

In high-income countries, the leading causes of death are noncommunicable diseases (NCDs), such as obesity, cancer, and cardiovascular disease. An important feature of most NCDs is inflammation-induced gut dysbiosis characterized by a shift in the microbial community structure from obligate to facultative anaerobes such as Proteobacteria. This microbial imbalance can contribute to disease pathogenesis by either a depletion in or the production of microbiota-derived metabolites. However, little is known about the mechanism by which inflammation-mediated changes in host physiology disrupt the microbial ecosystem in our large intestine leading to disease.
KEYWORDS: Enterobacteriaceae, intestinal epithelium, microbiota, noncommunicable diseases, obesity
ABSTRACT
In high-income countries, the leading causes of death are noncommunicable diseases (NCDs), such as obesity, cancer, and cardiovascular disease. An important feature of most NCDs is inflammation-induced gut dysbiosis characterized by a shift in the microbial community structure from obligate to facultative anaerobes such as Proteobacteria. This microbial imbalance can contribute to disease pathogenesis by either a depletion in or the production of microbiota-derived metabolites. However, little is known about the mechanism by which inflammation-mediated changes in host physiology disrupt the microbial ecosystem in our large intestine leading to disease. Recent work by our group suggests that during gut homeostasis, epithelial hypoxia derived from peroxisome proliferator-activated receptor γ (PPAR-γ)-dependent β-oxidation of microbiota-derived short-chain fatty acids limits oxygen availability in the colon, thereby maintaining a balanced microbial community. During inflammation, disruption in gut anaerobiosis drives expansion of facultative anaerobic Enterobacteriaceae, regardless of their pathogenic potential. Therefore, our research group is currently exploring the concept that dysbiosis-associated expansion of Enterobacteriaceae can be viewed as a microbial signature of epithelial dysfunction and may play a greater role in different models of NCDs, including diet-induced obesity, atherosclerosis, and inflammation-associated colorectal cancer.
INTRODUCTION
Infectious diseases, defined as those caused by microorganisms (e.g., bacteria, viruses, fungi, or parasites), were the most common cause of death worldwide in the early 20th century. However, deaths from infectious diseases dramatically declined by the end of the 20th century, resulting in large gains in life expectancy, which could be attributed to significant improvements in sanitation, disease prevention by vaccination, and antibiotic development and use as a first line of treatment for bacterial diseases (1).
Although the impact of infectious diseases has been reduced or eliminated in developed countries, the mortality rate from other causes, namely, noncommunicable diseases (NCDs), has increased significantly (2). NCDs are defined as noninfectious, nontransmissible diseases that may be caused by genetics or behavioral factors and generally have a slow progression and long duration (www.who.int/ncds/en). These include cardiovascular diseases, cancer, chronic respiratory diseases, diabetes, and neurodegenerative diseases such as Alzheimer’s disease, among others. The most recent data from the World Health Organization (WHO) state that NCDs kill 41 million people each year, which is equivalent to 71% of all deaths globally (www.who.int/ncds/en). Therefore, NCDs are considered the leading causes of death and disability globally and are estimated to cause a cumulative loss of $47 trillion between 2011 and 2030 (3). In the United States, the top two NCDs, cardiovascular disease and cancer, account for nearly 50% of all deaths. NCDs have long surpassed infectious diseases as the main cause of death in both male and female Americans (4) (Fig. 1A and B). As a result, NCDs are identified as one of the major health challenges for the 21st century.
FIG 1.

The major current causes of death in the United States are obesity associated noncommunicable diseases. In 2017, the major causes of death in the United States in males (A) and females (B) are cardiovascular disease, followed by cancer and chronic conditions. (Adapted from reference 15 with permission of the publisher.)
The risk factors associated with NCD development can be classified as modifiable behavioral (MB) risk factors and metabolic (ME) risk factors (www.who.int/ncds/en). The MB risk factors for NCDs are tobacco use, physical inactivity, unhealthy diet, and the harmful use of alcohol. ME risk factors arise either from genetic conditions or from MB risk factors and contribute to the four crucial metabolic changes that increase the risk of NCDs: (i) raised blood pressure, (ii) overweight status/obesity, (iii) hyperglycemia, and (iv) hyperlipidemia (2). Overweight status/obesity and elevated blood pressure are the leading ME risk factors responsible for NCD-attributable deaths worldwide (2). Thus, understanding the mechanisms by which major risk factors such as unhealthy diet and obesity contribute to the development and progression of NCDs is key for establishing effective preventive and treatment strategies to manage the 21st century NCD epidemic.
OBESITY IS A MAJOR RISK FACTOR FOR NONCOMMUNICABLE DISEASES
Obesity is a pressing public health problem worldwide, affecting more than 107.7 million children and 603.7 million adults around the globe (5). The incidence of obesity and overweight status has significantly increased in the past decades, and if the rising trends persist, global obesity prevalence is estimated to reach 18% in men and over 21% in women by 2025 (6). Strikingly, 20% of the world’s adult population is projected to have obese status by 2030 (7). In the United States, the prevalence of obesity surpasses the worldwide average, as the condition affects 39.8% of the adult population and ∼20% of school-age children, with an additional 31.8% of the population being considered overweight (www.cdc.gov/obesity/index.html). As a consequence, overweight and obesity are estimated to be linked to nearly 1 in 5 deaths (18.2%) among adults in the United States (8).
Obesity is a complex and multifactorial disease mainly attributed to genetic, behavioral, social, economic, and environmental factors (9). When assessed as an independent factor, the role of genetics in obesity pathogenesis is significantly less than that of the environment (10). Instead, genetic predisposition seems to increase the risk of weight gain if it interacts with other risk factors such as unhealthy diets and inactive lifestyle (11, 12). Indeed, the current global obesity epidemic can be largely attributed to significant changes in dietary habits, including increased consumption of “Western-style” diets, which are energy dense and rich in saturated fats and sugars. The establishment of diet-induced obesity (DIO) mouse models (13) using a high-fat (HF) and caloric-dense diet has been extremely helpful in understanding the mechanisms linking obesogenic dietary habits and the development of NCDs.
Overweight status and obesity strongly correlate with the incidence of several adverse comorbidities, including cardiovascular disease, cancer, and diabetes (14). The link between DIO and NCDs is particularly compelling in cardiovascular disease (CVD), the most common cause of death in the United States (Fig. 1A and B) (15). The majority of cardiac deaths (64%) are due to coronary heart disease secondary to atherosclerosis (16). Obese individuals are at a significantly higher risk of developing CVD (www.cdc.gov/obesity/index.html). Obesity and atherosclerosis share pathophysiological pathways, as both are chronic inflammatory conditions characterized by lipid storage imbalance and activation of the immune system (17, 18). Therefore, the study of the mechanisms linking DIO and CVD has a remarkable potential in aiding in the development of novel treatment strategies.
