

Clostridioides difficile is a Gram-positive, spore-forming, anaerobic bacterium that infects the human gastrointestinal tract, causing a wide range of disorders that vary in severity from mild diarrhea to toxic megacolon and/or death. Over the past decade, incidence, severity, and costs associated with C. difficile infection (CDI) have increased dramatically in both the pediatric and adult populations. The factors driving this rapidly evolving epidemiology remain largely unknown but are likely due in part to previously unappreciated host, microbiota, and environmental factors.
KEYWORDS: Clostridium difficile, microbiota, infectious disease
ABSTRACT
Clostridioides difficile is a Gram-positive, spore-forming, anaerobic bacterium that infects the human gastrointestinal tract, causing a wide range of disorders that vary in severity from mild diarrhea to toxic megacolon and/or death. Over the past decade, incidence, severity, and costs associated with C. difficile infection (CDI) have increased dramatically in both the pediatric and adult populations. The factors driving this rapidly evolving epidemiology remain largely unknown but are likely due in part to previously unappreciated host, microbiota, and environmental factors. In this review, we will cover the risks and challenges of CDI in adult and pediatric populations and examine asymptomatic colonization in infants. We will also discuss the emerging role of diet, pharmaceutical drugs, and pathogen-microbiota interactions in C. difficile pathogenesis, as well as the impact of host-microbiota interactions in the manifestation of C. difficile-associated disease. Finally, we highlight new areas of research and novel strategies that may shed light on this complex infection and provide insights into the future of microbiota-based therapeutics for CDI.
CLOSTRIDIOIDES DIFFICILE INFECTION
Clostridioides difficile (formerly known as Clostridium difficile) is a spore-forming anaerobic bacterium that infects the colon. In the United States, C. difficile is the most commonly reported nosocomial pathogen, and C. difficile infection (CDI) has emerged as an urgent public health threat worldwide (1). Over the past decade, the epidemiology of CDI has progressively evolved, and we are continuing to see increases in incidence, severity, and costs associated with infection (1, 2). Notably, non-antibiotic-associated CDI, most prominently in community-acquired cases, has been on the rise (2). In this review, we will discuss emerging concepts in C. difficile pathogenesis and epidemiology and provide insights into the potential factors contributing to the changes in epidemiology and rates of CDI.
C. DIFFICILE-ASSOCIATED DISEASE, RECURRENCE, AND CARRIAGE IN ADULTS AND THE ELDERLY
C. difficile-associated disease manifests as a wide spectrum of diseases that vary in severity from asymptomatic carriage to mild and moderate diarrhea to pseudomembranous colitis, toxic megacolon, and/or death (3). The primary risk factors for CDI are broad-spectrum antibiotic treatments, length of hospital stay, increasing age, and underlying comorbidities (4). CDI is most commonly reported in elderly hospitalized patients (3). Clindamycin, cephalosporins, ampicillin, and fluoroquinolones are the most highly associated classes of antibiotics with increased risk for CDI (5–7). C. difficile-associated diarrhea is the most common form of disease manifestation among patients (8). More severe presentations of CDI, such as pseudomembranous colitis, occur in a smaller subset of patients. Fulminant colitis, the most severe form of CDI-associated disease (9), occurs in approximately 3–8% of cases and accounts for the most serious complications of CDIs, including perforation of the epithelium, toxic megacolon, and death (10). Mortality rates for CDI have been on the rise over the past decade, which is correlated with the emergence of several epidemic strains of C. difficile (11, 12). These ribotype 027 (RT027) strains have been termed hypervirulent strains, but the link between these strains and increased disease severity has been questioned (13, 14). The emergence of RT027 is attributed to the acquisition of antibiotic resistance, specifically to fluoroquinolones (15). Within the adult population, the most susceptible group with the highest concurrent mortality rates is the elderly (11). This increase in risk is likely associated with decreased immune function, altered microbiota, and comorbidities.
One of the most substantial challenges in CDI is the high rate of recurrent infection. This is driven by the paradox that the primary risk factor for C. difficile, which is antibiotic treatment, is also the standard of care treatment for CDI. Approximately 25% of patients experience recurrent symptoms within 4 weeks after antibiotic therapy, and following each recurrence, rates of subsequent recurrent infection increase (1, 16, 17). In many cases, recurrence is attributed to infection with a different strain from the primary strain, suggesting continued microbiota perturbation following clearance, which allows for reinfection (18). However, most patients who relapse suffer from an infection with the same strain that caused the original episode (19, 20). Novel strategies for predicting and limiting recurrent infections are key for the future treatment of C. difficile.
Rates of colonization and asymptomatic carriage of C. difficile differ greatly between infants, adults, and the elderly population (21). In adults, carriage is quite rare and is associated with a myriad of risk factors, including prior antibiotic exposure, comorbidities, and age (21). Rates of carriage in nonhospitalized healthy adults are estimated to be between 0 and 15% of the population (22–27). These numbers creep to upwards of 0 to 50% when surveying the elderly population and cohorts in long-term health care facilities (17, 28–30). Extended hospital stays are also reported to be associated with increased rates of carriage, as continued exposure to spores and comorbidities likely increases the chance of transient colonization (27). It remains unclear if asymptomatic carriers of C. difficile are at a heightened risk for developing CDI following antibiotic exposure or if they represent a significant reservoir for C. difficile in the hospital or community settings.
C. DIFFICILE IN NEONATES AND INFANTS
A long-reported phenomenon in C. difficile epidemiology is the unusually high rates of asymptomatic colonization in neonates and infants. C. difficile was first isolated and characterized in 1935 from a healthy infant patient, emphasizing that this bacterium readily colonizes the infant gut (31). Studies vary in numbers, but it has been consistently reported that carriage rates in infants broadly range from 18 to 90% (32–36). This remarkably high rate of colonization is further convoluted by the fact that these infants harbor both toxigenic and nontoxigenic strains but rarely present with C. difficile-associated diarrhea. Surprisingly, neonates colonized with toxigenic strains of C. difficile harbor bacterial burdens at levels consistent with those in adults presenting pseudomembranous colitis (33, 37, 38). During the transition from neonate to infant, rates of C. difficile colonization decrease. Thirty-seven percent of infants younger than 1 month of age are colonized (33). Between 1 and 6 months of age, colonization decreases to 30%. During the period of 6 to 12 months, colonization decreases to 16%. After 2 years of age, the rates of asymptomatic colonization decrease to 10% (Fig. 1). The fact that the peak occurs during early life and decreases throughout development is indicative of ecological succession and maturation of the microbiota, as well as the maturation of host immunity.
FIG 1.
