

Supplemental Digital Content is available in the text.
Keywords: actomyosin, atherosclerosis, discoidin domain receptor, transdifferentiation, vascular calcification
Abstract
Objective:
Vascular calcification is a pathology characterized by arterial mineralization, which is a common late-term complication of atherosclerosis that independently increases the risk of adverse cardiovascular events by fourfold. A major source of calcifying cells is transdifferentiating vascular smooth muscle cells (VSMCs). Previous studies showed that deletion of the collagen-binding receptor, DDR1 (discoidin domain receptor-1), attenuated VSMC calcification. Increased matrix stiffness drives osteogenesis, and DDR1 has been implicated in stiffness sensing in other cell types; however, the role of DDR1 as a mechanosensor in VSMCs has not been investigated. Here, we test the hypothesis that DDR1 senses increased matrix stiffness and promotes VSMC transdifferentiation and calcification.
Approach and Results:
Primary VSMCs isolated from Ddr1+/+ (wild-type) and Ddr1−/− (knockout) mice were studied on collagen-I–coated silicon substrates of varying stiffness, culturing in normal or calcifying medium. DDR1 expression and phosphorylation increased with increasing stiffness, as did in vitro calcification, nuclear localization of Runx2 (Runt-related transcription factor 2), and expression of other osteochondrocytic markers. By contrast, DDR1 deficient VSMCs were not responsive to stiffness and did not undergo transdifferentiation. DDR1 regulated stress fiber formation and RhoA (ras homolog family member A) activation through the RhoGEF (rho guanine nucleotide exchange factor), Vav2. Inhibition of actomyosin contractility reduced Runx2 activation and attenuated in vitro calcification in wild-type VSMCs. Finally, a novel positive feedforward loop was uncovered between DDR1 and actomyosin contractility, important in regulating DDR1 expression, clustering, and activation.
Conclusions:
This study provides mechanistic insights into DDR1 mechanosignaling and shows that DDR1 activity and actomyosin contractility are interdependent in mediating stiffness-dependent increases in VSMC calcification.
Highlights.
DDR1 (discoidin domain receptor-1) promotes smooth muscle cell transdifferentiation and calcification in response to substrate stiffening.
DDR1 stimulates actin stress fiber formation via Vav2.
Inhibiting RhoA (ras homolog family member A) or actomyosin contractility reduced Runx2 (Runt-related transcription factor 2) activation and calcification.
DDR1 activation, clustering, and expression are regulated by RhoA and actomyosin contractility in a positive feedback loop.
Vascular calcification is a severe complication in patients with late-stage atherosclerosis and type 2 diabetes mellitus. It increases the risk of adverse cardiovascular events fourfold independent of other known cardiovascular risk factors.1 Vascular calcification contributes to vessel stiffening and increased risk of plaque rupture in atherosclerosis. It is characterized by the ectopic deposition of calcium-phosphate (hydroxyapatite) crystals in the vessel wall by osteochondrocytic cells and is an active process similar to physiological bone formation.2 A major source of osteochondrocytic cells is the transdifferentiation of vascular smooth muscle cells (VSMCs) to osteochondrocytic-like cells, which is mediated by the osteogenic transcription factor Runx2 (Runt-related transcription factor 2).3
The collagen-binding receptor tyrosine kinase, DDR1 (discoidin domain receptor-1), regulates cell differentiation and the pathogenesis of vascular calcification. In Ldlr−/− mice fed an atherogenic diet, knockout of DDR1 decreased plaque calcification in vivo.4 In vitro, primary murine DDR1 knockout VSMCs cultured in high glucose and phosphate calcifying media displayed reduced calcification.5 Nuclear localization of Runx2 was reduced in DDR1 knockouts both in vivo and in vitro.5 DDR1 deletion reduced phosphorylation of the Runx2-activating kinases Akt (protein kinase B) and p38 MAPK (mitogen-activated protein kinase) and prevented Runx2 nuclear transport via microtubules.5 However, the mechanisms by which DDR1 regulates VSMC transdifferentiation and calcification are not completely understood.
