

Osteocytes initiate four distinct pathways to accomplish adaptation of bone to changes in the mechanical environment.
Key Words: bone functional adaptation, mechanical loading, mechanostat, osteocytes, disuse, modeling, remodeling
Abstract
We review evidence supporting an updated mechanostat model in bone that highlights the central role of osteocytes within bone's four mechanoadaptive pathways: 1) formation modeling and 2) targeted remodeling, which occur with heightened mechanical loading, 3) resorption modeling, and 4) disuse-mediated remodeling, which occur with disuse. These four pathways regulate whole-bone stiffness in response to changing mechanical demands.
Key Points
In this review, we challenge the notion that bone remodeling is the primary adaptive physiologic response to exercise and disuse.
We propose an updated mechanostat model of bone functional adaptation that emphasizes the critical roles of both remodeling and modeling.
Bone remodeling and modeling can both occur in states of heightened loading and in periods of disuse — providing four bone mechanoadaptive pathways.
These four pathways are initiated by osteocyte activity and conclude with adaptations in whole-bone stiffness — providing a straightforward model from which the complex responses of bone to exercise and disuse can be framed.
Applying the proposed physiologic model can provide new insights into investigating long-standing problems of skeletal fragility during periods of heighted loading and disuse.
INTRODUCTION
Over 50 years ago, the orthopedic surgeon Harold Frost introduced a ground-breaking theory of bone functional adaptation, coined the “mechanostat.” The mechanostat theory outlined how postnatal load-bearing bones alter their mass and conformation in relation to changing mechanical demands. He further refined this concept and published his thesis in the two-volume anthology, The Utah Paradigm of Skeletal Physiology (1). Within his theoretical framework, Frost proposed the existence of two primary mechanoadaptive processes in bone — remodeling and modeling. He posited that bone remodeling, which involves the coupled actions of osteoclasts and osteoblasts resorbing and forming bone within a multicellular unit, occurs primarily when strains from mechanical loading fall below customary levels, as is observed in situations of disuse. When in a state of disuse, less bone is formed than was originally resorbed within each remodeling unit, resulting in decreased bone mass. On the other hand, when strains are greater than are customary due to heighted mechanical loading, a second adaptive process known as formation modeling occurs. In this process, Frost suggested that osteoblastic bone formation, independent of osteoclastic resorption, results in increased bone mass. Frost proposed that, together, these processes of bone remodeling and modeling regulate bone mass in response to changing mechanical demands.
In the decade-and-a-half since Frost's final publications, scientists in the field of bone mechanobiology have made major strides, particularly regarding how osteocytes, the primary mechanosensory and resident cells in bone, orchestrate the response of bone to changes in their mechanical environment. In this article, we review how these advances provide evidence for an updated mechanostat model — revealing osteocyte regulation of four distinct mechanoadaptive pathways, consisting of bone remodeling and modeling in both conditions of disuse and heightened mechanical loading. During heightened mechanical loading, both 1) formation modeling and 2) targeted remodeling can occur, whereas during disuse, both 3) resorption modeling and 4) disuse-mediated remodeling can occur. As we will review, each of these four pathways begins with stimulation of osteocytes and concludes with alterations in bone mechanical properties via osteoclastic and osteoblastic activity in the processes of bone modeling and remodeling. Collectively, these four mechanoadaptive pathways regulate whole-bone stiffness in response to novel mechanical stimuli and converge to provide a straightforward physiologic feedback model from which the complex responses of bones to mechanical loading and disuse can be framed. Recognition of the independent contributions of all four pathways challenges long-held concepts, such as the notion that bone remodeling, sometimes referred to as “bone turnover,” is the dominant mechanoadaptive response in bone and that the relative amount of bone formation to bone resorption within a remodeling unit is the primary determinant of bone health. Instead, we provide a model of bone functional adaptation that lends to a more comprehensive approach to investigating skeletal mechanobiology.
In this article, for simplicity, we focus primarily on the mechanical adaptations of cortical bone at the midshaft of long bones. Throughout the article, we aim to integrate long-standing concepts of bone functional adaptation with contemporary findings to provide evidence for the four mechanoadaptive pathways that comprise this theoretical model. We then discuss how application of this model provides new insights into investigating long-standing problems of skeletal fragility.
FOUNDATIONS OF THE MODEL
Bones continually adapt to changing mechanical demands by altering their mass and structure to maintain appropriate stiffness, while minimizing mass for efficiency in locomotion (2). The notion that bone is a living tissue able to sense and adapt to these competing mechanical requirements is not new, and increases in bone mass and strength in response to increased or novel mechanical loading are well documented (3,4). Conversely, conditions of disuse such as limb casting (5), hindlimb unloading (6), and exposure to microgravity (7) are accompanied by increased bone resorption and decreased bone mass and strength. The mechanisms that bones use to accomplish these adaptations to changing mechanical demands continue to be revealed, and although the roles of osteoblasts and osteoclasts in bone formation and resorption, respectively, have been appreciated for over a century, the role of osteocytes in coordinating the responses of these cells to mechanical stimuli has received increasing attention in the last two decades. Recent scientific contributions to the understanding of osteocyte mechanosensation and effector cell coordination support the updated theoretical framework of bone functional adaptation we present here.
