Obesity, Older adults
1. Introduction
Type 2 diabetes (T2D) and obesity are two largely preventable chronic conditions. Despite targeted public health interventions, the prevalence of both conditions has increased in recent years. These dual epidemics will likely continue to contribute to substantial morbidity and mortality and greater healthcare costs in the future (Dall et al., 2010; Dieren et al., 2010; Tobias et al., 2014; Wang et al., 2011; Withrow and Alter, 2011; Zhang et al., 2010).
Obesity and T2D both have strong genetic bases (Apovian, 2010; Das and Elbein, 2006; Lin and Sun, 2010; Walley et al., 2009). Environmental and lifestyle factors, such as diet and physical inactivity, are also critical to the pathogenesis of these conditions (Hu et al., 2001; Maes et al., 1997). Previous research investigating the complex interplay of factors contributing to risk has focused on interactions between genetic predisposition and health behaviors. For example, physical activity attenuates genetic vulnerability to obesity and T2D (Ahmad et al., 2013; Brito et al., 2009; Kilpelainen, 2009; Li et al., 2010), and eating foods associated with a Western dietary pattern exacerbates genetic risk on T2D (Cornelis and Hu, 2012). Our study extends this focus to education, a more upstream factor that is likely to moderate the effect of hereditary predispositions towards diabetes risk and obesity through several possible mechanisms.
As a fundamental cause of disease, education acts through multiple pathways to affect health and disease risks (Cutler and Lleras-Muney, 2008) related to differential access to resources (Johnson et al., 2011; Link and Phelan, 1995; Phelan et al., 2010). On an individual-level, education leads to increases in a person’s general capabilities, skills, knowledge, money and prestige (Becker, 1964). Education also increases a person’s ability to access societal resources such as healthy built environments and high-quality medical care (Fig. 1). It is this differential access to resources that individuals use to avoid disease risks or minimize the consequences of disease risks that cannot be directly modified, such as genetic background.
Fig. 1.
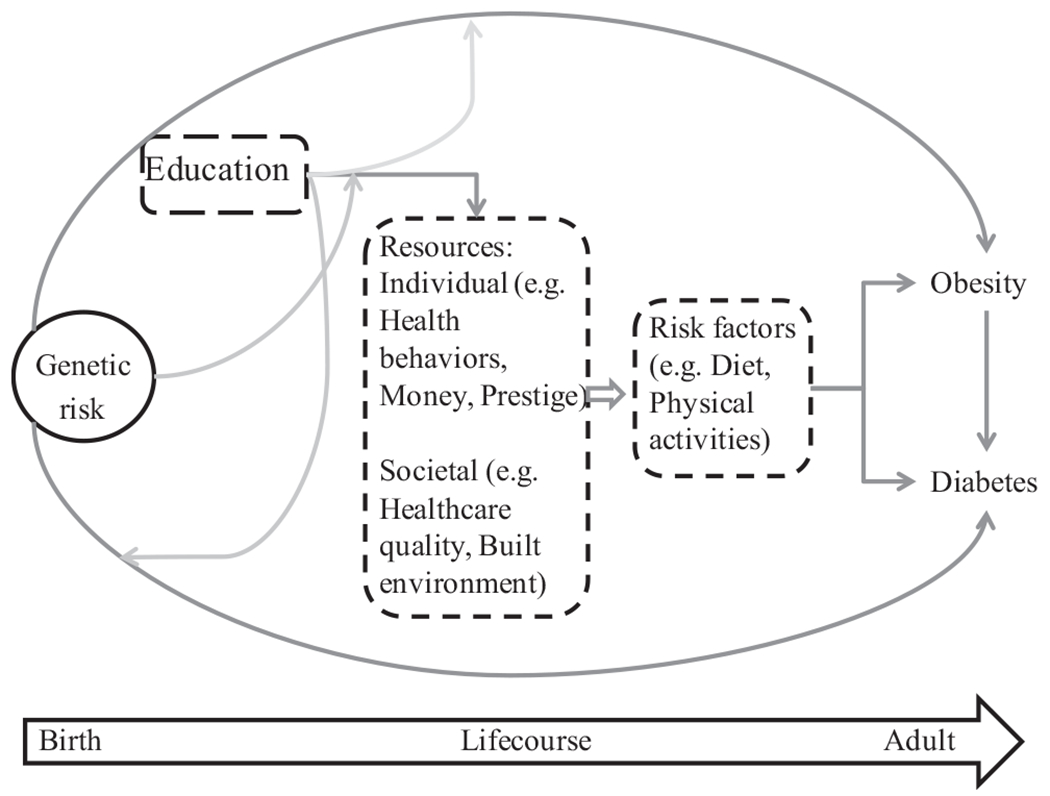
Conceptual framework illustrating the potential pathways linking genetic risk, education and obesity and Type 2 diabetes in later life. Circles indicated fixed risk factors present at birth and boxes with dashed lines indicate modifiable factors throughout the lifecourse.
In this study, we investigate whether education modifies genetic propensity for obesity or T2D. Under the social trigger framework, the presence or absence of contextual variable affects the phenotypic expression of a specific genotype (Shanahan and Hofer, 2005; Reiss et al., 2013). Educational attainment has consistently been shown to be inversely associated with obesity and T2D (Borrell et al., 2006; Cohen et al., 2013; McLaren, 2007). As a fundamental cause of disease, greater educational attainment may trigger mechanisms (e.g. increased psychosocial skills) and be used to attain tangible resources (e.g. high-quality medical care, higher income) that mitigate the impact of inherent genetic risks for these conditions. We hypothesize that attaining more education may enable individuals with higher genetic risk to overcome innate susceptibility to obesity and T2D. Although both obesity and T2D are affected by genetic risk, they are also highly influenced by modifiable risk factors. Education plays a major role in determining access to individual- and environmental-level factors that are protective against obesity and T2D (e.g. healthy food, less stressful occupational environments, residential neighborhoods more conducive to physical activity). Low educational attainment and the subsequent low socioeconomic status and psychological characteristics associated with it may act as a triggering mechanism that affects the expression of inherent genetic risk. For example, a person with high genetic risk for obesity and a college degree may never experience the individual-level and environmental conditions that will lead to greater BMI. However, if the same high-risk individual has less than a high school degree, he/she may have lower access to resources and, as a result, have a greater BMI. In this example, obesity may be “triggered” under the conditions of a high genetic risk and low educational attainment. We expect to find significant effect measure modification of inherent risk by education for T2D and obesity because these conditions have well-known prevention and disease management strategies that are associated with access to socioeconomic resources and more education. The links between HbA1c and BMI and health are not monotonic. Thus, we would expect that education should be associated with the largest reductions in BMI at high ends of the distribution.