Vascular disease pathogenesis in diabetes mellitus is confounded by stiffening of the matrix due to the formation of advanced glycation end products and fibrosis.6–8 This culminates in profound changes in the mechanical properties of the matrix, which is exacerbated by tissue mineralization.9,10 Matrix stiffness is an important determinant of cell differentiation in mesenchymal progenitor cells, with high matrix stiffness promoting osteogenesis.11 Matrix stiffness also influences the progression of valvular calcification.12 Cells respond to changes in matrix stiffness by mechanotransduction, whereby external physical forces or cues are translated into biochemical responses within the cell. Cells sense matrix stiffness through matrix-binding receptors like integrins and DDRs that tug locally at adhesion sites to deform the matrix.13 A given resistance is relayed back to the cell through the adhesion according to the matrix stiffness. The resisting force is transduced through the actin cytoskeleton and activates the Rho GTPase, RhoA, to promote bundling of actin and myosin into stress fibers.14 Recent studies have shown that in adipose stromal cells and fibroblasts, DDR1 responds to changes in matrix stiffness even in the absence of β1-integrins by interacting with the cytoskeletal protein NMMIIA (nonmuscle myosin IIA).15,16 Furthermore, DDR1 activates RhoA in T-cells during cell migration.17
In this study, we hypothesize that DDR1 regulates the transdifferentiation of VSMCs to osteochondrocytic cells by sensing matrix stiffening during disease progression and transmitting contractile forces through the actin cytoskeleton. Here, we use an in vitro model of VSMC calcification with cells grown on substrates of different stiffness and show that calcification increases with stiffness in a DDR1-dependent manner. Runx2 nuclear localization and expression of osteochondrocytic markers increase in a DDR1-dependent and substrate stiffness-dependent manner in wild-type (WT) VSMCs following long-term culture in calcifying media. Stiffness regulates RhoA activity and stress fiber formation through the RhoGEF (rho guanine nucleotide exchange factor), Vav2. We have made the novel observation that DDR1 activation and expression are positively regulated by stiffness and feedback regulation by actomyosin contractility downstream of RhoA activation. Finally, we observe that RhoA activity and actomyosin contractility are positive regulators of Runx2 nuclear localization and expression and also long-term calcification.
Materials and Methods
Data, materials, and methods will be made available to others by the corresponding author upon request for reproducibility purposes. Detailed methods are presented in the Data Supplement.
Cell Culture
Primary Ddr1+/+ (WT) or Ddr1−/− (knockout) VSMCs were isolated as previously described,18 and cultured in normal media (5.5 mmol/L glucose DMEM [11885084; Gibco], 10% FBS [12483020; Thermo Fisher], and 1% penicillin-streptomycin [15140122; Thermo Fisher]) or calcifying media (25 mmol/L glucose DMEM [11995065; Gibco], 3% heat-inactivated FBS [12483020], 1% penicillin-streptomycin [15140122], and 2.4 mmol/L inorganic phosphate). Silicon substrates were generated using the Sylgard 527 A&B Silicone Dielectric Gel Kit [1696742; Dow Corning] to obtain the desired elastic modulus (stiffness), as published previously by Calve and Simon.19 The silicon substrates were treated with 30 μg/cm2 PureCol bovine Col-I (collagen-I; 5005; Advanced BioMatrix) in Ca2+ and Mg2+-free PBS and allowed to coat overnight at 4°C.
Spin-coating silicon substrates on glass slides were performed as described under Methods in the Data Supplement. VSMCs were seeded on substrates of different stiffnesses in 6-well plates at 1563 cells/cm2 and grown to ≈70% to 80% confluency. Cells were cultured in calcifying media for 12 days, changing media every 2 days, to induce calcification. For long-term blebbistatin treatment, Blebbistatin was added at 25 μM with every media change. Calcification was assessed with Alizarin Red staining or o-Cresolphthalein colorimetric assay (Sigma Aldrich). Total calcium in μg was normalized to total protein in mg measured by detergent compatible protein assay (5000112; Bio-Rad). VSMCs were grown to 50% confluency before lipofectamine-mediated transfection with negative control (AM4611; Thermo Fisher) or Vav2 (AM16708; Thermo Fisher) siRNA (small interfering RNA) at a final concentration of 10 nM.
Immunoblotting, Subcellular fractionation, and Immunoprecipitation
Immunoblotting was performed as previously described,5 using antibodies identified in the Major Resources Table in the Data Supplement. Western blots were imaged using the ChemiDoc Touch Imaging System (Bio-Rad) and quantified with Bio-Rad Image Lab. For subcellular fractionation, 500 000 WT or knockout VSMCs were seeded on silicon substrates in 15 cm dishes and grown to confluency in normal media before culturing in calcifying media for 2 days. Subcellular fractionation was performed as previously described.5 For immunoprecipitation, serum-starved, confluent 15 cm dishes of WT VSMCs were treated with 10 μg/mL soluble Col-I for 3 hours before lysis with 1× RIPA (radioimmunoprecipitation assay) buffer (9806; Cell Signaling Technology) supplemented with EDTA-free mini complete protease inhibitor cocktail tablet (04693159001; Roche), 1 μM PMSF, and 200 nM NaF (sodium fluoride). DDR1 antibody (1:100; 5583) was added to 500 μg protein and incubated overnight at 4°C with rotation. Thirty microliters of 50% protein A agarose bead slurry (9863; Cell Signaling Technology) was added and rotated at 4°C for 3 hours. Samples were centrifuged at 5000×g for 1 minute to pellet beads and washed 3× by resuspending beads with 1× RIPA buffer and centrifuging at 5000×g for 1 minute or 3 minutes for the final wash. The beads were resuspended in 30 μL 2× sample buffer, and protein was eluted by boiling for 5 minutes.