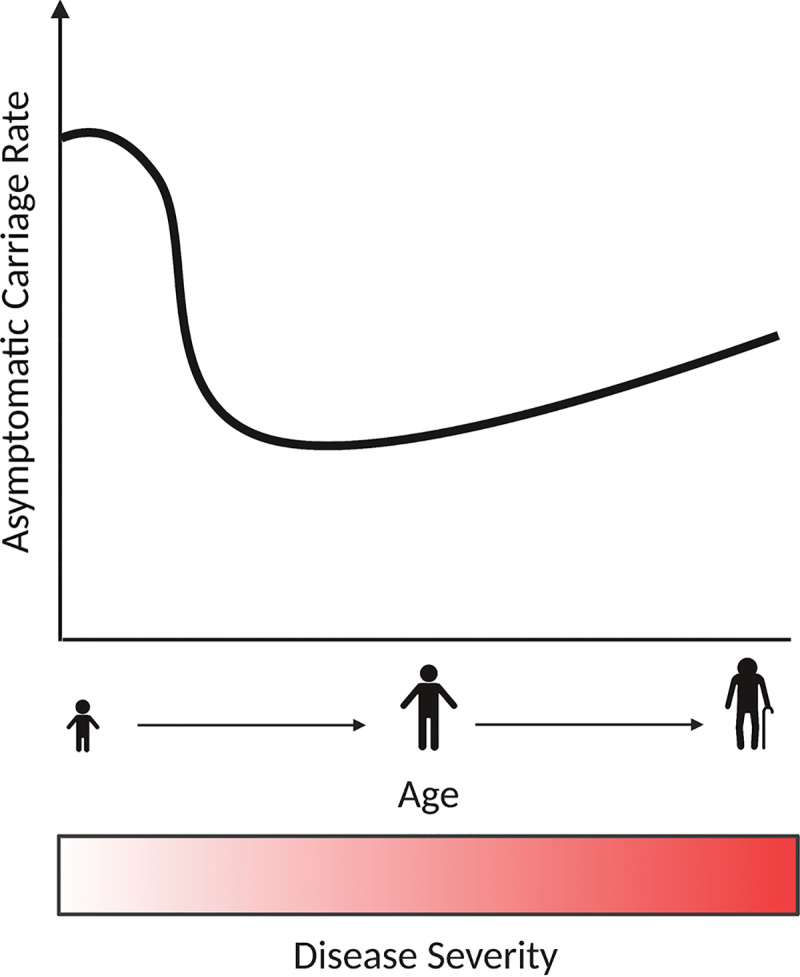
Relationship between asymptomatic colonization and disease severity of C. difficile with age. Higher rates of asymptomatic colonization are found in infants and neonates. As the microbiota and immune responses mature throughout development, changes in the bacterial community lead to a drop in colonization rates. In adults, colonization rates are low and increase in the elderly population. This is tightly associated with long-term stays in health facilities. Disease severity is also positively associated with age. While neonates and infants have higher carriage rates, they rarely suffer from infection. Meanwhile, adults and the elderly are the most susceptible population, and this is likely associated with alterations to the microbiota, decreased immune function, and comorbidities.
Early in life, communication between the developing immune system and commensal organisms sets the stage for a symbiotic and beneficial relationship (39, 40). The microbiota early in development is highly dynamic in nature, and emerging evidence suggests that damage to this ecosystem early in life can promote acute and chronic disorders later in life (41–44). Alterations in delivery method, introduction of antibiotics early in development, and shifts in nutrition have a marked impact on the community structure and homeostasis between host and microbiota (45–48). Due to the incredible complexity of the systems involved, we still know very little about the host, microbial, and nutritional factors that shape community assembly and pathogen colonization. It is likely that the early-life events in the microbiota play a key role in colonization and asymptomatic carriage of C. difficile, but more work is needed to shed light on the molecular mechanism associated with this phenomenon.
Although little is known regarding C. difficile-microbiota interactions at birth, the role of the microbiota in the transition from a carrier state to C. difficile clearance has been explored (36). In healthy babies, a reduction in C. difficile colonization at 1 year of age correlates with rising levels of Bacteroides and Eubacterium in the gut. Furthermore, Bifidobacterium and Lactobacillus are able to inhibit the growth of C. difficile in vitro, suggesting that early-life ecological events play a key role in reducing C. difficile colonization (49). It has also been suggested that breastfed babies may be more protected from C. difficile colonization than formula-fed babies, but the mechanisms by which breastfeeding prevents C. difficile colonization are unclear. Breastfed infants have a reduced mean fecal pH of 5.29 compared with pH 6.48 in bottle-fed infants, possibly related to the reduced buffering capacity of breast milk versus formula (50). Moreover, higher concentrations of IgA in breastfed infants can serve as a potent toxin A neutralizer (51, 52). In fact, a recent serological analysis of infants colonized with toxigenic strains of C. difficile revealed modest IgA and IgG response to toxins A and B, which could affect protection against infection later in life (53).
C. difficile carriage in neonates and infants represents a potential reservoir for C. difficile in both the hospital and community settings. Furthermore, asymptomatic colonization of infants with C. difficile could impact CDI risk, as carriers of toxigenic strains are at a higher risk for the development of infection than noncolonized patients (54). This is not clear-cut for infants, and data suggest even a potential benefit of asymptomatic colonization in infants, as antibody production can be protective against CDI (2, 55). Regardless, further research is needed to understand asymptomatic C. difficile colonization and if this acts protectively or represents a risk for developing CDI in infants.
FACTORS SHAPING C. DIFFICILE EPIDEMIOLOGY AND PATHOGENESIS
C. difficile disease falls on a wide spectrum of severity, and it is unclear what factors contribute to the severity of disease manifestation. Furthermore, the epidemiology of CDI has been rapidly changing over the past two decades, and the rates of both antibiotic-associated and non-antibiotic-associated CDIs are on the rise. Taken together, this suggests that previously unappreciated environmental factors may play a role in C. difficile infection and disease. In this section, we will highlight new evidence and emerging concepts that suggest a role for host, microbiota, and environmental factors in shaping CDI.
The microbiome and resistance to C. difficile.
Following birth, the human gastrointestinal (GI) tract is rapidly colonized by a diverse collection of microorganisms with rich metabolic potential (39). This complex microbial community, termed the gut microbiota, aids in digestion, stimulates the immune system, and provides essential vitamins and nutrients to the host (56). The microbiota can play a key role in the metabolism and efficacy of pharmaceutical drugs (57), and perturbation to the homeostasis between host and microbiota is associated with metabolic disease, cancer, inflammatory bowel disease, and GI infection (58). Importantly, the gut microbiota also serves as an important ecological barrier to invading pathogenic organisms, such as C. difficile (59). Thus, the primary risk factor for CDI is antimicrobial use, which perturbs the microbiota and decreases resistance to C. difficile. During health, the microbiota provides protection against pathogens like C. difficile by facilitating the production of a variety of antimicrobial factors, stimulating the immune system, and directly outcompeting pathogenic bacteria for resources and niches (60). Following antimicrobial use, large shifts in the microbiota lead to ecological changes in the community and significant metabolic alterations. These perturbations decrease competition for nutrients, alter levels of C. difficile germination factors, and modify the immune response (61).
Microbiota-mediated resistance against C. difficile is tightly associated with the metabolic state of the GI tract. One of the most important classes of metabolites linked to CDI is bile acids, which represent a key gatekeeper for C. difficile colonization (62). Bile acids are cholesterol-derived, water-soluble molecules that are synthesized in the liver by hepatocytes and are secreted into the small intestine to aid in nutrient absorption for the host (63). Many host-produced primary bile acids, which are bile acids conjugated to glycine or taurine, are the major inducers of C. difficile spore germination (64–66). These primary bile acids are readily metabolized by members of the microbiota, which harbor a collection of enzymes for deconjugation and metabolism of secreted bile acids (62, 67). Secondary bile acids, which are the product of this metabolism, have been shown to directly inhibit germination and possess potent antimicrobial properties against vegetative C. difficile cells (68–71). Disruption of the gut microbiota alters the balance of these metabolic processes and can markedly change the ratio of primary and secondary bile acids, leading to C. difficile germination and initiation of disease (68, 70).
Emerging research has begun to circle in on specific members of the microbiota that play a central role in bile acid metabolism and resistance to C. difficile (68). The ultimate goal is to harness the metabolic potential of the microbiota to combat C. difficile. One such member is Clostridium scindens, a low-abundance member of the microbiota that is negatively correlated with C. difficile colonization (Fig. 2). C. scindens expresses enzymes in the secondary bile acid biosynthesis pathway and can confer resistance to C. difficile through remodeling of the metabolic pool in the GI tract (66, 68). C. scindens and related Clostridia represent only a fraction of the taxa in the microbiota that possess the capacity to manipulate the bile acid pool (67). Future studies focused on identifying members of the microbiota that can be used to restore resistance to C. difficile via bile acid metabolism have incredible promise in the treatment of CDI.