Furthermore, we expect the relative contribution of genetic and environmental risk factors for T2D and obesity and their interaction to vary at higher versus lower points in the outcome distribution. Variation in the effects may reflect possible differences within the population that are not readily identifiable. Interaction effects of education and genetic risk score might be larger and negative at the high end of the HbA1c and BMI distribution, as these individuals may be at especially high risk for disease because of other unidentified characteristics. Differences in effects along the outcome distribution may thus reflect unmeasured sub-groupings in the population. Extreme BMI and HbA1c values, the underlying clinical indicators corresponding with risk of obesity and T2D respectively, are associated with elevated mortality risks (Aggarwal et al., 2012; Carson et al., 2010; Flegal et al., 2005) although the exact mechanisms are not always known. Estimates from standard models assuming uniform response will likely understate benefits of education and potentially entirely miss the interaction of education and genetic risk.
Finally, systematic inequalities in resources may also lead to gender-specific interaction effects between education and genetic risk of obesity and T2D. Gene-environment interactions differ by gender when the “environment” reflects gender inequality (Perry et al., 2013). Education differentially shapes the resources of men and women because of their differential access to resources. According to resource substitution theory (Ross and Mirowsky, 2006), females may be more reliant on education because they lack alternative resources to obtain comparable levels of socioeconomic status. Men in the US may have access to more alternative resources than women. For men, more education may not convey substantial additional benefits, because they already have other resources deriving from their physical capacity, inherited wealth, broader range of socially acceptable occupations and activities, and position in the social hierarchy. Women may have more limited options so higher education is necessary to be able to attain such benefits as high occupational prestige and socioeconomic status. Previous research has found stronger effects of education among women compared to men in health conditions as varied as disability, depression, and obesity (Brunello et al., 2013; Ross and Mirowsky, 2006, 2010). We extend this to investigate whether moderation of genetic risk by education is also stronger among women compared to men. In our study, this would imply that education has a stronger moderation effect for women, since females have fewer socioeconomic resources to plausibly offset genetic risk when compared with men. Men with low education levels may still have more opportunities than women with similar educational levels for maintaining a healthy weight or low diabetes risk.
In summary, little work has considered the simultaneous effects of genetic risk and education. Previous research has found that education reduces expression of genetic susceptibilities to health, but these studies used self-reported health outcomes and classical twin study designs (Johnson et al., 2011, 2010). The aim of this study is to investigate whether education can modify the consequences of genetic risk for diabetes and obesity as indicated by HbA1c and BMI. We hypothesize that highly educated individuals are better positioned to overcome genetic vulnerability to obesity and T2D, because these are conditions with well-established prevention or management strategies.
2. Methods
2.1. Sample
We estimated the associations of a polygenic risk score (GRS) specific to T2D and obesity using data from the Health and Retirement Study (HRS). HRS is a well-documented nationally representative sample of individuals 50 years of age or older and their spouses (Juster and Suzman, 1995). The first survey wave was collected in 1992, with biennial interviews (or proxy interviews for decedent participants) available through 2010. Genotype data were collected on a subset of HRS respondents in 2006 and 2008. From the 12,123 HRS participants for whom genotype data were available, we restricted the sample to individuals who self-identified as US-born, non-Hispanic White. Although previous research suggests genetic variation is greatest in African ancestry populations (Hindorff et al., 2009), there are limited large genome-wide association studies (GWAS) where risk variants for type 2 diabetes or obesity has been studied in African American populations. For that reason, we limited our sample to Non-Hispanic Whites. The final analytic sample for the models with BMI as the outcome comprised of 8374 respondents who contributed at least one BMI assessment with complete information on all covariates since entering the HRS study. The final analytical sample for the models with HbA1c as the outcome comprised 8207 individuals who had an HbA1c measurement in 2006 or 2008 with complete information on all covariates. The current analysis was determined exempt by the Harvard School of Public Health Institutional Review Board.
2.1.1. Genotyping
In 2006 and 2008, HRS invited participants to provide DNA samples. Eligible respondents were consented and provided saliva via a mouthwash technique (average age at DNA collection: 68 years). Genotyping was completed on the Illumina Omni-2.5 chip platform and imputed using the 1000G phase 1 reference panel. Genetic information for the first 12,507 participants was filed with the Database for Genotypes and Phenotypes (dbGaP, study accession number: phs000428.v1.p1) in April 2012. All SNPs were extracted from the 1000 Genome imputation sample and the imputation quality for all SNPs was higher than R2 = 0.95. Detailed information on the quality control procedures and the corresponding population eigenvectors is available via HRS and dbGaP (HRS. Health and Retirement Study (HRS) Genetic Data 2012; Available from: http://hrsonline.isr.umich.edu/gwas.). Principle components were used to identify and remove population outliers. Exact information on the QC procedures is available via HRS and dbGaP (study ID: phs000428.v1.p1) (Study, 2012). Correlation between the disease-specific GRS and the top six eigenvectors was negligible (range = −0.09 to 0.05 for T2D and range = −0.04 to 0.05 for obesity, respectively).
2.2. Exposures
Recent large-scale genome-wide association studies have identified multiple loci associated with body-mass index (BMI) and Type II diabetic risk. Each of the condition-specific polygenic risk scores constructed based on previously established genome-wide significant polymorphisms. The most current meta-analysis published reports 32 BMI single nucleotide polymorphisms (SNPs) as genome-wide significant predictors of BMI (Speliotes et al., 2010). Our GRS for obesity was calculated using 29 of those 32 SNPs, three of the 32 SNPs identified in the meta-analysis were not available in our GWAS data. These 29 single nucleotide polymorphisms (SNPs) were also previously associated with weight and waist circumference (Speliotes et al., 2010). We calculated the GRS for obesity for each individual i in our study sample as the sum of weighted risk alleles, with each allele SNP k weighted by the corresponding beta estimate previously reported (Speliotes et al., 2010), as shown in the equation below. We used weights to construct our genetic risk score because it is problematic to assume that all risk alleles confer the same risk. In addition, using a weighted genetic score has been shown to increase statistical power compared to an unweighted score (Burgess and Thompson, 2013). More information on the construction of our GRS is provided in the Supplementary Table S1.
The GRS for diabetes risk was calculated based on 39 known SNPs confirmed as genome-wide significant predictors of T2D, with meta-analyzed odds ratios reported in the GWAS central online database (http://www.genome.gov/gwastudies/). These previously reported β coefficients are from logistic regression models and correspond with the natural log of the OR. We used these β coefficients to calculate the OR for T2D associated with the genotype combination of each individual i in HRS (equation below).
We calculated the GRS for T2D as the log OR for each individual in the HRS cohort associated with his or her own genetic background (Supplementary information Table S2). Each polymorphism was weighted in proportion to its estimated effect on T2D risk and summed for each person across all of the polymorphisms known to be associated with T2D. The correlation between years of schooling and the disease-specific GRS was negligible (correlation coefficient = 0.02 for T2D and −0.004 for obesity, respectively). The GRS was constructed to estimate the expected difference in BMI or odds of T2D anticipated due to the individual’s genetic background, as compared to someone with the lowest possible genetically inferred risk.