RhoA Activity G-LISA and Total RhoA E-LISA
VSMCs were grown in normal media on Col-I–coated silicon substrates in 10 cm dishes or on Col-I–coated plastic to ≈70% to 80% confluency before serum starvation overnight. RhoA activity was stimulated with 10% FBS and 0.25 μg/mL ACT (Rho activator II; CN04; Cytoskeleton) for 3 hours. ACT deamidates Gln-63 (glutamine-63) of RhoA to block GTPase activity, thus preventing inactivation of endogenously activated RhoA, allowing for a more accurate assessment of RhoA activation after 3 hours of serum stimulation. Cells were lysed with G-LISA (GTPase-linked immunosorbent assay) lysis buffer from the RhoA Activity G-LISA kit (BK124; Cytoskeleton). RhoA activity was measured with the RhoA Activity G-LISA kit (BK124; Cytoskeleton) and normalized to total RhoA measured by RhoA E-LISA (enzyme-linked immunosorbent assay; BK150; Cytoskeleton) according to the manufacturer’s instructions.
Immunocytochemistry
To assess stiffness-dependent Runx2 nuclear localization and stress fiber formation, 100 000 VSMCs were seeded on silicon spin-coated slides and allowed to attach for 24 hours before 48 hours of culture in calcifying media. To examine Col-I–mediated stress fiber formation with Vav2 siRNA, VSMCs were seeded on uncoated or Col-I–coated coverslips and allowed to attach for 24 hours before control or Vav2 siRNA transfection for 24 hours. VSMCs were serum-starved overnight and serum-stimulated for 3 hours. For Runx2 immunostaining, WT VSMCs were seeded at 6000 cells per well in 8-well chamber slides (0030742079; Eppendorf) and allowed to attach for 24 hours before 48 hours of culture in calcifying media supplemented with 0.25 μg/mL ACT or 1 μg/mL C3 (C3 exoenzyme). Immunostaining was performed, as previously described,5 immunolabelling for Runx2 (1:100; 12556; Cell Signaling Technology), 1× AlexaFluor488 Phalloidin (A12379; Thermo Fisher), AlexaFluor488 goat anti-rabbit (1:200; A11008; Thermo Fisher), or AlexaFluor568 goat anti-rabbit secondary antibody (1:200; A11011; Thermo Fisher). Nuclei were stained with Hoechst-33342. VSMCs on spin-coated slides and stained for Runx2 were imaged with the Zeiss AxioObserver.Z1 confocal microscope. VSMCs transfected with siRNAs were imaged with the Nikon Eclipse Ci epifluorescence microscope. Zeiss Zen 3.0 blue was used to quantify total nuclear and cytoplasmic fluorescence intensity to calculate the nuclear to cytoplasmic ratio of Runx2. All samples were mounted with ProLong Gold Antifade mountant (P36930; Thermo Fisher).
Live Cell Imaging and Laser Cutting Stress Fibers
Briefly, WT VSMCs were seeded on Col-I–coated 35 mm2 glass-bottomed dishes and transfected with Ddr1b-YFP (full length Ddr1b isoform tagged to yellow fluorescent protein; gift from Dr Christopher McCulloch and Dr Nuno Coelho, University of Toronto) and Actin-mApple (gift from Dr Sergey Plotnikov, University of Toronto) plasmids with Lipofectamine 3000 (L3000008; Thermo Fisher) for 24 hours. Images were taken using a Revolution XD confocal microscope (Andor) with a 60× oil-immersion lens (numerical aperture [NA] 1.35; Olympus). Stress fibers were cut with a MicroPoint N2 laser (Andor) tuned to 365 nm, delivering 10 pulses at a diffraction-limited spot on the fiber. Images were acquired before and after cutting, and every 10 s henceforth up to 2 minutes postcut. Images were acquired with an iXon Ultra 897 camera (Andor) and Metamorph software (Molecular Devices). For quantification, 3 ROIs (regions of interest) were selected per cell: cut site (cut), away from cut site with clusters (control), and away from cut site without clusters (background). Fiducial markers were centered on each cluster at each time point using the image analysis software SIESTA.20 Using custom-written code in MATLAB, the average intensity of each cluster were normalized to the mean intensity of the background to correct for photobleaching. The resulting intensity curves were normalized to the initial (precut) intensity.
