

Supplemental Digital Content is available in the text.
Keywords: heart failure, inflammation, interferon, lymphangiogenesis, macrophages
Abstract
Objective:
Lymphatics play an essential pathophysiological role in promoting fluid and immune cell tissue clearance. Conversely, immune cells may influence lymphatic function and remodeling. Recently, cardiac lymphangiogenesis has been proposed as a therapeutic target to prevent heart failure after myocardial infarction (MI). We investigated the effects of gene therapy to modulate cardiac lymphangiogenesis post-MI in rodents. Second, we determined the impact of cardiac-infiltrating T cells on lymphatic remodeling in the heart.
Approach and Results:
Comparing adenoviral versus adeno-associated viral gene delivery in mice, we found that only sustained VEGF (vascular endothelial growth factor)-CC156S therapy, achieved by adeno-associated viral vectors, increased cardiac lymphangiogenesis, and led to reduced cardiac inflammation and dysfunction by 3 weeks post-MI. Conversely, inhibition of VEGF-C/-D signaling, through adeno-associated viral delivery of soluble VEGFR3 (vascular endothelial growth factor receptor 3), limited infarct lymphangiogenesis. Unexpectedly, this treatment improved cardiac function post-MI in both mice and rats, linked to reduced infarct thinning due to acute suppression of T-cell infiltration. Finally, using pharmacological, genetic, and antibody-mediated prevention of cardiac T-cell recruitment in mice, we discovered that both CD4+ and CD8+ T cells potently suppress, in part through interferon-γ, cardiac lymphangiogenesis post-MI.
Conclusions:
We show that resolution of cardiac inflammation after MI may be accelerated by therapeutic lymphangiogenesis based on adeno-associated viral gene delivery of VEGF-CC156S. Conversely, our work uncovers a major negative role of cardiac-recruited T cells on lymphatic remodeling. Our results give new insight into the interconnection between immune cells and lymphatics in orchestration of cardiac repair after injury.
Highlights.
Therapeutic lymphangiogenesis in the heart requires prolonged VEGF (vascular endothelial growth factor)-C expression as achieved by adeno-associated virus but not adenoviral gene therapy or systemic protein therapy.
Therapeutic lymphangiogenesis accelerates resolution of cardiac inflammation post-MI, with reduction of both T-cell and proinflammatory macrophage levels in the viable left ventricle, resulting in improved cardiac function.
Treatment with a VEGF-C/VEGF-D trap (soluble VEGFR3) acutely limits T-cell recruitment to the infarct, leading to delayed scar remodeling and reduced cardiac dysfunction postmyocardial infarction.
Cardiac-infiltrating T cells, both CD4+ and CD8+ subpopulations, exert deleterious effects on cardiac lymphatics postmyocardial infarction, in part through interferon-γ.
See accompanying editorial on page 1611
See cover image
The lymphatic vasculature is essential for maintenance of interstitial fluid homeostasis and for fine-tuning of immune responses by regulating immune cell, cytokine, and antigen tissue clearance.1,2 Additionally, lymphatics ensure the uptake of dietary lipids in chylomicrons as well as reverse cholesterol transport from tissues to the liver.3 Edema or increases in osmotic pressure lead to stimulation of lymphatic remodeling (lymphangiogenesis), in part via macrophage-driven VEGF (vascular endothelial growth factor)-C production,4 which acts to restore tissue fluid balance. In contrast, the interplay between lymphatic vessels and the immune system is more complex: although lymphatic function is reduced by many proinflammatory mediators,5,6 different immune cell populations impact lymphatic remodeling exerting either stimulatory or inhibitory effects on lymphatic endothelial cell growth and survival.7,8 Conversely, tissue clearance of immune cells depends on active lymphatic expression of chemokines to selectively recruit and drain different immune cell populations.9,10 Promisingly, whereas therapeutic lymphangiogenesis has been shown to resolve chronic inflammation, including innate immune responses in many organs,11,12 treatment with lymphangiogenic inhibitors limits adaptive immune responses and reduces graft rejection following organ transplantation.13,14
The cardiac immune response to ischemic injury is characterized by sequential waves of infiltrating immune cells, including not only neutrophils, monocytes/macrophages, and dendritic cells (DCs) but also B and T lymphocytes. Together these cells orchestrate the removal of dead cardiomyocytes, participate in granulation tissue formation required for stabilization and maturation of the scar, and regulate the subsequent cardiac repair and inflammation-resolution phase by secreting proangiogenic, prosurvival, and anti-inflammatory mediators.15,16 However, this immune cell infiltration represents a double-edged sword in that chronic inflammation in the heart, brought on by inefficient resolution of the inflammatory response, aggravates cardiac dysfunction by inducing coronary endothelial dysfunction and deleterious cardiac remodeling, including cardiomyocyte hypertrophy and interstitial fibrosis, leading to development of chronic heart failure.17 Improved resolution of cardiac inflammation thus represents a therapeutic goal in many cardiovascular diseases.
The adult heart is invested with a lymphatic network dense over the ventricles and sparser over the atria.18 Different from other lymphatic beds, the cardiac precollector lymphatics are essentially devoid of smooth muscle cells, as surrounding cardiomyocytes ensure lymphatic propulsion towards cardiac lymph nodes (Figure I and Movie I in the Data Supplement). Cardiac lymphatic remodeling occurs in many cardiovascular diseases characterized by edema and inflammation,19 including in patients with ischemic heart disease or terminal heart failure.20,21 Indeed, myocardial infarction (MI) leads to lymphatic remodeling in both infarct and viable infarct border zone in humans, pigs, rats, and mice.21–26 We previously showed that stimulation of cardiac lymphangiogenesis post-MI in rats, using targeted intramyocardial delivery of VEGFR3 (vascular endothelial growth factor receptor 3)-selective recombinant rat VEGF-CC152S protein, accelerated resorption of chronic myocardial edema and inflammation, reduced deleterious cardiac remodeling, and improved cardiac function.26 Here, we investigated the impact of gene therapy to modulate cardiac lymphangiogenesis post-MI in rodents. Second, we assessed the role of cardiac-infiltrating T lymphocytes in lymphatic remodeling post-MI with the aim to unravel the interplay between the immune system and the lymphatic vasculature during cardiac repair.
MaterialS and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Models and Therapeutic Agents
We investigated cardiac lymphatic remodeling in 20 to 22 g wild-type C57Bl/6J mice (Janvier Laboratories, France), in 20 to 22 g C57Bl/6 mice deficient for MHC (major histocompatibility complex) II (MHCIIΔ/Δ as a result of disruption of the IAβ gene) kindly provided by Dr J.P. van Meerwijk,27 and in 200 to 220 g Wistar rats (RccHan:WIST from Harlan/Envigo) following MI induced by permanent coronary artery ligation.28,29 Only female mice were used in our studies due to their superior survival rates post-MI, and only male rats were used as female rats display more rapid left ventricular (LV) dilation.30 Modulation of cardiac lymphangiogenesis was performed using either systemic growth factor therapy with recombinant human VEGF-CC156S protein (RnD system) as described,25 or using viral gene vectors encoding hVEGF-CC156S(human VEGF-CC156S) or sVEGFR3 (soluble VEGFR3)-IgG construct.29,31,32 Briefly, protein therapy consisted of repeated (day 0, 2, 3, 4, and 6 post-MI) intraperitoneal injections of 2 µg/mouse (0.1 μg/g) of rhVEGF-CC156S (recombinant human VEGF-C mutant selective for VEGFR3) or physiological saline in controls,25 adenoviral therapy consisted of a single intraperitoneal injection on day 0 of adenoviral-5 vector (5×108 viral particles) encoding hVEGF-CC156S or lacZ as a control, and adeno-associated viral (AAV) gene delivery consisted of a single intraperitoneal injection 7 days before MI of AAV-9 vector (1×1011 viral particles) encoding hVEGF-CC156S, sVEGFR3, or scrambled sequence as a control.33
Cardiac Functional, Histological, and Cellular Analyses
LV function was evaluated by echocardiography,28 and cardiac hypertrophy-to-LV dilatation index was calculated as the ratio of diastolic LV wall thickness to LV diastolic diameter. Cardiac sections were analyzed by histology and immunohistochemistry to determine infarct size, lymphatic and blood vessel densities and sizes, and immune cell infiltration levels (macrophages and T cells) as determined using Fiji.34 Cardiac whole mount-staining was performed26 followed by a modified iDISCO+ clearing protocol35 for imaging by lightsheet (ultramicroscope II, LaVision BioTec) and confocal laser scanning (Leica SP8, ×25) microscopy. For details see Data Supplement.
Flow-Cytometry
Cells isolated from blood and hearts of mice were analyzed by flow cytometry.36,37 For details, see Data Supplement. Results are expressed as % of parent population or as cells per mL blood or per mg cardiac tissue. Flow cytometric analyses were performed on an LSRFortessa (BD Biosciences) and analyzed with FlowJo software (TreeStar, Inc, San Carlos, CA).
Prevention of T-Cell Recruitment
Fingolimod (1 mg/kg, FTY-720, Sigma-Aldrich) intraperitoneal pharmacological treatment was initiated immediately after MI in mice with repeated injections on days 1 and 2 post-MI to prevent cardiac T-cell recruitment acutely post-MI. MI controls received physiological saline. Cardiac functional and cellular analyses were performed as described above. For depletion of specific T-cell populations or cytokines, InVivoMab antibodies were administered by repeated intraperitoneal injections on day 0 and 3 post-MI in mice according to the manufacturer’s instructions (BIOXCELL, NH). For details, see Data Supplement.
Study Approval
Animal experiments performed in this study were approved by the regional ethics review board in line with E.U, French and Finnish legislation (01181.01 / APAFIS [French Animal Experiment Ethical Commission] No. 8157-2016121311094625-v5 Normandy; B315557, Toulouse ENVT [École Nationale Vétérinaire de Toulouse]; ESAVI/6718/04.10.03/2012, Helsinki). A total of 250 C57Bl/6J female mice, 20 MHCIIΔ/Δ and matched wild-type C57Bl/6 female mice, and 33 male Wistar rats surviving coronary ligation or sham-operation were included in this study.