FIG 2.

Factors influencing susceptibility to and severity of C. difficile infection. The factors that act to shape C. difficile susceptibility and infection severity through the tripartite relationship between the host, commensal microbiota, and pathogen are multifactorial. Bystander members of the gut microbiome, as well as their associated metabolites, are known to both positively and negatively modulate C. difficile pathogenesis. This is the basis for the function and activity of fecal microbiota transplant (FMT) as a therapy for C. difficile infection. Dietary factors, which can act directly on C. difficile or mediate interactions through the host, are also known to affect infection severity. Recent studies suggest that common drugs such as nonsteroidal anti-inflammatory drugs (NSAIDs) could impact disease progression. Lastly, emerging data suggest that bidirectional transmission of C. difficile strains between humans and animal reservoirs could play an understudied role in C. difficile epidemiology.
In addition to bile acids, several other microbial-derived metabolites have been demonstrated to play a key role in CDI. For example, recent studies have demonstrated that antibiotic treatment leads to an enrichment in fermentable amino acids, such as proline, which are, in turn, utilized by C. difficile for energy in the GI tract (72). Additionally, it has been shown that upon antibiotic treatment, succinate levels are enriched in the GI tract of mice and associated with CDI (73). Specifically, it is postulated that perturbation to the microbiota leads to production of microbiota-derived succinate, which is utilized by C. difficile to enable expansion in the GI tract (Fig. 2). It has also been shown that sialic acid cleaved from the host mucus layer by members of the microbiota can be cross-fed to C. difficile to enhance expansion in the gut (74). These interactions further exemplify how pathogen-microbiota metabolic interactions and the metabolic state of the gut can have a profound impact on CDI.
Polymicrobial synergy during CDI.
In numerous infections, such as surgical wounds, otitis media, periodontal infections, and cystic fibrosis, interspecies interactions have been shown to drive physiological changes in bacteria that lead to the development of infection and induction of pathogenic states (75–78). This phenomenon is termed polymicrobial synergy, and these interactions can enhance pathogen persistence and exacerbate disease severity (78–80). Synergy between species can be driven by a number of mechanisms, including metabolite exchange, molecular signaling, or indirectly through the host. In periodontal infection, metabolite cross-feeding between the oral commensal Streptococcus gordonii and the opportunistic pathogen Aggregatibacter actinomycetemcomitans is critical for the establishment of infection and virulence (78).
During enteric infection, invading pathogens are exposed to a dynamic and rich polymicrobial environment. The bystander microbial community and polymicrobial interactions may play a key role in the outcome of CDI. As highlighted earlier, the microbiota plays an important role in antagonizing C. difficile, and our group has recently reviewed this topic in depth (126). However, our work and the work of others are beginning to explore how the antibiotic-perturbed microbiota may synergize with C. difficile to promote increased pathogenesis and negatively impact the outcome of infection. Specific taxa, including Enterococcus species and members of the Enterobacteriaceae, thrive in the C. difficile-infected gut and have been associated with susceptibility to infection (68, 81, 82). For example, following treatment with excess levels of zinc or different cocktails of antibiotics, Enterococcus becomes highly enriched in the microbiota of mice (68, 83). This enrichment of Enterococcus is correlated with increased susceptibility to infection and disease severity (68, 83). Despite these correlations, little work has been done to characterize the impact of bystander microbes on C. difficile pathogenesis, and the integration of microbiota-produced metabolic signals in disease outcome has not been thoroughly studied.
Immune response to C. difficile.
Following colonization of the GI tract, vegetative C. difficile outgrows and produces its potent toxins, TcdA and TcdB (84). The pathogenicity locus (PaLoc) that carries the toxin genes also encodes a positive regulator, TcdR, and a negative regulator, TcdC. Moreover, toxin production can also be controlled through nutrient availability via global transcriptional repressors CodY and CcpA (84–86). Manifestation of C. difficile-associated disease is largely driven by a hyperinflammatory immune response to the damage caused by the toxins (87). These large multidomain toxins target Rho and Ras family small GTPases, leading to inactivation, subsequent actin disassembly, and eventual cell death (84). Toxin-induced damage to the epithelium creates a hyperinflammatory and volatile environment in the gut, loss of barrier function, and bacterial and toxin translocation (87). Depending on concentrations of toxin, cell death can be apoptotic or necrotic in intestinal epithelial cells. In macrophages, intoxication of epithelial cells and activation of the inflammasome lead to robust production of proinflammatory chemokines and cytokines, such as interleukin-8 (IL-8), IL-1β, and IL-22 (87, 88). This proinflammatory response leads to the robust recruitment of neutrophils during infection, a hallmark of CDI and pseudomembranous colitis, activation of innate lymphoid cells, and the production of antimicrobial peptides (87, 89, 90). The consequential innate immune response to CDI is essential for protection from translocating microbes, healing of the epithelial and mucosal barrier, and clearance of infection. However, this hyperinflammatory response is also a primary cause of much of the damage associated with CDI and can hinder recovery. Early response by innate lymphoid cells (ILCs), specifically ILC1s, is also critical in defense against C. difficile (91). The role of the adaptive immune response in CDI is less well understood, but studies suggest that the humoral immune responses to the C. difficile toxins may provide protection against disease and recurrence (92–94). Moreover, neutralizing antibodies targeting TcdB, including the FDA-approved bezlotoxumab, have proven effective in treating CDI and blocking toxin activity (95–97). Despite this evidence, antibody responses do not seem to provide protection against colonization of C. difficile or alter the clinical course of CDI. Interestingly, clinical observations suggest that patients infected with C. difficile can develop disease that falls on a wide spectrum, ranging from asymptomatic colonization to severe colitis (3). It is hypothesized that differential immune responses to CDI likely play an important role in the outcome of infection. Understanding the factors that shape the immune response during CDI represents a major area of opportunity for the treatment of CDI.
Pharmaceutical drugs and CDI.
Recent studies have begun to shed light on the unexpected effects that pharmaceutical drugs have on the gut microbiota (98). With susceptibility and severity of CDI being so tightly linked to the structure and composition of the gut microbiota, it is likely that pharmaceutical drugs play an unappreciated role in CDI (Fig. 2). One such example is nonsteroidal anti-inflammatory drugs (NSAIDs), which are among the most highly prescribed and most widely consumed drugs in the United States, particularly among older adults, and have been implicated in causing spontaneous colitis in humans (99, 100). Recent epidemiological studies have established an association between NSAIDs and CDI (101). NSAIDs act by inhibiting cyclooxygenase (COX) enzymatic activity, which prevents the generation of prostaglandins (PGs) and alters the outcome of subsequent inflammatory events. Prostaglandins, especially PGE2, are important lipid mediators that are highly abundant at sites of inflammation and infection and that support gastrointestinal homeostasis. In the context of CDI, it has been shown that NSAIDs dramatically increase the mortality and intestinal pathology in mice. This is highlighted by alterations in the microbiota, prostaglandin dysregulation, altered proinflammatory profile, and decreased epithelial tight junction integrity (102, 103). Introduction of the stable PGE1 analogue misoprostol protects mice from severe CDI and reduces microbiota perturbations (104).
Several retrospective analyses over the past two decades have found that patients prescribed proton pump inhibitors (PPIs) have an increased risk of contracting C. difficile, especially if they are on concurrent high-risk antibiotics (Fig. 2) (105, 106). PPIs increase the pH of the gastrointestinal tract by suppressing acid production. To date, no mechanism has been rigorously shown to explain any association between PPI use and increased incidence of CDI. It is becoming an increasingly controversial hypothesis, as other studies have found limited associations (107–109). The role of PPIs in CDI remains to be clarified, and follow-up studies are needed.