Data and Statistical Analysis
Statistical analysis was performed with Graphpad Prism 5. Statistical significance was assessed by Student t test, 1-way ANOVA, or 2-way ANOVA with Bonferroni post hoc test. P<0.05 was considered statistically significant and denoted by asterisks. Image processing was done on Nikon NIS Elements with the Nikon Eclipse Ci epifluorescence microscope, Zen 3.0 Blue with the Zeiss AxioObserver.Z1 confocal microscope, and ImageJ with the Revolution XD confocal microscope.
Results
Increased Substrate Stiffness Promotes In Vitro Calcification and Osteochondrocytic Transdifferentiation in WT VSMCs but Not Knockout VSMCs
To determine whether VSMC calcification is dependent upon DDR1 and substrate stiffness, WT and knockout VSMCs were cultured on Col-I–coated silicon substrates or plastic plates for 12 days in calcifying media. Alizarin Red staining was used to assess hydroxyapatite deposition. WT VSMCs displayed no detectable Alizarin Red staining at 5 kPa and had increased staining with increased substrate stiffness (Figure 1A). By contrast, knockout VSMCs did not display increases in Alizarin Red staining with increased stiffness (Figure 1A). Matrix calcium content increased significantly at 100 compared with 5 kPa in WT but not in knockout VSMCs (Figure 1B). Increased matrix stiffness alone was insufficient to induce calcification in the absence of calcifying media (Figure I in the Data Supplement). Nuclear localization of the osteogenic transcription factor, Runx2, was assessed to determine whether VSMCs transdifferentiated to the osteochondrocytic phenotype. WT and knockout VSMCs were cultured on 5, 50, or 100 kPa substrates in calcifying media for 2 days before subcellular fractionation and immunoblot. Runx2 increased in the nucleus and cytoplasm with increasing substrate stiffness in WT VSMCs but was not stiffness-responsive and was attenuated above 50 in knockout VSMCs compared to WT VSMCs (Figure 1C through E). Similarly, Runx2 nuclear to cytoplasmic ratio was higher at 100 than at 5 kPa in WT VSMCs but was not stiffness-responsive in knockout VSMCs (Figure 1F and 1G). To assess whether DDR1 affects mechanosensitive changes of pro-osteogenic signaling in other cell types, C3H10T1/2 mesenchymal stem cells were grown on 5 or 100 kPa substrates in osteogenic media supplemented with the DDR1 inhibitor, D1in1 (DDR1-in-1), which binds to the kinase domain and prevents autophosphorylation. D1in1 prevented stiffness-mediated enhancement of osteogenesis (Figure II in the Data Supplement). Taken together, this data suggests that DDR1 regulates osteogenic signaling in response to substrate stiffness.
