

Abstract
Chronic wounds include, but are not limited to, radiation ulcers, pressure ulcers, vascular ulcers and diabetic foot ulcers. These chronic wounds can persist for years without healing and severe ulcers may lead to amputation. Unfortunately, the underlying pathologies of refractory chronic wounds are not fully characterized, and new treatments are urgently needed. Recently, increasing evidence has indicated that cell senescence plays an important role in the development of chronic wounds, and preventing cell senescence or removing senescent cells holds promise as a new therapeutic strategy. In this review, we aim to probe these latest findings to promote the understanding of cellular senescence in the pathological process and potential management of chronic wounds.
Keywords: Chronic wounds; Cell senescence; Therapeutic agent; Skin, Mucosa
Background
Chronic wounds do not progress in a timely manner during the healing process, causing a huge financial and medical burden on the health system [1, 2]. Chronic wounds can be classified as radiation ulcers and non-radiation ulcers, including pressure ulcers, diabetic foot ulcers and vascular ulcers (including venous and arterial ulcers) [3]. These chronic wounds can last for several months to years and often recur, leading to functional loss of skin or mucosa and decreased life quality [4]. Various prevention and treatment approaches, such as anti-inflammatory drugs, growth factors, local anaesthetics, extracellular matrix (ECM) treatment, negative-pressure wound therapy and engineered skin have been used to cure chronic wounds, but many of these therapies are less effective [5, 6]. Therefore, new, valid agents or treatments are urgently needed.
The common features of these wounds include persistent infection, prolonged or exaggerated inflammation, failure of epidermal and/or dermal cells to respond to repair stimuli and the formation of biofilms caused by resistant microorganisms [3, 7]. Thus, these pathophysiological phenomena contribute to the failure of wound healing, but the underlying pathologies are numerous or even unclear in different chronic wounds. Recent evidence has shown that senescent cells accumulate in some chronic wounds, promoting the development of poorly healing wounds [8–12]. Furthermore, removing senescent cells or preventing cell senescence has been reported to mitigate chronic wounds [11, 12]. Based on these new advances, we hypothesize that cellular senescence is a promising target for chronic wounds.
Review
Clinical challenges of chronic ulcers
Wound healing is among the most complex processes in the human body [13, 14]. At the cellular level, wound healing requires the participation of many cell types, including fibroblasts, keratinocytes, macrophages, endothelial cells and platelets that are timely coordinated in space [15]. The physiological process of wound healing can be divided into four phases: haemostasis, inflammation, proliferation and remodelling [16, 17]. After injury, the clotting cascade is activated immediately and haemostasis occurs, preventing blood loss and providing a temporary matrix for cell migration [18]. During this process, immune cells, fibroblasts and endothelial cells are attracted by several growth factors, such as platelet-derived growth factor (PDGF), transforming growth factor-β and epidermal growth factor (EGF), which are secreted by platelets and can activate the healing process. Meanwhile, inflammatory cells migrate to the wound site and remove bacteria or necrotic tissues. Next, macrophages can release many growth factors and cytokines that can initiate the formation of granulation tissue. Next, fibroblast growth factor (FGF) and vascular endothelial growth factor stimulate endothelial cells to proliferate; FGF, transforming growth factor-α and EGF promote epithelial cells to proliferate and migrate, increasing the formation of blood vessels and epithelialization. Finally, organized collagen bundles are remodelled by the provisional matrix when the wound has closed, keratinocytes begin to differentiate and stratify and scar remodelling appears; this phase may last for 1–2 years or longer [19].
In most clinical settings, wound healing can be properly executed with highly organized and coordinated cellular processes, and many acute wounds can heal [6]. However, if healing cannot progress in an orderly and timely manner, chronic wounds may form [20]. The nonhealing state results in the loss of function and morbidity and has a huge impact on life quality [13]. The 5-year mortality rate after amputation is approximately 50% [21, 22]. In general, chronic wounds stall in the inflammatory phase, leading to persistent inflammation. Although the aetiology is different at the molecular level, chronic wounds have some prevalent characteristics, such as increased proteases, pro-inflammatory cytokines, harmful reactive oxygen species (ROS), prolonged infection, accumulation of senescent cells and dysfunctional stem cells. Additionally, PDGF and microorganisms stimulate the constant influx of immune cells that contribute to the amplified and persistent pro-inflammatory cytokine cascade with high levels of protease. In chronic wounds, the level of proteases is too high and cannot be suppressed to normal levels, resulting in ECM destruction and promotion of the degradation of growth factors and their receptors. However, in acute wounds, the inhibitors of proteases can strictly control ECM levels. When the ECM is proteolytically destroyed, the wound fails to move on to the proliferative phase and attracts many inflammatory cells, amplifying the inflammatory cycle (Figure 1) [23].