Evaluation of patients with depression who were on antidepressant medications demonstrated that utilization of certain antidepressant medications, such as mirtazapine and fluoxetine, is associated with CDI risk (110). This effect was independent of antibiotic exposure and particularly significant in patients taking both of these medications in combination. Taken together, these studies demonstrate the role of pharmaceutical drugs in risk and severity of CDI and highlight the potential role of these drugs in C. difficile epidemiology.
Diet and nutrition in CDI.
Diet plays an essential role in shaping the microbiota, and interactions between the microbiota and dietary nutrients have been shown to be associated with numerous diseases (56, 111–114). Thus, it is not surprising that the impact of diet on CDI has become an emerging area of research in recent years. For example, a recent study demonstrated that a single micronutrient, zinc, given in excess to mice, dramatically alters the microbiota, increases susceptibility to CDI, and exacerbates disease (83). It is postulated that excess Zn alters the ecology of the microbial community, permitting C. difficile and other pathogenic microbes to thrive and cause severe C. difficile-associated disease (115). In an additional study, it was demonstrated that microbiota-accessible carbohydrates suppress C. difficile in the gastrointestinal tract by enriching for taxa that antagonize C. difficile. The availability of these carbohydrates leads to the production of metabolic end products, such as acetate, butyrate, and propionate, that decrease C. difficile fitness in the GI tract (116). Because of C. difficile’s ability to ferment amino acids as a nutrient source, dietary protein may also play a role in C. difficile pathogenesis. A recent study found that a high-fat/high-protein diet intensified C. difficile proliferation and virulence in a mouse model of infection (117). This study also supported the protective role of a high-carbohydrate diet. Finally, recent studies demonstrated that the dietary additive trehalose is an important mediator of the emergence of epidemic ribotype 027 (RT027) as well as ribotype 078 (RT078) strains of C. difficile (118, 119). Specifically, point mutations acquired by these highly virulent epidemic strains of C. difficile increased sensitivity to trehalose, allowing for utilization of this resource at low levels and selecting for emergence. Taken together, these studies have begun to shed light on the role diet can have on susceptibility to infection, the ecology of the C. difficile-infected gut, and pathogenesis and behavior of C. difficile in the GI tract.
C. difficile and One Health.
Epidemic RT027 strains of C. difficile have classically been associated with hospital-acquired infections, while an apparent role for RT078 in community-acquired infections is emerging (120, 121). Community-acquired C. difficile is on the rise, and though reservoirs such as asymptomatic carriers and animals are known, causal linkages have yet to be shown for the acquisition of C. difficile outside a hospital setting. However, the prevalence of C. difficile in domesticated animals, particularly agricultural animals, has sparked interest in how One Health (https://www.cdc.gov/onehealth/index.html) concepts can be applied to C. difficile (121). RT078, in particular, has a demonstrably high disease severity and attributable mortality at least as high as RT027 strains (122). Additionally, the identification of C. difficile RT078 strains and closely related lineages in the sequence type 11 group in domesticated animals such as pigs and cows adds a new level of complexity to the fight against antibiotic resistance (123). A recent study analyzing 247 C. difficile RT078 genomes from distinct geographic locations and hosts revealed a strong bidirectional correlation between human and animal strains with very little geographic clustering, indicating a high degree of intercontinental transmission (122). Data from a study in the Netherlands support this concept of interspecies transmission with the finding of clonal RT078 strains between pigs and farmers (124). Hospital-associated epidemics of certain C. difficile strains tend to emerge when they acquire resistance to high-risk antibiotics such as clindamycin and fluoroquinolones. However, with respect to RT078, there is an emergence of tetracycline resistance due to the heavy use of this antibiotic in agriculture (123). Another study, which sampled 400 RT078 genomes from across North America, Europe, and the United Kingdom, found that tetracycline resistance due to the tetM gene was by far the most abundant antimicrobial resistance marker among all the isolates, with resistance rates as high as 77.5% (123). These data highlight the emergence of antibiotic resistance in pathogens of human interest outside a hospital environment.
PERSPECTIVES
In this review, we highlighted recent evidence for the role of the pathogen, host, microbiota, and environment in the outcome of C. difficile infection and disease. Despite this growing evidence, defining the multidimensional interactions and molecular mechanisms in this complex ecosystem has proven to be incredibly challenging and has not moved far beyond simple associations. This gap in knowledge is particularly striking when one considers the broad and significant impact that C. difficile has on human health and health care systems worldwide. Furthermore, the continued rise of C. difficile incidence, high rates of recurrence, and emergence of hypervirulent strains highlight the need for identification of drug targets and the development of novel therapeutic strategies to treat this infection.
Several major focuses of the field, moving forward, center around further describing the role of bystander microbiota on C. difficile virulence and behavior during infection. Moreover, understanding the role of pharmaceutical drugs, diet, and nutritional status in susceptibility to and severity of CDI is paramount. A major theme that has emerged in C. difficile research and will continue to be a focus of our group and others is metabolic cross talk between the host, microbiota, and C. difficile. Metabolism and metabolic interactions at the subcellular level form the basis for the function, survival, and behavior of all living cells (125). The role of metabolism and the importance of metabolic state are shared throughout the tree of life, and the building blocks for these behaviors are communal between immune cells, commensal bacteria, and invading pathogens. Thus, it is not surprising that metabolic cross talk between cells, species, and kingdoms has emerged as a key component of human health and disease. At no area in the body is this cross talk more evident than the GI tract. C. difficile interfaces with metabolites from the host, microbiota, environment, and diet, and each may directly impact this pathogen differently. Furthermore, metabolites produced by C. difficile and the microbiota can be sensed by the host, eliciting a myriad of cellular responses. It is well-known that microbiota-produced metabolites can even stimulate systemic responses in the host, including in distant tissues like the brain, but it is unclear how many of these metabolites may shape immunity to C. difficile during infection (58). Understanding how metabolites from the microbiota impact C. difficile and host immunity remains a major focus for the field moving forward.
In conclusion, it is clear that numerous variables impact susceptibility to this pathogen and the outcome of CDI. Future work will need to consider each facet of the tripartite interaction between the host, pathogen, and microbiota during infection. This will take continued development of novel methods that incorporate each of these variables, such as organoid models, intestine-on-a-chip systems, and advanced gnotobiotics. Furthermore, with the emergence of fecal microbiota transplantation as an incredibly successful treatment for CDI, it is clear that harnessing the ecological and metabolic potential of the microbiota will be at the forefront of therapeutic potential (Fig. 2).
ACKNOWLEDGMENTS
J.P.Z. is supported by K22AI7220 (NIH/NIAID). A.B.S. is supported by T32GM07229.
Figures were created with Biorender.com.
We have no relevant conflict of interest to disclose.
Biographies

Alexander B. Smith earned a Bachelor of Science in biochemistry and molecular biology in 2018 from the Pennsylvania State University. While at Penn State, he studied the genetic determinants of Gram-negative bacterial membrane modifications in the laboratory of Dr. Timothy Meredith. Due to his interest in bacteriology and microbial ecology, he began his graduate training in the Microbiology, Virology, and Parasitology graduate group within the Cell and Molecular Biology program at the University of Pennsylvania. In 2019, Alexander joined the laboratory of Dr. Joseph Zackular. In the Zackular laboratory, Alexander studies synergistic microbial interactions between the enteric pathogen Clostridioides difficile and other members of the human gut microbiota.