Numerous cancers are associated with excess body weight, and the American Cancer Society suggests that obesity is responsible for about 8% of all cancers in the United States, as well as about 7% of all cancer-related deaths (19). Obese individuals are at higher risk of developing colorectal cancer, breast cancer, and cancer of the endometrium, esophagus, kidney, and pancreas (20). High-income countries, including the United States, have reported a significant rise in the incidence of early-onset colorectal cancer (21–23), a trend that could be partially attributed to the obesity epidemic (23). Experimental studies using preclinical mouse models indicate that obesity and Western-style high-fat diet (HFD) accelerate the multistage transition from normal tissue to invasive malignancy and metastatic disease (24). Taken together, these studies point to the urgency of establishing new prevention and treatment measurements for the current obesity epidemic and obesity-associated cancers.
A new player in the development and progression of NCDs is the intestinal microbiota, also referred to as the “microbial organ” (25, 26). Recent data suggest that gut microbes and their metabolites can affect disease progression through multiple mechanisms, including altering the immune response (reviewed in reference 27), changing host-cell metabolic state (28), and even affecting response to immunotherapy (29). Undeniably, the potential causative role of gut microbiota in obesity represents one of the most extraordinary findings of the past decade. Therefore, the impact of changes in the intestinal microbial community in the pathogenesis of obesity-related NCDs is an extremely relevant and emerging field (30). However, we are only just beginning to understand the mechanisms by which risk factors associated with NCDs promote changes in the intestinal physiology and gut microbiota and how these changes may contribute to NCD pathogenesis.
An important feature of most NCD is inflammation-induced disruption of the intestinal microbiota (dysbiosis), characterized by a shift in the microbial community structure from obligate to facultative anaerobes such as Enterobacteriaceae (31) (Fig. 2). In this review, we will explore the potential mechanisms causing Enterobacteriaceae expansion in the inflamed gut and during diet-induced obesity. Moreover, we will discuss the role of facultative anaerobic bacteria intestinal bloom in the pathogenesis of obesity-associated NCDs, namely, cardiovascular disease and colorectal cancer.
FIG 2.
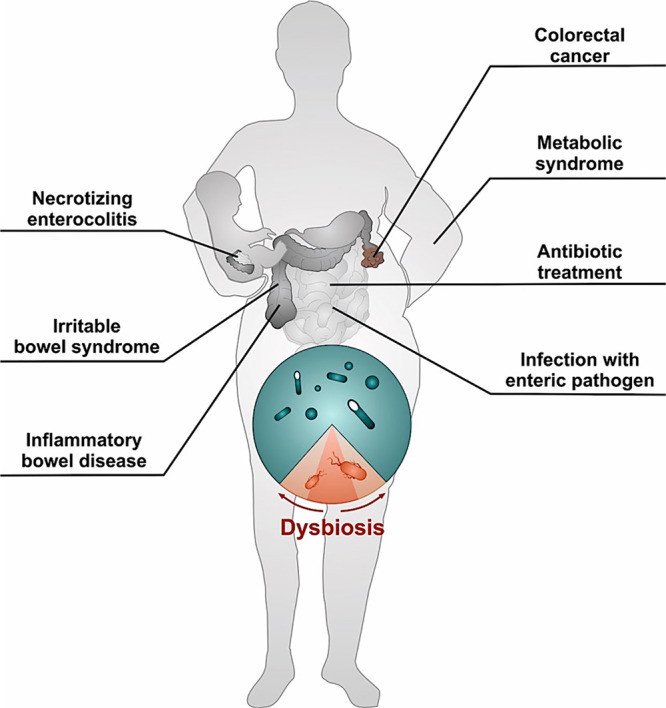
Noncommunicable diseases are linked to intestinal dysbiosis. In the healthy gut, the intestinal microbiota is dominated by obligate anaerobic bacteria (teal). An important feature of most noncommunicable diseases is inflammation-induced gut dysbiosis characterized by a shift in the microbial community structure from obligate to facultative anerobic bacteria (red) such as Enterobacteriaceae.
ROLE OF ENTEROBACTERIACEAE EXPANSION IN THE PATHOGENESIS OF OBESITY-ASSOCIATED NCDs
The human large intestine is home to a large and complex bacterial ecosystem, composed mostly of anaerobic organisms. This balanced microbial community (microbiota) performs multiple beneficial functions for the host such as immune education, nutrition, and protection against invasion by enteric pathogens (32).
In the healthy large intestine, Enterobacteriaceae is a minor constituent of microbiota (33). However, a wide range of human NCDs are associated with a severe disruption of the balanced gut microbial ecosystem, often characterized by an expansion of facultative anaerobic Enterobacteriaceae (31) (Fig. 2). Indeed, a disturbance of the intestinal microbial community by antibiotic treatment results in a dysbiotic outgrowth of facultative anaerobic Enterobacteriaceae in humans (34, 35) and murine models (36–38). Intestinal inflammation triggered by genetic predisposition, chemicals, or infection with enteric pathogens causes an uncontrolled luminal expansion of Enterobacteriaceae in mouse models (39–43). An intestinal bloom of Enterobacteriaceae is also observed in humans with severe intestinal inflammation, including patients with inflammatory bowel disease (44–47), colorectal cancer (48, 49), or necrotizing enterocolitis (50), or during conditions of low-level intestinal inflammation, such as irritable bowel syndrome (51, 52). Taken together, these studies suggest that Enterobacteriaceae expansion may play an important role in the pathogenesis of NCDs (31).
Role of Enterobacteriaceae expansion in the pathogenesis of cardiovascular disease.
Obesity is considered a key underlying risk factor for many NCDs, including heart disease (53, 54). A Western-style HFD is thought to increase the risk for cardiovascular disease due to systemic hyperlipidemia characterized by increased circulating levels of low-density lipoprotein (LDL) and cholesterol (55). Additionally, Western-style HFD-mediated changes in intestinal microbiota composition have been linked to cardiovascular disease (56, 57). For example, several studies have demonstrated the role of HFD-induced elevated plasma lipopolysaccharide (LPS) levels, a major component of the Gram-negative bacterial outer membrane, in promoting atherosclerosis (58–60). Activation of Toll-like receptor 4 (TLR4) by the lipid A portion of LPS in endothelial cells results in the recruitment of inflammatory monocytes. Once inside the subendothelial space, the recruited inflammatory monocytes become activated macrophages, partially via an LPS/TLR4-dependent mechanism, and help promote the development of the atherosclerotic plaque (61).
Microbiota-derived metabolites may also contribute to the pathogenesis of cardiovascular disease. Recent studies have described the ability of members of the gut microbiota to catabolize dietary choline into trimethylamine (TMA) and acetaldehyde (62). TMA is absorbed in the intestine and oxidized in the liver to trimethylamine N-oxide (TMAO), a metabolite that promotes atherosclerosis via the formation of foam cells and atherogenic plaque (63, 64) through poorly identified mechanisms. Interestingly, the cut operon containing the gene cluster responsible for choline utilization and TMA production is commonly found in facultative anaerobic Enterobacteriaceae such as Proteus mirabilis and Escherichia coli (62, 65, 66). Choline degradation is thought to occur under anaerobic conditions in a bacterial microcompartment, a protein shell that encloses enzymes and protects the bacterial cell from toxicity of aldehyde intermediates such as acetaldehyde in the case of choline metabolism (66). Strikingly, outgrowth of facultative anaerobic Enterobacteriaceae is also described in obese individuals exposed to HFDs (67–70), raising the possibility that HFD-mediated expansion of TMA-producing Enterobacteriaceae may play a role in the pathogenesis of obesity-associated cardiovascular disease.