Figure 1.
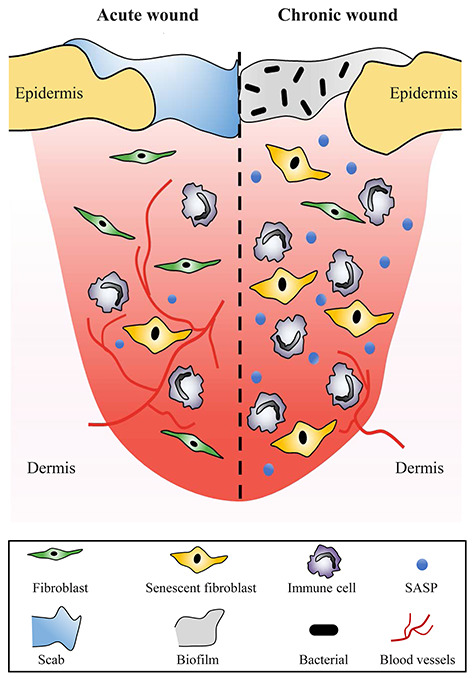
Molecular and cellular differences between chronic and acute wounds. The transient inflammatory response is initiated in the healing process of acute wounds and provides a beneficial environment for re-epithelialization and regeneration. However, chronic wounds stall in the inflammatory phase, leading to persistent inflammation. Chronic wounds exhibit the accumulation of senescent cells and an increased senescence-associated secretory phenotype (SASP) with poor blood vessel infiltration
Numerous topical dressings and antimicrobials are available for clinicians. However, few prospective studies have favoured their effectiveness in promoting chronic wound repair, and doctors tend to use strategies based on personal experience. Many products or therapies, such as the Oasis wound matrix, Promogran, Renasys, Regranex, OxyHeal1000 and Integra, have been applied to cure chronic wounds but the effect is not very ideal and the treatment duration is relatively long. Bioengineered substitutes containing living cells (including Grafix, Dermagraft and Apligraf) have been developed to increase the effect on the skin [13]. Additionally, cellular therapies are effective and safe in treating chronic wounds for people with diabetes and other impaired conditions. However, despite using the best care, 15–20% of all chronic wound sufferers show a poor response to the therapies mentioned above.
It is crucial not only to improve the symptoms of the wound, such as pus and pain, but also to ameliorate potential metabolic and systemic disorders, such as peripheral arterial disease and infections. More importantly, the choice of treatments should be based on the available evidence to ensure the highest possible efficacy. Many challenges exist to study the mechanisms and main contributing factors of chronic wounds, including complex fundamental changes and various processes or cell types involved in chronic wounds, as well as the lack of a specific target on which to focus interventions in this multifactorial system. Additionally, it is essential to further understand the physiological perturbations and underlying molecular mechanisms in nonhealing wounds; however, it would be challenging to establish an optimal animal model to replicate their complexity [24].
Characteristics of cell senescence and the senescence-associated secretory phenotype
Cell senescence is a regulatory response to multiple types of cellular stress, such as DNA damage, telomere erosion, oncogene activation, oxidative damage, protein misfolding and exposure to extracellular signals (like mitogens and cytokines), which may occur at any point in the cell’s life cycle [25, 26]. At the molecular level, p53 and p16INK4a/Rb are two core senescence-regulating pathways in cellular growth arrest [19]. During this process, cells undergo a series of phenotypic transformations with prolonged cell cycle arrest. There is increased production of ROS, persistent DNA damage foci (containing DNA damage sensors such as gamma histone variant H2AX and serine/threonine-kinase Ataxia Telangiectasia Mutated (ATM)-like protein, which are dependent on the stimulus and frequency) and epigenetic rearrangements in senescent cells. After injury, excessive ROS in the wounds can destroy proteins, lipids and nucleic acids, contributing to impaired stem cell function and cell senescence [27]. These cells fail to activate and expand, undergoing accelerated entry into a full senescence state, even in a youthful environment [28]. Additionally, telomere shortening can limit the proliferation of primary cells to a finite number of divisions after injury, resulting in replicative senescence; meanwhile, the deficiency of growth factors also contributes to premature senescence at the wound site [29, 30]. Moreover, mechanical trauma or radiation promotes DNA double- and single-strand breaks, which are known inducers of cell cycle arrest signals [31]. Therefore, cell senescence is a common phenomenon following injury.