Joshua Soto Ocana graduated from the University of Puerto Rico at Mayagüez in 2018 with a B.S. in Industrial Microbiology, a B.S. in Biology, and a minor in Biochemistry. During his undergraduate experience he worked on different areas of research which included investigating the role of insulin amyloid fibrils in type II diabetes, developing a green approach for the synthesis of indoles, and understating the role of IL-13 signaling in soft tissue sarcoma. Despite his undergraduate research experiences, Joshua knew he wanted to continue his graduate studies in microbiology. For that reason, he joined the Microbiology, Virology and Parasitology graduate group under the Cell and Molecular Biology Program at the University of Pennsylvania. In 2019, he joined Dr. Joseph Zackular’s laboratory due to his interest in host-pathogen interactions. In Dr. Zackular’s laboratory, Joshua is interested in understanding the role of pharmaceutical drugs on Clostridioides difficile infection, specifically looking at how nonsteroidal anti-inflammatory drugs can impact C. difficile pathogenesis.

Joseph P. Zackular, Ph.D., is an Assistant Professor in the Department of Pathology and Laboratory Medicine at the University of Pennsylvania and the Children’s Hospital of Philadelphia. He received his Ph.D. from the University of Michigan where he studied the role of the gut microbiota in colorectal cancer in the laboratory of Dr. Patrick Schloss. He joined the laboratory of Dr. Eric Skaar at Vanderbilt University Medical Center for his postdoctoral fellowship where he studied the role of dietary metals and nutritional immunity in Clostridium difficile infection. In August of 2018, he started his laboratory in the Children’s Hospital of Philadelphia. The Zackular laboratory is focused on understanding how interactions between the host, gut microbiota, and pathogenic microbes impact human health and disease. The lab’s recent efforts center on understanding how the important nosocomial pathogen C. difficile interacts with resident gut microbiota during infection and how interspecies cross-talk impacts growth, behavior, and virulence of this pathogen. Research in the Zackular lab draws from a number of diverse fields including microbial ecology, bacterial pathogenesis, biochemistry, host-pathogen interactions, and microbiota research.
REFERENCES
- 1.Lessa FC, Winston LG, McDonald LC, Emerging Infections Program C. difficile Surveillance Team. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:2369–2370. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leffler DA, Lamont JT. 2015. Clostridium difficile infection. N Engl J Med 373:287–288. doi: 10.1056/NEJMra1403772. [DOI] [PubMed] [Google Scholar]
- 3.Kelly CP, LaMont JT. 1998. Clostridium difficile infection. Annu Rev Med 49:375–390. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 4.Seekatz AM, Young VB. 2014. Clostridium difficile and the microbiota. J Clin Invest 124:4182–4189. doi: 10.1172/JCI72336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown KA, Khanafer N, Daneman N, Fisman DN. 2013. Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection. Antimicrob Agents Chemother 57:2326–2332. doi: 10.1128/AAC.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DD, Sferra TJ, Hernandez AV, Donskey CJ. 2013. Community-associated Clostridium difficile infection and antibiotics: a meta-analysis. J Antimicrob Chemother 68:1951–1961. doi: 10.1093/jac/dkt129. [DOI] [PubMed] [Google Scholar]
- 7.Teng C, Reveles KR, Obodozie-Ofoegbu OO, Frei CR. 2019. Clostridium difficile infection risk with important antibiotic classes: an analysis of the FDA adverse event reporting system. Int J Med Sci 16:630–635. doi: 10.7150/ijms.30739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wenisch JM, Schmid D, Kuo HW, Simons E, Allerberger F, Michl V, Tesik P, Tucek G, Wenisch C. 2012. Hospital-acquired Clostridium difficile infection: determinants for severe disease. Eur J Clin Microbiol Infect Dis 31:1923–1930. doi: 10.1007/s10096-011-1522-5. [DOI] [PubMed] [Google Scholar]
- 9.Sailhamer EA, Carson K, Chang Y, Zacharias N, Spaniolas K, Tabbara M, Alam HB, DeMoya MA, Velmahos GC. 2009. Fulminant Clostridium difficile colitis: patterns of care and predictors of mortality. Arch Surg 144:433–439. doi: 10.1001/archsurg.2009.51. [DOI] [PubMed] [Google Scholar]
- 10.Adams SD, Mercer DW. 2007. Fulminant Clostridium difficile colitis. Curr Opin Crit Care 13:450–455. doi: 10.1097/MCC.0b013e3282638879. [DOI] [PubMed] [Google Scholar]
- 11.Kelly CP, LaMont JT. 2008. Clostridium difficile–more difficult than ever. N Engl J Med 359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 12.Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan VK, Johnson S, Gerding DN, Vedantam G. 2010. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol 192:4904–4911. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson PE, Walk ST, Bourgis AE, Liu MW, Kopliku F, Lo E, Young VB, Aronoff DM, Hanna PC. 2013. The relationship between phenotype, ribotype, and clinical disease in human Clostridium difficile isolates. Anaerobe 24:109–116. doi: 10.1016/j.anaerobe.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walk ST, Micic D, Jain R, Lo ES, Trivedi I, Liu EW, Almassalha LM, Ewing SA, Ring C, Galecki AT, Rogers MA, Washer L, Newton DW, Malani PN, Young VB, Aronoff DM. 2012. Clostridium difficile ribotype does not predict severe infection. Clin Infect Dis 55:1661–1668. doi: 10.1093/cid/cis786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald LC, Killgore GE, Thompson A, Owens RC, Kazakova SV, Sambol SP, Johnson S, Gerding DN. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 16.Lessa FC, Gould CV, McDonald LC. 2012. Current status of Clostridium difficile infection epidemiology. Clin Infect Dis 55(Suppl 2):S65–S70. doi: 10.1093/cid/cis319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belmares J, Johnson S, Parada JP, Olson MM, Clabots CR, Bettin KM, Peterson LR, Gerding DN. 2009. Molecular epidemiology of Clostridium difficile over the course of 10 years in a tertiary care hospital. Clin Infect Dis 49:1141–1147. doi: 10.1086/605638. [DOI] [PubMed] [Google Scholar]
- 18.Barbut F, Jones G, Eckert C. 2011. Epidemiology and control of Clostridium difficile infections in healthcare settings: an update. Curr Opin Infect Dis 24:370–376. doi: 10.1097/QCO.0b013e32834748e5. [DOI] [PubMed] [Google Scholar]
- 19.Cho J, Cunningham S, Pu M, Lennon RJ, Dens Higano J, Jeraldo P, Sampathkumar P, Shannon S, Kashyap P, Patel R. 17 February 2020. Clostridioides difficile whole genome sequencing differentiates relapse with the same strain from reinfection with a new strain. Clin Infect Dis doi: 10.1093/cid/ciaa159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figueroa I, Johnson S, Sambol SP, Goldstein EJ, Citron DM, Gerding DN. 2012. Relapse versus reinfection: recurrent Clostridium difficile infection following treatment with fidaxomicin or vancomycin. Clin Infect Dis 55(Suppl 2):S104–S109. doi: 10.1093/cid/cis357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furuya-Kanamori L, Marquess J, Yakob L, Riley TV, Paterson DL, Foster NF, Huber CA, Clements AC. 2015. Asymptomatic Clostridium difficile colonization: epidemiology and clinical implications. BMC Infect Dis 15:516. doi: 10.1186/s12879-015-1258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viscidi R, Willey S, Bartlett JG. 1981. Isolation rates and toxigenic potential of Clostridium difficile isolates from various patient populations. Gastroenterology 81:5–9. doi: 10.1016/0016-5085(81)90644-2. [DOI] [PubMed] [Google Scholar]