Like all structures subjected to mechanical loading, bones experience strains that can occur in shear, compression, or tension. The characteristics of the mechanical loads placed on the bone (Fig. 1.1) and the mechanical properties of the bone itself, such as whole-bone stiffness (Fig. 1.2), dictate the characteristics of the strains experienced within the tissue (Fig. 1.3) (8,9). Observations that several strain characteristics influence the osteogenic potential of mechanical loading led to the adoption of the phrase, “strain stimulus” (10) (Fig. 1.3). The potency of the strain stimulus is determined by the magnitude, duration, rate, and distribution of the strain throughout the mineralized matrix, as well as by the amount of rest inserted between loading cycles (10).
Figure 1.
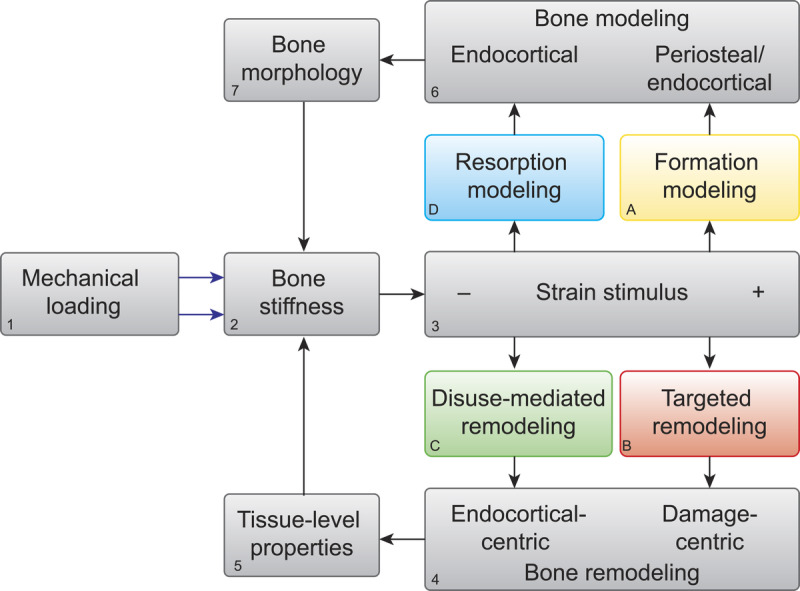
Theoretical framework of bone functional adaptation at the diaphysis. Mechanical loading [1] on a bone of a given stiffness [2] will produce a strain stimulus [3], which, if greater than customary, may elicit bone formation modeling [A] and targeted remodeling [B]. Formation modeling deposits new bone, primarily on the endocortical and periosteal surfaces within the diaphysis, whereas targeted remodeling aims to remove bone damaged by fatigue loading. In the case of disuse and a lower than customary strain stimulus, disuse-mediated remodeling [C] and resorption modeling [D] may occur. Both processes begin with bone resorption centered on or near the endocortical surface of the diaphysis. In the case of disuse-mediated remodeling, resorption is coupled with formation. In the case of modeling, resorption is independent of formation. Thus, bone modeling and remodeling occur on either end of the strain stimulus spectrum. Remodeling [4] primarily alters tissue-level mechanical properties [5], whereas modeling [6] alters the bone morphology [7]. Bone morphology and tissue-level properties collectively determine whole-bone stiffness [2], which then influences the strain response to subsequent bouts of mechanical loading.
When strain stimuli are appreciably greater or lower than that to which bone is accustomed, two physiologic processes can be initiated — bone modeling and bone remodeling. Distinguishing between these processes is a key tenet of the proposed framework. Bone remodeling, as reviewed above, refers to the sequential and coupled action of osteoclasts and osteoblasts working in teams known as bone multicellular units, wherein osteoclasts first resorb a defined region of bone, followed by osteoblast recruitment to the same site and deposition of new bone tissue — effectively removing and refilling small portions of bone tissue (11). Remodeling replaces mature bone with new, less mineralized bone matrix (12) and can therefore result in increased porosity and decreased matrix mineral density until osteoblastic infilling and secondary mineralization are complete (13). Remodeling (Fig. 1.4), therefore, can be thought of as largely altering porosity and the tissue-level properties of bone (e.g., the degree of matrix mineralization and collagen cross-linking) (Fig. 1.5). Not all bone remodeling ultimately results in the same amount of bone tissue formed as was originally removed, and an overall net bone loss can ensue, particularly in cases of disuse, as we will review below.
Bone modeling (Fig. 1.6) is an altogether different process and refers to the independent actions of either osteoblasts forming bone on a surface or osteoclasts resorbing bone on a surface (14,15), thereby altering the size and structure, or morphology, of the bone (Fig. 1.7). Prolonged bone formation on a surface such as the periosteum can lead to periosteal expansion and widening of the bone. This de novo bone formation by osteoblasts is referred to as formation modeling. On the other hand, when bone is resorbed extensively, such as on the endocortical surface of the diaphysis of a long bone, this can result in excavation of the marrow cavity and thinning of the cortices. This type of modeling is referred to as resorption modeling (14). Together, bone modeling and remodeling, through altering bone morphology and tissue-level properties, respectively, determine whole-bone mechanical properties such as bone stiffness (Fig. 1.2) (16).
There is strong evidence that the processes of modeling and remodeling that occur on either end of the strain stimulus spectrum are initiated by osteocytes (17,18). When the osteocyte-specific responses to mechanical stimuli are connected with the surface-specific effector responses, four primary mechanoadaptive pathways emerge — two that occur with greater than customary strain stimuli (formation modeling and targeted remodeling) (Figs. 1–5A, B) and two with lower than customary strain stimuli (resorption modeling and disuse-mediated remodeling) (Figs. 1–5C, D).
Figure 5.