Cell senescence is another fate besides apoptosis when cells are exposed to irreparable or excessive cellular and genotoxic stress, and senescent cells can undergo apoptosis resistance [25]. Furthermore, senescent cells are highly metabolically active in tissues; they can secrete high levels of senescence-associated secretory phenotype (SASP) components, including cytokines, matrix remodelling proteins and growth factors [32]. These molecules can change the microenvironment and play an important role in a wide range of biological processes from physiology to pathology [33, 34]. Additionally, these secreted factors cause inflammation, which may be crucial for the removal of senescent cells by phagocytosis, at least in some cases; for example, inflammation can drive the recruitment and activation of immune cells, including monocytes/macrophages, natural killer cells and T-cells, leading to the subsequent elimination of senescent cells [35–37]. SASP components also trigger growth arrest and dysfunction in neighbouring cells via a mechanism that generates DNA damage and ROS in a paracrine manner [38–40]. There is a distinct hierarchy among SASP factors—some of them are necessary for maintenance, and others are used to induce a secretory phenotype. The expression of interleukin-1α can activate the C/EBPβ and NF-κB pathways, which cooperatively regulate SASP components in various senescence contexts and result in the induction of the SASP [38, 41, 42]. Other factors, such as interleukin-6 and chemokine receptor 2-binding chemokines, can form positive feedback loops that reinforce the expression of the SASP as well as growth arrest [42, 43]. Therefore, the SASP has powerful paracrine and autocrine activities, which could create an inflammatory and profibrotic microenvironment.
Cell senescence in wound healing and regeneration
The role of cell senescence has been mainly limited to cellular damage or stress. However, cell senescence has been observed in human, chicken, mouse and quail embryo development [44–46], suggesting that it is a conservative characteristic of vertebrate embryonic development. In addition to embryonic development, cell senescence also occurs in adults in physiologically programmed ways; particularly, placental syncytiotrophoblasts and normal megakaryocytes undergo cell senescence as part of the natural maturity programme [47, 48]. Remarkably, however, senescent cells were ultimately eliminated in both normal development and physiology processes that involve delayed infiltration of macrophages and compensatory apoptosis [36, 37, 44, 46, 49]. Additionally, potent and rapid activation of cell senescence in adult animals has been identified in multiple-wound-healing models.
Activation of the p16INK4a promoter is observed within 2–3 days in injured tissues, peaks between 4 and 7 days and then resolves over 2–3 weeks using p16INK4a reporter mice [50]. Significant induction of cellular senescence occurs during salamander limb regeneration, but rapid and effective mechanisms of senescent cell clearance operate in normal and regenerating tissues. Cellular senescence is a normal process during salamander limb regeneration and it is subject to dynamic regulation [51]. The expression of SASP cytokines and NF-κB activation are found at the wound sites and the clearance of p16INK4a-positive cells delays wound closure with increased fibrosis, suggesting cell senescence is crucial for optimal healing [50]. After the stage of cell proliferation and ECM deposition, myofibroblasts from the wound become senescent, with cell cycle arrest and upregulation of the ECM-degrading enzyme, emphasizing the importance of cell senescence as a limiting mechanism of fibrosis in wound healing [50]. However, animals deficient in p16INK4a show no defects in the healing process, indicating that not p16INK4a, but some feature of p16INK4a-expressing cells (senescent cells), promotes tissue remodelling in the wound [52]. Senescent endothelial cells and fibroblasts are induced instantaneously at the wound site, where they promote wound closure by inducing myofibroblast differentiation by secreting platelet-derived growth factor alpha polypeptide a (PDGF AA); therefore, it is probable that SASP components are candidates for this effect [50]. Furthermore, matricellular protein cellular communication network factor 1 is dynamically expressed following injury and could activate the ROS-dependent p16INK4a/pRb pathway, contributing to the expression of antifibrotic genes and cellular senescence in the wound, where the accumulation of senescent fibroblasts in granulation tissues and expression of antifibrotic genes in the healing cutaneous wounds are observed [53]. Maintaining the integrity of the tissues around the wound is a key aspect of wound healing and this process depends on ECM deposition, which should be strictly controlled; otherwise, it will lead to fibrosis and scarring [54]. More importantly, there is convincing evidence that cells undergoing injury-induced senescence are usually cleared through immune-mediated removal in the late wound-healing process [36, 37, 49]. Taken together, the results of these studies show that cell senescence promotes skin development, repair and regeneration in the early stages of wound healing.