Prior research finds that the functional form linking education and health may depend on the specific health outcome under consideration, with some outcomes following a dose–response relationship with years of education and other outcomes primarily moving with credential or level of education. We therefore considered two approaches to characterizing education, based on years of schooling and degree attainment. Although the average attainment in our sample was 13 years, we centered this variable at 12 years of schooling, because this corresponds to the typical length of time needed to complete an HS degree. A quadratic term for years of schooling was initially included but it was not statistically significant and not included in the final models. Educational attainment was also characterized by reported highest degree attained – less than HS, HS graduate/GED, or college degree.
2.3. Outcomes
Our study used objective clinical markers, BMI and HbA1c level, as our outcomes. We characterized the outcomes as continuous measures because this allowed us to examine whether effects were heterogeneous along the outcome distribution. We used the BMI averaged across all waves of available data to maximize sample size. Analysis using wave-specific BMI as an alternative to BMI averaged across all waves yielded similar estimates and statistical significance (results not shown). BMI was constructed from self-reports as weight in kilograms divided by the square of height in meters (kg/m2). Self-reported and measured weight in HRS are highly correlated with a reported correlation coefficient = 0.98 (Sutin, 2013). Diabetes risk was assessed using measured HbA1c levels collected from a subset of HRS participants in 2006 or 2008 (n = 9411). The American Diabetes Association recommends HbA1c as a diagnostic test for diabetes (Verbrugge and Sevak, 2002). HbA1c measures the percentage of hemoglobin, a protein that transports oxygen in red blood cells, that is coated with sugar. It reflects a person’s average blood sugar level over the last six to twelve weeks. Elevated HbA1c level indicates poorer blood sugar control. For people without diabetes, the normal range for HbA1c is between 4% and 5.6% and HbA1c at or above 6.5% is considered to indicate diabetes.
2.4. Covariates
All models included age at time of outcome assessment, a quadratic term for age at time of outcome assessment, year of outcome assessment (2006 vs. 2008), gender (male vs. female), and six eigenvector variables reflecting differences in population distribution of risk. Allele frequencies and, by extension, disease risk, vary across populations of different genetic ancestry. If unaccounted for, these systematic differences may lead to spurious associations between genetic risk markers and disease.
The eigenvector variables in our sample were created using principal components analysis (PCA). The PCA method is currently the recommended approach to control for population stratification (Patterson et al., 2006). PCA identifies a small number of components that explain the maximum covariance between different SNPs across the genome. These eigenvectors variables reflect differences in allele frequencies in the population and help control for the population structure when they are included in a statistical model. Following standard recommendations for HRS data, we included six of the provided eigenvector variables as covariates.
2.5. Analysis
We first modeled the association between genetic risk and BMI and HbA1c using ordinary least squares regression. An interaction term was included to examine possible effect measure moderation by education. We then used quantile regression to assess whether estimates differed along the 10th, 25th, 50th, 75th, and 90th percentiles of the outcome distribution. Quantile regression allows us to model the associations of interest through the entire outcome distribution without specifying the exact cutoffs for “abnormal” values and without specifying a reference category. We estimated effects at multiple quantiles because we do not expect the effects of education will be similar at high versus low quantiles of these outcomes. Quantile regression minimizes the sum of absolute deviations under a specified quantile, allowing the explanatory variables’ marginal effects to vary over different quantiles of the outcome distribution and appropriately accounting for any heteroskedasticity (Johnson et al., 2010). Coefficients from an ordinary least squares regression model are interpreted as the conditional difference in the mean of the dependent variable associated with a unit difference in the independent variable. Coefficients from a quantile regression model are interpreted as the conditional difference for the particular quantile of the outcome distribution (e.g. median) associated with a unit difference in the independent variable.
We examined whether the interaction of GRS and education differed by gender by conducting the following subanalyses: 1) re-running the models outlined above stratified by gender; and 2) re-running the models outlined above including two-way interaction effects (Education × Gender, GRS × Gender, GRS × Education) and three-way interaction effects (GRS × Education × Gender) using a pooled sample.
3. Results
Among this cohort of older White Americans, approximate 64% were 65 or older and 42% were male. On average, the study respondents reported 13 years of schooling (range 0–17). Over 60% reported an HS diploma as their highest level of educational attainment and a quarter had a four-year college degree or more. Almost half of the sample self-reported a doctor’s diagnosis of diabetes, 11% had a measured HbA1c level of 6.5 or higher, and 65% were overweight/obese (Table 1). Genetic risk scores for T2D and BMI were approximately normally distributed (Fig. 2), although the GRS distribution for individuals with HbA1c above 6.5 was slightly skewed.
Table 1.
Socio-demographics of US-born, Non-Hispanic White HRS study respondents.
| N | 8374 |
| Age 65 and over (%) | 64 |
| Male (%) | 42 |
| Mean years of schooling (range) | 13 (0–17) |
| Mean genetic risk score | |
| Diabetes (standard deviation) | 4.1 (0.5) |
| Obesity (standard deviation) | 3.9 (0.5) |
| Highest education attained | |
| Less than HS (%) | 13 |
| HS degree (%) | 62 |
| College degree (%) | 25 |
| Adult health | |
| Mean HbA1c (standard deviation) | 5.8 (0.8) |
| HbA1c ≥ 6.5 (%)a | 10 |
| Mean BMI (standard deviation) | 27.5 (5.1) |
| BMI ≥ 25 (%) | 65 |
| Ever diagnosed with diabetes (%)a | 49 |
Variables reporting HbA1c level used a smaller sample with available HbA1c levels (n = 8207). The socio-demographics for the sample using HbA1c as an outcome was similar to the results given in the table above.
Fig. 2.
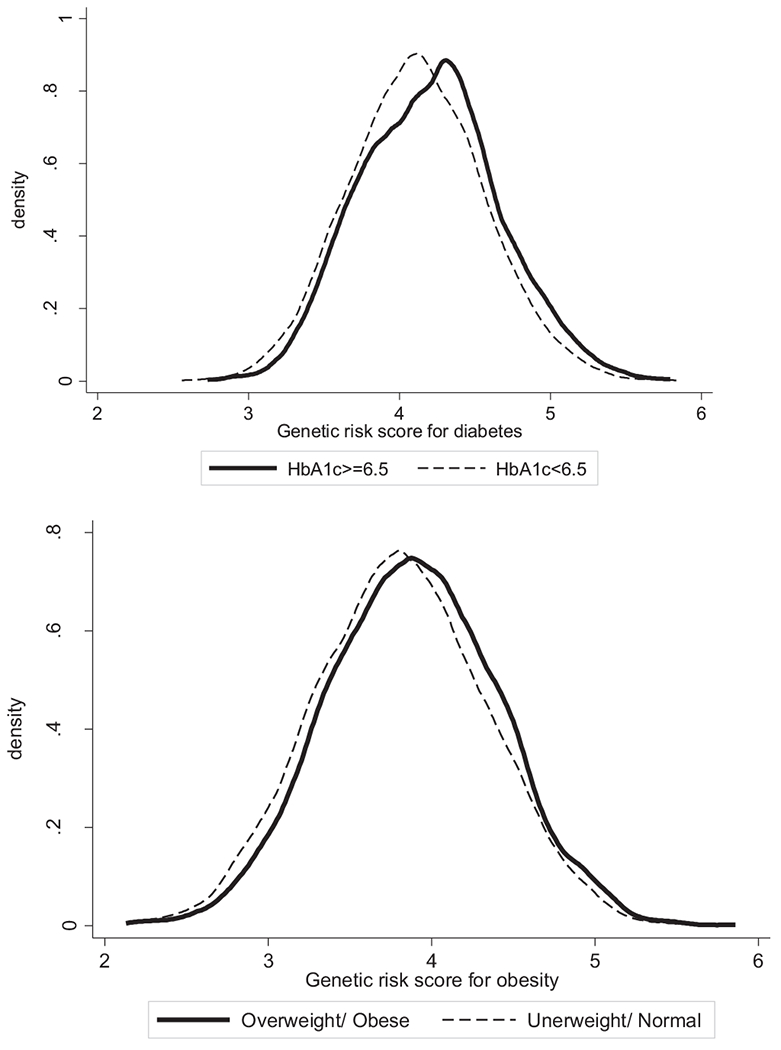
Distribution of genetic risk score for T2D by HbA1c levels and distribution of genetic risk score for obesity by overweight/obesity status.