Statistics
Data are presented as mean±SEM. Comparisons were performed to determine (1) impact of pathology (healthy versus MI controls); (2) effect of treatment (treated versus MI controls). Statistical analyses for comparisons of 2 independent groups were performed using either Student 2-tailed t test for groups with normal distribution or alternatively by Mann Whitney U test for samples where normality could not be ascertained based on D’Agostino and Pearson omnibus normality test. For comparisons of ≥3 groups, either 1-way ANOVA followed by Bonferroni posthoc (for parameters with n>7 with normal distribution) or alternatively Kruskal-Wallis nonparametric analysis followed by Dunn posthoc multiple comparison (for parameters with n<8, or with n>7 but with nongaussian distribution) were performed. Finally, longitudinal echocardiography studies in rats were analyzed by paired 2-way ANOVA followed by Bonferroni posthoc. Outlier samples were identified as an individual value exceeding group mean±4SD in groups with n≧7. All analyses were performed using GraphPad Prism software.
Results
Sustained VEGF-C Delivery Is Required for Therapeutic Lymphangiogenesis
We previously showed that cardiac lymphatic remodeling after MI in rats included expansion of lymphatic capillaries accompanied by precollector slimming and rarefaction of open lymphatic vessels, used as a proxy of cardiac precollectors, both in the infarct and in the viable LV wall, leading to poor lymphatic transport capacity during the first months post-MI.26 To stimulate cardiac lymphangiogenesis post-MI previous studies have used protein therapy with VEGFR3-selective VEGF-C, either by intramyocardial injection, coupled to biopolymers for spatiotemporally controlled growth factor release, in rats,26 or by intraperitoneal repeated injections of VEGF-CC156S protein in mice.25,38
We set out to compare the efficacy of systemic intraperitoneal protein therapy versus gene therapy with either adenoviral or AAV vectors to deliver VEGFR3-selective human VEGF-CC156S31,32 with the aim to stimulate cardiac lymphangiogenesis following MI induced by permanent coronary artery ligation in mice. Importantly, we confirmed that VEGFR3 expression was essentially restricted to lymphatic vessels, and not blood vessels, both in healthy and post-MI mouse hearts (Figure IIA in the Data Supplement). Control MI mice were injected with saline, adenoviral-lacZ, or noncoding scrambled sequence AAV control vectors, respectively. First, we found that intraperitoneal delivery of recombinant human VEGF-CC156S protein failed to increase circulating VEGF-C levels in mice (Figure IIIA in the Data Supplement) and did not increase cardiac lymphangiogenesis (Figure IIIB through IIID in the Data Supplement). Second, we found that adenoviral gene delivery led to a potent, but transient, increase in VEGF-C plasma levels (Figure IVA in the Data Supplement), in line with the expected antiadenoviral immune response that rapidly eliminates transduced cells. Such transient elevation of circulating VEGF-C levels was not sufficient to stimulate lymphatic expansion. Indeed, cardiac lymphatic proliferation and densities were not increased in adenoviral-VEGF-CC156S–treated mice, as compared to adenoviral-lacz MI controls (Figure IVB and IVC in the Data Supplement). In contrast, AAV–VEGF-CC156S gene therapy induced a potent and sustained increase in plasma levels (Figure IVA in the Data Supplement), as well as elevated and prolonged cardiac expression of hVEGF-CC156S (Figure 1A, Figure IVD in the Data Supplement). This resulted in a 3-fold increase in lymphatic endothelial proliferation rates and lymphatic densities already by 7 days post-MI, as compared to AAV-scrambled sequence MI controls (Figure IVB, IVC, and IVE in the Data Supplement). Surprisingly, despite persistently elevated circulating and cardiac hVEGF-C levels in the AAV–VEGF-CC156S group, cardiac lymphatic proliferation (Figure 1B), but not density (Figure 1C), in the viable LV returned to baseline by 21 days post-MI, indicating that factors other than VEGF-C bioavailability contribute to cardiac lymphatic expansion. For our subsequent studies, we thus focused on the AAV gene delivery approach to determine the functional impact of cardiac lymphangiogenesis post-MI.
Figure 1.
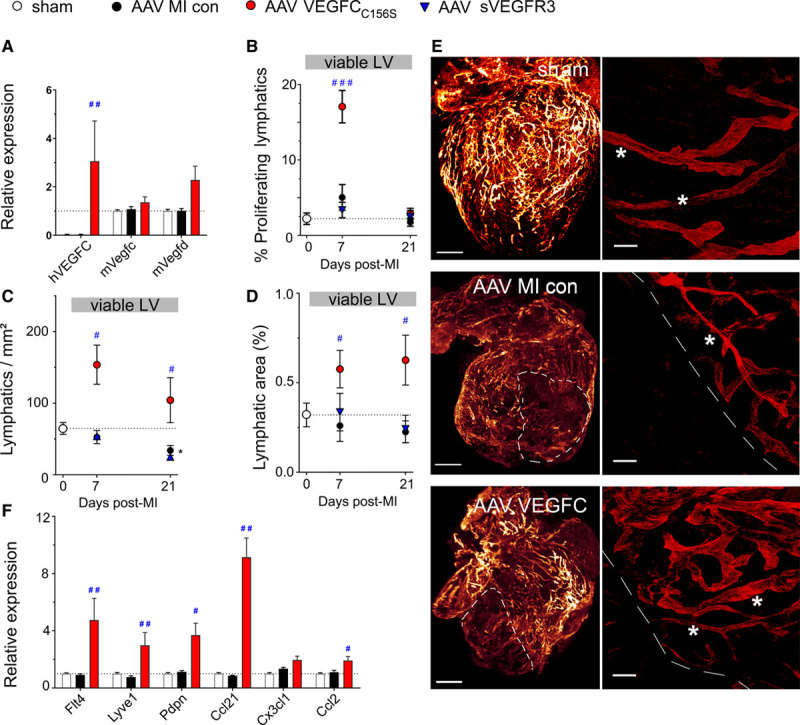
Adeno-associated viral (AAV)-VEGF (vascular endothelial growth factor)-CC156S therapy stimulates cardiac lymphangiogenesis postmyocardial infarction (MI). Mice were treated with AAV-hVEGF (human VEGF)-CC156S gene therapy (red circles, n=8), sVEGFR3 (vascular endothelial growth factor receptor 3; AAV-sVEGFR3, blue triangles, n=5–7), or AAV-scrambled virus (MI controls, black circles, n=8), and sham-operated mice (white circles, n=7–9) served as healthy controls. Cardiac gene expression of lymphangiogenic factors at 21 days post-MI (A). Lymphangiogenesis in the viable left ventricular (LV) wall bordering the infarct was evaluated as % proliferating lymphatic vessels (B), lymphatic densities (C), and lymphatic area at 7 or 21 days post-MI (D). Examples of light sheet imaging (E, left) of Lyve1+ (lymphatic vessel endothelial receptor 1) cardiac lymphatics in healthy mice, in AAV-MI controls, and VEGFC-treated (AAV–VEGF-CC156S) mice at 21 d post-MI. Infarct outlined by white dashed lines. Scale bar=1 mm; ×0.8. Confocal imaging (E, right) of viable LV areas in the same samples. Cardiac precollectors indicated by white asterisk. Scale bar=50 µm; ×25. Relative cardiac expression of lymphatic-related genes in sham (n=8), MI controls (AAV-scrambled virus, n=8), and VEGFC-treated (AAV-hVEGF-CC156S, n=5) at 21 days post-MI (F). Kruskal-Wallis followed by Dunn posthoc test. *P<0.05 vs sham; #P<0.05; ##P<0.01; ###P<0.001 vs AAV-MI controls. con indicates control.
AAV–VEGF-CC156S Gene Delivery Prevents Cardiac Lymphatic Rarefaction Post-MI in Mice
In AAV-MI control mice, following initial destruction of lymphatics in the infarct zone (Figure 1E, Figure 2B) and rarefaction of lymphatic vessels in the viable LV bordering the infarct (Figure 1C through E), the subsequent endogenous lymphangiogenic response appeared weaker than in rats. Indeed, VEGF-C expression was not increased in the viable LV at either 7 days (Figure IVD in the Data Supplement) or 21 days post-MI (Figure 1A). In contrast, in the infarct, VEGF-C–expressing macrophages were observed at 7 days (Figure IVD in the Data Supplement). Accordingly, lymphatic proliferation only tended to increase in the infarct (Figure 2A) and not in the viable LV (Figure 1B). Similar as in rats, there was severe rarefaction and slimming of open lymphatics in the viable LV (Figure 1D, Figure VA through VD in the Data Supplement) and in the infarct (Figure 2B), and loss of button-like junctions in lymphatic capillaries, together suggestive of lymphatic transport dysfunction (Figure VE in the Data Supplement).
Figure 2.
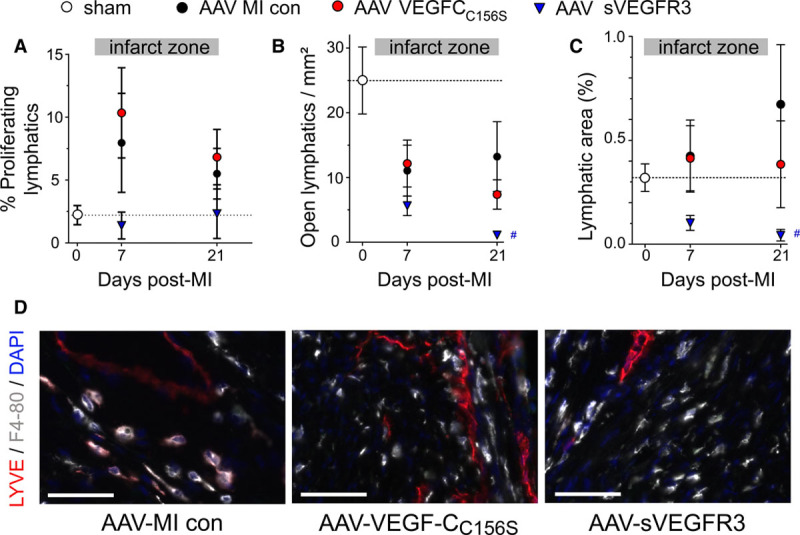
Adeno-associated viral (AAV)-sVEGFR3 (soluble vascular endothelial growth factor receptor 3) therapy inhibits infarct lymphangiogenesis postmyocardial infarction (MI). Mice were treated with AAV-hVEGF (human VEGF)-CC156S gene therapy (red circles, n=8), sVEGFR3 (AAV-sVEGFR3, blue triangles, n=5–7), or AAV-scrambled virus (MI controls, black circles, n=8), and sham-operated mice (white circle, n=7–9) served as healthy controls. Lymphangiogenesis in the infarct scar was evaluated as % proliferating lymphatic vessels (A), open lymphatic densities (B), and lymphatic area at 7 or 21 days post-MI (C). Examples (D) at 21 days post-MI of infarct zone left ventricular lymphatics (Lyve1 [lymphatic vessel endothelial receptor 1], red), macrophages (F4-80, gray), and cell nuclei (DAPI, blue); ×20 magnification, scale bar=50 µm. Note LYVE1-expressing macrophages close to lymphatic vessels. con indicates control.