Figure 1.
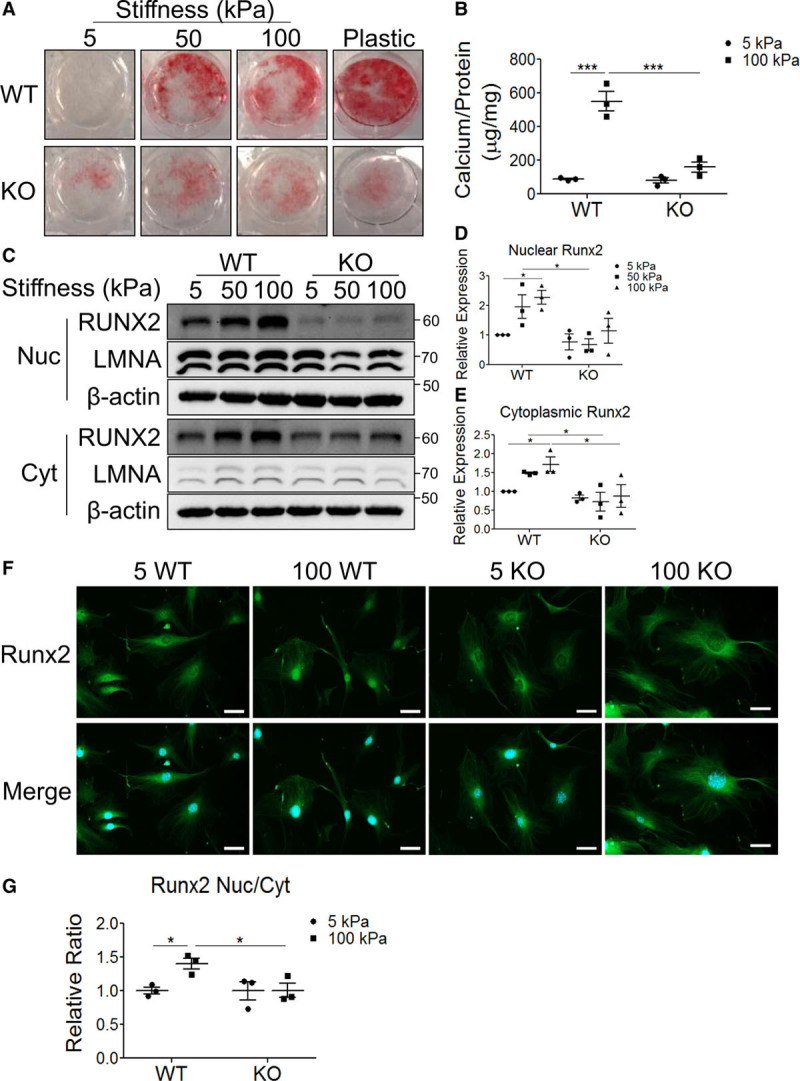
Increased stiffness promotes in vitro calcification and Runx2 (Runt-related transcription factor 2) nuclear localization in wild-type (WT) vascular smooth muscle cells (VSMCs). A and B, Ddr1+/+ (WT) or Ddr1−/− (knockout [KO]) VSMCs cultured in calcifying media were (A) stained with the calcium stain Alizarin Red (n=3) or (B) quantified for deposited calcium by o-cresolphthalein assay (n=3) normalized to total protein quantified by Bradford assay. C, Cytoplasmic (Cyt) and nuclear (Nuc) fractions from WT and KO VSMCs cultured in calcifying media for 2 d were immunoblotted for Runx2 (n=3). D, Nuclear Runx2 and (E) Cyt Runx2 were quantified by densitometry and normalized to Lamin A/C (LMNA) and β-actin respectively. F, WT and KO VSMCs cultured in calcifying media for 2 d were immunostained for Runx2. Scale bar represents 50 μm. G, Runx2 Nuc to Cyt ratio was quantified and expressed relative to 5 kPa WT. *P<0.05, ***P<0.001, bars represent means±SEM. B, D, E, and G, Statistical analysis was performed by 2-way ANOVA with Bonferroni post hoc test. DDR1 indicates discoidin domain receptor-1.
To assess the extent of VSMC osteochondrocytic transdifferentiation with long-term calcification, WT and knockout VSMCs were cultured in calcifying media for 12 days on 5 or 100 kPa substrates before immunoblotting for DDR1 and the osteochondrocytic markers Runx2, Sox9 (SRY [sex-determining region Y]-box 9), BMP-2 (bone morphogenetic protein-2), and OPN (osteopontin) (Figure 2A). We noted that DDR1 expression increased with increased stiffness (Figure 2A and 2B). At 5 kPa, Runx2 expression was higher in WT VSMCs than knockout VSMCs, although the expression of the other osteochondrocytic markers was not significantly different (Figure 2A, 2C through 2F). Expression of all markers increased with increased stiffness in WT but not knockout VSMCs (Figure 2A, 2C through 2F). These results suggest that DDR1 expression is correlated with the stiffness-sensitive osteochondrocytic transdifferentiation of VSMCs.
Figure 2.
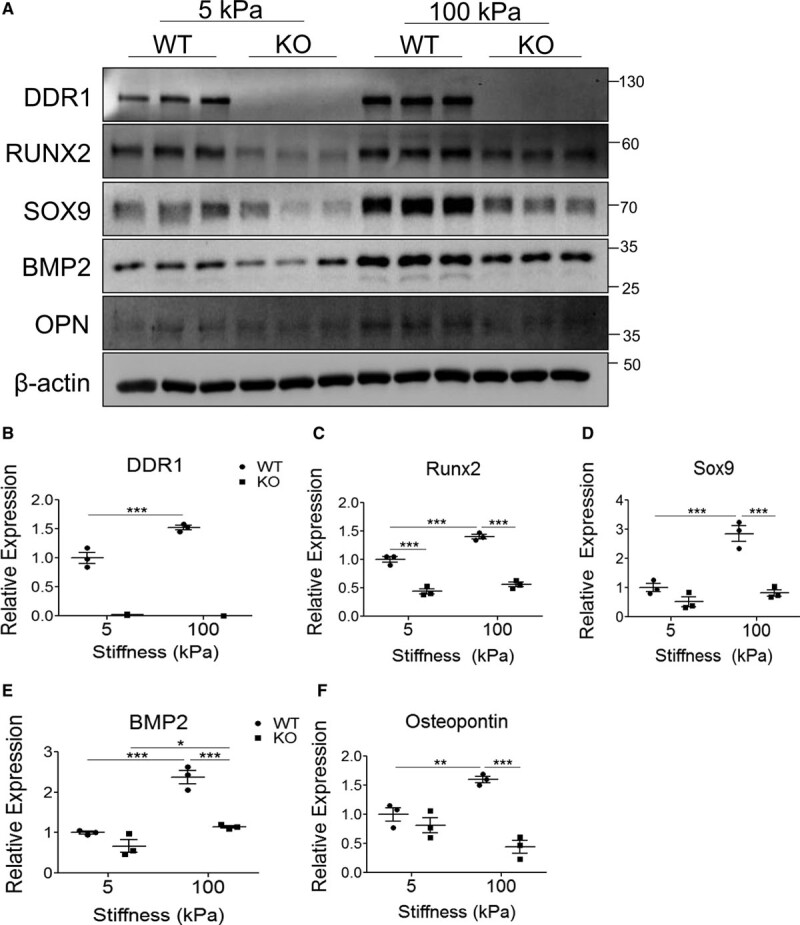