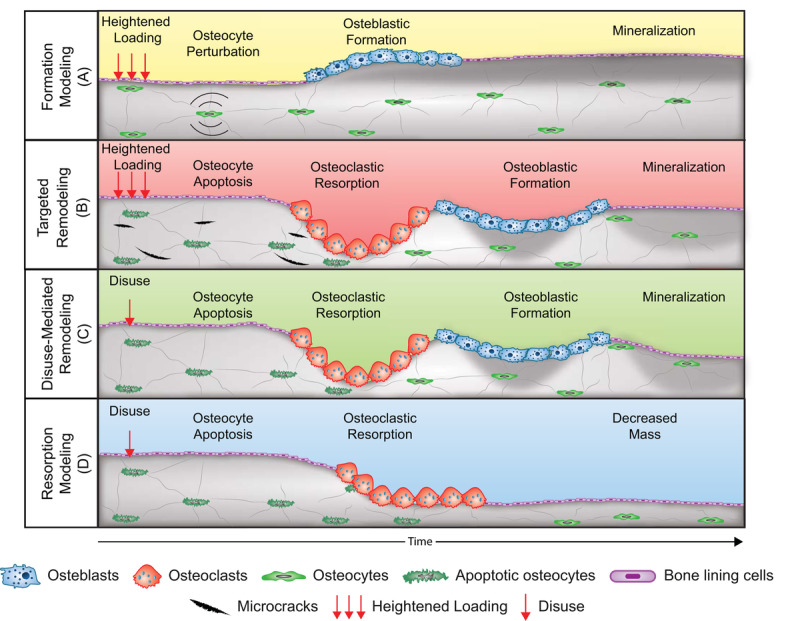
Schematic of the four mechanoadaptive responses at the cellular level. Formation modeling [A]: osteocyte perturbation by mechanical loading induces osteoblastic bone formation on a surface. Targeted remodeling [B]: microcracks generated during loading stimulate osteocyte apoptosis and targeted removal of bone by osteoclasts and subsequent formation of bone by osteoblasts. Disuse-mediated remodeling [C]: osteocyte apoptosis with disuse stimulates bone resorption and coupled formation. Also depicted is the negative bone balance within each remodeling unit that can accompany disuse-mediated remodeling. Resorption modeling [D]: disuse-mediated osteocyte apoptosis stimulates osteoclastic bone resorption on a surface. Note: Trabecular, trench-like bone remodeling is depicted for simplicity rather than centripetal bone formation around a vascular canal as occurs in remodeling of cortical bone.
Formation Modeling and Targeted Remodeling
Formation modeling (Fig. 2A) is initiated when mechanical loading of bone above customary levels results in deformation of the mineralized matrix, which is hypothesized to produce interstitial fluid pressure gradients within the network of caves and tunnels that are home to osteocytes and their cytoplasmic processes — the lacunar-canalicular system (19). Theoretical and experimental models suggest that rapid movement of interstitial fluid throughout the lacunar-canalicular system stimulates osteocytes (Fig. 2A.1) via shear stress generated along their cell membranes (19) and hoop strains where integrins tether osteocyte cytoplasmic processes to the surrounding bone matrix (20). Osteocyte surface molecules and structures such as integrins, primary cilia, G protein-coupled receptors, and ion channels are proposed to act in concert to sense these mechanical cues and convert the mechanical stimuli into cellular signals that alter gene expression (18). In response to mechanical stimuli, osteocyte intracellular calcium signaling is initiated, along with secretion of pro-osteoblast paracrine factors (Fig. 2A.2a), including, but not limited to, prostaglandins, nitric oxide (NO), insulin-like growth factor 1 (IGF-1), and adenosine triphosphate (21–23). These factors promote the osteoblast recruitment, proliferation, and differentiation necessary to mount a bone formation response (Fig. 2A.3) (24).
Figure 2.
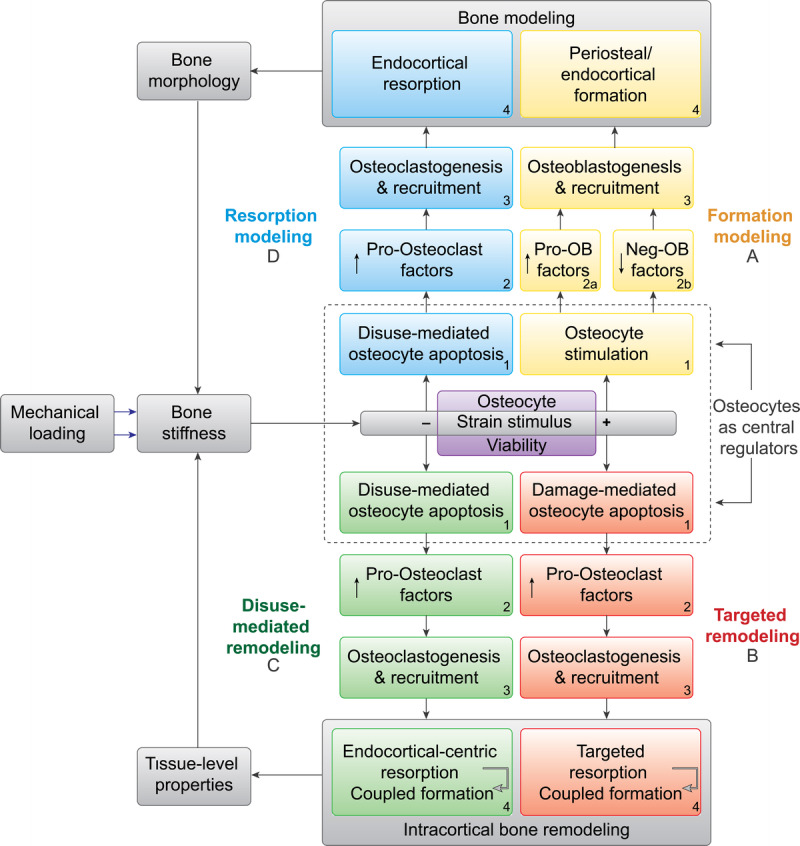
Expanded theoretical framework of bone functional adaptation at the diaphysis. This expanded framework highlights the central role of osteocytes in regulating bone functional adaptation. Neg-OB factors, negative osteoblast factors; Pro-OB factors, pro-osteoblast factors.