As mentioned above, AAV–VEGF-CC156S gene delivery substantially increased lymphangiogenesis in viable LV (Figure 1B through 1D), but it did not increase infarct lymphangiogenesis as compared with AAV-MI controls (Figure 2A through 2D). Cardiac whole-mount staining further revealed an expanded lymphatic network in the viable LV bordering the infarct in AAV–VEGF-CC156S–treated mice at 21 days post-MI, including regeneration of straight, valved precollector segments reaching towards the base of the aorta (Figure 1E). In agreement with potent stimulation of cardiac lymphatic growth in viable LV of AAV–VEGF-CC156S–treated mice, cardiac expression analyses revealed increased levels of lymphatic-selective markers Lyve1 (lymphatic vessel endothelial receptor 1), Flt4 (Vegfr3), and podoplanin (Pdpn) at 21 days post-MI as compared to AAV-MI controls (Figure 1F). Together, these findings indicated that persistent therapeutic stimulation of VEGFR3 signaling in the heart sufficed to accelerate lymphatic capillary expansion but also to limit MI-induced loss of open lymphatic vessels in the viable LV, similar to our previous findings with cardiac-targeted lymphangiogenic protein therapy in rats.26 Furthermore, we found that the stimulation of lymphangiogenesis was restricted to the heart following AAV-9 VEGF-CC156S gene therapy because although transgene expression also occurred in the liver, it was not sufficient to stimulate liver lymphangiogenesis in the absence of other triggers such as inflammation or edema (data not shown). Finally, AAV–VEGF-CC156S gene therapy did not influence infarct scar size, heart-to-body weight ratio, cardiomyocyte hypertrophy, or cardiac angiogenesis and arteriolar remodeling as compared to AAV-MI controls (Figure VIA through VIG in the Data Supplement).
Therapeutic Lymphangiogenesis Limits Cardiac Inflammation and Dysfunction Post-MI in Mice
We next set out to investigate the functional impact of therapeutic lymphangiogenesis on cardiac inflammation in the acute (7 days) and chronic (21 days) phases of repair and recovery after MI. We hypothesized that expansion of cardiac lymphatics, as induced by AAV–VEGF-CC156S therapy, would accelerate resolution of inflammation by expediting cardiac efflux of proinflammatory immune cells.
First, we analyzed the kinetics and profile of cardiac inflammation in the viable LV bordering the infarct in saline-injected MI control mice using immunohistochemistry and flow cytometry. We found that cardiac CD68+ macrophage density increased rapidly during the first-week post-MI (Figure VIIA and VIID in the Data Supplement). These macrophages were initially mostly of classical M1 type (negative for CD206, and positive for CCR2 [chemokine receptor 2]; Figure VIIB, VIIE, VIIG in the Data Supplement). Furthermore, CD3+ total T-cell densities peaked already by day 3 in the viable LV, with persistent elevation of CD8+ T cells observed at 7 days post-MI (Figure VIIC, VIIF, VIIH, VIII in the Data Supplement).
Second, we investigated cardiac immune cell population dynamics in both the acute and chronic phases post-MI in our AAV-treated groups. In AAV-MI control mice, similar as in saline-treated MI mice, the cardiac levels of proinflammatory immune cells, including M1 macrophages and CD3+ T cells, were transiently elevated in both infarct (Figure 3E through 3G) and viable LV (Figure 3A and 3B). At 7 days, the cardiac T-lymphocyte population included 48±3% CD4+ and 41±2% CD8+ cells among all CD3+ T cells in the noninfarcted LV wall in AAV-MI controls. In contrast, cardiac-infiltrating Tregs were very sparse at all time points analyzed (1.8±0.5 cells/mm2 observed in infarct at 7 days post-MI, representing only 2% to 5% of all CD4+ T cells).
Figure 3.
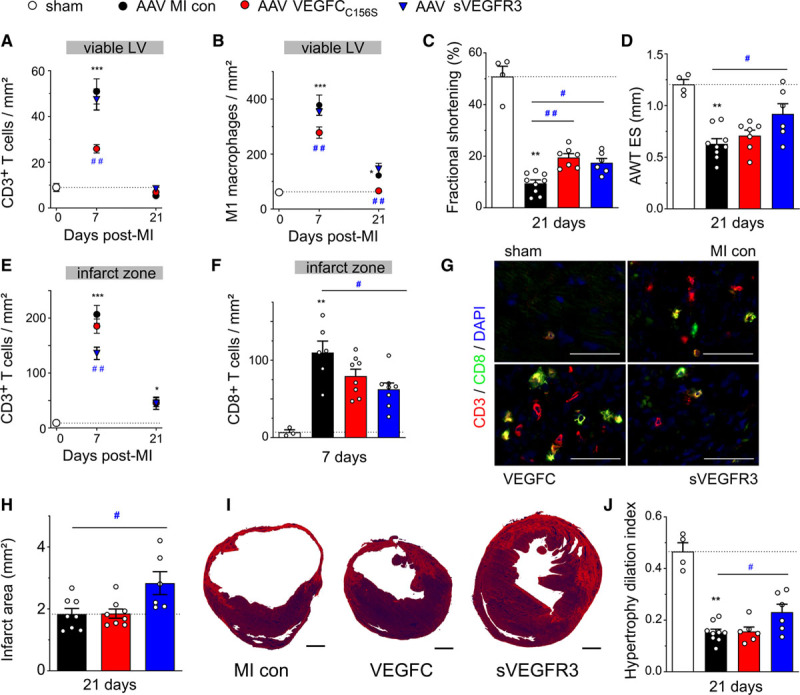
Adeno-associated viral (AAV) delivery of VEGF (vascular endothelial growth factor)-CC156S reduces cardiac inflammation and improves cardiac function, while AAV-sVEGFR3 (soluble VEGF receptor 3) therapy reduces infarct scar T-cell levels and wall thinning leading to improved cardiac function in mice postmyocardial infarction (MI). Cardiac densities in the viable left ventricular (LV) of CD3+ T cells (A) and M1 pro-inflammatory macrophages (CD68+/CD206− cells), (B) were determined by immunohistochemistry in MI controls (AAV-scramble, black circles, n=8), and VEGFC (AAV–VEGF-CC156S, red circles, n=7–8) or sVEGFR3 (AAV-sVEGFR3, blue triangles, n=5–7) treated mice at 7 and 21 d post-MI. Healthy sham levels (n=7–9) are indicated by white circles / bars. Cardiac function and remodeling were analyzed by echocardiography at 21 days post-MI to determine LV fractional shortening (C) and LV anterior wall thickness in end-systole (AWT ES, D). Infarct densities of CD3+ total T cells (E) and CD8+ T-cell subpopulation (F). Examples of CD8+ T-cell density in the infarct at 7 d post-MI (G). CD3 (red), CD8 (green), DAPI (blue), ×20 magnification, scale bar=50 µm. Infarct scar remodeling evaluated in Sirius red-stained histological sections as absolute infarct area (H) at 21 days post-MI in MI controls (AAV-scramble, n=8), VEGFC-(AAV-hVEGF-CC156S, n=7–8) and sVEGFR3-(AAV-sVEGFR3, n=5–7) treated mice. Examples of infarct scar remodeling. Scale bar=1 mm (I). LV hypertrophy/dilatation index was calculated from echocardiographic parameters at 21 days post-MI (J). Kruskal-Wallis, Dunn posthoc test. *P<0.05; **P<0.01; ***P<0.001 vs sham; #P<0.05; ##P<0.01 vs MI control. con indicates control.
AAV–VEGF-CC156S lymphangiogenic therapy led to a substantial reduction in both T-cell and macrophage densities in the viable LV (Figure 3A and 3B). Importantly, this decrease was restricted to M1 proinflammatory macrophages, as the numbers of M2-like regenerative macrophages did not differ from AAV-MI controls (5±2 versus 5±1 M2-like cells/mm2 at 21 days). In contrast, in the infarct zone, total CD3+ and CD8+ T-cell densities were unaltered (Figure 3E through 3G). These data revealed expedited resolution of inflammation by expanded and activated cardiac lymphatics only in the viable LV, but not in the infarct zone where lymphatics remain severely altered despite the lymphangiogenic therapy (Figure 1E, Figure 2B). In addition to lymphatic vessel expansion, AAV–VEGF-CC156S–treated mice displayed increased cardiac expression of the LEC-derived chemokine Ccl21 (Figure 1F), which may mediate lymphatic clearance of cardiac-infiltrating T cells, notably naive/central memory cells expressing the cognate CCR7 receptor. Similarly, cardiac Ccl2 levels were also increased, whereas Cx3cl1 levels were unaltered (Figure 1). Taken together, we found that stimulation of lymphangiogenesis with AAV–VEGF-CC156S reduced T-cell densities only in the viable LV and not in the infarct where lymphatics remained highly dysfunctional and presumably incapable of mediating immune cell exit.