Increased stiffness promotes DDR1 (discoidin domain receptor-1) expression and osteochondrocytic differentiation in wild-type (WT) vascular smooth muscle cells (VSMCs) with long-term in vitro calcification. A, WT and knockout (KO) VSMCs grown on 5 or 100 kPa substrates were cultured in calcifying media for 12 d before lysis and immunoblot (n=3). B–F, Densitometry quantification of (B) DDR1 and (C–F) osteochondrocytic marker expression. *P<0.05, **P<0.01, ***P<0.001. Bars represent means±SEM. B–F, Statistical analysis was performed by 2-way ANOVA with Bonferroni post hoc test. BMP-2 indicates bone morphogenetic protein-2; OPN osteopontin; Runx2, Runt-related transcription factor 2; and Sox, SRY (sex-determining region Y)-box 9.
Increased Stiffness Promotes DDR1 Expression and Phosphorylation, and RhoA Activity and Stress Fiber Formation in WT VSMCs
To determine whether DDR1 is a mechanosensor, we measured receptor phosphorylation and expression in response to changes in substrate stiffness. WT VSMCs were allowed to attach on 5 or 100 kPa Col-I–coated substrates for 3 hours in normal media before immunoblot. pDDR1/DDR1 (phosphorylated DDR1 to total DDR1) ratio was increased at 100 compared with at 5 kPa (Figure 3A and 3B). To assess DDR1 expression in response to increased substrate stiffness, WT VSMCs were grown on substrates of different stiffness for 3 days in normal media and we found that DDR1 protein expression increased with substrate stiffness (Figure 3C). We reasoned that if DDR1 is a mechanosensor, it should regulate stress fiber formation in response to changes in stiffness. Stress fiber formation was visualized in WT and knockout VSMCs that were seeded on 5 or 100 kPa substrates and cultured in calcifying media for 2 days. Stress fiber formation increased with increased substrate stiffness in WT VSMCs but was not stiffness-responsive and was attenuated at 100 kPa in knockout VSMCs compared with WT VSMCs (Figure 3D). Actin polymerization, stress fiber formation, and enhanced myosin contraction of stress fibers are regulated by the small Rho GTPase, RhoA. RhoA G-LISA assay was performed to measure activity. VSMCs were cultured on Col-I–coated silicon substrates or plastic in normal media until ≈70% to 80% confluency. Cells were serum-starved overnight, then serum-stimulated to activate RhoA, and treated with ACT to maintain RhoA in its active state. RhoA activity increased with increasing stiffness in WT VSMCs but was not stiffness-responsive and was attenuated above 50 kPa in knockout VSMCs compared to WT VSMCs (Figure 3E).
Figure 3.