Role of Enterobacteriaceae expansion in colorectal cancer pathogenesis.
Colorectal cancer is the third most common cancer and the second most common cause of cancer-related death worldwide (71). A recent report from the American Cancer Society states that the incidence of colorectal cancer has significantly increased in young adults (23), especially those who consume HFDs. Strikingly, only about 20% of colorectal cancer cases can be genetically attributed to familial history (72), suggesting that environmental factors such as obesogenic HFDs may play an important role in promoting tumorigenesis.
Overweight status and obesity may contribute to colorectal cancer pathogenesis through multiple concurrent mechanisms, including (i) stimulation of low-level intestinal inflammation; (ii) increased reactive oxygen species (ROS), which may play a role in DNA damage and mutagenesis; and (iii) changes in levels of growth-promoting factors such as insulin and insulin-like growth factor (IGF-1), which are secondary to obesity-associated metabolic syndrome (24, 73). However, despite the advantages in deciphering how obesity may contribute to cancer pathogenesis, the exact mechanisms underlying HFD-induced colorectal cancer risk and recurrence remain unclear.
In addition to the mechanisms described above, HFD-induced intestinal dysbiosis may be an important missing piece of the obesity-colorectal cancer puzzle. A wide body of literature has suggested that the gut microbiota can enhance colorectal cancer development through its impact on tumor-associated inflammation. Recognition of microbial components (e.g., LPS and flagellin) by the innate and adaptive immune system leads to production of proinflammatory cytokines and other inflammatory products which exert the neoplastic effect (74–77).
The carcinogenic effects of commensal gut bacteria can also result from direct effects of microbially derived products. Members of the Enterobacteriaceae family are able to produce toxins with carcinogenic properties, including colibactin, a polyketide-derived genotoxin which is able to cause intestinal epithelial cell double-strand breaks (DSBs) and DNA alkylation, which leads to cell cycle arrest and activation of DNA repair pathways, resulting in increased carcinogenesis (78–82). Indeed, members of the Enterobacteriaceae family, in particular, E. coli (phylogroups B2 and D), Klebsiella spp., and P. mirabilis, are frequently detected and overrepresented in the microbiota of colorectal cancer (CRC) patients (47–49). Benign polyps developing early in life of patients with familial adenomatous polyposis are covered by patchy bacterial biofilms containing colibactin-producing E. coli (83). Additionally, the expression of colibactin biosynthesis genes is highly induced in biopsy specimens from human CRC patients (84), suggesting a key role of the expansion of colibactin-producing Enterobacteriaceae in tumor induction.
RESPIRATION AS A STRATEGY FOR ENTEROBACTERIACEAE EXPANSION IN THE GUT
A major goal of every organism in the intestinal microbial community is to ensure long-term survival in the gut lumen and to thrive in a nutritionally competitive environment. Therefore, members of the microbiota have evolved a wide range of metabolic pathways, with each microbe employing a different “winning strategy” for nutrient acquisition and utilization (85). Considering the strong evidence of inflammation-associated dysbiosis, one would assume that inflammation may cause a significant change in the metabolic landscape of the gut, leading to accumulation of a novel set of nutrients for which the microbes that inhabit the intestinal lumen will need to compete. Although several studies have started to explore this concept in the context of both infectious and noninfectious inflammatory diseases (Table 1), we are still only beginning to understand the microbial metabolic adaption during inflammation-induced gut dysbiosis.
TABLE 1.
Nutrient sources available to Enterobacteriaceae during intestinal dysbiosis
| Nutrient | Source | Electron acceptor | Species | Reference(s) |
|---|---|---|---|---|
| Ethanolamine | Host | Tetrathionate | S. Typhimurium | 86 |
| Lactate | Host | Oxygen | S. Typhimurium | 87 |
| Glucarate/galactarate | Microbiota | Oxygen and tetrathionate | S. Typhimurium, commensal E. coli | 88 |
| 1,2-propanediol | Microbiota | Oxygen, nitrate, tetrathionate | S. Typhimurium | 90 |
| Succinate | Microbiota | Oxygen, nitrate, tetrathionate | S. Typhimurium | 91 |
| l-Serine | Diet | Not determined | Adherent-invasive E. coli, C. rodentium | 92, 102 |
Carbon sources are key for microorganisms to build biomass and thrive in an environment. A strong body of literature suggests that intestinal inflammation causes changes in both host physiology and microbiota composition, which, in turn, generates a unique set of carbon sources that can be used by Enterobacteriaceae to outgrow the resident microbiota (Table 1). During inflammation, intestinal epithelial damage is a source of ethanolamine (86) and lactate (87) for the enteric pathogen Salmonella enterica serovar Typhimurium. In addition, an influx of inflammatory cells in response to antibiotic treatment can lead to an increased abundance of sugar oxidation products, such as glucarate or galactarate, and utilization of these carbon sources drives a postantibiotic expansion of E. coli and S. Typhimurium (88).
Metabolites generated by the microbiota also contribute to dysbiotic Enterobacteriaceae expansion. Bacteroidia, an abundant member of the intestinal microbial community, is able to break down complex carbohydrates and release monosaccharides like rhamnose and fucose (89). Such monosaccharides can be further fermented into 1,2-propanediol (89), which, in turn, is used by S. Typhimurium to grow during inflammation (90). Pathogenic Enterobacteriaceae can expand in the gut by taking advantage of additional products from Bacteroidia glycan metabolism, such as the poorly fermentable dicarboxylic acid succinate (91).
Recent work by Kitamoto et al. shows that intestinal inflammation alters the amino acid availability in the gut lumen (92). As a consequence, pathogenic Enterobacteriaceae, such as adherent invasive E. coli and Citrobacter rodentium, adapt to gut inflammation by reprogramming their metabolism toward amino acid catabolism (92). This work suggests that amino acids derived from diet, the host, or the intestinal microbiota may be a key resource for Enterobacteriaceae survival during intestinal dysbiosis. Future work in further exploring the mechanisms by which pathogenic and commensal bacteria may take advantage of this inflammation-dependent amino acid availability to gain a growth advantage in the inflamed gut will be of great interest.
At first glance, it is not obvious why an increased availability of carbon sources specifically favors the growth of Enterobacteriaceae over Clostridia or Bacteroidia during gut dysbiosis. It is important to remember that Enterobacteriaceae are facultative anaerobes that can utilize oxygen (O2) better than obligate anaerobes Clostridia or Bacteroidia (93). Therefore, elevated availability of oxygen can potentially increase the abundance of facultative anaerobic Enterobacteriaceae within the gut-associated microbial community and, at the same time, inhibit growth of highly oxygen-sensitive commensals, also known as “the oxygen hypothesis” (94). Recent work has confirmed that pathogenic and commensal Enterobacteriaceae use oxygen to bloom in the gut during infectious and noninfectious colitis (38, 87, 95–97). This body of work provides experimental evidence that Enterobacteriaceae takes advantage of the ability to perform aerobic respiration to outcompete the commensal microbiota in the inflamed gut because respiration generates more energy from the catabolism of carbon sources than fermentation (98).