Cell senescence in the impaired healing of chronic wounds
Senescent cells secrete a series of SASPs that regulate the surrounding microenvironment, which directly or indirectly affects various processes of regeneration, including angiogenesis, matrix remodelling, cell plasticity and growth [42, 43, 55]. Senescent cells are then cleared by macrophage-dependent immunosurveillance. However, senescent cells are not removed from chronic wounds, causing persistently elevated secretion of cytokines and decreased proliferation [56]. There is a shift to type M2 macrophages during ageing and trauma that correlates with a reduced immune response, tumour promotion and impaired phagocytosis and chemotaxis [57, 58]; and it is speculated that reduced chemotaxis of macrophages in chronic wounds is involved in the impaired capacity to migrate to the places where senescent cells accumulate (e.g. impaired response to SASP factors), contributing to the accumulation of senescent cells [59]. Because macrophages are key immune cells for the clearance of senescent cells, their absence should lead to high levels of senescence markers (unless other compensatory pathways are activated) and SASPs with sustained inflammation (Figure 2). Additionally, fibroblasts from chronic wounds are less able to respond to growth factors that usually stimulate mitotic responses. Studies have shown that the responses to FGF, EGF and PDGF are reduced in senescent cells, activities that are not related to the reduction in the number of receptors but could be due to the inactivation of intracellular signals [60]. Elevated matrix metalloproteinase levels are observed in chronic wounds and have been implicated in the degradation of growth factors and delays to wound healing [9]. More importantly, to identify accurate therapeutic strategies to remove the senescent cells and their products, differences in the SASP between healing wounds and chronic wounds need to be investigated.
Figure 2.
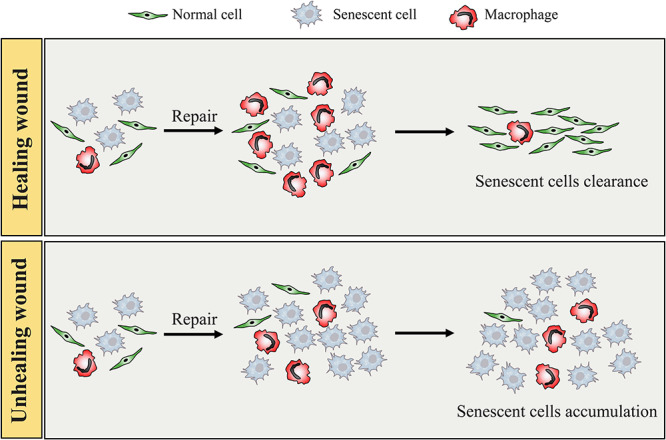
Cell senescence acts as a double-edged sword in wound healing. Cell senescence is crucial for the optimal healing process of acute wounds at the early stage, and then senescent cells are cleared by macrophage-dependent immunosurveillance. However, reduced chemotaxis of macrophages in chronic wounds is involved in the impaired capacity to migrate to the sites where senescent cells accumulate (e.g. impaired response to senescence-associated secretory phenotype factors), contributing to the accumulation of senescent cells. Additionally, senescent cells induce a pro-senescent and pro-inflammatory environment, and the process of cell senescence is constantly being amplified in chronic wounds
In the radiation-induced chronic wound model, we reported that senescent cells and DNA damage accumulate in radiation-induced ulcers in both animal and human tissues [11, 12], and the development of radiation ulcers is accelerated when senescent cells are injected subcutaneously into the irradiated area [11, 12]. Skin injected with senescent cells shows an accelerated process of redness, swelling, hair loss and ulceration. In non-radiation-induced chronic wounds, senescent fibroblasts, endothelial cells and keratinocytes have been reported to accumulate at the wound sites [9, 61–63]. Senescent cells with a prolonged inflammatory response, niche disruption or progenitor depletion have been reported in non-healing pressure ulcers, resulting in impaired wound healing [64]. In venous hypertension, premature cell senescence was observed to result in venous ulcers with delayed healing: approximately 15% of senescent cells were isolated from the wound sites and the rate of wound healing was negatively correlated with the number of senescent cells [65]. The SASP in chronic wounds can result in oxidative stress, which contributes to abnormal metabolic changes and DNA damage in patients with diabetes [9, 66]. Additionally, a long-term inflammatory response may have adverse effects on wound closure. Long-term exposure to chronic wound fluid may also decrease cell activity in the wound and lead to cell senescence. Moreover, prolonged inflammation and cell senescence may have adverse effects on the efficacy of topical biologics (including growth factors) by creating an environment with fewer receptors for growth factors [8]. Those phenomena indicate that cell senescence plays a vital role in both radiation-induced and non-radiation-induced chronic ulcer development.