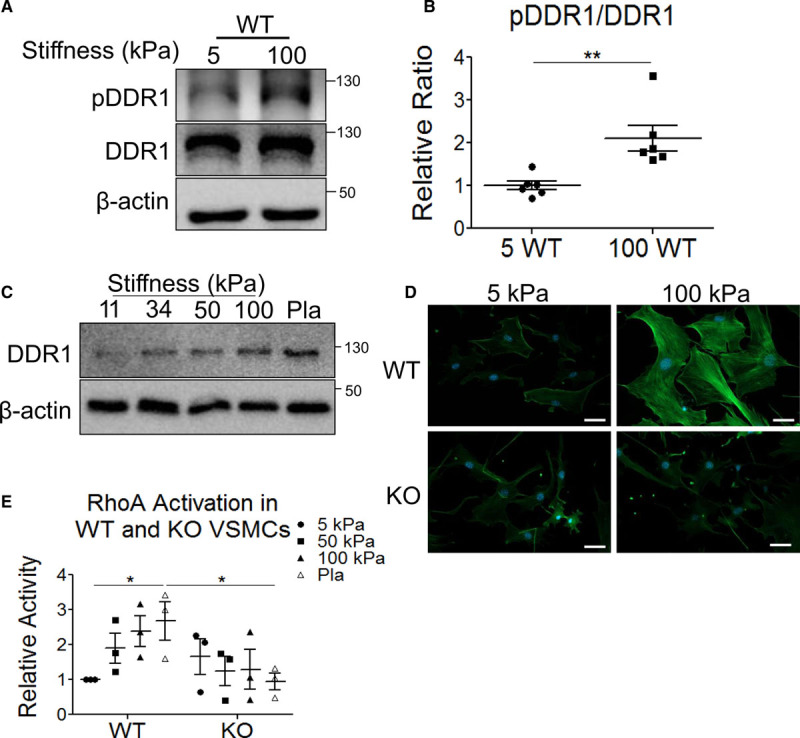
Increased stiffness promotes DDR1 (discoidin domain receptor-1) phosphorylation and expression as well as RhoA (ras homolog family member A) activity and stress fiber formation in wild-type (WT) vascular smooth muscle cells (VSMCs). A, WT VSMCs were seeded on 5 or 100 kPa substrates and lysed after 3 h before immunoblotting (n=6). B, pDDR1/DDR1 (phosphorylated DDR1 to total DDR1) ratios were quantified by densitometry and expressed relative to 5 kPa. C, DDR1 expression in WT VSMCs cultured on substrates of varying stiffnesses (n=3). D, WT and knockout (KO) VSMCs on 5 or 100 kPa substrates were stained with phalloidin to visualize F-actin stress fibers (n=3). Phalloidin staining was imaged at ×200 magnification. Scale bar represents 50 μm. E, Active to total RhoA ratios were calculated by G-LISA (GTPase-linked immunosorbent assay) and E-LISA (enzyme-linked immunosorbent assay) for WT and KO VSMCs cultured on different substrate stiffnesses, and expressed relative to 5 kPa WT (n=3). *P<0.05, **P<0.01, bars represent means±SEM. B, Statistical analysis was performed by Student t test. E, Statistical analysis was performed by 2-way ANOVA with Bonferroni post hoc test.
Vav2 Promotes Col-I–Dependent Stress Fiber Formation
To investigate the link between DDR1 and RhoA activation, the RhoGEF, Vav2, was examined. Vav2 can activate RhoA21 and interacts with pY484 in the cytoplasmic region of DDR1.22 First, we determined whether Vav2 was stiffness-responsive. The pVav2/Vav2 ratio increased at 100 compared with 5 kPa in WT VSMCs but was not stiffness-responsive and was attenuated at 100 kPa in knockout VSMCs compared with WT VSMCs (Figure 4A and 4B). To determine whether DDR1 and Vav2 interacted, WT VSMCs cultured on plastic were treated with soluble Col-I before immunoprecipitation of DDR1. Col-I stimulation promoted phosphorylation of DDR1 and increased Vav2 pulldown with DDR1 (Figure 4C).
Figure 4.
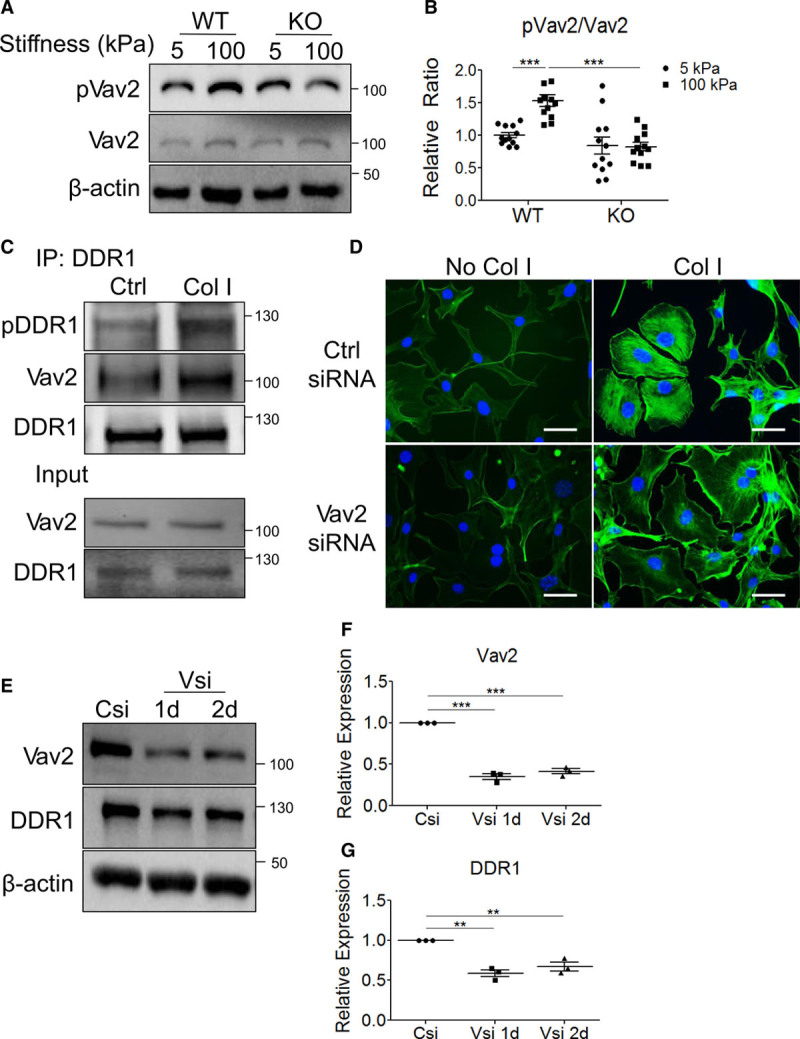
Vav2 promotes Col-I (collagen-I)–dependent stress fiber formation. A, Wild-type (WT) or knockout (KO) vascular smooth muscle cells (VSMCs) were seeded on 5 or 100 kPa substrates and lysed after 3 h before immunoblotting (n=12). B, pVav2/Vav2 (phosphorylated Vav2 to total Vav2) ratios were quantified by densitometry and expressed relative to 5 kPa WT. C, DDR1 (discoidin domain receptor-1) was immunoprecipitated (IP) using WT VSMCs with or without Col-I stimulation (n=3). D, WT VSMCs transfected with control (Ctrl) siRNA (small interfering RNA; Csi) or Vav2 siRNA (Vsi) were serum-stimulated for 3 h on uncoated coverslips