A groundbreaking work by Winter et al. was the first to reveal that gut inflammation leads to the generation of alternative electron acceptors, which promote anaerobic respiration of Enterobacteriaceae (99). In this study, the authors showed ROS generated by the host inflammatory response oxidized thiosulfate (S2O32–) into tetrathionate (S4O62–), which, in turn, could be used by S. Typhimurium to expand during colitis. Additional work solidified the concept that Enterobacteriaceae expands within the microbiota when electron acceptors for anaerobic respiration become available (Table 1). An elevated mucosal synthesis of inducible nitric oxide synthase (iNOS) triggered during pathogen or chemically-induced colitis in mice leads to the production of nitric oxide (NO), which reacts to form nitrate (NO3−) in the gut lumen, thereby driving an uncontrolled expansion of commensal E. coli or pathogenic S. Typhimurium by nitrate respiration (43, 100).
The impact of the respiration-dependent bloom of Enterobacteriaceae in disease pathogenesis has been assessed in mouse models of inflammatory bowel disease and colitis-associated colorectal cancer (CAC). Notably, the use of tungstate to selectively inhibit microbial respiratory pathways, operational only during episodes of inflammation, significantly blunted dysbiotic expansion of colitis and CAC-associated E. coli and ameliorated signs of disease (101, 102).
Collectively, these studies give rise to the possibility that respiration plays an important role in Enterobacteriaceae intestinal expansion that contributes to the pathogenesis of diet-induced obesity and associated NCDs. Additionally, selectively blocking microbial metabolic pathways that are only active during disease may be an unexplored and very attractive treatment strategy for NCDs.
COLONOCYTE METABOLISM AS A KEY DRIVER OF DYSBIOSIS-ASSOCIATED ENTEROBACTERIACEAE BLOOM
An important benefit of the obligate anaerobic microbes that inhabit our large bowel is their ability to digest complex dietary carbohydrates (fiber) into fermentation products that are absorbed by the host (103), contributing to host nutrition (104), immune development (105–108), and niche protection against enteric pathogens (38, 95). In contrast, facultative anaerobic bacteria, such as Enterobacteriaceae, do not provide such benefits and may be capable of affecting host nutrition by metabolizing fermentation products to carbon dioxide when oxygen is present (90, 91, 109), as discussed above. Thus, it is very likely that the host has developed strategies to help maintain a diverse intestinal microbial community dominated by obligate anaerobic bacteria that provide benefit by generating fermentation products from fiber, a strategy also known as “microbiota-nourishing immunity” (25, 110).
Recent studies propose that colonic epithelial cells (colonocytes) play a central role in shaping a beneficial microbiota (38, 95) and promoting microbiota-nourishing immunity (Fig. 3). Colonocyte maturation and differentiation require peroxisome proliferator-activated receptor γ (PPAR-γ) (111), a nuclear receptor highly expressed in differentiated colonic epithelial cells of mice and humans (112). PPAR-γ activates mitochondrial β-oxidation of long-chain and short-chain fatty acids, resulting in O2 consumption through oxidative phosphorylation of fatty acids (113–115). As a consequence, mature colonocytes must consume high levels of O2 to maintain their oxidative metabolic state, resulting in an O2 partial pressure of less than 7.6 mm Hg (<1% oxygen), a condition known as physiologic epithelial hypoxia (116). Therefore, the highly oxidative metabolism of mature colonocytes limits the amount of O2 diffusing from the mucosal surface, which helps to maintain an anaerobic environment in the lumen of the large bowel (Fig. 3) (38, 93). Through this mechanism, the colonic epithelium ensures a dominance of beneficial anaerobic microorganisms, thereby maintaining gut homeostasis (93).
FIG 3.

Intestinal epithelial dysfunction contributes to dysbiosis associated expansion of Enterobacteriaceae. (A and B, left panels) During gut homeostasis, β-oxidation of microbiota-derived butyrate causes epithelial hypoxia, which supports an anaerobic environment in the lumen of the large intestine. As a consequence, the lack of luminal oxygen drives a dominance of beneficial obligate anaerobic bacteria (green) in the gut microbiota. (A and B, right panels) During gut dysbiosis, colonocytes decrease their oxidative capacity, either due to antibiotic mediated decrease in butyrate-dependent PPAR-γ signaling (A) or due to HFD-induced mitochondrial dysfunction. The resulting epithelial dysfunction disrupts anaerobiosis in the lumen and increases the availability of alternative electron acceptors, driving an expansion of facultative anaerobic Enterobacteriaceae by aerobic and anaerobic respiration. SCFAs, short-chain fatty acids.
The considerations described above suggest that an imbalance in the intestinal microbiota could be caused by an underlying defect in epithelial metabolic functions that maintain homeostasis in the colon (25). The initial studies into mechanisms of gut homeostasis disruption used antibiotic models of microbiota disruption (117), which alters epithelial metabolism via the depletion of the microbial-derived short-chain fatty acids butyrate, propionate, and acetate (38). Butyrate activates PPAR-γ signaling in human epithelial cells (118) to drive the metabolism of surface colonocytes toward mitochondrial β-oxidation of fatty acids (113–115), which is important for maintaining physiologic hypoxia (38). Additionally, short-chain fatty acids inhibit intestinal inflammation by maintaining the regulatory T cell pool in mucosa via the activation of G-coupled receptors (105–108). As a result, antibiotic treatment increases the inflammatory tone of the colonic mucosa (119) by downregulating epithelial PPAR-γ signaling (38) and decreasing the number of regulatory T cells in the colonic mucosa (105–108). The resulting upregulation of inflammatory signals shifts the metabolism of differentiated colonocytes toward anaerobic glycolysis, a metabolism characterized by low oxygen consumption, high glucose consumption, and high lactate release (38, 87), leading to loss of epithelial hypoxia (117). An important consequence of elevated epithelial oxygenation is an increase in the amount of O2 emanating from the mucosal surface, providing a key resource for an expansion of facultative anaerobic bacteria by aerobic respiration (Fig. 3A) (38, 120). Importantly, PPAR-γ can inhibit transcription of proinflammatory genes, including the iNOS gene (Nos2) (121). Thus, downregulation of PPAR-γ signaling in epithelial cells also results in the elevated synthesis of iNOS, which generates NO to form nitrate (NO3−) in the gut lumen, thereby promoting Enterobacteriaceae expansion via anaerobic nitrate respiration (38).
Insights into the role of the intestinal epithelium in maintaining gut homeostasis have also come from infectious colitis models (96, 122). C. rodentium (family Enterobacteriaceae), a mouse enteric pathogen, uses its virulence factors to intimately attach to the colonic surface, creating a favorable niche for competition with the gut microbiota (39, 123). Interestingly, epithelial injury caused by C. rodentium virulence factors induces excessive epithelial repair responses, leading to colonic crypt hyperplasia and accumulation of undifferentiated transit-amplifying cells at the mucosal surface (122), which rely on glycolysis for energy production (124). The resulting loss of differentiated colonic epithelial cells increases the amount of O2 emanating from the mucosal surface and drives growth of C. rodentium through aerobic respiration (96).