Stem cell proliferation and signal transduction occur throughout each stage of wound healing; thus, stem cell dysfunction can lead to chronic wounds [67]. Cell-based therapy is a distinct and reasonable step to treat chronic wounds, and its clinical application may be beneficial because stem cells can directly interact with the environment in multifactorial and complex ways at the wound sites and they can directly differentiate and replace components of the lost tissues or cells, such as fibroblasts, keratinocytes and skin appendages. Additionally, stem cells possess powerful immunomodulatory properties and can activate various cytoprotective genes in target tissues [68]. Furthermore, mesenchymal stem cells have been characterized to play a vital role in the healing process [69, 70]; when injury occurs, they can be recruited into the circulation and engraft into the remodelling microvasculature. However, the function of stem cells in chronic wounds is defective [69, 71, 72]. Endothelial progenitor cells from patients with diabetes can adhere to tumour necrosis factor-activated endothelial cells and show damaged migration capacity to wound sites [67]. The currently available evidence also indicates that persistent senescent cells delay the healing of chronic wounds; senescent cells also induce a DNA damage response and cell senescence in neighbouring cells via processes involving ROS and gap junction-mediated cell–cell contact [40]. Senescent cells were transplanted into the skeletal skin and muscle of immunocompromised neuron-specific gene (NSG) mice and, 3 weeks after the last transplantation, the dermal fibroblasts and myofibres expressed various senescence markers around the area where senescent cells were transplanted but not in the area with non-senescent or no cells injected [73]. Therefore, resident senescent cells can result in the dysfunction of stem cells in the healing process of chronic wounds. In this regard, senescent cells may contribute to the dysfunction of stem cells and delay the healing process of chronic wounds. To conclude, abnormal wound healing is closely linked to an impaired microenvironment, biofilm deposition and cell function, and cell senescence is detrimental in the healing process of chronic wounds.
Senescent cells as an emerging therapeutic target for chronic wounds
Very recently, several studies have shown that the clearance of endogenous senescent cells or the prevention of cell senescence could be a beneficial repair process in chronic wounds. To screen candidate compounds that can ameliorate cell senescence and prevent radiation ulcers, we have established a cell senescence model induced by radiation in vitro using fibroblasts, because they play a crucial role in ulcer development [11, 12]. We also established three ulcer models, for skin ulcers, intestine ulcers and oral mucositis, to verify the effectiveness of the screened drug [11]. Next, we identified a natural nucleoside analogue compound, cordycepin, which can prevent cell senescence and radiation ulcer effectively using the small-molecule library we established before [11]. Additionally, dasatinib + quercetin (DQ) has been reported to selectively promote the apoptosis of senescent cells [74, 75]. We identified that senescent cells are removed by DQ by inducing senescent cell apoptosis directly in vivo and in vitro; not surprisingly, DQ treatment also alleviates radiation-induced ulcers [12]. Moreover, our findings suggest that cordycepin can directly bind to adenosine 5′-monophosphate-activated protein kinase (AMPK) near the autoinhibitory domain at the α1 and γ1 subunits, relieving the autoinhibition of AMPK and promoting the translocation of nuclear factor E2-related factor 2 (NRF2) to the nucleus [11]. Furthermore, activation of NRF2 or AMPK can be a therapeutic target to prevent cell senescence and radiation ulcers, providing a reference for future drug development. Similarly, it was reported that rapamycin can prevent epithelial stem cell senescence by inhibiting the mammalian target of rapamycin and protecting against radiation-induced mucositis [76]. In non-radiation-induced chronic wounds, a link between ageing and fibrosis has also been discovered during skeletal muscle injury, and inactivation of the endocytic adapter Numb in mice leads to sustained p53-dependent senescence of myofibroblasts after severe injury, leading to reduced regeneration potential [77]. The regenerative capacity in Numb mutants is functionally rescued and the levels of cell senescence markers are reduced to normal levels by p53 ablation or antioxidant treatment [77]. However, it should be noted that irradiation induces ROS and DNA damage in cells, resulting in cell apoptosis or senescence, which is more complicated than non-radiation-induced cell senescence. The therapy of senescent cell clearance and prevention in radiation-induced chronic wounds is more challenging than that in non-radiation-induced chronic wounds. We should identify accurate therapeutic strategies or agents to prevent cell senescence and clear senescent cells for different wounds. Thus, preventing cell senescence and removing senescent cells are emerging therapeutic strategies for chronic wounds.