- 23.Miyajima F, Roberts P, Swale A, Price V, Jones M, Horan M, Beeching N, Brazier J, Parry C, Pendleton N, Pirmohamed M. 2011. Characterisation and carriage ratio of Clostridium difficile strains isolated from a community-dwelling elderly population in the United Kingdom. PLoS One 6:e22804. doi: 10.1371/journal.pone.0022804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozaki E, Kato H, Kita H, Karasawa T, Maegawa T, Koino Y, Matsumoto K, Takada T, Nomoto K, Tanaka R, Nakamura S. 2004. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J Med Microbiol 53:167–172. doi: 10.1099/jmm.0.05376-0. [DOI] [PubMed] [Google Scholar]
- 25.Kato H, Kita H, Karasawa T, Maegawa T, Koino Y, Takakuwa H, Saikai T, Kobayashi K, Yamagishi T, Nakamura S. 2001. Colonisation and transmission of Clostridium difficile in healthy individuals examined by PCR ribotyping and pulsed-field gel electrophoresis. J Med Microbiol 50:720–727. doi: 10.1099/0022-1317-50-8-720. [DOI] [PubMed] [Google Scholar]
- 26.Galdys AL, Nelson JS, Shutt KA, Schlackman JL, Pakstis DL, Pasculle AW, Marsh JW, Harrison LH, Curry SR. 2014. Prevalence and duration of asymptomatic Clostridium difficile carriage among healthy subjects in Pittsburgh, Pennsylvania. J Clin Microbiol 52:2406–2409. doi: 10.1128/JCM.00222-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RL, Donskey CJ. 2007. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis 45:992–998. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 28.Campbell RR, Beere D, Wilcock GK, Brown EM. 1988. Clostridium difficile in acute and long-stay elderly patients. Age Ageing 17:333–336. doi: 10.1093/ageing/17.5.333. [DOI] [PubMed] [Google Scholar]
- 29.Bender BS, Bennett R, Laughon BE, Greenough WB, Gaydos C, Sears SD, Forman MS, Bartlett JG. 1986. Is Clostridium difficile endemic in chronic-care facilities? Lancet 2:11–13. doi: 10.1016/s0140-6736(86)92559-6. [DOI] [PubMed] [Google Scholar]
- 30.Furuya-Kanamori L, Clements ACA, Foster NF, Huber CA, Hong S, Harris-Brown T, Yakob L, Paterson DL, Riley TV. 2017. Asymptomatic Clostridium difficile colonization in two Australian tertiary hospitals, 2012–2014: prospective, repeated cross-sectional study. Clin Microbiol Infect 23:48.e1–48.e7. doi: 10.1016/j.cmi.2016.08.030. [DOI] [PubMed] [Google Scholar]
- 31.Hall IC, O’Toole E. 1935. Intestinal flora in new-born infants: with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child 49:390–402. doi: 10.1001/archpedi.1935.01970020105010. [DOI] [Google Scholar]
- 32.Al-Jumaili IJ, Shibley M, Lishman AH, Record CO. 1984. Incidence and origin of Clostridium difficile in neonates. J Clin Microbiol 19:77–78. doi: 10.1128/JCM.19.1.77-78.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jangi S, Lamont JT. 2010. Asymptomatic colonization by Clostridium difficile in infants: implications for disease in later life. J Pediatr Gastroenterol Nutr 51:2–7. doi: 10.1097/MPG.0b013e3181d29767. [DOI] [PubMed] [Google Scholar]
- 34.Sammons JS, Toltzis P, Zaoutis TE. 2013. Clostridium difficile infection in children. JAMA Pediatr 167:567–573. doi: 10.1001/jamapediatrics.2013.441. [DOI] [PubMed] [Google Scholar]
- 35.Stoesser N, Eyre DW, Quan TP, Godwin H, Pill G, Mbuvi E, Vaughan A, Griffiths D, Martin J, Fawley W, Dingle KE, Oakley S, Wanelik K, Finney JM, Kachrimanidou M, Moore CE, Gorbach S, Riley TV, Crook DW, Peto TEA, Wilcox MH, Walker AS, Modernising Medical Microbiology Informatics Group (MMMIG). 2017. Epidemiology of Clostridium difficile in infants in Oxfordshire, UK: risk factors for colonization and carriage, and genetic overlap with regional C. difficile infection strains. PLoS One 12:e0182307. doi: 10.1371/journal.pone.0182307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rousseau C, Levenez F, Fouqueray C, Doré J, Collignon A, Lepage P. 2011. Clostridium difficile colonization in early infancy is accompanied by changes in intestinal microbiota composition. J Clin Microbiol 49:858–865. doi: 10.1128/JCM.01507-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stark PL, Lee A, Parsonage BD. 1982. Colonization of the large bowel by Clostridium difficile in healthy infants: quantitative study. Infect Immun 35:895–899. doi: 10.1128/IAI.35.3.895-899.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naaber P, Klaus K, Sepp E, Björksten B, Mikelsaar M. 1997. Colonization of infants and hospitalized patients with Clostridium difficile and lactobacilli. Clin Infect Dis 25:S189–S190. doi: 10.1086/516183. [DOI] [PubMed] [Google Scholar]
- 39.Sommer F, Bäckhed F. 2013. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol 11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 40.Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, Zárate Rodriguez JG, Rogers AB, Robine N, Loke P, Blaser MJ. 2014. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gregory KE, Samuel BS, Houghteling P, Shan G, Ausubel FM, Sadreyev RI, Walker WA. 2016. Influence of maternal breast milk ingestion on acquisition of the intestinal microbiome in preterm infants. Microbiome 4:68. doi: 10.1186/s40168-016-0214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, Li H, Chung J, Sohn J, Kim S, Gao Z, Barber C, Kim J, Ng S, Rogers AB, Sumner S, Zhang XS, Cadwell K, Knights D, Alekseyenko A, Bäckhed F, Blaser MJ. 2016. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol 1:16140. doi: 10.1038/nmicrobiol.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ. 2012. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trasande L, Blustein J, Liu M, Corwin E, Cox LM, Blaser MJ. 2013. Infant antibiotic exposures and early-life body mass. Int J Obes (Lond) 37:16–23. doi: 10.1038/ijo.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, Kumar N, Stares MD, Rodger A, Brocklehurst P, Field N, Lawley TD. 2019. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574:117–121. doi: 10.1038/s41586-019-1560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumann-Dudenhoeffer AM, D'Souza AW, Tarr PI, Warner BB, Dantas G. 2018. Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nat Med 24:1822–1829. doi: 10.1038/s41591-018-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, D Lieber A, Wu F, Perez-Perez GI, Chen Y, Schweizer W, Zheng X, Contreras M, Dominguez-Bello MG, Blaser MJ. 2016. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8:343ra82. doi: 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, Tarr PI, Warner BB, Dantas G. 2019. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat Microbiol 4:2285–2297. doi: 10.1038/s41564-019-0550-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trejo FM, Pérez PF, De Antoni GL. 2010. Co-culture with potentially probiotic microorganisms antagonises virulence factors of Clostridium difficile in vitro. Antonie Van Leeuwenhoek 98:19–29. doi: 10.1007/s10482-010-9424-6. [DOI] [PubMed] [Google Scholar]
- 50.Tonooka T, Sakata S, Kitahara M, Hanai M, Ishizeki S, Takada M, Sakamoto M, Benno Y. 2005. Detection and quantification of four species of the genus Clostridium in infant feces. Microbiol Immunol 49:987–992. doi: 10.1111/j.1348-0421.2005.tb03694.x. [DOI] [PubMed] [Google Scholar]
- 51.Dallas SD, Rolfe RD. 1998. Binding of Clostridium difficile toxin A to human milk secretory component. J Med Microbiol 47:879–888. doi: 10.1099/00222615-47-10-879. [DOI] [PubMed] [Google Scholar]
- 52.Rolfe RD, Song W. 1995. Immunoglobulin and non-immunoglobulin components of human milk inhibit Clostridium difficile toxin A-receptor binding. J Med Microbiol 42:10–19. doi: 10.1099/00222615-42-1-10. [DOI] [PubMed] [Google Scholar]