DIO causes low-grade intestinal inflammation characterized by loss of differentiated epithelial cells and induction of endoplasmic reticulum stress response in colonocytes (125). Systemic hyperglycemia contributes to obesity-associated impairment in intestinal epithelium barrier functions (126). Additionally, consumption of an obesogenic HFD and increased saturated fatty acids may directly affect intestinal epithelial oxidative capacity, impairing mitochondrial bioenergetics by inducing hydrogen peroxide production in the mitochondria (127, 128). Importantly, recent studies suggest that mitochondria-derived ROS may play a role in reducing gut microbiota diversity (129) through unknown mechanisms. Collectively, these findings raise the possibility that the Enterobacteriaceae expansion seen in obesity-driven NCDs may be a result of HFD-induced deterioration of the intestinal epithelial ability to maintain anaerobiosis-driven gut homeostasis (Fig. 3B).
Taken together, the studies described above suggest that an imbalance in the microbial community could be caused by an underlying defect in epithelial immune functions that maintain homeostasis in the colon (25, 130). This concept is particularly important when understanding the mechanisms by which Enterobacteriaceae may expand in the gut lumen in a wide range of human diseases (Fig. 2). We now know that the population of facultative anaerobic bacteria blooms in the intestinal lumen during dysbiosis due to a disruption of epithelial physiologic hypoxia, which, in turn, increases the amount of oxygen emanating from the colonic epithelium (131). Additionally, changes in colonic epithelium physiology lead to increased levels of electron acceptors that can be used by Enterobacteriaceae for anaerobic respiration (38). Therefore, the colonic expansion of facultative anaerobic bacteria associated with many human NCDs might be caused by a common underlying driver: colonocyte dysfunction.
CONCLUSIONS
Recent research has demonstrated that the gut microbiota, the largest microbial community inhabiting our body, plays a key role in the pathogenesis of a variety of NCDs, especially those associated with obesity. A hallmark of most NCDs is inflammation-induced gut dysbiosis characterized by a shift in the microbial community structure from obligate to facultative anaerobes such as Enterobacteriaceae, which may contribute to NCD pathogenesis. However, little is known about how environmental and metabolic factors contribute to obesity-associated dysbiosis. Therefore, further studies on this topic should be of great interest.
The picture emerging from recent studies is that the colonic intestinal epithelium plays a key role in modulating gut microbiota composition, and changes in colonocyte metabolism may be a common driver of disease-associated dysbiosis in the large bowel. Additional work is needed to investigate if changes in the intestinal expansion of Enterobacteriaceae observed in individuals consuming a Western-style HFD (67, 69) are driven by an underlying defect in colonic epithelial metabolic function. Nevertheless, the view that colonocyte metabolism plays a key role in balancing the gut microbiota may provide a novel target for therapies to modulate the colonization by members of the microbiota (e.g., colibactin-producing E. coli and TMA-producing Enterobacteriaceae) that increase the risk for obesity-associated NCDs.
ACKNOWLEDGMENT
We acknowledge Sandy Pernitzsch for her help in preparing the figures for the manuscript.
Biographies

Catherine D. Shelton earned a bachelor of science degree in biochemistry in 2016 from Western Washington University. During her undergraduate studies, she investigated the inhibition of prokaryotic translation factors in the lab of Dr. Clint Spiegel. After graduation, she joined the Tuberculosis Discovery Program at the Infectious Disease Research Institute in Seattle, where she assisted in the identification of potential therapeutics. Catherine began her graduate studies in 2018 in the Interdisciplinary Graduate Program at Vanderbilt University. In 2019, she joined the lab of Dr. Mariana Byndloss and the Microbe-Host Interactions program. Under Dr. Byndloss, Catherine is studying how perturbations to the gut microbiota alter intestinal epithelial function and promote obesity.

Mariana X. Byndloss, D.V.M., Ph.D., earned her D.V.M. and M.Sc. in veterinary pathology from Universidade Federal de Minas Gerais (UFMG) in Brazil. Her doctoral work performed at UFMG and University of California, Davis (UC Davis) was awarded the Brazilian National Prize for best Ph.D. thesis in veterinary medicine. She performed her postdoctoral training in Andreas Bäumler’s laboratory at UC Davis, studying the link between endoplasmic reticulum (ER) stress and innate immunity as well as the interactions between the host and intestinal microbiota during dysbiosis. Currently, she is an assistant professor in the Pathology, Immunology, and Microbiology Department at Vanderbilt University Medical Center. Dr. Byndloss has extensive experience in studying the host-microbe interactions in gastrointestinal diseases and has authored and coauthored over 60 scientific publications. Dr. Byndloss is particularly interested in how inflammation-mediated changes in gut epithelial metabolism lead to gut dysbiosis and increased risk of noncommunicable diseases, namely, inflammatory bowel disease, obesity, cardiovascular disease, and colon cancer.
REFERENCES
- 1.Centers for Disease Control and Prevention. 1999. Control of infectious diseases. MMWR Morb Mortal Wkly Rep 48:621–629. [PubMed] [Google Scholar]
- 2.GBD 2015 Risk Factors Collaborators. 2016. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388:1659–1724. doi: 10.1016/S0140-6736(16)31679-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S, Kuhn M, Prettner K, Bloom DE. 2018. The macroeconomic burden of noncommunicable diseases in the United States: estimates and projections. PLoS One 13:e0206702. doi: 10.1371/journal.pone.0206702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Global Burden of Disease Collaborative Network. 2017. Global Burden of Disease Study 2016 (GBD 2016) results. Institute for Health Metrics and Evaluation (IHME), Seattle, WA. [Google Scholar]
- 5.Afshin A, GBD 2015 Obesity Collaborators, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, Marczak L, Mokdad AH, Moradi-Lakeh M, Naghavi M, Salama JS, Vos T, Abate KH, Abbafati C, Ahmed MB, Al-Aly Z, Alkerwi A, Al-Raddadi R, Amare AT, Amberbir A, Amegah AK, Amini E, Amrock SM, Anjana RM, Ärnlöv J, Asayesh H, Banerjee A, Barac A, Baye E, Bennett DA, Beyene AS, Biadgilign S, Biryukov S, Bjertness E, Boneya DJ, Campos-Nonato I, Carrero JJ, Cecilio P, Cercy K, Ciobanu LG, Cornaby L, Damtew SA, Dandona L, Dandona R, Dharmaratne SD, Duncan BB, Eshrati B, Esteghamati A, Feigin VL, et al. 2017. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med 377:13–27. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.NCD Risk Factor Collaboration (NCD-RisC). 2016. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 387:1377–1396. doi: 10.1016/S0140-6736(16)30054-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly T, Yang W, Chen CS, Reynolds K, He J. 2008. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 32:1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 8.Masters RK, Reither EN, Powers DA, Yang YC, Burger AE, Link BG. 2013. The impact of obesity on US mortality levels: the importance of age and cohort factors in population estimates. Am J Public Health 103:1895–1901. doi: 10.2105/AJPH.2013.301379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh S, Bouchard C. 2017. Convergence between biological, behavioural and genetic determinants of obesity. Nat Rev Genet 18:731–748. doi: 10.1038/nrg.2017.72. [DOI] [PubMed] [Google Scholar]