Conclusions
Chronic wounds are a huge challenge for wound-care researchers and clinicians. The use of advanced treatment modalities, such as tissue replacement and growth factors, may offer a strategy to accelerate wound closure in chronic wounds; however, some wounds still show no response to these treatments. Senescent cells accumulate in chronic wounds, creating an environment of prolonged inflammation and contributing to the dysfunction of stem cells. Increasing evidence has shown that preventing cell senescence or removing senescent cells can mitigate chronic wounds, and cellular senescence can be a promising target for chronic wounds. Although some clues have been provided in this review, the underlying mechanism of senescent cells contributing to the development of chronic wounds requires further investigation. More importantly, more research is needed regarding cell senescence in chronic ulcers, as well as the evaluation of the clinical significance of this strategy.
Abbreviations
ECM: extracellular matrix; PDGF: platelet-derived growth factor; EGF: epidermal growth factor; FGF: fibroblast growth factor; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; DQ: dasatinib + quercetin; AMPK: adenosine 5′-monophosphate-activated protein kinase; NRF2: nuclear factor E2-related factor 2
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research and Development Program (2016YFC1000805), the University Innovation Team Building Program of Chongqing (CXTDG201602020) and intramural research project grants (AWS17J007 and 2018-JCJQ-ZQ-001).
Availability of data and materials
Not applicable.
Authors’ contributions
CS and ZW wrote the manuscript. ZW created the figures.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare that they have no competing interests.
References
- 1. Brownrigg JR, Apelqvist J, Bakker K, Schaper NC, Hinchliffe RJ. Evidence-based management of PAD & the diabetic foot. Eur J Vasc Endovasc Surg. 2013;45:673–81. [DOI] [PubMed] [Google Scholar]
- 2. Richmond NA, Maderal AD, Vivas AC. Evidence-based management of common chronic lower extremity ulcers. Dermatol Ther. 2013;26:187–96. [DOI] [PubMed] [Google Scholar]
- 3. Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care (New Rochelle). 2015;4:560–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rice JB, Desai U, Cummings AK, Birnbaum HG, Skornicki M, Parsons NB. Burden of diabetic foot ulcers for medicare and private insurers. Diabetes Care. 2014;37:651–8. [DOI] [PubMed] [Google Scholar]
- 5. Gurtner GC, Chapman MA. Regenerative medicine: charting a new course in wound healing. Adv Wound Care (New Rochelle). 2016;5:314–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Han G, Ceilley R. Chronic wound healing: a review of current management and treatments. Adv Ther. 2017;34:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones RE, Foster DS, Longaker MT. Management of Chronic Wounds-2018. JAMA. 2018;320:1481–2. [DOI] [PubMed] [Google Scholar]
- 8. Mulder GD, Vande Berg JS. Cellular senescence and matrix metalloproteinase activity in chronic wounds. Relevance to debridement and new technologies. J Am Podiatr Med Assoc. 2002;92:34–7. [DOI] [PubMed] [Google Scholar]
- 9. Telgenhoff D, Shroot B. Cellular senescence mechanisms in chronic wound healing. Cell Death Differ. 2005;12:695–8. [DOI] [PubMed] [Google Scholar]
- 10. Stanley A, Osler T. Senescence and the healing rates of venous ulcers. J Vasc Surg. 2001;33:1206–11. [DOI] [PubMed] [Google Scholar]
- 11. Wang Z, Chen Z, Jiang Z, Luo P, Liu L, Huang Y, et al. . Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat Commun. 2019;10:2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Wang Z, Huang Y, Zhou Y, Sheng X, Jiang Q, et al. . Senolytics (DQ) mitigates radiation ulcers by removing senescent cells. Front Oncol. 2020;9:1576. doi: 10.3389/fonc.2019.01576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: a cellular perspective. Physiol Rev. 2019;99:665–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dekoninck S, Blanpain C. Stem cell dynamics, migration and plasticity during wound healing. Nat Cell Biol. 2019;21:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Darwin E, Tomic-Canic M. Healing chronic wounds: current challenges and potential solutions. Curr Dermatol Rep. 2018;7:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014. doi: 10.1126/scitranslmed.3009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun BK, Siprashvili Z, Khavari PA. Advances in skin grafting and treatment of cutaneous wounds. Science. 2014;346:941–5. [DOI] [PubMed] [Google Scholar]