We next evaluated the functional cardiac impact at 21 days post-MI by echocardiography. We found that AAV-MI control mice displayed similar cardiac dysfunction and remodeling as MI mice injected with saline (Table 1, Table I in the Data Supplement), indicating that AAV gene delivery does not adversely affect the functional cardiac response to injury.
Table 1.
Functional Cardiac Evaluation at 21 Days Post-MI in Mouse AAV Study
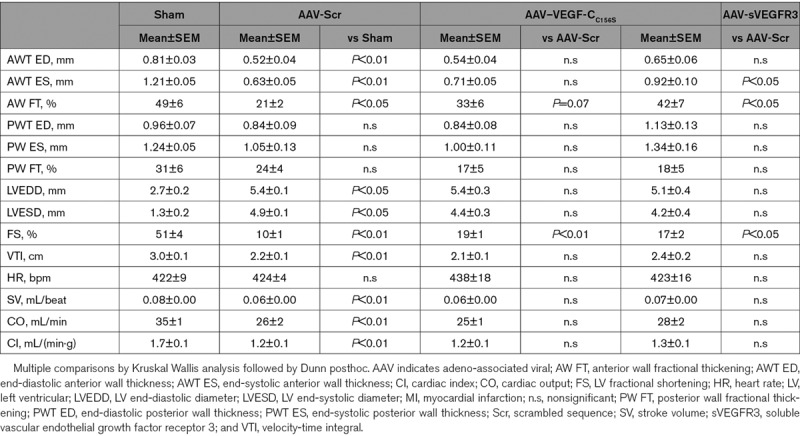
AAV–VEGF-CC156S therapy improved cardiac function, including LV fractional shortening (Figure 3C) and LV anterior wall thickening fraction (33±6 versus 21±2%. LV anterior wall thickness, P=0.07, Table 1) as compared to AAV-MI controls. In contrast, it did not alter LV wall thickness or scar maturation (Figure 3D, 3H through 3I). This promisingly reveals that selective stimulation of cardiac lymphangiogenesis, and the associated accelerated resolution of cardiac inflammation in the viable LV, suffices to improve cardiac function after MI.
Soluble VEGFR3 Inhibits Infarct Lymphangiogenesis Post-MI in Mice
To investigate the impact of the endogenous lymphangiogenic response post-MI, we next applied a VEGF-C/VEGF-D trap (sVEGFR3), delivered by an AAV vector, to block VEGF-C/-D signaling in the heart. Due to the low endogenous level of VEGF-C and VEGF-D expression and poor lymphangiogenesis in mice post-MI, notably in the viable LV (Figure 1A and 1B), AAV-sVEGFR3 inhibition of lymphatic proliferation did not reach significance (AAV-sVEGFR3: 1.4±1.0 versus AAV-MI control: 8.0±4.0% proliferating vessels in the infarct at 7 days post-MI, P=0.12, Figure 2A) nor did it reduce total lymphatic densities (AAV-sVEGFR3: 27±15 versus AAV-MI control: 58±12 vessels/mm2 in the infarct at 21 days post-MI, P=0.12). However, open lymphatic vessel density and total lymphatic area in the infarct zone was markedly reduced at 21 days (Figure 2B through 2D), revealing a key role for endogenous VEGF-C and VEGF-D in limiting MI-induced rarefaction of open lymphatic vessels in the infarct. Although AAV-sVEGFR3 did not influence infarct scar size, heart-to-body weight ratio, cardiomyocyte hypertrophy, or angiogenesis (Figure VIA through VID in the Data Supplement), arteriogenesis was reduced in the viable LV at 21 days post-MI, as compared to AAV-MI controls, although arterial sizes tended to be increased (Figure VIE through VIG in the Data Supplement). This indirectly suggests that endogenous VEGF-C and VEGF-D ligands may participate in regulation of arteriogenesis post-MI acting via VEGFR2 expressed by blood vascular endothelial cells, as previously published,39 as we did not detect VEGFR3 expression in cardiac blood vessels post-MI (Figure II in the Data Supplement).
Soluble VEGFR3 Limits Infarct T-Cell Levels and Reduces Deleterious Cardiac Remodeling
Investigating next the functional cardiac impact of the VEGF-C/VEGF-D trap, we expected that the treatment would not influence cardiac immune cell levels nor cardiac function post-MI, given its limited effects on cardiac lymphangiogenesis beyond the infarct. Indeed, AAV-sVEGFR3 treatment, which, as mentioned above, did not reduce total lymphatic density as compared to AAV-MI controls, did not aggravate cardiac inflammation in the viable LV and both T-cell and M1 macrophage densities were found to be unaltered (Figure 3A and 3B). Surprisingly, at 7 days post-MI in the infarct, where AAV-sVEGFR3 aggravated MI-induced loss of open lymphatic vessels (Figure 2C), we found a marked reduction of cardiac T-cell levels, both total CD3+ T-cell and CD8+ T-cell subpopulation (Figure 3E through 3G), as compared to AAV-MI controls. A similar effect of the VEGF-C/VEGF-D trap on limiting cardiac T-cell infiltration has been described previously in a mouse model of allograft transplantation,13 although its mechanistic explanation awaits further studies.
Echocardiographic analyses revealed unexpectedly that sVEGFR3 treatment improved cardiac function as compared to AAV-MI controls. Indeed, both fractional shortening and LV wall thickness (Figure 3C and 3D), notably of the anterior myocardial segment, were improved. Given that LV cardiomyocyte hypertrophy was not increased (Figure VIC in the Data Supplement), this prevention of MI-induced LV wall thinning observed in sVEGFR3-treated mice likely reflected increased scar thickness/strength linked to reduced infarct T-cell levels. Indeed, previous studies have shown improved infarct healing in the absence of T cells.40 In support of this mechanism, infarct thinning was found to be reduced, and scar area increased at 21 days post-MI by sVEGFR3 treatment (Figure 3H and 3I). As a direct hemodynamic consequence of reduced wall thinning, cardiac hypertrophy-to-LV dilatation index was improved (Figure 3J), explaining the improvement of cardiac function observed in sVEGFR3-treated mice.
To confirm the beneficial effects of sVEGFR3 on cardiac function post-MI, we extended our studies to a rat MI model, again using AAV gene delivery of either sVEGFR3 or a scrambled sequence in MI controls. We again found that sVEGFR3 reduced lymphangiogenesis in the infarct, but had no effect on angiogenesis (Figure 4A through 4C). Serial echocardiography revealed that cardiac function, notably fractional shortening, was improved at 28 days post-MI (Figure 4D). Furthermore, sVEGFR3 reduced LV dilation (Figure 4E) and interventricular septal wall thinning (Figure 4F) and, as a consequence, improved cardiac hypertrophy-to-LV dilatation index, as compared to AAV-MI controls (Figure 4G, Table 2). This suggested, similar to our findings in mice, that cardiac function was improved by sVEGFR3 treatment due to reduced LV wall thinning linked to delayed infarct scar remodeling. Taken together, our studies suggest that sVEGFR3 treatment reduced infarct thinning by limiting cardiac infiltration of T cells. This led to prevention of deleterious LV wall remodeling post-MI resulting in improved cardiac function.
Figure 4.
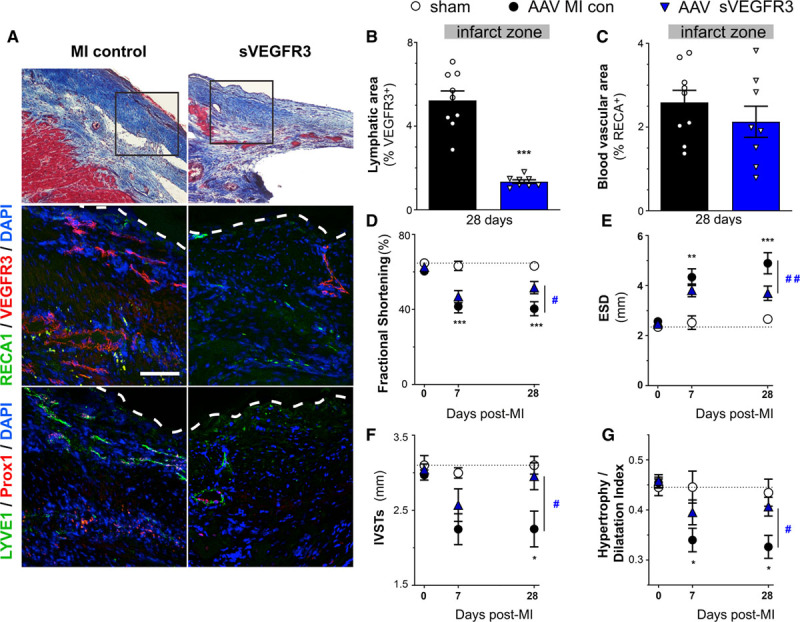
Adeno-associated viral (AAV)-sVEGFR3 (soluble vascular endothelial growth factor receptor 3) therapy suppresses infarct lymphangiogenesis and improves cardiac function and remodeling in rats postmyocardial infarction (MI). Lymphatic remodeling was investigated in cardiac sections from MI controls (AAV-scramble, black circles, n=8) and sVEGFR3-treated (AAV-sVEGFR3, blue triangles, n=9) rats. Examples of blood vessels and lymphatics in the infarct zone at 28 d post-MI (A): middle panel: lymphatics (VEGFR3, red), and blood vessels (RECA1, green); bottom panel: lymphatics (Lyve1 [lymphatic vessel endothelial receptor 1], green; Prox1 [homeobox gene homologue Drosophilia prospero gene], red). ×20, scale bar=50 µm. White dashed lines outline infarct epicardium. Quantification of VEGFR3+ lymphatic area (B) and RECA1+ blood vascular area (C) in the infarct at 28 days post-MI. Comparison by Student 2-tailed t test: ***P<0.001 vs MI control. Cardiac function was investigated by serial echocardiography in healthy sham rats (open circles, n=4), MI controls (AAV-scramble, black circles, n=13), and sVEGFR3-treated (AAV-sVEGFR3, blue triangles, n=10) MI rats. Left ventricular (LV) fractional shortening (D), end-systolic diameter (ESD, E), systolic interventricular septal wall thickness (IVSTs, F) were measured, and LV hypertrophy/dilatation index (G) calculated. Two-way ANOVA, followed by Bonferroni posthoc: *P<0.05; **P<0.01; ***P<0.001 vs sham; #P<0.05, ##P<0.01 vs MI control. RECA indicates rat endothelial cell antigen.