- 10.Meldrum DR, Morris MA, Gambone JC. 2017. Obesity pandemic: causes, consequences, and solutions-but do we have the will? Fertil Steril 107:833–839. doi: 10.1016/j.fertnstert.2017.02.104. [DOI] [PubMed] [Google Scholar]
- 11.Kilpeläinen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, Demerath E, Ahmad T, Mora S, Kaakinen M, Sandholt CH, Holzapfel C, Autenrieth CS, Hyppönen E, Cauchi S, He M, Kutalik Z, Kumari M, Stančáková A, Meidtner K, Balkau B, Tan JT, Mangino M, Timpson NJ, Song Y, Zillikens MC, Jablonski KA, Garcia ME, Johansson S, Bragg-Gresham JL, Wu Y, van Vliet-Ostaptchouk JV, Onland-Moret NC, Zimmermann E, Rivera NV, Tanaka T, Stringham HM, Silbernagel G, Kanoni S, Feitosa MF, Snitker S, Ruiz JR, Metter J, Larrad MTM, Atalay M, Hakanen M, Amin N, Cavalcanti-Proença C, Grøntved A, Hallmans G, Jansson J-O, et al. 2011. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med 8:e1001116. doi: 10.1371/journal.pmed.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell JA, Church TS, Rankinen T, Earnest CP, Sui X, Blair SN. 2010. FTO genotype and the weight loss benefits of moderate intensity exercise. Obesity (Silver Spring) 18:641–643. doi: 10.1038/oby.2009.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. 1988. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 14.Haslam DW, James WP. 2005. Obesity. Lancet 366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 15.Heron M. 2019. Deaths: leading causes for 2017. Natl Vital Stat Rep 68:1–76. [PubMed] [Google Scholar]
- 16.Dalen JE, Alpert JS, Goldberg RJ, Weinstein RS. 2014. The epidemic of the 20(th) century: coronary heart disease. Am J Med 127:807–812. doi: 10.1016/j.amjmed.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Ross R. 1999. Atherosclerosis–an inflammatory disease. N Engl J Med 340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil GS. 2006. Inflammation and metabolic disorders. Nature 444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 19.Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I, Flanders WD, Brawley OW, Gapstur SM, Jemal A. 2018. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin 68:31–54. doi: 10.3322/caac.21440. [DOI] [PubMed] [Google Scholar]
- 20.Lauby-Secretan B, International Agency for Research on Cancer Handbook Working Group, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. 2016. Body fatness and cancer–viewpoint of the IARC Working Group. N Engl J Med 375:794–798. doi: 10.1056/NEJMsr1606602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel RL, Fedewa SA, Anderson WF, Miller KD, Ma J, Rosenberg PS, Jemal A. 2017. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 109:djw322. doi: 10.1093/jnci/djw322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young JP, Win AK, Rosty C, Flight I, Roder D, Young GP, Frank O, Suthers GK, Hewett PJ, Ruszkiewicz A, Hauben E, Adelstein BA, Parry S, Townsend A, Hardingham JE, Price TJ. 2015. Rising incidence of early-onset colorectal cancer in Australia over two decades: report and review. J Gastroenterol Hepatol 30:6–13. doi: 10.1111/jgh.12792. [DOI] [PubMed] [Google Scholar]
- 23.Sung H, Siegel RL, Rosenberg PS, Jemal A. 2019. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health 4:e137–e147. doi: 10.1016/S2468-2667(18)30267-6. [DOI] [PubMed] [Google Scholar]
- 24.Berger NA. 2018. Young adult cancer: influence of the obesity pandemic. Obesity (Silver Spring) 26:641–650. doi: 10.1002/oby.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byndloss MX, Baumler AJ. 2018. The germ-organ theory of non-communicable diseases. Nat Rev Microbiol 16:103–110. doi: 10.1038/nrmicro.2017.158. [DOI] [PubMed] [Google Scholar]
- 26.Baquero F, Nombela C. 2012. The microbiome as a human organ. Clin Microbiol Infect 18(Suppl 4):2–4. doi: 10.1111/j.1469-0691.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 27.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin AM, Sun EW, Rogers GB, Keating DJ. 2019. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front Physiol 10:428. doi: 10.3389/fphys.2019.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 30.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 31.Shin NR, Whon TW, Bae JW. 2015. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Brestoff JR, Artis D. 2013. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vollaard EJ, Clasener HA, Janssen AJ. 1992. Co-trimoxazole impairs colonization resistance in healthy volunteers. J Antimicrob Chemother 30:685–691. doi: 10.1093/jac/30.5.685. [DOI] [PubMed] [Google Scholar]
- 35.Kieser S, Sarker SA, Berger B, Sultana S, Chisti MJ, Islam SB, Foata F, Porta N, Betrisey B, Fournier C, Descombes P, Mercenier A, Sakwinska O, Brussow H. 2018. Antibiotic treatment leads to fecal Escherichia coli and coliphage expansion in severely malnourished diarrhea patients. Cell Mol Gastroenterol Hepatol 5:458–460.e6. doi: 10.1016/j.jcmgh.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohnhoff M, Drake BL, Miller CP. 1954. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc Soc Exp Biol Med 86:132–137. doi: 10.3181/00379727-86-21030. [DOI] [PubMed] [Google Scholar]
- 37.Saito K. 1961. Studies on the habitation of pathogenic Escherichia coli in the intestinal tract of mice. I. Comparative experiments on the habitation of each type of resistant pathogenic Escherichia coli under an administration of streptomycin. Paediatr Jpn 65:385–393. (In Japanese.) [PubMed] [Google Scholar]
- 38.Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, Litvak Y, Lopez CA, Xu G, Napoli E, Giulivi C, Tsolis RM, Revzin A, Lebrilla CB, Bäumler AJ. 2017. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357:570–575. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, Hardt WD. 2007. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, Gordon JI, Onderdonk AB, Glimcher LH. 2010. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haag LM, Fischer A, Otto B, Plickert R, Kuhl AA, Gobel UB, Bereswill S, Heimesaat MM. 2012. Intestinal microbiota shifts towards elevated commensal Escherichia coli loads abrogate colonization resistance against Campylobacter jejuni in mice. PLoS One 7:e35988. doi: 10.1371/journal.pone.0035988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, Popova IE, Parikh SJ, Adams LG, Tsolis RM, Stewart VJ, Baumler AJ. 2013. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Doré J. 2003. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. 2006. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol 44:4136–4141. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. 1998. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 48.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. 2004. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 49.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. 1998. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 115:281–286. doi: 10.1016/s0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 50.Hunter CJ, Upperman JS, Ford HR, Camerini V. 2008. Understanding the susceptibility of the premature infant to necrotizing enterocolitis (NEC). Pediatr Res 63:117–123. doi: 10.1203/PDR.0b013e31815ed64c. [DOI] [PubMed] [Google Scholar]