- 18. Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. Keratins and the keratinocyte activation cycle. J Invest Dermatol. 2001;116:633–40. [DOI] [PubMed] [Google Scholar]
- 19. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. [DOI] [PubMed] [Google Scholar]
- 20. Stadelmann WK, Digenis AG, Tobin GR. Physiology and healing dynamics of chronic cutaneous wounds. Am J Surg. 1998;176:26S–38. [DOI] [PubMed] [Google Scholar]
- 21. Armstrong DG, Wrobel J, Robbins JM. Guest editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J. 2007;4:286–7. [DOI] [PubMed] [Google Scholar]
- 22. Moffatt CJ, Franks PJ, Doherty DC, Smithdale R, Steptoe A. Psychological factors in leg ulceration: a case-control study. Br J Dermatol. 2009;161:750–6. [DOI] [PubMed] [Google Scholar]
- 23. McCarty SM, Percival SL. Proteases and delayed wound healing. Adv Wound Care (New Rochelle). 2013;2:438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pastar I, Wong LL, Egger AN, Tomic-Canic M. Descriptive vs mechanistic scientific approach to study wound healing and its inhibition: is there a value of translational research involving human subjects? Exp Dermatol. 2018;27:551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Childs BG, Baker DJ, Kirkland JL, Campisi J, Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, et al. . Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int Wound J. 2017;14:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. . Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–21. [DOI] [PubMed] [Google Scholar]
- 29. Ogrodnik M, Salmonowicz H, Jurk D, Passos JF. Expansion and cell-cycle arrest: common denominators of cellular senescence. Trends Biochem Sci. 2019;44: 996–1008. [DOI] [PubMed] [Google Scholar]
- 30. Li J, Song S, Li X, Zhu J, Li W, Du B, et al. . Down-regulation of fibroblast growth factor 2 (FGF2) contributes to the premature senescence of mouse embryonic fibroblast. Med Sci Monit. 2020;26:e920520. doi: 10.12659/MSM.920520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, et al. . Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol. 2004;24:5459–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14:617–28. [DOI] [PubMed] [Google Scholar]
- 36. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. . Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoenicke L, Zender L. Immune surveillance of senescent cells—biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012;33:1123–6. [DOI] [PubMed] [Google Scholar]
- 38. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. . A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging. 2012;4:932–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. . A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. . The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, Doorn R, Desmet CJ, et al. . Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. [DOI] [PubMed] [Google Scholar]
- 43. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. . Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. [DOI] [PubMed] [Google Scholar]
- 44. Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, et al. . Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18. [DOI] [PubMed] [Google Scholar]
- 45. Nacher V, Carretero A, Navarro M, Armengol C, Llombart C, Rodriguez A, et al. . The quail mesonephros: a new model for renal senescence? J Vasc Res. 2006;43:581–6. [DOI] [PubMed] [Google Scholar]
- 46. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. . Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–30. [DOI] [PubMed] [Google Scholar]
- 47. Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, et al. . A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. 2010;8. doi: 10.1371/journal.pbio.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al. . Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27:2356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. . Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. [DOI] [PubMed] [Google Scholar]