As shown in Table 2, condition-specific genetic risk scores were associated with higher levels of HbA1c and BMI, respectively (Model 1 and Model 3, Table 2). BMI and HbA1c were both inversely associated with years of schooling. Less than HS was associated with higher BMI and HbA1c levels and college degree was associated with lower BMI and HbA1c compared with those in HS. Models including a quadratic term for years of schooling did not indicate a better fit (results not shown). Including BMI as a covariate in the models with HbA1c did not lead to any substantial changes in the effect estimates (results not shown).
Table 2.
Adjusted associations between genetic risk score and education for HbA1c and BMI, HRS.a
| HbA1c |
Average BMI |
|||||||
|---|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | Model 4 | Model 1 | Model 2 | Model 3 | Model 4 | |
| GRS | 0.18b (0.14–0.22) | 0.20b (0.16–0.25) | 0.18b (0.14–0.22) | 0.17b (0.12–0.22) | 1.04b (0.84–1.25) | 1.05b (0.83–1.27) | 1.05b (0.85–1.25) | 1.03b (0.780 −1.28) |
| Years of schooling | −0.03b (−0.04 to −0.02) | −0.03b (−0.04 to −0.02) | – | – | −0.22b (−0.26 to −0.17) | −0.22b (−0.26 to −0.17) | – | – |
| GRS*years of schooling | – | −0.02b (−0.04 to −0.01) | – | – | – | 0.00 (−0.08 to 0.08) | – | – |
| Degree: Less than HS | – | – | 0.13b (0.08 −0.19) | 0.12b (0.06–0.18) | – | – | 0.54b (0.21–0.86) | 0.58b (0.24–0.92) |
| Degree: GED/HS | – | – | Reference | Reference | – | – | Reference | Reference |
| Degree: College | – | – | −0.13b (−0.17 to 0.08) | −0.12b (−0.17 to −0.08) | – | – | −1.19b (−1.44 to −0.93) | −1.20b (−1.46 to −0.94) |
| Less than HS*GRS | – | – | – | 0.14b (0.02–0.26) | – | – | – | 0.32 (−0.31 to 0.94) |
| College degree*GRS | – | – | – | −0.06 (−0.15 to 0.04) | – | – | – | −0.09 (−0.57 to 0.40) |
| Constant | 7.19 | 7.18 | 7.21 | 7.21 | 35.96 | 35.96 | 36.12 | 36.14 |
| n | 8207 | 8207 | 8207 | 8207 | 8374 | 8374 | 8374 | 8374 |
Models included age at time of outcome assessment as a continuous variable, a quadratic term for age, a binary variable for year of HbA1c outcome assessment (2006 vs. 2008), gender and indicator variables reflecting differences in population distribution of risk. BMI was averaged across all the waves the respondent was in the HRS study.
p < 0.05.
In the second set of models we included two-way interaction terms between GRS and education. The interaction terms between GRS and education were not statistically significant in any of the models predicting BMI. The interaction term between GRS and years of schooling was statistically significant for the model predicting HbA1c (Model 2, Table 2). The negative value for the GRS and years of schooling interaction term implies increasing years of schooling attenuated the effect of GRS on HbA1c (Fig. 3, Panel A). In the models including two-way interaction terms for GRS and degree attainment, the interaction term between less than HS and GRS was statistically significant. The slope for predicted HbA1c was significantly steeper as genetic risk score increased for those with less than HS degree compared to HS degree-holders (Fig. 3, Panel B). The interaction between college degree and GRS was not statistically significant. To account for possible complex relationships between BMI and diabetes, we reran all the above models including BMI as a potential confounder in the models with HbA1c as the outcome (results not shown). Including BMI as a potential confounder in the HBA1c models attenuated the coefficient associated with the education variables while slightly increasing the coefficients associated with GRS. However, none of these small changes in the estimated coefficients made any meaningful difference in statistical significance or interpretation. Including an indicator variable for diabetes medication (insulin or otherwise) substantially reduced the regression coefficients for the diabetes genetic risk score, the education variable and the interaction term between GRS and education (Supplementary Table 3). For example, the regression coefficient associated with college degree decreased from −0.12 (95% CI = −0.17, −0.08) to −0.08 (95% CI = −0.12, −0.04) and the regression coefficient associated with College*GRS decreased from −0.6 (−0.15 to 0.04) to −0.01 (−0.09, 0.07).
Fig. 3.
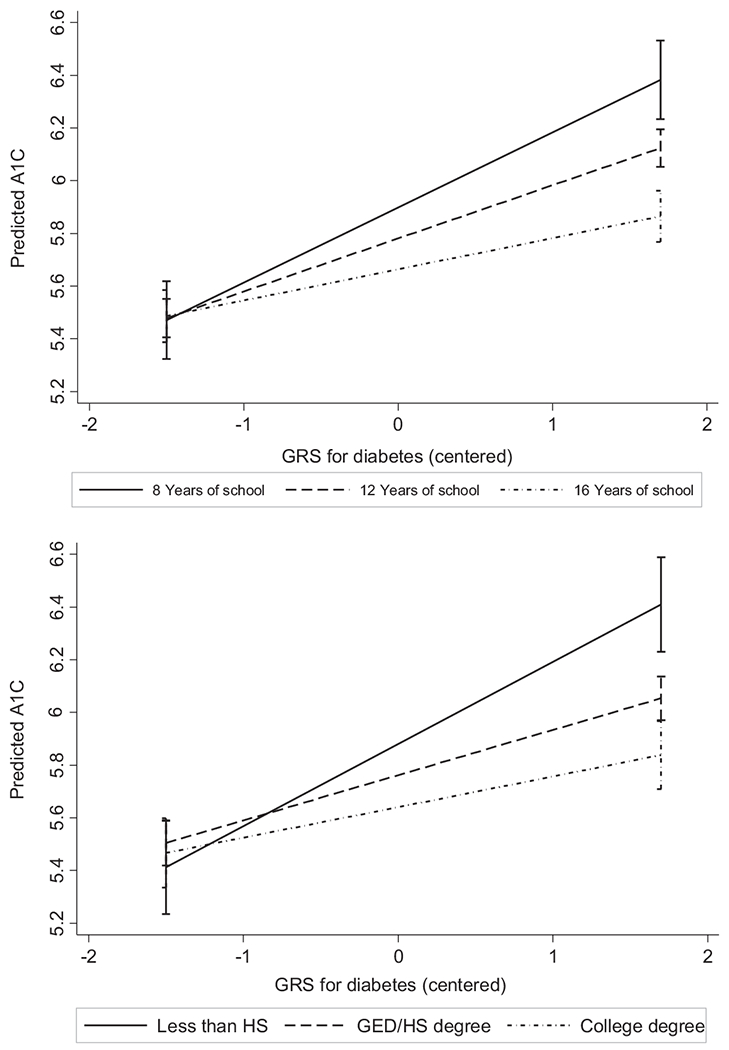
Adjusted predictions HbA1c for given values of GRS and education.