- 51.Krogius-Kurikka L, Lyra A, Malinen E, Aarnikunnas J, Tuimala J, Paulin L, Makivuokko H, Kajander K, Palva A. 2009. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol 9:95. doi: 10.1186/1471-230X-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gobert AP, Sagrestani G, Delmas E, Wilson KT, Verriere TG, Dapoigny M, Del'homme C, Bernalier-Donadille A. 2016. The human intestinal microbiota of constipated-predominant irritable bowel syndrome patients exhibits anti-inflammatory properties. Sci Rep 6:39399. doi: 10.1038/srep39399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L, INTERHEART Study Investigators. 2004. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 54.Yusuf S, Vaz M, Pais P. 2004. Tackling the challenge of cardiovascular disease burden in developing countries. Am Heart J 148:1–4. doi: 10.1016/j.ahj.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 55.Korakas E, Dimitriadis G, Raptis A, Lambadiari V. 2018. Dietary composition and cardiovascular risk: a mediator or a bystander? Nutrients 10:1912. doi: 10.3390/nu10121912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battson ML, Lee DM, Weir TL, Gentile CL. 2018. The gut microbiota as a novel regulator of cardiovascular function and disease. J Nutr Biochem 56:1–15. doi: 10.1016/j.jnutbio.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Chan YK, Brar MS, Kirjavainen PV, Chen Y, Peng J, Li D, Leung F-C, El-Nezami H. 2016. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE−/− mice. BMC Microbiol 16:264. doi: 10.1186/s12866-016-0883-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Curtiss LK, Tobias PS. 2009. Emerging role of Toll-like receptors in atherosclerosis. J Lipid Res 50:S340–S345. doi: 10.1194/jlr.R800056-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wick MC, Mayerl C, Backovic A, Van der Zee R, Jaschke W, Dietrich H, Wick G. 2008. In vivo imaging of the effect of LPS on arterial endothelial cells: molecular imaging of heat shock protein 60 expression. Cell Stress Chaperones 13:275–285. doi: 10.1007/s12192-008-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. 2004. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A 101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowman JD, Surani S, Horseman MA. 2017. Endotoxin, Toll-like receptor-4, and atherosclerotic heart disease. Curr Cardiol Rev 13:86–93. doi: 10.2174/1573403X12666160901145313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. 2015. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 6:e00042-15. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. 2013. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. 2011. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu Y, Jameson E, Crosatti M, Schafer H, Rajakumar K, Bugg TD, Chen Y. 2014. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc Natl Acad Sci U S A 111:4268–4273. doi: 10.1073/pnas.1316569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herring TI, Harris TN, Chowdhury C, Mohanty SK, Bobik TA. 2018. A bacterial microcompartment is used for choline fermentation by Escherichia coli 536. J Bacteriol 200:e00764-17. doi: 10.1128/JB.00764-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. 2012. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fei N, Zhao L. 2013. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 7:880–884. doi: 10.1038/ismej.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, Darfeuille-Michaud A, Barnich N. 2014. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63:116–124. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 70.Anitha M, Reichardt F, Tabatabavakili S, Nezami BG, Chassaing B, Mwangi S, Vijay-Kumar M, Gewirtz A, Srinivasan S. 2016. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. Cell Mol Gastroenterol Hepatol 2:328–339. doi: 10.1016/j.jcmgh.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, Znaor A, Bray F. 2019. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 72.Rustgi AK. 2007. The genetics of hereditary colon cancer. Genes Dev 21:2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 73.Berger NA. 2014. Obesity and cancer pathogenesis. Ann N Y Acad Sci 1311:57–76. doi: 10.1111/nyas.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, Gonzalez A, Clemente JC, Cullender TC, Barnich N, Darfeuille-Michaud A, Vijay-Kumar M, Knight R, Ley RE, Gewirtz AT. 2012. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gronbach K, Flade I, Holst O, Lindner B, Ruscheweyh HJ, Wittmann A, Menz S, Schwiertz A, Adam P, Stecher B, Josenhans C, Suerbaum S, Gruber AD, Kulik A, Huson D, Autenrieth IB, Frick JS. 2014. Endotoxicity of lipopolysaccharide as a determinant of T-cell-mediated colitis induction in mice. Gastroenterology 146:765–775. doi: 10.1053/j.gastro.2013.11.033. [DOI] [PubMed] [Google Scholar]
- 76.Kang M, Martin A. 2017. Microbiome and colorectal cancer: unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol 32:3–13. doi: 10.1016/j.smim.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 77.Pickard JM, Zeng MY, Caruso R, Nunez G. 2017. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev 279:70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. 2013. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, Knight P, Codling C, Marchesi JR, Winstanley C, Hall N, Rhodes JM, Campbell BJ. 2014. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 63:761–770. doi: 10.1136/gutjnl-2013-304739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Putze J, Heniquin C, Nougayrede JP, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W, Oelschlaeger TA, Oswald E, Forestier C, Hacker J, Dobrindt U. 2009. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun 77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallee A, Dechelotte P, Darcha C, Pezet D, Bonnet R, Bringer MA, Darfeuille-Michaud A. 2014. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol 20:6560–6572. doi: 10.3748/wjg.v20.i21.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carra A, Brennan CA, Chun E, Ngo L, Samson LD, Engelward BP, Garrett WS, Balbo S, Balskus EP. 2019. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363:eaar7785. doi: 10.1126/science.aar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, Anders RA, Giardiello FM, Wick EC, Wang H, Wu S, Pardoll DM, Housseau F, Sears CL. 2018. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359:592–597. doi: 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dutilh BE, Backus L, van Hijum SA, Tjalsma H. 2013. Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer. Best Pract Res Clin Gastroenterol 27:85–99. doi: 10.1016/j.bpg.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 85.Winter SE, Baumler AJ. 2014. Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut Microbes 5:71–73. doi: 10.4161/gmic.27129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. 2011. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A 108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gillis CC, Hughes ER, Spiga L, Winter MG, Zhu W, Furtado de Carvalho T, Chanin RB, Behrendt CL, Hooper LV, Santos RL, Winter SE. 2018. Dysbiosis-associated change in host metabolism generates lactate to support Salmonella growth. Cell Host Microbe 23:570. doi: 10.1016/j.chom.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Faber F, Tran L, Byndloss MX, Lopez CA, Velazquez EM, Kerrinnes T, Nuccio SP, Wangdi T, Fiehn O, Tsolis RM, Baumler AJ. 2016. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature 534:697–699. doi: 10.1038/nature18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Badia J, Ros J, Aguilar J. 1985. Fermentation mechanism of fucose and rhamnose in Salmonella typhimurium and Klebsiella pneumoniae. J Bacteriol 161:435–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Faber F, Thiennimitr P, Spiga L, Byndloss MX, Litvak Y, Lawhon S, Andrews-Polymenis HL, Winter SE, Bäumler AJ. 2017. Respiration of microbiota-derived 1,2-propanediol drives Salmonella expansion during colitis. PLoS Pathog 13:e1006129. doi: 10.1371/journal.ppat.1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spiga L, Winter MG, Furtado de Carvalho T, Zhu W, Hughes ER, Gillis CC, Behrendt CL, Kim J, Chessa D, Andrews-Polymenis HL, Beiting DP, Santos RL, Hooper LV, Winter SE. 2017. An oxidative central metabolism enables Salmonella to utilize microbiota-derived succinate. Cell Host Microbe 22:291–301.e6. doi: 10.1016/j.chom.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kitamoto S, Alteri CJ, Rodrigues M, Nagao-Kitamoto H, Sugihara K, Himpsl SD, Bazzi M, Miyoshi M, Nishioka T, Hayashi A, Morhardt TL, Kuffa P, Grasberger H, El-Zaatari M, Bishu S, Ishii C, Hirayama A, Eaton KA, Dogan B, Simpson KW, Inohara N, Mobley HLT, Kao JY, Fukuda S, Barnich N, Kamada N. 4 November 2019. Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol doi: 10.1038/s41564-019-0591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Litvak Y, Byndloss MX, Tsolis RM, Baumler AJ. 2017. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol 39:1–6. doi: 10.1016/j.mib.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 94.Rigottier-Gois L. 2013. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J 7:1256–1261. doi: 10.1038/ismej.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rivera-Chávez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, Bäumler AJ. 2016. Depletion of butyrate-producing Clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19:443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, Chávez-Arroyo A, Lokken KL, Tsolis RM, Winter SE, Bäumler AJ. 2016. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 353:1249–1253. doi: 10.1126/science.aag3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cevallos SA, Lee JY, Tiffany CR, Byndloss AJ, Johnston L, Byndloss MX, Baumler AJ. 2019. Increased epithelial oxygenation links colitis to an expansion of tumorigenic bacteria. mBio 10:e02244-19. doi: 10.1128/mBio.02244-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Olsan EE, Byndloss MX, Faber F, Rivera-Chávez F, Tsolis RM, Bäumler AJ. 2017. Colonization resistance: the deconvolution of a complex trait. J Biol Chem 292:8577–8581. doi: 10.1074/jbc.R116.752295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lopez CA, Rivera-Chávez F, Byndloss MX, Bäumler AJ. 2015. The periplasmic nitrate reductase NapABC supports luminal growth of Salmonella enterica serovar Typhimurium during colitis. Infect Immun 83:3470–3478. doi: 10.1128/IAI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Büttner L, de Lima Romão E, Behrendt CL, Lopez CA, Sifuentes-Dominguez L, Huff-Hardy K, Wilson RP, Gillis CC, Tükel Ç, Koh AY, Burstein E, Hooper LV, Bäumler AJ, Winter SE. 2018. Precision editing of the gut microbiota ameliorates colitis. Nature 553:208–211. doi: 10.1038/nature25172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu W, Miyata N, Winter MG, Arenales A, Hughes ER, Spiga L, Kim J, Sifuentes-Dominguez L, Starokadomskyy P, Gopal P, Byndloss MX, Santos RL, Burstein E, Winter SE. 2019. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J Exp Med 216:2378–2393. doi: 10.1084/jem.20181939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. 2013. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54:2325–2340. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velazquez OC, Lederer HM, Rombeau JL. 1997. Butyrate and the colonocyte, p 123–134. In Kritchevsky D, Bonfield C (ed), Dietary fiber in health and disease. Springer, New York, NY. [Google Scholar]
- 105.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. 2011. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H. 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 108.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. 2013. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bronner DN, Faber F, Olsan EE, Byndloss MX, Sayed NA, Xu G, Yoo W, Kim D, Ryu S, Lebrilla CB, Bäumler AJ. 2018. Genetic ablation of butyrate utilization attenuates gastrointestinal Salmonella disease. Cell Host Microbe 23:266–273.e4. doi: 10.1016/j.chom.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Byndloss MX, Litvak Y, Baumler AJ. 2019. Microbiota-nourishing immunity and its relevance for ulcerative colitis. Inflamm Bowel Dis 25:811–815. doi: 10.1093/ibd/izz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tylichová Z, Straková N, Vondráček J, Vaculová AH, Kozubík A, Hofmanová J. 2017. Activation of autophagy and PPARγ protect colon cancer cells against apoptosis induced by interactive effects of butyrate and DHA in a cell type-dependent manner: the role of cell differentiation. J Nutr Biochem 39:145–155. doi: 10.1016/j.jnutbio.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 112.Lefebvre M, Paulweber B, Fajas L, Woods J, McCrary C, Colombel JF, Najib J, Fruchart JC, Datz C, Vidal H, Desreumaux P, Auwerx J. 1999. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol 162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 113.Duszka K, Oresic M, Le May C, König J, Wahli W. 2017. PPARγ modulates long chain fatty acid processing in the intestinal epithelium. Int J Mol Sci 18:2559. doi: 10.3390/ijms18122559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, Bultman SJ. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13:517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roediger W. 1980. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 21:793–798. doi: 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. 2001. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. 2015. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, Koppen A, Mudde K, Haenen D, Al-Lahham S, Roelofsen H, Houtman R, van der Burg B, Mandrup S, Bonvin AMJJ, Kalkhoven E, Müller M, Hooiveld GJ, Kersten S. 2013. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor γ. Mol Cell Biol 33:1303–1316. doi: 10.1128/MCB.00858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spees AM, Wangdi T, Lopez CA, Kingsbury DD, Xavier MN, Winter SE, Tsolis RM, Bäumler AJ. 2013. Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. mBio 4:e00430-13. doi: 10.1128/mBio.00430-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reese AT, Cho EH, Klitzman B, Nichols SP, Wisniewski NA, Villa MM, Durand HK, Jiang S, Midani FS, Nimmagadda SN, O'Connell TM, Wright JP, Deshusses MA, David LA. 2018. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. Elife 7:e35987. doi: 10.7554/eLife.35987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li M, Pascual G, Glass CK. 2000. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 123.Kamada N, Kim Y-G, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. 2012. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fan Y-Y, Davidson LA, Callaway ES, Wright GA, Safe S, Chapkin RS. 2015. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am J Physiol Gastrointest Liver Physiol 309:G1–G9. doi: 10.1152/ajpgi.00052.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, Denman S, Begun J, Florin TH, Perkins A, Cuív P, McGuckin MA, Hasnain SZ. 2016. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep 6:28990. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, Ben-Zeev R, Lehavi-Regev D, Katz MN, Pevsner-Fischer M, Gertler A, Halpern Z, Harmelin A, Aamar S, Serradas P, Grosfeld A, Shapiro H, Geiger B, Elinav E. 2018. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 359:1376–1383. doi: 10.1126/science.aar3318. [DOI] [PubMed] [Google Scholar]
- 127.Kakimoto PA, Tamaki FK, Cardoso AR, Marana SR, Kowaltowski AJ. 2015. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol 4:375–380. doi: 10.1016/j.redox.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cardoso AR, Kakimoto PA, Kowaltowski AJ. 2013. Diet-sensitive sources of reactive oxygen species in liver mitochondria: role of very long chain acyl-CoA dehydrogenases. PLoS One 8:e77088. doi: 10.1371/journal.pone.0077088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yardeni T, Tanes CE, Bittinger K, Mattei LM, Schaefer PM, Singh LN, Wu GD, Murdock DG, Wallace DC. 2019. Host mitochondria influence gut microbiome diversity: a role for ROS. Sci Signal 12:eaaw3159. doi: 10.1126/scisignal.aaw3159. [DOI] [PubMed] [Google Scholar]
- 130.Litvak Y, Byndloss MX, Baumler AJ. 2018. Colonocyte metabolism shapes the gut microbiota. Science 362:eaat9076. doi: 10.1126/science.aat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rivera-Chávez F, Lopez CA, Bäumler AJ. 2017. Oxygen as a driver of gut dysbiosis. Free Radic Biol Med 105:93–101. doi: 10.1016/j.freeradbiomed.2016.09.022. [DOI] [PubMed] [Google Scholar]