Table 2.
Functional Evaluation by Echocardiography at 7 and 28 Days Post-MI in Rat AAV Study
Cardiac-Infiltrating CD4+ and CD8+ T Cells Suppress Lymphangiogenesis Post-MI
We next sought to investigate the cellular mechanisms orchestrating the cardiac lymphatic response to ischemic injury. Previous studies have revealed that immune cells contribute to regulation of lymphangiogenesis in many tissues.7,8,41 However, whether cardiac-infiltrating immune cells influence lymphangiogenesis post-MI has not been previously investigated. As mentioned above, our data in AAV-MI controls indicated that macrophages present in the infarct at day 7 may have contributed to scar lymphangiogenesis by locally producing VEGF-C (Figure IVD in the Data Supplement). We next turned our attention to T lymphocytes, given their well-documented acute deleterious effects on infarct remodeling, including both direct and indirect effects on cardiomyocytes, fibroblast, and vascular cells.27,42,43 We hypothesized that CD4+ helper or CD8+ cytotoxic T cells additionally may contribute to the poor endogenous cardiac lymphangiogenic response observed acutely post-MI in our mouse model. To prevent T-cell mobilization and cardiac entry after MI, we treated mice with an S1P (sphingosine-1-phosphate) receptor agonist, Fingolimod, as described.33 Due to the angiogenic and lymphangiogenic effects associated with chronic S1P stimulation,44,45 we restricted treatment to the first 48 hours following MI to selectively target acute-phase cardiac T-cell recruitment. We found, as expected, that Fingolimod potently reduced circulating and cardiac T-cell levels (Figure 5A through 5C, Figure VIIIA in the Data Supplement). However, by 21 days post-MI, cardiac T cells in the viable LV of control MI mice returned to healthy sham levels, and no difference was observed in the Fingolimod group at this later time point. Similarly, in the infarct, where MI controls displayed persistently elevated T-cell levels, acute-phase Fingolimod treatment did not suppress chronic phase T-cell levels. Interestingly, Fingolimod-induced prevention of cardiac T-cell recruitment at 3 days post-MI led to a significant reduction of M1 proinflammatory macrophage densities in the viable LV at 7 days post-MI (Figure 5D).
Figure 5.
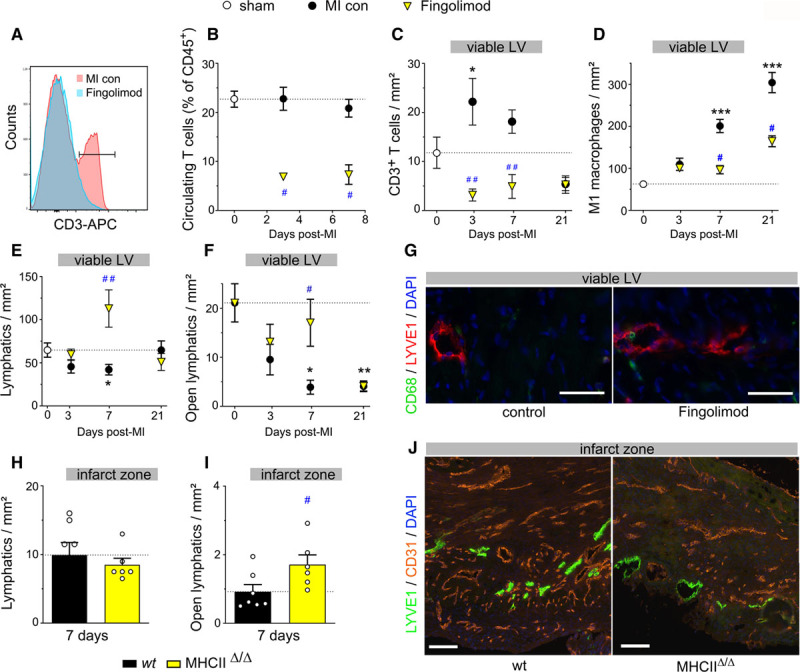
Cardiac-infiltrating T cells suppress lymphangiogenesis in mice postmyocardial infarction (MI). Circulating T-cell levels (A and B) were determined by flow cytometry in saline-injected MI controls (black circles, n=4–5) and Fingolimod-treated wild-type (wt) MI mice (yellow triangles, n=4–5) at 3 and 7 days post-MI and healthy sham mice (white circles, n=4). Cardiac sections were analyzed to determine densities of CD3+ total T cells (C), M1 proinflammatory macrophages (D), lymphatic vessels (E), and open lymphatic vessels (F) in the viable left ventricular (LV) of saline-injected MI controls (black circles, n=4–13) and Fingolimod-treated MI mice (yellow triangles, n=7–9) at 3, 7 and 21 days post-MI. Healthy sham levels (n=5–10) indicated by white circle. One-way ANOVA, Dunn posthoc test. *P<0.05; **P<0.01; ***P<0.001 vs sham; #P<0.05; ##P<0.01 vs MI controls. Examples of cardiac lymphatics (Lyve1 [lymphatic vessel endothelial receptor 1], red) in the viable LV at 7 days post-MI (G). ×20, scale bar=50 µm. Note peri-lymphatic macrophages (CD68, green). Analysis of lymphatic remodeling in wt (black, n=7) vs MHC (major histocompatibility complex) IIΔ/Δ CD4+ T cell-deficient (yellow, n=6) mice at 7 days post-MI assessed as densities of total lymphatic vessel (H) and open lymphatic vessels (I and J) in the infarct zone. Kruskal-Wallis followed by Dunn posthoc test. #P<0.05 vs wt MI controls. Examples of Lyve1+ (green) lymphatics and CD31+ blood vessels (red) in the infarct zone at 7 days post-MI (J). ×10, scale bar=50 µm. APC indicates antigen-presenting cell.
Prevention of cardiac T-cell recruitment by Fingolimod strikingly reduced MI-induced lymphatic rarefaction, with increased density of total lymphatics (Figure 5E) as well as open lymphatics observed in the viable LV (Figure 5F and 5G), as compared to MI controls. Although cardiac LEC proliferation rates were not increased by Fingolimod (Fingolimod: 15.1±2.3 versus MI controls: 13.2±1.6% proliferating lymphatics at 3 days), cardiac lymphatic density at 7 days post-MI was inversely correlated with cardiac T-cell density (Figure VIIIC in the Data Supplement). Remarkably, the results observed at 7 days in the viable LV of Fingolimod-treated mice (Figure 5E and 5F) closely mirror our findings with the AAV–VEGF-CC156S therapy (Figure 1C, Figure VC in the Data Supplement), indicating that cardiac-recruited T cells mediate adverse lymphatic remodeling and rarefaction after MI. However, different from lymphangiogenic gene therapy, the lymphatic density was reduced by 21 days post-MI in the Fingolimod-treated group (Figure 5E and 5F), although T-cell levels at this later stage were no longer elevated in the viable LV (Figure 5C). This indicates that beyond stimulation of initial LEC growth or survival post-MI, the maintenance of newly formed lymphatic vessels requires prolonged expression of lymphangiogenic factors. This may explain why AAV therapy, but not adenoviral therapy, induced cardiac lymphatic expansion post-MI.
In the infarct, Fingolimod-induced acute suppression of T cells similarly increased lymphatic density by 7 days (Figure VIIIB in the Data Supplement). However, the MI-induced early slimming and rarefaction of open lymphatic vessels was not prevented (Figure VIIID through VIIIF in the Data Supplement), likely due to incomplete inhibition of T-cell invasion by Fingolimod in the infarct (Figure VIIIA in the Data Supplement). Surprisingly, by 21 days post-MI, both total lymphatic density and open vessel density were increased in the infarct in Fingolimod-treated mice, as compared to MI controls, despite the return of T cells to control MI levels. This indicates that only T-cell activity in the acute post-MI phase limits lymphangiogenesis. In contrast, blood vessel rarefaction, observed during the first-week post-MI, was not significantly altered by Fingolimod (Figure VIIIH in the Data Supplement). Furthermore, in agreement with previous findings,46 at 21 days post-MI, although infarct size was not altered (Figure VIIIG in the Data Supplement), cardiac function and remodeling were improved in Fingolimod-treated mice as compared to MI controls (Figure VIIII through VIIIK and Table I in the Data Supplement). Indeed, Fingolimod treatment, similar to sVEGFR3 treatment, reduced LV wall thinning and improved cardiac hypertrophy-to-LV dilatation index (Table I and Figure VIIIK in the Data Supplement). Taken together, our data demonstrate that T cells recruited to the heart in the acute-phase post-MI contribute to lymphatic loss and deleterious remodeling after MI.
To determine which subset of T cells was responsible for the deleterious impact on cardiac lymphatics post-MI, we next investigated lymphatic remodeling in MHCIIΔ/Δ mice that lack CD4+ T cells. We found that infarct sizes were similar between wild-type mice and MHCIIΔ/Δ mice at 7 days post-MI (Figure IXA in the Data Supplement). Cardiac lymphatic densities and total lymphatic areas were similar in CD4+-deficient mice as compared to wild-type mice both at baseline (before MI) and at 7 days post-MI (Figure 5H, Figure IXB in the Data Supplement). In contrast, open lymphatic vessel density in the infarct was significantly increased at 7 days post-MI in mice lacking CD4+ T cells (Figure 5I and 5J), consistent with the data from Fingolimod-treated mice. Furthermore, in the absence of cardiac-infiltrating CD4+ T cells in MHCIIΔ/Δ mice, flow cytometric evaluation revealed increased cardiac macrophage density but decreased levels of proinflammatory M1 (CD206−/ CD86+) macrophages at 7 days post-MI (Figure IXC and IXD in the Data Supplement), similar as our findings with Fingolimod (Figure 5D).