- 50. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. . An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yun MH, Davaapil H, Brockes JP. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife. 2015;4:e05505:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang T, Rivera-Perez JA. Senescence-associated beta-galactosidase activity marks the visceral endoderm of mouse embryos but is not indicative of senescence. Genesis. 2014;52:300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim KH, Won JH, Cheng N, Lau LF. The matricellular protein CCN1 in tissue injury repair. J Cell Commun Signal. 2018;12:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, et al. . Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348:aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. . Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schultz GS, Sibbald RG, Falanga V, Ayello EA, Dowsett C, Harding K, et al. . Wound bed preparation: a systematic approach to wound management. Wound Repair Regen. 2003;11:S1–28. [DOI] [PubMed] [Google Scholar]
- 57. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017. doi: 10.1186/s12915-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. 2016;139:653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kobbe C. Cellular senescence: a view throughout organismal life. Cell Mol Life Sci. 2018;75:3553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Agren MS, Steenfos HH, Dabelsteen S, Hansen JB, Dabelsteen E. Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J Invest Dermatol. 1999;112:463–9. [DOI] [PubMed] [Google Scholar]
- 61. Bourguignon LY. Matrix hyaluronan-activated CD44 signaling promotes keratinocyte activities and improves abnormal epidermal functions. Am J Pathol. 2014;184:1912–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cook H, Davies KJ, Harding KG, Thomas DW. Defective extracellular matrix reorganization by chronic wound fibroblasts is associated with alterations in TIMP-1, TIMP-2, and MMP-2 activity. J Invest Dermatol. 2000;115:225–33. [DOI] [PubMed] [Google Scholar]
- 63. Wall IB, Moseley R, Baird DM, Kipling D, Giles P, Laffafian I, et al. . Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J Invest Dermatol. 2008;128:2526–40. [DOI] [PubMed] [Google Scholar]
- 64. Vande Berg JS, Rose MA, Haywood-Reid PL, Rudolph R, Payne WG, Robson MC. Cultured pressure ulcer fibroblasts show replicative senescence with elevated production of plasmin, plasminogen activator inhibitor-1, and transforming growth factor-beta1. Wound Repair Regen. 2005;13:76–83. [DOI] [PubMed] [Google Scholar]
- 65. Thomas CA, Holdstock JM, Harrison CC, Price BA, Whiteley MS. Healing rates following venous surgery for chronic venous leg ulcers in an independent specialist vein unit. Phlebology. 2013;28:132–9. [DOI] [PubMed] [Google Scholar]
- 66. Bitar MS. The GSK-3beta/Fyn/Nrf2 pathway in fibroblasts and wounds of type 2 diabetes: on the road to an evidence-based therapy of non-healing wounds. Adipocyte. 2012;1:161–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coalson E, Bishop E, Liu W, Feng Y, Spezia M, Liu B, et al. . Stem cell therapy for chronic skin wounds in the era of personalized medicine: from bench to bedside. Genes Dis. 2019;6:342–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maranda EL, Rodriguez-Menocal L, Badiavas EV. Role of Mesenchymal stem cells in dermal repair in burns and diabetic wounds. Curr Stem Cell Res Ther. 2017;12:61–70. [DOI] [PubMed] [Google Scholar]
- 69. Ennis WJ, Sui A, Bartholomew A. Stem cells and healing: impact on inflammation. Adv Wound Care (New Rochelle). 2013;2:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang X, Chen Z, Luo S, Jin T, Wang Y, Chen F, et al. . Development of therapeutic small-molecule Fluorophore for cell transplantation. Adv Funct Mater. 2016;26:8397–407. [Google Scholar]
- 71. Cianfarani F, Toietta G, Di Rocco G, Cesareo E, Zambruno G, Odorisio T. Diabetes impairs adipose tissue-derived stem cell function and efficiency in promoting wound healing. Wound Repair Regen. 2013;21:545–53. [DOI] [PubMed] [Google Scholar]
- 72. Rodriguez-Menocal L, Salgado M, Ford D, Van Badiavas E. Stimulation of skin and wound fibroblast migration by mesenchymal stem cells derived from normal donors and chronic wound patients. Stem Cells Transl Med. 2012;1:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, et al. . The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 2019;18:e12848. doi: 10.1111/acel.12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. . Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24:1246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. . Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, et al. . mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Le Roux I, Konge J, Le Cam L, Flamant P, Tajbakhsh S. Numb is required to prevent p53-dependent senescence following skeletal muscle injury. Nat Commun. 2015;6:8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.