Estimates from the quantile regression models consistently indicate stronger associations for years of schooling and GRS at the higher end of the outcome distribution, where individuals are at actual risk for T2D and obesity. For example, in the fully adjusted models the regression coefficient for the genetic risk score ranged from 0.07 (95% CI = 0.03, 0.12) at the 10th percentile to 0.38 (95% CI = 0.27, 0.49) at the 90th percentile of HbA1c distribution (Table 3). A year of school was associated with only a −0.01 (95% CI = −0.02, 0.00) decrease in HbA1c level at the 10th percentile and a point estimate five times as large at the 90th percentile (beta coefficient = −0.05, 95% CI = −0.07, −0.03). For the models predicting HbA1C, the interaction between years of schooling and GRS was larger at higher values of HbA1c. At the 10th percentile of HbA1c levels in our sample, the interaction term for GRS and years of schooling was only −0.01 (95% CI = −0.03, 0.00) compared to −0.03 (95% CI = −0.08, 0.00) at the 90th percentile.
Table 3.
Estimates from quantile regression models with interaction term for GRS and years of schooling, corresponding th the 10th, 25th, 50th, 75th, and 90th percentiles.a
| P10 | P25 | P50 | P75 | P90 | |
|---|---|---|---|---|---|
| HbA1c | |||||
| GRS | 0.07b (0.03 −0.12) | 0.11b (0.08 −0.15) | 0.16b (0.13 −0.20) | 0.21b (0.15 −0.26) | 0.38b (0.27 −0.49) |
| Years of schooling | −0.01b (−0.02 to 0.00) | −0.01b (−0.02 to −0.01) | −0.02b (−0.02 to −0.01) | −0.03b (−0.03 to −0.02) | −0.05b (−0.07 to −0.03) |
| GRS*years of schooling | −0.01 (−0.03 to 0.00) | −0.01b (−0.03 to 0.00) | −0.02b (−0.03 to −0.01) | −0.02b (−0.04 to −0.01) | −0.03 (−0.07 to 0.00) |
| Constant | 5.60 | 5.86 | 6.57 | 7.70 | 8.94 |
| n | 8207 | 8207 | 8207 | 8207 | 8207 |
| BMI | |||||
| GRS | 0.58b (0.36 −0.81) | 0.81b (0.58 −1.04) | 0.95b (0.69 −1.21) | 1.17b (0.85 −1.50) | 1.29b (0.77 −1.82) |
| Years of schooling | −0.11b (−0.16 to −0.06) | −0.16b (−0.20 to −0.12) | −0.20b (−0.25 to −0.15) | −0.25b (−0.32 to −0.19) | −0.34b (−0.45 to −0.23) |
| GRS*years of schooling | −0.02 (−0.10 to 0.06) | −0.03 (−0.11 to 0.04) | 0.02 (−0.07 to 0.11) | −0.02 (−0.13 to 0.09) | −0.01 (−0.19 to 0.18) |
| Constant | 32.70 | 33.07 | 34.51 | 37.16 | 40.23 |
| n | 8.374 | 8374 | 8374 | 8374 | 8398 |
Models included age at time of outcome assessment as a continuous variable, a quadratic term for age, a binary variable for year of outcome assessment (2006 vs. 2008), gender and six eigenvector variables reflecting differences in population distribution of risk.
p < 0.05.
For models predicting BMI, the absolute effect of GRS and education increased across the BMI distribution. However, the interaction term for GRS and years of schooling was approximately zero throughout the BMI distribution and not statistically significant.
In the quantile regression models using degree attainment in lieu of years of schooling, results were similar. Regression coefficients associated with “less than HS” and the interaction term between “Less than HS” and GRS increased across the HbA1c distribution but were not always statistically significant (Table 4). In the models which included an indicator variable for diabetes medication, regression coefficients generally followed the same pattern of being larger at the higher quantiles (Supplementary Tables 4 and 5). However, the interaction term for GRS and education was no longer statistically significant once we accounted for diabetes medication.
Table 4.
Estimates from quantile regression models with interaction term for GRS and degree.a
| P10 | P25 | P50 | P75 | P90 | |
|---|---|---|---|---|---|
| HbA1c | |||||
| GRS | 0.05b (0.00 −0.12) | 0.08b (0.04 −0.13) | 0.13b (0.09 −0.17) | 0.16b (0.10 −0.22) | 0.30b (0.19 −0.42) |
| Degree: Less than HS | −0.03 (−0.02 to 0.09) | −0.05b (0.01–0.10) | 0.04 (−0.01 to 0.08) | 0.09b (0.00 −0.17) | 0.13 (−0.05 to 0.31) |
| Degree: HS/GED | Reference | Reference | Reference | Reference | Reference |
| Degree: College | −0.02 (−0.06 to 02.02) | −0.05b (−0.08 to 0.02) | −0.09b (−0.12 to −0.05) | −0.12b (−0.17 to −0.08) | −0.24b (−0.33 to −0.15) |
| GRS*Less than HS | 0.09 (−0.03 to 0.22) | 0.12b (0.03 −0.21) | 0.11b (0.02 −0.19) | 0.18 (−0.01 to 0.37) | 0.26 (−0.04 to 0.56) |
| GRS*College | −0.02 (−0.11 to 0.07) | −0.01b (−0.08 to 0.05) | −0.04b (−0.12 to 0.03) | −0.04 (−0.14 to 0.06) | −0.06 (−0.24 to 0.13) |
| Constant | 5.60 | 5.86 | 6.57 | 7.70 | 8.94 |
| n | 8207 | 8207 | 8207 | 8207 | 8207 |
| BMI | |||||
| GRS | 0.05b (0.25 −0.83) | 0.87b (0.62 −1.12) | 1.04b (0.73 −1.35) | 1.20b (0.84 −1.56) | 1.24b (0.62 −1.85) |
| Degree: Less than HS | 0.51 (0.19 −0.84) | 0.18b (−0.12 to 0.48) | 0.25 (−0.13 to 0.64) | 0.64b (0.12 −1.17) | 1.25b (0.45 −2.06) |
| Degree: HS/GED | Reference | Reference | Reference | Reference | Reference |
| Degree: College | −0.43b (−0.69 to −0.16) | −1.01b (−1.22 to −0.80) | −1.34b (−1.68 to −1.01) | −1.39b (−1.78 to −1.00) | −1.46b (−2.01 to −0.91) |
| GRS*Less than HS | 0.18 (−0.39 to 0.74) | 0.02 (−0.53 to 0.58) | 0.09 (−0.60 to 0.77) | 0.33 (−0.53 to 1.20) | 0.88 (−0.61 to 2.37) |
| GRS*College | −0.09 (−0.56 to 0.37) | −0.36 (−0.76 to 0.05) | −0.17 (−0.67 to 0.33) | −0.15 (−0.76 to 0.46) | 0.09 (−1.01 to 1.19) |
| Constant | 33.17 | 33.03 | 34.53 | 37.35 | 42.07 |
| n | 8374 | 8374 | 8374 | 8374 | 8374 |
Models included age at time of outcome assessment as a continuous variable, a quadratic term for age, a binary variable for year of HbA1c outcome assessment (2006 vs. 2008), gender and six eigenvector variables reflecting differences in population distribution of risk.