Mechanical loading of osteocytes not only stimulates pro-osteoblastic paracrine signaling but also suppresses osteocyte secretion of sclerostin, a Wnt antagonist and negative regulator of osteoblastic bone formation (25). Accordingly, in response to mechanical stimulation, osteocytes promote new bone formation through both secretion of pro-osteoblast factors (e.g., NO and IGF-1) and suppression of negative regulators of bone formation (e.g., sclerostin) (Fig. 2A.2b). This new bone formation, as a result of heightened mechanical loading, occurs independently of prior bone resorption (26,27) and is therefore a bone modeling response (26). This formation modeling can increase the thickness of existing trabecular elements (27,28) and increase cortical thicknesses at the diaphysis of long bones through bone deposition on the endocortical and periosteal surfaces (Fig. 2A.4, 3A) (4).
Figure 3.
Schematic of the four mechanoadaptive responses at the long-bone diaphysis. A and B: Novel mechanical loading can stimulate bone formation on the endocortical surface or periosteal surface (depicted here) and targeted removal of microdamage through remodeling. Together, these responses can result in a wider bone that is temporarily more porous. C and D: Disuse can lead to increased intracortical remodeling and resorption of bone around the endocortical surface. These different responses to disuse result in increased intracortical porosity and thinner cortices, respectively. Note: Cross sections of the long-bone diaphysis depicted as tubular for simplicity (diaphyseal morphology is normally heterogeneous around the marrow cavity, with varying cortical thicknesses).
Formation modeling is not the only response to greater than customary strain stimuli. Bone, like other materials that experience repetitive loading, can accrue fatigue damage. In bone, this damage can appear as linear microcracks or diffuse, sublamellar tissue damage (29). However, unlike inert materials, bone is able to detect tissue damage and replace it through targeted remodeling (Fig. 2B). Targeted remodeling, like formation modeling, appears to be orchestrated by osteocytes, with the initiating stimulus attributed to osteocyte apoptosis (Fig. 2B.1) (30). Osteocyte apoptosis can be triggered as early as 24 h after fatigue loading and microdamage induction (31), followed by secretion of pro-osteoclastic factors (Fig. 2B.2), osteoclast recruitment (Fig. 2B.3), and intracortical resorption approximately 10 to 14 d later, focused primarily at regions containing linear microcracks (Fig. 2B.4) (32). The critical regulatory role that osteocytes play in the targeted remodeling process was demonstrated when the intracortical remodeling response after microdamage induction was abolished by inhibition of osteocyte apoptosis via administration of a pan-caspase inhibitor (33).
In targeted remodeling, the apoptotic osteocytes are not themselves the main source of pro-osteoclastic factors necessary for osteoclastogenesis. Rather, the neighboring viable osteocytes, within 100 and 300 μm from damage foci, secrete the pro-osteoclastic cytokines, vascular endothelial growth factor, and receptor activator of NF-κB ligand (RANKL), and downregulate the latter's decoy receptor, osteoprotegerin (OPG). These factors promote the osteoclastogenesis necessary for initiating intracortical remodeling. In turn, the osteoclastic resorption and subsequent formation by osteoblasts (Fig. 2B.4) replace fatigue-damaged tissue (32) and promote extension of the fatigue life of the skeletal structure (34).
Collectively, the targeted remodeling and formation modeling that accompany greater than customary strain stimuli can result in a wider bone due to periosteal expansion, thicker cortices due to endocortical deposition, and increased intracortical porosity due to removal of fatigue damage (Figs. 3A, B, and 4A, B).
Disuse-Mediated Remodeling and Resorption Modeling
Profound bone resorption can accompany inadequate mechanical stimulation, with reports from animal studies of 12% cortical bone loss after 8 wk of limb isolation in turkeys (35), and in a classic experiment, decrements of almost half of baseline metaphyseal bone mass in dogs after 60 wk of limb casting (36). Furthermore, introduction of the partial weight bearing model in rodents has revealed bone loss proportional to the degree of unloading (6).
Similar to the bone loss that precedes bone formation in the process of targeted remodeling, bone loss with disuse appears to be initiated by osteocyte apoptosis (Figs. 2C.1, D.1) (37). Studies of hindlimb unloading in murine models indicate that resorption is spatially and temporally associated with osteocyte apoptosis (37–39), and again in parallel with targeted remodel-ing, osteocyte apoptosis with disuse triggers both pro-osteoclastic RANKL expression (37,38) and downregulation of OPG (38) (Figs. 2C.2, D.2). The importance of osteocyte apoptosis in regulating osteoclastic resorption (Fig. 2C.3, D3) with disuse is made apparent by studies reporting resistance to unloading-induced bone resorp-tion via targeted ablation of osteocytes (40) and with maintenance of osteocyte viability through administration of a pan-caspase inhibitor (37).