To confirm the specific involvement of cardiac-infiltrating T cells, we next applied in vivo depleting antibodies to acutely block CD4+ or CD8+ T-cell subsets immediately post-MI. Additionally, given that IFN (interferon)-γ has been proposed as a major T-cell–derived lymphangiogenic inhibitor,8,47 we evaluated the impact of neutralizing antibodies against this Th1 cytokine. Flow cytometry at 2 and 8 days post-MI confirmed selective knockdown of circulating CD4+ or CD8+ T cells (Figure 6A and 6B). Immunohistochemistry of cardiac sections at 8 days post-MI similarly confirmed selective depletion of cardiac T-cell subsets (Figure 6C and 6D). We found that CD4+ or CD8+ depletion prevented lymphatic rarefaction post-MI (Figure 6E through 6G), although it did not reduce lymphatic precollector slimming (Figure XD in the Data Supplement). Interestingly, whereas neutralization of IFNγ did not alter circulating or cardiac T- or B-cell levels (Figure 6A through 6C, Figure XA and XB in the Data Supplement), it partially blocked MI-induced deleterious lymphatic remodeling, including loss of open lymphatic vessels in the heart (Figure XC and XE in the Data Supplement). In conclusion, our data reveal that cardiac-infiltrating CD4+ and CD8+ T cells exert potent deleterious effects on cardiac lymphatics and contribute, in part, through IFNγ, to rarefaction of both lymphatic capillaries and precollectors in the acute-phase post-MI.
Figure 6.
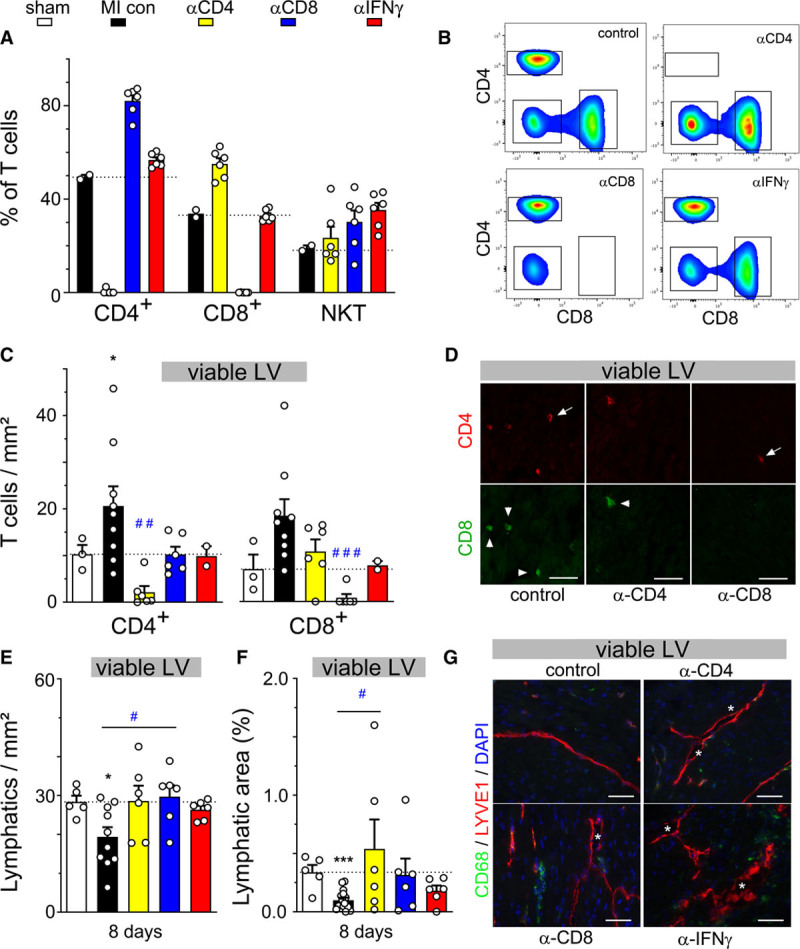
Depletion of CD4 and CD8, but not NKT, T-cell subpopulations prevents rarefaction of cardiac lymphatics acutely postmyocardial infarction (MI). Assessment by flow cytometry at 8 days post-MI of circulating T-cell populations (A and B) in control MI mice (black, n=2), CD4-depleted mice (yellow, n=6), CD8-depleted mice (blue, n=6), and mice treated with an IFN (interferon) γ–neutralizing antibody (red, n=6). Evaluation by immunohistochemistry of viable left ventricular (LV) at 8 days post-MI of CD4+ and CD8+ cardiac T-cell densities (C) and total lymphatic density (E) and lymphatic area (F) in healthy sham mice (white, n=3–5) control MI mice (black, n=10), CD4-depleted mice (yellow, n=6), CD8-depleted mice (blue, n=6), and anti-IFNγ–treated mice (red, n=7). Examples of cardiac T-cell densities (D) and lymphatic densities (G) in viable LV at 8 days post-MI. CD4+ cells indicated by arrow, CD8+ cells by arrowhead, and open lymphatics indicated by asterisk. ×20, scale bar=50 µm. Kruskal-Wallis followed by Dunn posthoc test. *P<0.05; ***P<0.001 vs sham; #P<0.05; ###P<0.001 vs MI controls.
Discussion
In this study, we demonstrated that therapeutic stimulation of lymphatic vessel growth in the heart required sustained elevated expression of lymphangiogenic growth factors, as achieved by AAV but not adenoviral gene therapy. Moreover, although we previously demonstrated that lymphangiogenesis may be induced by intramyocardial sustained-release of VEGF-C protein, we found that systemic delivery of recombinant human VEGF-CC156S protein, as proposed recently,25,38 was inefficient to increase cardiac lymphangiogenesis. Indeed, although our study revealed no increase in either plasma VEGF-C levels or lymphatic proliferation in the heart in response to repeated intraperitoneal injections of VEGF-CC156S protein, the previous studies, based on the same approach, notably did not provide any evidence of increased circulating or cardiac VEGF-C levels.25,38 Consequently, our data support the concept of strict time- and dose-dependence of VEGF-C delivery to achieve therapeutic lymphangiogenesis in the heart. Promisingly, our study revealed that therapeutic expansion of lymphatic vessels accelerated resolution of cardiac inflammation with reduction of both cardiac T lymphocytes and M1 proinflammatory macrophages in the viable LV. This selective improvement of immune cell clearance, without modification of angiogenesis or cardiomyocyte hypertrophy, significantly reduced cardiac dysfunction. To further advance our understanding of how cardiac lymphangiogenesis is regulated after MI, with the aim to develop better therapies, future studies may benefit from assessment not only of VEGF-C/VEGF-D but also of the molecular partners essential for their proteolytic processing, CCBE1 (collagen- and calcium-binding EGF domains 1) and ADAMTS3 (a disintegrin and metalloproteinase with a thrombospondin type I motif 3).48
Our findings of the cardiac functional benefit with sVEGFR3 treatment in both mouse and rat MI models were completely unexpected. The absence of aggravation of cardiac inflammation was likely due to the low endogenous level of lymphangiogenesis observed in the viable LV post-MI, leading to limited functional impact of antilymphangiogenic therapy. However, in both mice and rats, we found reduced LV wall thinning with sVEGFR3 due to delayed infarct scar remodeling. These beneficial functional cardiac effects of sVEGFR3 were observed in female mice. Interestingly, a recent study also investigating sVEGFR3 treatment in male mice reported increased post-MI mortality linked to cardiac rupture.49 This apparent discrepancy with our findings may be explained by the sex-dependence of extracellular matrix remodeling post-MI, with weaker scars formed in male mice.50 It is likely that the sVEGFR3-induced delay in infarct maturation, due to prevention of T-cell infiltration, is beneficial only in female mice, as in males, the scar will rupture if not rapidly matured. Importantly, we observed functional cardiac benefit of sVEGFR3 treatment in male rats, which have thicker scars than mice. This indicates that both sex and species-differences in post-MI wall strength may influence the risk/benefit of acute-phase T-cell–modulating therapies. This issue will require further mechanistic studies. Taken together, our data revealed that whereas stimulation of cardiac lymphangiogenesis with VEGF-CC156S was beneficial after MI, due to accelerated resolution of inflammation in the viable LV wall, treatment with the VEGF-C/VEGF-D trap also attenuated cardiac remodeling due to reduced infarct thinning by suppression of recruitment/expansion/activation of T cells in the infarct. These lymphangiogenesis-independent immunomodulatory effects of sVEGFR3 remain to be further explored. However, the beneficial effects previously reported with sVEGFR3 therapy in another model of cardiac inflammation are noteworthy in this context: in a mouse cardiac allograft model adenoviral delivery of sVEGFR3 reduced cardiac CD8+ T-cell levels.13 This was proposed to be linked to decreased cardiac-derived DC homing to the spleen, due to reduced cardiac lymphatic expression of Ccl21, a major lymphatic chemokine for DCs and T cells. In addition, it is possible that endogenous VEGF-C and VEGF-D may directly influence cardiac recruitment, maturation, and activation of cardiac DCs that express both VEGFR2 and VEGFR3.51 In agreement, our preliminary data indicate significantly reduced mature CD103+ CD11c+ DC levels in the heart of sVEGFR3-treated mice as compared to AAV-MI controls (sVEGFR3: 18±5 versus AAV-MI control: 64±19 mature DCs/mm2, P<0.01). This suggests that treatment with sVEGFR3 may have reduced antigen presentation by cardiac DCs post-MI, which would limit recruitment or expansion of T cells in the infarct. However, increased cardiac DCs levels in the viable LV has been proposed to protect the remodeling postischemic heart by limiting M1 proinflammatory macrophage recruitment, inducing a beneficial inflammatory-to-reparative macrophage shift but also by stimulating Treg expansion in cardiac-draining lymph nodes. Of note, this mechanism has been proposed to be crucial for maintenance of lymphocyte tolerance to self-antigens following cardiac injury.52–54 Future studies are needed to address the potential impact of lymphangiogenesis on cardiac immune tolerance after injury.