p < 0.05.
In the gender-stratified analysis, genetic risk score was directly associated with HbA1c levels and years of schooling was inversely associated with HbA1c levels (Appendix Table A). The GRS × years of schooling term for HbA1c was statistically significant for females but not for males (beta coefficient for females = −0.03 (95% CI = −0.05, −0.01) vs. −0.01 (95% CI = −0.04, 0.01) for males). The two-way interaction term between genetic risk and years of schooling was the only statistically significant interaction term in the models using the entire sample with HbA1c as the outcome (Appendix Table B). Similarly, the two-way interaction between genetic risk and less than HS education was the only statistically significant interaction term in the model with degree attainment. In the models with BMI as the outcome, only the interaction term between years of schooling and gender was statistically significant.
4. Conclusion
Common diseases such as T2D and obesity are likely to arise from the interaction of multiple genetic and environmental risk factors. The multitudes of components that determine an individual’s health status suggest that genetic susceptibility may be affected by larger social factors, which may be highly germane to policy. Previous gene and environment research focused on the aspects of the physical environment. However, it is also important to recognize features in the social environment that can modify genetic risk. We investigated whether genetic susceptibility to diabetes and obesity is moderated by education in a cohort of older White Americans. The interaction for education and genetic risk score was statistically significant in models with HbA1c as an outcome. Years of schooling offsets the genetic risk for HbA1c and this effect increases at the higher end of the HbA1c distribution. Similarly, having less than an HS degree augmented genetic risk for HbA1c and this moderation also seems to increase at the higher end of the HbA1c distribution. The interaction term for education and GRS was no longer statistically significant once we adjusted for diabetes medication. Diabetes medication may be a proxy for healthcare access and health knowledge, resources associated with greater educational attainment. These results suggest education may be an important source of heterogeneity in responses to genetic vulnerability to T2D.
Type 2 diabetes develops when our physiological response to chronic fuel excess is inadequate (e.g. low insulin production and increased glucagon secretion). As a result, diabetic individuals develop insulin resistance, abnormal blood nutrient concentrations and metabolic stress, which eventually may lead to damage in key body organs (Nolan et al., 2013). Experts have long argued that the current Type 2 diabetes epidemic results from environmental and social characteristics triggering inherent T2D susceptibility (Ershow, 2009; Franks et al., 2013; Zimmet et al., 2001). Low education levels may be especially deleterious for individuals with high genetic T2D risks because these respondents are unable to access compensatory mechanisms. Having less than an HS degree or low years of schooling is associated with individual-level characteristics (e.g. low socioeconomic status, low income) and other contextual factors (e.g. neighborhoods with limited healthy food options, health norms) that may be triggers for genetic influences for T2D.
According to the social trigger model, certain types of inherited health risk may be responsive to social environments and become more readily apparent under specific circumstances. Our finding is in line with a recent study which found an interaction between APOE-E4 and education, with APOE-E4 having worse effects on memory for those with less than 8 years of education (McArdle and Prescott, 2010). Our study joins an emerging body of literature that suggests education and other upstream social and contextual factors may offset or attenuate genetic health risk (Boardman et al., 2008).
Our finding that the individual and joint effects of genetic vulnerability and schooling are larger at the higher end of the HbA1c distribution reflects that the largest effects may be among individuals with high health risk. Individuals at the higher ends of the HbA1c distribution are likely to have underlying physiological problems (e.g. insulin resistance, hypertension) that may be more sensitive to the social environment. Alternatively, there may be threshold-level of physiological risk before moderation occurs. In either scenario, ordinary least squares regression that estimates the conditional mean of the outcome leads to underestimations of the effect in the most relevant, high-risk group.
In addition, we found the individual effects of genetic vulnerability and schooling to be larger at higher ends of the BMI distribution. This finding is similar to results from a previous study, which found an increasing effect size for BMI genetic risk score along the distribution of fat mass among children (Riedel et al., 2013). However, we did not find any statistically significant interactions between genetic risk and education for BMI. Results from commonly used mean-based regression models may be misleading, because they underestimated the effects of risk factors in general. Furthermore, they do not conceptually reflect the effect in the subpopulation of interest (i.e., those at risk for disease).
Although we hypothesized that the effects of GRS and education would differ by gender, our results were inconclusive. Our gender-stratified findings provide some support for the resource substitution theory of education, suggesting that gender differences in other resources may lead to differentially larger effect measure modification among women. However, investigating effect measure modification by gender using interaction terms did not show any statistically significant interactions between education, genetic risk and gender. More research is needed to better understand possible gender differences with regard to the effect of education on health outcomes.
Alternatively, our results may reflect selective mortality. Selective mortality would explain the gendered pattern of our results if a much smaller fraction of men with low education and high genetic vulnerability to diabetes survived to participate in our study compared to the fraction of similar women who survived. Although we cannot conclusively rule this out, we believe that gender differences in cumulative mortality are insufficient to introduce substantial biases unless the effects of education and genetic risk are extraordinarily large.
This study has several limitations. Our GRS was calculated based on the most current and comprehensive available information but has relatively modest associations with each of the phenotypes. The modest effects are reflected in the 95% confidence intervals provided for all our estimates, which are often consistent with both the null and substantively important effect estimates. As indicated by the wide confidence intervals, in some cases we did not have enough power to rule out important effects, even when associations were not statistically significant (Colegrave and Ruxton, 2003).
The focus of our study was on creating a genetic risk score that would allow maximum statistical power in identifying differential associations of genotype depending on social environment. A limitation of this approach is its inability to identify the specific biological pathways that interact with the social environment. Knowledge of the function of genes have allowed prior studies examining main effects to subdivide gene scores based on whether SNPs are related to genes for satiety (Llewellyn et al., 2014), apidogenesis (Arner et al., 2011) or other specific biological pathways. Future work based on combined data sets may provide adequate statistical power to investigate such interactions to increase our knowledge of the specific biological pathways through which social factors modify genetic risks.