In conditions of disuse, patterns of bone loss include thinning or complete removal of trabecular elements, increased intracortical porosity, expansion of the marrow space, and thinning of the cortices (5,6,41). As with heightened loading, periods of unloading of the skeleton can result in both a remodeling (Fig. 2C.4) and a modeling response (Fig. 2D.4). The first response, disuse-mediated remodeling (Fig. 2C), results in increased intracortical porosity due to an imbalance between osteoclast and osteoblast activity within each remodeling unit. The results of this imbalance are larger osteons with less infill and more residual porous space (42) and overall net bone loss within the skeleton. This disuse-mediated remodeling and resulting porosity preferentially occur at the endocortical region in diaphyseal bone (Figs. 3C, 4C) (35). The other resorption pattern, resorption modeling (Fig. 2D), involves independent osteoclast activity around the marrow cavity that ultimately expands the marrow cavity and thins the cortices (Figs. 3D, 4D) (35,43). Novel image registration techniques were used to demonstrate that the endocortical expansion by osteoclasts with unloading occurs where osteoblasts are least active on existing endocortical surfaces (43). The potential extent of endocortical-specific resorption modeling has been demonstrated in animal studies, with reports of over 9% endocortical volume expansion after 3 wk of muscle paralysis (43) and 12% reduction in cortical cross-sectional area, due primarily to endocortical expansion, after 12 wk of limb isolation (35).
Figure 4.
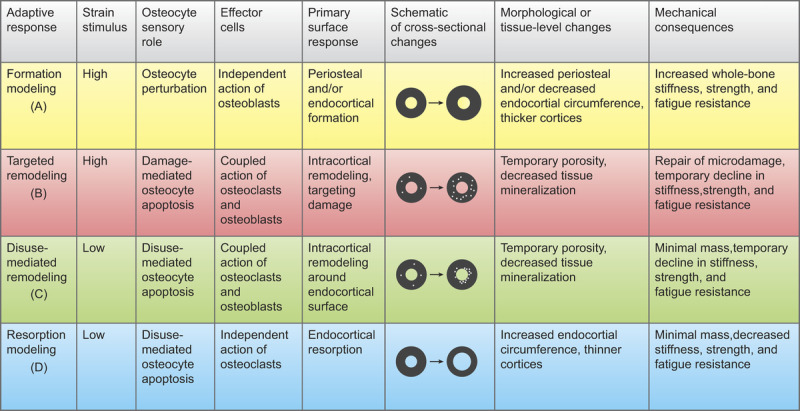
Summary of the strain stimulus, osteocyte stimulus, effector cell response, primary surface response, morphological/tissue-level adaptations, and mechanical consequences of the four mechanoadaptive pathways: formation modeling [A], targeted remodeling [B], disuse-mediated remodeling [C], and resorption modeling [D]. Bone cross-sectional schematics are presented as circular for simplicity; actual cross-sections are heterogeneous in shape and have varying cortical thicknesses around the marrow cavity.
Together, the disuse-mediated remodeling that results in in-tracortical porosity, and the resorption modeling responsible for marrow expansion and thinning of the cortices (Figs. 3C, D) rid the bone of excess mass and decrease the stiffness of the structure in response to the new, lower than customary strain stimuli (Fig. 4C, D).
Regulation of Bone Stiffness by the Mechanoadaptive Pathways
As we reviewed, when strain stimuli fall below or rise above customary levels, both bone modeling and remodeling can be initiated, with osteocytes playing important roles in coordinating the responses. When remodeling is initiated, whether targeted at microdamage or in response to disuse, we propose that the predominant effect is altered tissue-level mechanical properties (Fig. 1.5). When modeling is initiated, whether in the formation or resorption mode, we propose that the predominant effect is to alter bone morphology (Fig. 1.7). Collectively, these processes, through regulating bone morphology and tissue-level properties, regulate whole-bone stiffness in response to changing mechanical demands (Fig. 1.2).
Within this physiologic framework, we focus on regulation of whole-bone stiffness as the target for bone mechanoadaptation rather than the more traditionally referenced “bone strength.” The argument can be made that the primary mechanical function of bone is not to be as strong as possible for all potential loading conditions, but rather to be as stiff as necessary to resist excessive deformation under routine loading while also maintaining minimal mass (2,9,16). Naturally, it follows that the goal of bone functional adaptation throughout an organism’s lifespan is to uphold these principles.
By applying this model of regulation of bone stiffness in response to changing mechanical demands to problems of skeletal fragility and by recognizing the distinct mechanoadaptive pathways and central regulatory role of osteocytes, new approaches to investigating the influences of mechanical loading and disuse on skeletal properties can be revealed. Below, we discuss how applying the proposed model can reframe research efforts aimed at preventing bone fragility in a condition of heightened loading and in a condition of disuse.