Finally, in this study, we uncovered a dominant-negative role of cardiac-infiltrating T cells, both CD4+ helper and CD8+ cytotoxic T cells, on the endogenous cardiac lymphangiogenic response acutely post-MI. There are many previous reports demonstrating the deleterious short- and long-term cardiac effects of T cells, especially CD4+ helper cells, on cardiomyocytes, macrophages, and fibroblasts.27,42,43 Our data revealed that, in addition, infiltrating CD4+ and CD8+ T cells reduce cardiac lymphatic vessel survival acutely post-MI, in part via IFNγ secretion, rendering the vessels less responsive to endogenous lymphangiogenic stimuli. A similar direct negative impact of CD4+ T cells on lymphatics has been described in both inflamed skin and lymph nodes.8,41 However, it is noteworthy that cardiac lymphatic slimming (reduced lymphatic diameters) was not influenced by Fingolimod or IFNγ neutralization, nor by AAV–VEGF-CC156S therapy or sVEGFR3 treatment, suggesting involvement of factors other than T-cell–derived cytokines or the VEGF-C/VEGF-D/VEGFR3 axis.55 Interestingly, adrenomedullin was recently shown to increase vessel diameters in female mice post-MI.56 Additionally, it has been reported that apelin-deficient male mice display increased endogenous lymphangiogenesis post-MI, potentially linked to elevated cardiac recruitment of VEGF-C-secreting B cells.57 Conversely, apelin overexpression by gene therapy limited cardiac inflammation (macrophage density and IL [interleukin]-1β levels) post-MI and reduced lymphangiogenic responses. Hence inflammation may both drive and oppose cardiac lymphangiogenesis. The tipping point remains to be determined and may relate to local T- and B-cell balance in the heart, similar as suggested for remodeling lymph nodes.7,8
A key weakness of our study is that we opted for a pretreatment approach with the AAV gene therapy, with the aim to achieve similar post-MI acute-phase expression kinetics of VEGF-CC156S as with adenoviral gene therapy to compare these treatments. It thus remains to be determined whether AAV-mediated therapeutic gene delivery after MI will be equally efficient to stimulate lymphatic repair, reduce cardiac inflammation, and improve cardiac function. Given that our AAV therapy led to long-term growth factor delivery, it is likely that this approach will be suitable to stimulate cardiac lymphangiogenesis also when administered post-MI. Moreover, it will be important to assess whether the chronic, low-grade inflammation that occurs months after MI in patients prone to heart failure development is similarly amenable to improvement by lymphangiogenic therapy. Furthermore, in our model, there was no significant fibrosis in the viable LV at 21 days post-MI, and it thus remains to be determined whether lymphangiogenic gene therapy may reduce interstitial cardiac fibrosis post-MI similar, as we reported previously in rats. Finally, the effects on cardiac remodeling and function of combined stimulation of cardiac angiogenesis and lymphangiogenesis post-MI remain to be investigated.
Taken together, our findings revealed that cardiac inflammation and lymphangiogenic responses are closely interconnected. We propose that while therapeutic stimulation of lymphatic remodeling by gene therapy may provide a novel approach to limit cardiac inflammation in cardiovascular diseases, T-cell–targeted therapy, including anti-IFNγ treatment,58 may act in part by enhancing endogenous lymphangiogenic responses post-MI leading to improved lymphatic drainage with accelerated cardiac fluid and immune cell clearance, both essential for optimal cardiac functional recovery and repair.
Acknowledgments
M. Houssari and E. Brakenhielm designed and carried out protein and gene therapies in mice; M. Houssari and A. Dumesnil performed and analyzed immunohistochemistry and histology. V. Tardif, A. Dumesnil, and J. Rondeaux performed and analyzed flow cytometry together with G. Riou, V. Tardif, and S. Adriouch designed the antibody-depletion experiments. I. Boukhalfa performed and analyzed echocardiography in mice; R. Kivelä, K.A. Hemanthakumar, and K. Alitalo designed, performed, and analyzed the rat myocardial infarction (MI) study; N. Pizzinat and M. Bizou designed, performed, and analyzed the MHCIIΔ/Δ mouse MI study and edited the final article; D. Godefroy and D. Schapman carried out and postprocessed lightsheet and confocal imaging, respectively; J.P. Henry carried out mouse MI model; M. Houssari, S. Fraineau, and S. Renet designed and carried out cardiac gene expression analyses in mice; Y. Anouar contributed reagents/materials/analysis tools; R. Kivelä and K. Alitalo designed and produced adenoviral and adeno-associated viral (AAV) gene vectors and edited the final article, P. Mulder guided echocardiography analyses and development of gene therapy protocols, and P. Mulder and V. Richard edited the final article; E. Brakenhielm and M. Houssari prepared the article draft. All authors approved the final version of the article. We thank Morgane Grosset and Paul Rouault at Normandy University for assistance with the mouse MI model, Päivi Leinikka, and Eero Mervaala at Helsinki University for carrying out the rat MI model, and the AAV Gene Transfer and Cell Therapy Core Facility and Biomedicum imaging unit at the University of Helsinki for their expert help.
Sources of Funding
The research leading to these results received funding from the ERA-CVD Research Programme, which is a transnational R&D programme jointly funded by national funding organizations (ANR-16-ECVD-0004) within the framework of the ERA-NET ERA-CVD. The project also benefitted from funds from the FHU REMOD-VHF (Inserm U1096 laboratory), and generalized institutional funds (Inserm U1096 laboratory) from French Inserm and the Normandy Region together with the European Union: Europe gets involved in Normandie with European Regional Development Fund (ERDF): CPER/FEDER 2015 (DO-IT) and CPER/FEDER 2016 (PACT-CBS).
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- AAV
- adeno-associated virus
- DCs
- dendritic cells
- IFN
- interferon
- IL
- interleukin
- INF
- interferon
- LV
- left ventricle
- MI
- myocardial infarction
- Pdpn
- podoplanin
- S1P
- sphingosine-1-phosphate
- sVEGFR3
- soluble VEGF receptor 3
- VEGF
- vascular endothelial growth factor
For Sources of Funding and Disclosures, see page 1736.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314370.
References
- 1.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 2.Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP. The Lymphatic System: Integral Roles in Immunity. Annu Rev Immunol. 2017;35:31–52. doi: 10.1146/annurev-immunol-041015-055354. doi: 10.1146/annurev-immunol-041015-055354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124:929–935. doi: 10.1172/JCI71610. doi: 10.1172/JCI71610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiig H, Schröder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, Boschmann M, Goss J, Bry M, Rakova N, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123:2803–2815. doi: 10.1172/JCI60113. doi: 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, Ruddle NH, Jain RK, Fukumura D, Padera TP. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci U S A. 2011;108:18784–18789. doi: 10.1073/pnas.1116152108. doi: 10.1073/pnas.1116152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scallan JP, Zawieja SD, Castorena-Gonzalez JA, Davis MJ. Lymphatic pumping: mechanics, mechanisms and malfunction: Lymphatic pumping mechanisms. J Physiol. 2016;594(20):5749–5768. doi: 10.1113/JP272088. doi:10.1113/JP272088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angeli V, Ginhoux F, Llodrà J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity. 2006;24:203–215. doi: 10.1016/j.immuni.2006.01.003. doi: 10.1016/j.immuni.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, Gollamudi S, Kim YK, Lee SH, Koh GY. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity. 2011;34:96–107. doi: 10.1016/j.immuni.2010.12.016. doi: 10.1016/j.immuni.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 9.Förster R, Braun A, Worbs T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012;33:271–280. doi: 10.1016/j.it.2012.02.007. doi: 10.1016/j.it.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Aebischer D, Iolyeva M, Halin C. The inflammatory response of lymphatic endothelium. Angiogenesis. 2014;17:383–393. doi: 10.1007/s10456-013-9404-3. doi: 10.1007/s10456-013-9404-3. [DOI] [PubMed] [Google Scholar]
- 11.Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med. 2010;207:2255–2269. doi: 10.1084/jem.20100559. doi: 10.1084/jem.20100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, Genua M, Arena V, Spinelli A, Peyrin-Biroulet L, et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. 2014;124:3863–3878. doi: 10.1172/JCI72189. doi: 10.1172/JCI72189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nykänen AI, Sandelin H, Krebs R, Keränen MA, Tuuminen R, Kärpänen T, Wu Y, Pytowski B, Koskinen PK, Ylä-Herttuala S, et al. Targeting lymphatic vessel activation and CCL21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation. 2010;121:1413–1422. doi: 10.1161/CIRCULATIONAHA.109.910703. doi: 10.1161/CIRCULATIONAHA.109.910703. [DOI] [PubMed] [Google Scholar]
- 14.Dieterich LC, Seidel CD, Detmar M. Lymphatic vessels: new targets for the treatment of inflammatory diseases. Angiogenesis. 2014;17:359–371. doi: 10.1007/s10456-013-9406-1. doi: 10.1007/s10456-013-9406-1. [DOI] [PubMed] [Google Scholar]
- 15.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaht-Zabost A, Gula G, Ciszek B, Czarnowska E, Jankowska-Steifer E, Madej M, Niderla-Bielińska J, Radomska-Leśniewska D, Ratajska A. Cardiac mouse lymphatics: developmental and anatomical update. Anat Rec Hoboken NJ 2007. 2014;297(6):1115–1130. doi: 10.1002/ar.22912. doi:10.1002/ar.22912. [DOI] [PubMed] [Google Scholar]
- 19.Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol. 2019;16:56–68. doi: 10.1038/s41569-018-0087-8. doi: 10.1038/s41569-018-0087-8. [DOI] [PubMed] [Google Scholar]