Emerging research suggests that behavioral and environmental characteristics can moderate inherent genetic health risks (Boardman et al., 2008; McArdle and Prescott, 2010). Our findings provide some support for the social trigger model, which speculates that the social environment can attenuate or exacerbate inherent genetic risks (Shanahan and Hofer, 2005). Furthermore, it suggests social stratification may shape how genetic vulnerability is expressed. Social hierarchies based on socioeconomic status determine the health status of individuals (House et al., 1994). According to fundamental cause theory, policies and interventions must address social factors directly to have a population-level impact on disease risk (Link and Phelan, 1995). Our results show how education, a fundamental cause of health and disease, can serve as a valuable resource that offsets even innate biological risk. Education increases an individual’s ability to adapt, modify, and use surrounding resources (Becker, 1964; Cutler and Lleras-Muney, 2008; Ross and Mirowsky, 2006). As such, polices that reduce disparities in education may help offset underlying genetic risk.
Supplementary Material
Acknowledgements
Funding was partially provided by American Heart Association grant 10SDG2640243 and NIH R21 grant R21AG034385.
Appendix Table A.
Gender-stratified analysis for HbA1c and BMI.a
| HbA1c |
BMI |
|||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| GRS | 0.20b (0.13 −0.28) | 0.20b (0.15 −0.25) | 1.05b (0.74, 1.35) | 1.04b (0.73, 1.35) |
| Years of schooling | −0.03b (−0.04 to −0.02) | −0.03b (−0.04 to −0.02) | −0.15b (−0.21, −0.10) | −0.28b (−0.34, −0.21) |
| GRS*years of schooling | −0.01 (−0.04, 0.01) | −0.03b (−0.05 to −0.01) | 0.02 (−0.08, 0.12) | −0.02 (−0.14, 0.11) |
| Constant | 7.37 | 7.13 | 34.20 | 38.00 |
| n | 3415 | 4792 | 3493 | 4881 |
Models included age at time of outcome assessment as a continuous variable, a quadratic term for age, a binary variable for year of HbA1c outcome assessment (2006 vs. 2008), gender and six eigenvector variables reflecting differences in population distribution of risk.
p < 0.05.
Appendix Table B.
Estimates from GRS × Years of schooling × Gender regression modelsa.
| HbA1C | BMI | |||
|---|---|---|---|---|
| GRS | 0.20b (0.15 −0.26) | 0.16b (0.10 −0.23) | 1.05 (0.77 −1.34) | 0.99b (0.67 −1.31) |
| Years of schooling | −0.03b (−0.05 to −0.01) | – | −0.28b (−0.34 to −0.22) | – |
| GRS* Years of schooling | −0.03b (−0.05 to −0.01) | – | −0.02 (−0.13 to 0.09) | – |
| GRS*Male | 0.00 (−0.09 to 0.09) | 0.02 (−0.09 to 0.12) | −0.02 (−0.47 to 0.43) | 0.11 (−0.42 to 0.63) |
| Years of schooling*Male | 0.00 (−0.02 to 0.01) | – | 0.13b (0.04 −0.22) | – |
| GRS*Years of schooling*Male | 0.02 (−0.01 to 0.05) | – | 0.03 (−0.12 to 0.19) | – |
| Degree: Less than HS | – | 0.11b (0.03 −0.18) | – | 0.78b (0.34 −1.22) |
| Degree: GED/HS | Reference | Reference | Reference | Reference |
| Degree: College | – | −0.13b (−0.19 to −0.07) | – | −1.43b (−1.79 to −1.07) |
| Male*Less than HS | – | 0.03 (−0.08 to 0.14) | – | −0.47 (−0.06 to 0.99) |
| Male*College | – | 0.02 (−0.07 to 0.11) | – | 0.58 (−0.24 to 1.40) |
| GRS*Less than HS | – | 0.18b (0.02 −0.34) | – | −0.12 (−0.80 to 0.56) |
| GRS*College | – | −0.08 (−0.21 to 0.05) | – | −0.67 (−1.94 to 0.60) |
| GRS*Less than HS*Male | – | −0.10 (−0.34 to 0.14) | – | 0.03 (−0.95 to 1.01) |
| GRS*College*Male | – | 0.04 (−0.14 to 0.23) | – | 0.03 (−0.95 to 1.01) |
| Constant | 7.19 | 7.22 | 36.01 | 36.11 |
| n | 8207 | 8207 | 8374 | 8374 |
Models included age at time of outcome assessment as a continuous variable, a quadratic term for age, a binary variable for year of HbA1c outcome assessment (2006 vs. 2008) and six eigenvector variables reflecting differences in population distribution of risk.
p < 0.05.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.socscimed.2014.09.009.