APPLICATION OF THE MODEL
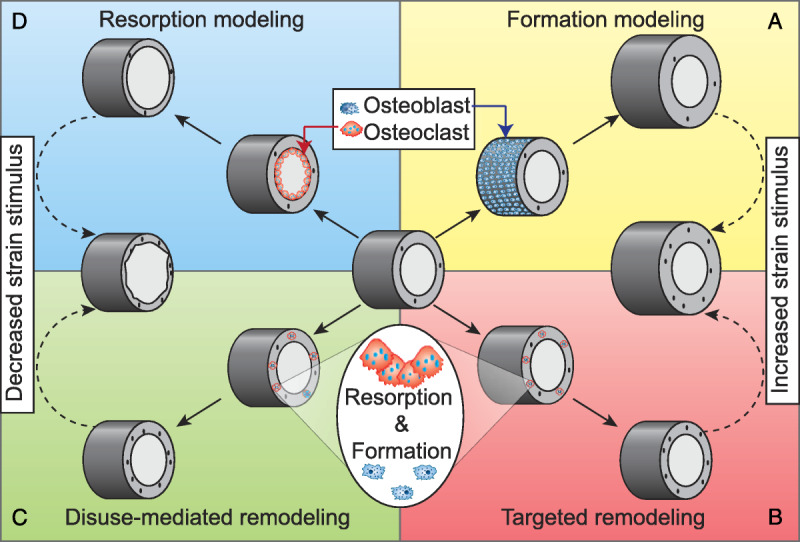
A primary tenet of the proposed model is that precise interpretations of the responses of bone to increased mechanical loading and disuse require relinquishing long-held axioms such as the notion that bone remodeling is the only process that occurs in the adult skeleton and the assertion that bone health depends on the balance between bone formation and resorption — with resorption resulting in negative consequences and formation resulting in favorable ones. On the contrary, as depicted in Figure 5, both osteoblastic bone formation and osteoclastic bone resorption are required to alter bone properties to meet novel mechanical demands on either end of the strain stimulus spectrum. Observations such as increased bone formation after disuse (44) (Fig. 5C) or increases in serum markers of bone resorption after initiation of novel mechanical loading (28) (Fig. 5B) are understandable when the cellular-level responses to these novel stimuli are considered.
Considering both effector responses (modeling and remodeling) during increased loading may reveal new insights into the pathophysiology and prevention of bone stress injuries such as stress fractures. These repetitive loading injuries are common in endurance athletes and military trainees and are prime examples of when both the remodeling and modeling responses play a role in the pathophysiology of injury (45). In the case of stress fractures, the temporary porosity and decreased matrix mineralization that accompanies targeted removal of fatigue damage (Fig. 3B) may be offset by formation modeling on the periosteum (Fig. 3A) in a manner that conserves bone stiffness and the fatigue life of the structure (3). It is possible, therefore, that in situations with heightened repetitive loading, interventions that focus on promoting formation modeling may be more effective than those that aim to inhibit the targeted bone remodeling that is needed to replace fatigue-damaged bone. During times of intensive physical training, low concentrations of circulating biochemical markers of bone formation likely signal that conditions are not favorable for an optimal bone formation response that may help prevent stress fractures (46). Accordingly, interventions to reduce bone stress injuries during heightened physical activity may include appropriate nutrition and sleep needed to promote a bone formation modeling response, as well as avoiding therapies, such as nonsteroidal anti-inflammatory drugs, that may inhibit bone formation modeling (45,47). Studies that test these potential interventions need to be conducted to provide evidence-based recommendations for injury prevention.
Although stress fractures are common in people exposed to periods of heightened mechanical loading, astronauts are susceptible to the skeletal fragility that can accompany diminished mechanical loading. Bone loss in astronauts exposed to microgravity is a major concern and a barrier to long-duration manned spaceflight such as a mission to Mars, which could require over 2 yr in space. The concern that microgravity could result in progressive bone loss and microstructural deterioration, as demonstrated by casting studies in animals (5), affirms the importance of countermeasures to make long-duration manned space exploration physiologically tenable. Unraveling the mechanisms of bone loss in conditions of disuse could reveal new approaches to preventing bone loss in microgravity. As denoted by the dotted line in Figure 2, osteocyte activity is foundational in initiating all four mechanoadaptive pathways. With disuse, osteocyte apoptosis initiates both disuse-mediated intracortical remodeling and bone resorption modeling. Thus, it follows that promoting osteocyte viability (Fig. 2, purple box) should be a primary focus of research aimed at preventing bone loss in microgravity.
Irisin is a protein secreted by muscle during exercise and has been shown to promote osteocyte survival and inhibit osteoclast activity (48). Interestingly, treatment with recombinant irisin prevents bone loss that accompanies hindlimb unloading in mice (49). This and other osteocyte-preserving interventions, although requiring further investigation, could hold the key to preventing disuse-mediated bone loss in astronauts and in other populations at risk for bone fragility.
KNOWLEDGE GAPS AND SUMMARY
Although this physiologic framework provides an updated model of bone functional adaptation and a foundation for interpretation of skeletal responses to exercise and disuse, the simplicity of the model requires some generalization at the cost of addressing finer details and accentuating the true exquisiteness of bone functional adaptation. For example, this model does not address the localized remodeling of the osteocyte's lacunar environment, known as perilacunar remodeling (50), which may occur with altered mechanical loading, nor does it account for the nontargeted, hormonally mediated remodeling that can be stimulated by declining serum calcium concentrations during exercise (51). Finally, expansion of the framework to focus more closely on trabecular bone and account for potential influences of pharmacological and nutritional interactions, as well as changes in reproductive hormone status, will also improve the model's application to diverse situations.
In summary, advances in our understanding of the fundamental mechanisms of bone functional adaptation allow for recognition of four primary mechanoadaptive physiologic pathways. These pathways are initiated by osteocyte activity and are carried out by bone modeling and remodeling in situations of disuse and loading. The four adaptive pathways collectively regulate bone stiffness in a manner that prepares bone to meet new mechanical challenges. Consideration of this physiologic framework and further investigations into underlying mechanisms may reveal novel approaches to solving long-standing problems of skeletal fragility and provide insight into how to build a resilient skeleton that can endure varying mechanical demands throughout life.