- 53.Kociolek LK, Espinosa RO, Gerding DN, Hauser AR, Ozer EA, Budz M, Balaji A, Chen X, Tanz RR, Yalcinkaya N, Conner ME, Savidge T, Kelly CP. 2019. Natural Clostridioides difficile toxin immunization in colonized infants. Clin Infect Dis doi: 10.1093/cid/ciz582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zacharioudakis IM, Zervou FN, Pliakos EE, Ziakas PD, Mylonakis E. 2015. Colonization with toxinogenic C. difficile upon hospital admission, and risk of infection: a systematic review and meta-analysis. Am J Gastroenterol 110:381–390; quiz, 391. doi: 10.1038/ajg.2015.22. [DOI] [PubMed] [Google Scholar]
- 55.Viscidi R, Laughon BE, Yolken R, Bo-Linn P, Moench T, Ryder RW, Bartlett JG. 1983. Serum antibody response to toxins A and B of Clostridium difficile. J Infect Dis 148:93–100. doi: 10.1093/infdis/148.1.93. [DOI] [PubMed] [Google Scholar]
- 56.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 57.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. 2019. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570:462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaiss CA, Zmora N, Levy M, Elinav E. 2016. The microbiome and innate immunity. Nature 535:65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 59.Britton RA, Young VB. 2014. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146:1547–1553. doi: 10.1053/j.gastro.2014.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sorbara MT, Pamer EG. 2019. Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol 12:1–9. doi: 10.1038/s41385-018-0053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Winston JA, Theriot CM. 2016. Impact of microbial derived secondary bile acids on colonization resistance against Clostridium difficile in the gastrointestinal tract. Anaerobe 41:44–50. doi: 10.1016/j.anaerobe.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ridlon JM, Kang DJ, Hylemon PB. 2006. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Wilson KH. 1983. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J Clin Microbiol 18:1017–1019. doi: 10.1128/JCM.18.4.1017-1019.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson KH, Kennedy MJ, Fekety FR. 1982. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J Clin Microbiol 15:443–446. doi: 10.1128/JCM.15.3.443-446.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sorg JA, Sonenshein AL. 2010. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol 192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foley MH, O'Flaherty S, Barrangou R, Theriot CM. 2019. Bile salt hydrolases: gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog 15:e1007581. doi: 10.1371/journal.ppat.1007581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weingarden AR, Chen C, Zhang N, Graiziger CT, Dosa PI, Steer CJ, Shaughnessy MK, Johnson JR, Sadowsky MJ, Khoruts A. 2016. Ursodeoxycholic acid inhibits Clostridium difficile spore germination and vegetative growth, and prevents the recurrence of ileal pouchitis associated with the infection. J Clin Gastroenterol 50:624–630. doi: 10.1097/MCG.0000000000000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Theriot CM, Bowman AA, Young VB. 2016. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere 1:e00045-15. doi: 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Francis MB, Allen CA, Sorg JA. 2013. Muricholic acids inhibit Clostridium difficile spore germination and growth. PLoS One 8:e73653. doi: 10.1371/journal.pone.0073653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Battaglioli EJ, Hale VL, Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, Rekdal VM, Till LM, Huq L, Smits SA, Moor WJ, Jones-Hall Y, Smyrk T, Khanna S, Pardi DS, Grover M, Patel R, Chia N, Nelson H, Sonnenburg JL, Farrugia G, Kashyap PC. 2018. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci Transl Med 10:eaam7019. doi: 10.1126/scitranslmed.aam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. 2014. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16:770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Armbruster CR, Wolter DJ, Mishra M, Hayden HS, Radey MC, Merrihew G, MacCoss MJ, Burns J, Wozniak DJ, Parsek MR, Hoffman LR. 2016. Staphylococcus aureus protein A mediates interspecies interactions at the cell surface of Pseudomonas aeruginosa. mBio 7:e00538-16. doi: 10.1128/mBio.00538-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Armbruster CE, Hong W, Pang B, Weimer KE, Juneau RA, Turner J, Swords WE. 2010. Indirect pathogenicity of Haemophilus influenzae and Moraxella catarrhalis in polymicrobial otitis media occurs via interspecies quorum signaling. mBio 1:e00102-10. doi: 10.1128/mBio.00102-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DeLeon S, Clinton A, Fowler H, Everett J, Horswill AR, Rumbaugh KP. 2014. Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro wound model. Infect Immun 82:4718–4728. doi: 10.1128/IAI.02198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stacy A, Everett J, Jorth P, Trivedi U, Rumbaugh KP, Whiteley M. 2014. Bacterial fight-and-flight responses enhance virulence in a polymicrobial infection. Proc Natl Acad Sci U S A 111:7819–7824. doi: 10.1073/pnas.1400586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramsey MM, Rumbaugh KP, Whiteley M. 2011. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLoS Pathog 7:e1002012. doi: 10.1371/journal.ppat.1002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murray JL, Connell JL, Stacy A, Turner KH, Whiteley M. 2014. Mechanisms of synergy in polymicrobial infections. J Microbiol 52:188–199. doi: 10.1007/s12275-014-4067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zackular JP, Moore JL, Jordan AT, Juttukonda LJ, Noto MJ, Nicholson MR, Crews JD, Semler MW, Zhang Y, Ware LB, Washington MK, Chazin WJ, Caprioli RM, Skaar EP. 2016. Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat Med 22:1330–1334. doi: 10.1038/nm.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandrasekaran R, Lacy DB. 2017. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 41:723–750. doi: 10.1093/femsre/fux048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 86.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 87.Abt MC, McKenney PT, Pamer EG. 2016. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol 14:609–620. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Lacy DB. 2016. Clostridium difficile toxins TcdA and TcdB cause colonic tissue damage by distinct mechanisms. Infect Immun 84:2871–2877. doi: 10.1128/IAI.00583-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poxton IR, McCoubrey J, Blair G. 2001. The pathogenicity of Clostridium difficile. Clin Microbiol Infect 7:421–427. doi: 10.1046/j.1198-743x.2001.00287.x. [DOI] [PubMed] [Google Scholar]
- 90.Souza MH, Melo-Filho AA, Rocha MF, Lyerly DM, Cunha FQ, Lima AA, Ribeiro RA. 1997. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology 91:281–288. doi: 10.1046/j.1365-2567.1997.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, Ling L, Leiner I, Pamer EG. 2015. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe 18:27–37. doi: 10.1016/j.chom.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kyne L, Warny M, Qamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357:189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 93.Kyne L, Warny M, Qamar A, Kelly CP. 2000. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med 342:390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 94.Islam J, Taylor AL, Rao K, Huffnagle G, Young VB, Rajkumar C, Cohen J, Papatheodorou P, Aronoff DM, Llewelyn MJ. 2014. The role of the humoral immune response to Clostridium difficile toxins A and B in susceptibility to C. difficile infection: a case-control study. Anaerobe 27:82–86. doi: 10.1016/j.anaerobe.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gerding DN, Kelly CP, Rahav G, Lee C, Dubberke ER, Kumar PN, Yacyshyn B, Kao D, Eves K, Ellison MC, Hanson ME, Guris D, Dorr MB. 2018. Bezlotoxumab for prevention of recurrent Clostridium difficile infection in patients at increased risk for recurrence. Clin Infect Dis 67:649–656. doi: 10.1093/cid/ciy171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kufel WD, Devanathan AS, Marx AH, Weber DJ, Daniels LM. 2017. Bezlotoxumab: a novel agent for the prevention of recurrent Clostridium difficile infection. Pharmacotherapy 37:1298–1308. doi: 10.1002/phar.1990. [DOI] [PubMed] [Google Scholar]