References
- Aggarwal V, Schneider ALC, Selvin E, 2012. Low hemoglobin A1c in nondiabetic adults: an elevated risk state? Diabetes Care 35, 2055–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Rukh G, Varga T, Ali A, Kurbasic A, et al. 2013. Gene × physical activity interactions in obesity: combined analysis of 111,421 individuals of European ancestry. PLoS Genet. 9, e1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apovian CM, 2010. The causes, prevalence, and treatment of obesity revisited in 2009: what have we learned so far? Am. J. Clin. Nutr 91, 277S–279S. [DOI] [PubMed] [Google Scholar]
- Arner P, Arner E, Hammarstedt A, Smith U, 2011. Genetic predisposition for type 2 diabetes, but not for overweight/obesity, is associated with a restricted adipogenesis. PLoS One 6, e18284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker GS, 1964. Human Capital. Columbia University Press, New York. [Google Scholar]
- Boardman J, Saint Onge J, Haberstick B, Timberlake D, Hewitt J, 2008. Do schools moderate the genetic determinants of smoking? Behav. Genet 38, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell LN, Dallo FJ, White K, 2006. Education and diabetes in a racially and ethnically diverse population. Am. J. Public Health 96, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito EC, Lyssenko V, Renström F, Berglund G, Nilsson PM, Groop L, et al. 2009. Previously associated type 2 diabetes variants may interact with physical activity to modify the risk of impaired glucose regulation and type 2 diabetes: a study of 16,003 Swedish adults. Diabetes 58, 1411–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunello G, Fabbri D, Fort M, 2013. The causal effect of education on body mass: evidence from Europe. J. Labor Econ. 31, 195–223. [Google Scholar]
- Burgess S, Thompson SG, 2013. Use of allele scores as instrumental variables for Mendelian randomization. Int. J. Epidemiol 42, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson AP, Fox CS, McGuire DK, Levitan EB, Laclaustra M, Mann DM, et al. 2010. Low hemoglobin A1c and risk of all-cause mortality among US adults without diabetes. Circ. Cardiovasc. Qual. Outcomes 3, 661–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AK, Rehkopf DH, Deardorff J, Abrams B, 2013. Education and obesity at age 40 among American adults. Soc. Sci. Med 78, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrave N, Ruxton GD, 2003. Confidence intervals are a more useful complement to nonsignificant tests than are power calculations. Behav. Ecol 14, 446–44. [Google Scholar]
- Cornelis MC, Hu FB, 2012. Gene-environment interactions in the development of type 2 diabetes: recent progress and continuing challenges. Annu. Rev. Nutr 32, 245–259. [DOI] [PubMed] [Google Scholar]
- Cutler D, Lleras-Muney A (Eds.), 2008. Education and Health: Evaluating Theories and Evidence. Russell Sage Foundation, New York. [Google Scholar]
- Dall TM, Zhang Y, Chen YJ, Quick WW, Yang WG, Fogli J, 2010. The economic burden of diabetes. Health Aff. 29, 297–303. [DOI] [PubMed] [Google Scholar]
- Das SK, Elbein SC, 2006. The genetic basis of type 2 diabetes. Cellscience 2, 100–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieren S.v., Beulens JWJ, Schouw Y.T.v.d., Grobbee DE, Nealb B, 2010. The global burden of diabetes and its complications: an emerging pandemic. Eur. J. Cardiovasc. Prev. Rehabil 17, s3–s8. [DOI] [PubMed] [Google Scholar]
- Ershow AG, 2009. Environmental influences on development of type 2 diabetes and obesity: challenges in personalizing prevention and management. J. Diabetes Sci. Technol. 3, 727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Graubard BI, Williamson DF, Gail MH, 2005. Excess deaths associated with underweight, overweight, and obesity. J. Am. Med. Assoc 293, 1861–1867. [DOI] [PubMed] [Google Scholar]
- Franks PW, Pearson E, Florez JC, 2013. Gene-environment and gene-treatment interactions in type 2 diabetes: progress, pitfalls, and prospects. Diabetes Care 36, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci 106, 9362–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House JS, Lepkowski JM, Kinney AM, Mero RP, Kessler RC, Herzog AR, 1994. The social stratification of aging and health. J. Health Soc. Behav 35, 213–234. [PubMed] [Google Scholar]
- Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, et al. 2001. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med 345, 790–797. [DOI] [PubMed] [Google Scholar]
- Johnson W, Kyvik K, Skytthe A, Deary IJ, Sørensen TIA, 2011. Education modifies genetic and environmental influences on BMI. PLoS One 6 (1), e16290 10.1371/journal.pone.0016290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W, Kyvik KO, Mortensen EL, Skytthe A, Batty GD, Deary IJ, 2010. Education reduces the effects of genetic susceptibilities to poor physical health. Int. J. Epidemiol 39, 406–414. [DOI] [PubMed] [Google Scholar]
- Juster FT, Suzman R, 1995. An overview of the health and retirement study. J. Hum. Resour 30, S7–S56. [Google Scholar]
- Kilpelainen T, 2009. Physical Activity, Genetic Variation and Type 2 Diabetes Medical Sciences. Kuopio University, Finalnd, pp. 1–126. [Google Scholar]
- Li S, Zhao JH, Luan J.a., Ekelund U, Luben RN, Khaw K-T, et al. 2010. Physical activity attenuates the genetic predisposition to obesity in 20,000 men and women from EPIC-Norfolk prospective population study. PLoS Med. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Sun Z, 2010. Current views on type 2 diabetes. J. Endocrinol 204,1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link BG, Phelan JC, 1995. Social conditions as fundamental causes of disease. J. Health Soc. Behav Spec No. 80–94. [PubMed] [Google Scholar]
- Llewellyn CH, Trzaskowski M, van Jaarsveld CM, Plomin R, Wardle J, 2014. Satiety mechanisms in genetic risk of obesity. JAMA Pediatr. 168, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes HM, Neale M, Eaves L, 1997. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet 27, 325–351. [DOI] [PubMed] [Google Scholar]
- McArdle JJ, Prescott CA, 2010. Contemporary modeling of gene × environment effects in Randomized Multivariate Longitudinal studies. Perspect. Psychol. Sci 5, 606–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren L, 2007. Socioeconomic status and obesity. Epidemiol. Rev 29, 29–48. [DOI] [PubMed] [Google Scholar]
- Nolan CJ, Damm P, Prentki M, 2013. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 2011, 169–181. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price A, Reich D, 2006. Population Structure and Eigen analysis. PLoS Genet. 2, e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry BL, Pescosolido BA, Bucholz K, Edenberg H, Kramer J, Kuperman S, et al. 2013. Gender-specific gene-environment interaction in alcohol dependence: the impact of daily life events and GABRA2. Behav. Genet 43, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JC, Link BG, Tehranifar P, 2010. Social conditions as fundamental causes of health inequalities: theory, evidence, and policy implications. J. Health Soc. Behav 51 (Suppl.), S28–S40. [DOI] [PubMed] [Google Scholar]
- Reiss D, Leve LD, Neiderhiser JM, 2013. How genes and the social environment moderate each other. Am. J. Public Health 103, S111–S121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C, von Kries R, Fenske N, Strauch K, Ness AR, Beyerlein A, 2013. Interactions of genetic and environmental risk factors with respect to body fat mass in children: results from the ALSPAC study. Obes. Silver Spring 21, 1238–1242. [DOI] [PubMed] [Google Scholar]
- Ross C, Mirowsky J, 2006. Sex differences in the effect of education on depression: resource multiplication or resource substitution? Soc. Sci. Med 63, 1400–1413. [DOI] [PubMed] [Google Scholar]
- Ross C, Mirowsky J, 2010. Gender and the health benefits of education. Sociol. Q 51, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan MJ, Hofer SM, 2005. Social context in gene-environment interactions: retrospect and prospect. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci 60, 65–76. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. 2010. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet 42, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study H.a.R., 2012. Quality Control Report for Genotypic Data. University of Washington. [Google Scholar]
- Sutin A, 2013. Optimism, pessimism and bias in self-reported body weight among older adults. Obesity 21, E508–E512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias DK, Pan A, Jackson CL, O’Reilly EJ, Ding EL, Willett WC, et al. 2014. Body-mass index and mortality among adults with incident type 2 diabetes. N. Engl. J. Med 370, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbrugge LM, Sevak P, 2002. Use, type, and efficacy of assistance for disability. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci 57, S366–S379. [DOI] [PubMed] [Google Scholar]
- Walley AJ, Asher JE, Froguel P, 2009. The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet 10, 431–442. [DOI] [PubMed] [Google Scholar]
- Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M, 2011. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 378, 815–825. [DOI] [PubMed] [Google Scholar]
- Withrow D, Alter DA, 2011. The economic burden of obesity worldwide: a systematic review of the direct costs of obesity. Obes. Rev 12, 131–141. [DOI] [PubMed] [Google Scholar]
- Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, et al. 2010. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 293–301. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti KGMM, Shaw J, 2001. Global and societal implications of the diabetes epidemic. Nature 414, 782–787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.