Acknowledgments
We would like to acknowledge Dr Ego Seeman, Dr Susan Proctor, and Dr Patrick Hughes for their thoughtful feedback and critical review of the manuscript.
This research was supported by funding from the U.S. Army Medical Research and Development Command, Defense Health Program, Joint Program Committee 5 (W811XWH-15-C-0024), U.S. Department of Defense (DOD). The views, opinions, and/or findings in this report are those of the authors and should not be construed as an official Department of the Army position, policy, or decision, unless so designated by other official documentation.
This research was also supported in part by an appointment to the DOD Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the DOD (CC and KP). ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this article are the author's and do not necessarily reflect the policies and views of the DOD, DOE, or ORAU/ORISE.
Footnotes
Associate Editor: Nancy I. Williams, Sc.D., FACSM.
References
- 1.Frost HM. The Utah Paradigm of Skeletal Physiology. Greece: International Society of Musculoskeletal and Nueronal Interactions; 2004. 444 p. [Google Scholar]
- 2.Martin RB. The importance of mechanical loading in bone biology and medicine. J. Musculoskelet Neuronal Interact. 2007; 7(1):48–53. [PubMed] [Google Scholar]
- 3.Warden SJ, Hurst JA, Sanders MS, Turner CH, Burr DB, Li J. Bone adaptation to a mechanical loading program significantly increases skeletal fatigue resistance. J. Bone Miner Res. 2005; 20(5):809–16. [DOI] [PubMed] [Google Scholar]
- 4.Robling AG, Hinant FM, Burr DB, Turner CH. Improved bone structure and strength after long-term mechanical loading is greatest if loading is separated into short bouts. J. Bone Miner Res. 2002; 17(8):1545–54. [DOI] [PubMed] [Google Scholar]
- 5.Uthoff HK, Jaworski ZF. Bone loss in response to long-term immobilisation. J. Bone Joint Surg Br. 1978; 60B:420–9. [DOI] [PubMed] [Google Scholar]
- 6.Ellman R, Spatz J, Cloutier A, Palme R, Christiansen BA, Bouxsein ML. Partial reductions in mechanical loading yield proportional changes in bone density, bone architecture, and muscle mass. J. Bone Miner Res. 2013; 28(4):875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkhart K, Allaire B, Anderson DE, Lee D, Keaveny TM, Bouxsein ML. Effects of long-duration spaceflight on vertebral strength and risk of spine fracture. J. Bone Miner Res. 2019; 35:269–76. [DOI] [PubMed] [Google Scholar]
- 8.Bouxsein ML, Seeman E. Quantifying the material and structural determinants of bone strength. Best Pract. Res. Clin. Rheumatol. 2009; 23(6):741–53. [DOI] [PubMed] [Google Scholar]
- 9.Currey JD. Bones: Structure and Mechanics. Princeton: Princeton University Press; 2002. [Google Scholar]
- 10.Skerry TM. One mechanostat or many? Modifications of the site-specific response of bone to mechanical loading by nature and nurture. J. Musculoskelet. Neuronal Interact. 2006; 6(2):122–7. [PubMed] [Google Scholar]
- 11.Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone. 2002; 30(1):5–7. [DOI] [PubMed] [Google Scholar]
- 12.Fuchs RK, Allen MR, Ruppel ME, et al. In situ examination of the time-course for secondary mineralization of Haversian bone using synchrotron Fourier transform infrared microspectroscopy. Matrix Biol. 2008; 27(1):34–41. [DOI] [PubMed] [Google Scholar]
- 13.Heaney RP. The bone-remodeling transient: implications for the interpretation of clinical studies of bone mass change. J. Bone Miner Res. 1994; 9(10):1515–23. [DOI] [PubMed] [Google Scholar]
- 14.Seeman E. Bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Expr. 2009; 19(3):219–33. [DOI] [PubMed] [Google Scholar]
- 15.Szulc P, Seeman E. Thinking inside and outside the envelopes of bone: dedicated to PDD. Osteoporos Int. 2009; 20(8):1281–8. [DOI] [PubMed] [Google Scholar]
- 16.Jepsen KJ. Functional interactions among morphologic and tissue quality traits define bone quality. Clin. Orthop. Relat. Res. 2011; 469(8):2150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaffler MB, Cheung WY, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif. Tissue Int. 2014; 94(1):5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uda Y, Azab E, Sun N, Shi C, Pajevic PD. Osteocyte mechanobiology. Curr. Osteoporos. Rep. 2017; 15(4):318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinbaum S, Cowin SC, Zeng Y. A model for the excitation of osteocytes by mechanical loading–induced bone fluid shear stresses. J. Biomech. 1994; 27(3):339–60. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, McNamara LM, Schaffler MB, Weinbaum S. Strain amplification and integrin based signaling in osteocytes. J. Musculoskelet Neuronal Interact. 2008; 8(4):332–4. [PMC free article] [PubMed] [Google Scholar]
- 21.Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J. Cell. Physiol. 2007; 212(1):207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pathak JL, Bravenboer N, Luyten FP, et al. Mechanical loading reduces inflammation-induced human osteocyte-to-osteoclast communication. Calcif. Tissue Int. 2015; 97(2):169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzger CE, Brezicha JE, Elizondo JP, Narayanan SA, Hogan HA, Bloomfield SA. Differential responses of mechanosensitive osteocyte proteins in fore- and hindlimbs of hindlimb-unloaded rats. Bone. 2017; 105:26–34. [DOI] [PubMed] [Google Scholar]