- 97.Kroh HK, Chandrasekaran R, Zhang Z, Rosenthal K, Woods R, Jin X, Nyborg AC, Rainey GJ, Warrener P, Melnyk RA, Spiller BW, Lacy DB. 2018. A neutralizing antibody that blocks delivery of the enzymatic cargo of Clostridium difficile toxin TcdB into host cells. J Biol Chem 293:941–952. doi: 10.1074/jbc.M117.813428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR, Bork P, Typas A. 2018. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555:623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cryer B, Barnett MA, Wagner J, Wilcox CM. 2016. Overuse and misperceptions of nonsteroidal anti-inflammatory drugs in the United States. Am J Med Sci 352:472–480. doi: 10.1016/j.amjms.2016.08.028. [DOI] [PubMed] [Google Scholar]
- 100.Tonolini M. 2013. Acute nonsteroidal anti-inflammatory drug-induced colitis. J Emerg Trauma Shock 6:301–303. doi: 10.4103/0974-2700.120389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Permpalung N, Upala S, Sanguankeo A, Sornprom S. 2016. Association between NSAIDs and Clostridium difficile-associated diarrhea: a systematic review and meta-analysis. Can J Gastroenterol Hepatol 2016:7431838. doi: 10.1155/2016/7431838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Muñoz-Miralles J, Trindade BC, Castro-Córdova P, Bergin IL, Kirk LA, Gil F, Aronoff DM, Paredes-Sabja D. 2018. Indomethacin increases severity of Clostridium difficile infection in mouse model. Future Microbiol 13:1271–1281. doi: 10.2217/fmb-2017-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maseda D, Zackular JP, Trindade B, Kirk L, Roxas JL, Rogers LM, Washington MK, Du L, Koyama T, Viswanathan VK, Vedantam G, Schloss PD, Crofford LJ, Skaar EP, Aronoff DM. 2019. Nonsteroidal anti-inflammatory drugs alter the microbiota and exacerbate Clostridium difficile colitis while dysregulating the inflammatory response. mBio 10:e02282-18. doi: 10.1128/mBio.02282-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zackular JP, Kirk L, Trindade BC, Skaar EP, Aronoff DM. 2019. Misoprostol protects mice against severe Clostridium difficile infection and promotes recovery of the gut microbiota after antibiotic perturbation. Anaerobe 58:89–94. doi: 10.1016/j.anaerobe.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cunningham R, Dale B, Undy B, Gaunt N. 2003. Proton pump inhibitors as a risk factor for Clostridium difficile diarrhoea. J Hosp Infect 54:243–245. doi: 10.1016/s0195-6701(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 106.Dial S, Delaney JA, Barkun AN, Suissa S. 2005. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA 294:2989–2995. doi: 10.1001/jama.294.23.2989. [DOI] [PubMed] [Google Scholar]
- 107.Caffrey AR, Timbrook TT, Ali SR, Nizet V, Sakoulas G. 2019. Proton-pump inhibitors do not influence clinical outcomes in patients with Staphylococcus aureus bacteremia. Therap Adv Gastroenterol 12:1756284819834273. doi: 10.1177/1756284819834273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Faleck DM, Salmasian H, Furuya EY, Larson EL, Abrams JA, Freedberg DE. 2016. Proton pump inhibitors do not increase risk for Clostridium difficile infection in the intensive care unit. Am J Gastroenterol 111:1641–1648. doi: 10.1038/ajg.2016.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tomkovich S, Lesniak NA, Li Y, Bishop L, Fitzgerald MJ, Schloss PD. 2019. The proton pump inhibitor omeprazole does not promote Clostridioides difficile colonization in a murine model. mSphere 4:e00693-19. doi: 10.1128/mSphere.00693-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rogers MA, Greene MT, Young VB, Saint S, Langa KM, Kao JY, Aronoff DM. 2013. Depression, antidepressant medications, and risk of Clostridium difficile infection. BMC Med 11:121. doi: 10.1186/1741-7015-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turnbaugh PJ, Gordon JI. 2009. The core gut microbiome, energy balance and obesity. J Physiol 587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. 2009. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zackular JP, Skaar EP. 2018. The role of zinc and nutritional immunity in Clostridium difficile infection. Gut Microbes 9:469–476. doi: 10.1080/19490976.2018.1448354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hryckowian AJ, Van Treuren W, Smits SA, Davis NM, Gardner JO, Bouley DM, Sonnenburg JL. 2018. Microbiota-accessible carbohydrates suppress Clostridium difficile infection in a murine model. Nat Microbiol 3:662–669. doi: 10.1038/s41564-018-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mefferd CC, Bhute SS, Phan JR, Villarama JV, Do DM, Alarcia S, Abel-Santos E, Hedlund BP. 2020. A high-fat/high-protein, Atkins-type diet exacerbates Clostridioides (Clostridium) difficile infection in mice, whereas a high-carbohydrate diet protects. mSystems 5:e00765-19. doi: 10.1128/mSystems.00765-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, Auchtung JM, Britton RA. 2018. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 553:291–294. doi: 10.1038/nature25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Collins J, Danhof H, Britton RA. 2018. The role of trehalose in the global spread of epidemic Clostridium difficile. Gut Microbes 10:204–209. doi: 10.1080/19490976.2018.1491266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smits WK. 2013. Hype or hypervirulence: a reflection on problematic C. difficile strains. Virulence 4:592–596. doi: 10.4161/viru.26297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Knight DR, Elliott B, Chang BJ, Perkins TT, Riley TV. 2015. Diversity and evolution in the genome of Clostridium difficile. Clin Microbiol Rev 28:721–741. doi: 10.1128/CMR.00127-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knetsch CW, Kumar N, Forster SC, Connor TR, Browne HP, Harmanus C, Sanders IM, Harris SR, Turner L, Morris T, Perry M, Miyajima F, Roberts P, Pirmohamed M, Songer JG, Weese JS, Indra A, Corver J, Rupnik M, Wren BW, Riley TV, Kuijper EJ, Lawley TD. 2018. Zoonotic transfer of Clostridium difficile harboring antimicrobial resistance between farm animals and humans. J Clin Microbiol 56:e01384-17. doi: 10.1128/JCM.01384-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dingle KE, Didelot X, Quan TP, Eyre DW, Stoesser N, Marwick CA, Coia J, Brown D, Buchanan S, Ijaz UZ, Goswami C, Douce G, Fawley WN, Wilcox MH, Peto TEA, Walker AS, Crook DW. 2019. A role for tetracycline selection in recent evolution ofagriculture-associated Clostridium difficile PCR ribotype 078. mBio 10:e02790-18. doi: 10.1128/mBio.02790-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Knetsch CW, Connor TR, Mutreja A, van Dorp SM, Sanders IM, Browne HP, Harris D, Lipman L, Keessen EC, Corver J, Kuijper EJ, Lawley TD. 2014. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill 19:20954. doi: 10.2807/1560-7917.ES2014.19.45.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russell DG, Huang L, VanderVen BC. 2019. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol 19:291–304. doi: 10.1038/s41577-019-0124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abbas A, Zackular JP. 2020. Microbe-microbe interactions during Clostridioides difficile infection. Curr Opin Microbiol 53:19–25. doi: 10.1016/j.mib.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]