- 20.Dashkevich A, Bloch W, Antonyan A, Fries JU, Geissler HJ. Morphological and quantitative changes of the initial myocardial lymphatics in terminal heart failure. Lymphat Res Biol. 2009;7:21–27. doi: 10.1089/lrb.2008.1010. doi: 10.1089/lrb.2008.1010. [DOI] [PubMed] [Google Scholar]
- 21.Kholová I, Dragneva G, Cermáková P, Laidinen S, Kaskenpää N, Hazes T, Cermáková E, Steiner I, Ylä-Herttuala S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. 2011;41:487–497. doi: 10.1111/j.1365-2362.2010.02431.x. doi: 10.1111/j.1365-2362.2010.02431.x. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa Y, Akishima-Fukasawa Y, Ito K, Akasaka Y, Tanaka M, Shimokawa R, Kimura-Matsumoto M, Morita H, Sato S, Kamata I, et al. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology. 2007;51:345–353. doi: 10.1111/j.1365-2559.2007.02785.x. doi: 10.1111/j.1365-2559.2007.02785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perin EC, Tian M, Marini FC, III, Silva GV, Zheng Y, Baimbridge F, Quan X, Fernandes MR, Gahremanpour A, Young D, et al. Imaging long-term fate of intramyocardially implanted mesenchymal stem cells in a porcine myocardial infarction model. PLoS One. 2011;6:e22949. doi: 10.1371/journal.pone.0022949. doi: 10.1371/journal.pone.0022949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun QN, Wang YF, Guo ZK. Reconstitution of myocardial lymphatic vessels after acute infarction of rat heart. Lymphology. 2012;45:80–86. [PubMed] [Google Scholar]
- 25.Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62–67. doi: 10.1038/nature14483. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Lévy F, Henry JP, Dumesnil A, Boukhalfa I, Banquet S, et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation. 2016;133:1484–97. doi: 10.1161/CIRCULATIONAHA.115.020143. discussion 1497. doi: 10.1161/CIRCULATIONAHA.115.020143. [DOI] [PubMed] [Google Scholar]
- 27.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124. doi: 10.1161/CIRCULATIONAHA.113.007101. doi: 10.1161/CIRCULATIONAHA.113.007101. [DOI] [PubMed] [Google Scholar]
- 28.Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, Mulder P, Brakenhielm E, Thuillez C, Germain S, et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014;28:3351–3361. doi: 10.1096/fj.13-245753. doi: 10.1096/fj.13-245753. [DOI] [PubMed] [Google Scholar]
- 29.Kivelä R, Bry M, Robciuc MR, Räsänen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mäyränpää MI, et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307–321. doi: 10.1002/emmm.201303147. doi: 10.1002/emmm.201303147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YF, Redetzke RA, Sivertson RM, Coburn TS, Cypher LR, Gerdes AM. Post-myocardial infarction left ventricular myocyte remodeling: are there gender differences in rats? Cardiovasc Pathol. 2011;20:e189–e195. doi: 10.1016/j.carpath.2010.09.007. doi: 10.1016/j.carpath.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visuri MT, Honkonen KM, Hartiala P, Tervala TV, Halonen PJ, Junkkari H, Knuutinen N, Ylä-Herttuala S, Alitalo KK, Saarikko AM. VEGF-C and VEGF-C156S in the pro-lymphangiogenic growth factor therapy of lymphedema: a large animal study. Angiogenesis. 2015;18:313–326. doi: 10.1007/s10456-015-9469-2. doi: 10.1007/s10456-015-9469-2. [DOI] [PubMed] [Google Scholar]
- 32.Antila S, Karaman S, Nurmi H, Airavaara M, Voutilainen MH, Mathivet T, Chilov D, Li Z, Koppinen T, Park JH, et al. Development and plasticity of meningeal lymphatic vessels. J Exp Med. 2017;214:3645–3667. doi: 10.1084/jem.20170391. doi: 10.1084/jem.20170391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anisimov A, Alitalo A, Korpisalo P, Soronen J, Kaijalainen S, Leppänen VM, Jeltsch M, Ylä-Herttuala S, Alitalo K. Activated forms of VEGF-C and VEGF-D provide improved vascular function in skeletal muscle. Circ Res. 2009;104:1302–1312. doi: 10.1161/CIRCRESAHA.109.197830. doi: 10.1161/CIRCRESAHA.109.197830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belle M, Godefroy D, Couly G, Malone SA, Collier F, Giacobini P, Chédotal A. Tridimensional Visualization and Analysis of Early Human Development. Cell. 2017;169:161–173.e12. doi: 10.1016/j.cell.2017.03.008. doi: 10.1016/j.cell.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Barbay V, Houssari M, Mekki M, Banquet S, Edwards-Lévy F, Henry JP, Dumesnil A, Adriouch S, Thuillez C, Richard V, et al. Role of M2-like macrophage recruitment during angiogenic growth factor therapy. Angiogenesis. 2015;18:191–200. doi: 10.1007/s10456-014-9456-z. doi: 10.1007/s10456-014-9456-z. [DOI] [PubMed] [Google Scholar]
- 37.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieira JM, Norman S, Villa Del Campo C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, Johnson LA, Greaves DR, Carr CA, Jackson DG, et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest. 2018;128:3402–3412. doi: 10.1172/JCI97192. doi: 10.1172/JCI97192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, Kholová I, Kauppinen RA, Achen MG, Stacker SA, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. 2003;92:1098–1106. doi: 10.1161/01.RES.0000073584.46059.E3. doi: 10.1161/01.RES.0000073584.46059.E3. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 41.Gousopoulos E, Proulx ST, Bachmann SB, Scholl J, Dionyssiou D, Demiri E, Halin C, Dieterich LC, Detmar M. Regulatory T cell transfer ameliorates lymphedema and promotes lymphatic vessel function. JCI Insight. 2016;1:e89081. doi: 10.1172/jci.insight.89081. doi: 10.1172/jci.insight.89081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 43.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail. 2017;10:e003688. doi: 10.1161/CIRCHEARTFAILURE.116.003688. doi: 10.1161/CIRCHEARTFAILURE.116.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon CM, Hong BS, Moon HG, Lim S, Suh PG, Kim YK, Chae CB, Gho YS. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood. 2008;112:1129–1138. doi: 10.1182/blood-2007-11-125203. doi: 10.1182/blood-2007-11-125203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, Frank AC, Scholich K, Pierre S, Syed SN, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. 2017;214:2695–2713. doi: 10.1084/jem.20160392. doi: 10.1084/jem.20160392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann U, Hu K, Walter F, Burkard N, Ertl G, Bauersachs J, Ritter O, Frantz S, Bonz A. Pharmacological pre- and post-conditioning with the sphingosine-1-phosphate receptor modulator FTY720 after myocardial ischaemia-reperfusion. Br J Pharmacol. 2010;160:1243–1251. doi: 10.1111/j.1476-5381.2010.00767.x. doi: 10.1111/j.1476-5381.2010.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zampell JC, Avraham T, Yoder N, Fort N, Yan A, Weitman ES, Mehrara BJ. Lymphatic function is regulated by a coordinated expression of lymphangiogenic and anti-lymphangiogenic cytokines. Am J Physiol Cell Physiol. 2012;302:C392–C404. doi: 10.1152/ajpcell.00306.2011. doi: 10.1152/ajpcell.00306.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppänen VM, Holopainen T, Kivelä R, Ortega S, Kärpanen T, Alitalo K. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation. 2014;129:1962–1971. doi: 10.1161/CIRCULATIONAHA.113.002779. doi: 10.1161/CIRCULATIONAHA.113.002779. [DOI] [PubMed] [Google Scholar]
- 49.Vuorio T, Ylä-Herttuala E, Laakkonen JP, Laidinen S, Liimatainen T, Ylä-Herttuala S. Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci Rep. 2018;8:16709. doi: 10.1038/s41598-018-34770-4. doi: 10.1038/s41598-018-34770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang L, Gao XM, Moore XL, Kiriazis H, Su Y, Ming Z, Lim YL, Dart AM, Du XJ. Differences in inflammation, MMP activation and collagen damage account for gender difference in murine cardiac rupture following myocardial infarction. J Mol Cell Cardiol. 2007;43:535–544. doi: 10.1016/j.yjmcc.2007.06.011. doi: 10.1016/j.yjmcc.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Hajrasouliha AR, Funaki T, Sadrai Z, Hattori T, Chauhan SK, Dana R. Vascular endothelial growth factor-C promotes alloimmunity by amplifying antigen-presenting cell maturation and lymphangiogenesis. Invest Ophthalmol Vis Sci. 2012;53:1244–1250. doi: 10.1167/iovs.11-8668. doi: 10.1167/iovs.11-8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234–1245. doi: 10.1161/CIRCULATIONAHA.111.052126. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 53.Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, Koh YS, Kim E, Seung KB, Park C, et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation. 2017;135:1444–1457. doi: 10.1161/CIRCULATIONAHA.116.023106. doi: 10.1161/CIRCULATIONAHA.116.023106. [DOI] [PubMed] [Google Scholar]
- 54.Van der Borght K, Scott CL, Nindl V, Bouché A, Martens L, Sichien D, Van Moorleghem J, Vanheerswynghels M, De Prijck S, Saeys Y, et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017;18:3005–3017. doi: 10.1016/j.celrep.2017.02.079. doi: 10.1016/j.celrep.2017.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaahtomeri K, Karaman S, Mäkinen T, Alitalo K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev. 2017;31:1615–1634. doi: 10.1101/gad.303776.117. doi: 10.1101/gad.303776.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trincot CE, Xu W, Zhang H, Kulikauskas MR, Caranasos TG, Jensen BC, Sabine A, Petrova TV, Caron KM. Adrenomedullin Induces Cardiac Lymphangiogenesis After Myocardial Infarction and Regulates Cardiac Edema Via Connexin 43. Circ Res. 2019;124:101–113. doi: 10.1161/CIRCRESAHA.118.313835. doi: 10.1161/CIRCRESAHA.118.313835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tatin F, Renaud-Gabardos E, Godet A-C, Hantelys F, Pujol F, Morfoisse F, Calise D, Viars F, Valet P, Masri B, et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight. 2017;2:93887. doi: 10.1172/jci.insight.93887. doi:10.1172/jci.insight.93887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levick SP, Goldspink PH. Could interferon-gamma be a therapeutic target for treating heart failure? Heart Fail Rev. 2014;19:227–236. doi: 10.1007/s10741-013-9393-8. doi: 10.1007/s10741-013-9393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.