- 24.Brady RT, O'Brien FJ, Hoey DA. Mechanically stimulated bone cells secrete paracrine factors that regulate osteoprogenitor recruitment, proliferation, and differentiation. Biochem Biophys Res Commun. 2015; 459(1):118–23. [DOI] [PubMed] [Google Scholar]
- 25.Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 2008; 283(9):5866–75. [DOI] [PubMed] [Google Scholar]
- 26.Barak MM. Bone modeling or bone remodeling: that is the question. Am. J. Phys. Anthropol. 2019. Epub 2019/11/12. doi: 10.1002/ajpa.23966. PubMed PMID: 31710704. [DOI] [PubMed] [Google Scholar]
- 27.Birkhold AI, Razi H, Duda GN, Weinkamer R, Checa S, Willie BM. The influence of age on adaptive bone formation and bone resorption. Biomaterials. 2014; 35(34):9290–301. [DOI] [PubMed] [Google Scholar]
- 28.Hughes JM, Gaffney-Stomberg E, Guerriere KI, et al. Changes in tibial bone microarchitecture in female recruits in response to 8 weeks of U.S. Army Basic Combat Training. Bone. 2018; 113:9–16. [DOI] [PubMed] [Google Scholar]
- 29.Seref-Ferlengez Z, Kennedy OD, Schaffler MB. Bone microdamage, remodeling and bone fragility: how much damage is too much damage? Bonekey Rep. 2015; 4:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy OD, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone. 2014; 64:132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J. Bone Miner Res. 2000; 15(1):60–7. [DOI] [PubMed] [Google Scholar]
- 32.Bentolila V, Boyce TM, Fyhrie DP, Drumb R, Skerry TM, Schaffler MB. Intracortical remodeling in adult rat long bones after fatigue loading. Bone. 1998; 23(3):275–81. [DOI] [PubMed] [Google Scholar]
- 33.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner Res. 2009; 24(4):597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor D, Kuiper JH. The prediction of stress fractures using a ‘stressed volume’ concept. J. Orthop. Res. 2001; 19(5):919–26. [DOI] [PubMed] [Google Scholar]
- 35.Gross TS, Rubin CT. Uniformity of resorptive bone loss induced by disuse. J. Orthop. Res. 1995; 13(5):708–14. [DOI] [PubMed] [Google Scholar]
- 36.Jaworski ZF, Uhthoff HK. Reversibility of nontraumatic disuse osteoporosis during its active phase. Bone. 1986; 7(6):431–9. [DOI] [PubMed] [Google Scholar]
- 37.Cabahug-Zuckerman P, Frikha-Benayed D, Majeska RJ, et al. Osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J. Bone Miner Res. 2016; 31:1356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plotkin LI, Gortazar AR, Davis HM, et al. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor kappaB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J. Biol. Chem. 2015; 290(31):18934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguirre JI, Plotkin LI, Stewart SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner Res. 2006; 21(4):605–15. [DOI] [PubMed] [Google Scholar]
- 40.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007; 5(6):464–75. [DOI] [PubMed] [Google Scholar]
- 41.Sugiyama T, Meakin LB, Browne WJ, Galea GL, Price JS, Lanyon LE. Bones' adaptive response to mechanical loading is essentially linear between the low strains associated with disuse and the high strains associated with the lamellar/woven bone transition. J. Bone Miner Res. 2012; 27(8):1784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexandre C, Vico L. Pathophysiology of bone loss in disuse osteoporosis. Joint Bone Spine. 2011; 78(6):572–6. [DOI] [PubMed] [Google Scholar]
- 43.Ausk BJ, Huber P, Poliachik SL, Bain SD, Srinivasan S, Gross TS. Cortical bone resorption following muscle paralysis is spatially heterogeneous. Bone. 2012; 50(1):14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas T, Vico L, Skerry TM, et al. Architectural modifications and cellular response during disuse-related bone loss in calcaneus of the sheep. J. Appl. Physiol. 1996; 80(1):198–202. [DOI] [PubMed] [Google Scholar]
- 45.Hughes JM, Popp KL, Yanovich R, Bouxsein ML, Matheny RW., Jr The role of adaptive bone formation in the etiology of stress fracture. Exp. Biol. Med. (Maywood). 2017; 242(9):897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes JM, Smith MA, Henning PC, et al. Bone formation is suppressed with multi-stressor military training. Eur. J. Appl. Physiol. 2014; 114(11):2251–9. [DOI] [PubMed] [Google Scholar]
- 47.Hughes JM, McKinnon CJ, Taylor KM, et al. Nonsteroidal anti-inflammatory drug prescriptions are associated with increased stress fracture diagnosis in the US Army population. J. Bone Miner Res. 2019; 34(3):429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, Wrann CD, Jedrychowski M, et al. Irisin mediates effects on bone and fat via αV integrin receptors. Cell. 2019; 178(2):507–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storlino G, Colaianni G, Sanesi L, et al. Irisin prevents disuse-induced osteocyte apoptosis. J. Bone Miner Res. 2019; 35:766–75. [DOI] [PubMed] [Google Scholar]
- 50.Bach-Gansmo FL, Wittig NK, Bruel A, Thomsen JS, Birkedal H. Immobilization and long-term recovery results in large changes in bone structure and strength but no corresponding alterations of osteocyte lacunar properties. Bone. 2016; 91:139–47. [DOI] [PubMed] [Google Scholar]
- 51.Kohrt WM, Wherry SJ, Wolfe P, et al. Maintenance of serum ionized calcium during exercise attenuates parathyroid hormone and bone resorption responses. J. Bone Miner Res. 2018; 33(7):1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]