or Col-I–coated coverslips, then stained with phalloidin (n=3). Phalloidin staining was imaged at 400x magnification. Scale bar represents 50 μm. E, WT VSMCs were transfected with Csi or Vsi and lysed after 1 or 2 d (n=3). Quantification of immunoblots in (E) is shown for (F) Vav2 and (G) DDR1, with values expressed relative to Csi. B, F, and G, **P<0.01, ***P<0.001, bars represent means±SEM, and statistics were done by (F and G) 1-way or (B) 2-way ANOVA with Bonferroni post hoc tests.
To determine whether Col-I promotes Vav2-dependent stress fiber formation, serum-starved WT VSMCs on uncoated or Col-I–coated coverslips transfected with control or Vav2 siRNA (small interfering RNA) were serum-stimulated for 3 hours to induce stress fiber formation before phalloidin staining. Col-I enhanced stress fiber formation was blocked by Vav2 knockdown (Figure 4D). Vav2 siRNA decreased Vav2 protein level by 60% up to 2 days post-transfection compared to control siRNA, thus validating knockdown efficacy and Vav2 knockdown at the time of phalloidin staining (Figure 4E and 4F). Vav2 knockdown also decreased DDR1 protein level by 40% up to 2 days post-transfection (Figure 4E and 4G). This suggests potential Vav2-dependent regulation of DDR1 expression.
RhoA, Cell Contractility, and Actin Dynamics Mediate Feedback Regulation of DDR1 Expression, Phosphorylation, and Clustering
Because DDR1 protein expression was changed by increased substrate stiffness and in response to Vav2 knockdown, we postulated that a positive feedback loop may exist to regulate DDR1 expression, as illustrated in Figure III in the Data Supplement. Little is known about the regulation of DDR1 expression, but one previous study reported that prolonged Col-I treatment increased DDR1 expression in fibroblasts, supporting potential feedback regulation.23 To investigate this, serum-starved WT VSMCs grown in normal media were treated with soluble Col-I or D1in1 to activate and inhibit DDR1, respectively, and ACT or C3 to activate and inhibit RhoA, respectively, before cell lysis and immunoblotting for DDR1.
First, we determined whether RhoA was involved in the regulation of DDR1 protein expression. RhoA activation (ACT) or inhibition (C3) increased or decreased DDR1 expression respectively (Figure 5A and 5B). DDR1 inhibition (D1in1) abolished the ACT-mediated increase in DDR1 (Figure 5A and 5B). Treatment with C3 reduced the stimulation of DDR1 expression by Col-1. This data suggests that a feedback loop exists between DDR1 and RhoA with RhoA activation by DDR1 resulting in DDR1 upregulation (Figure III in the Data Supplement).
Figure 5.
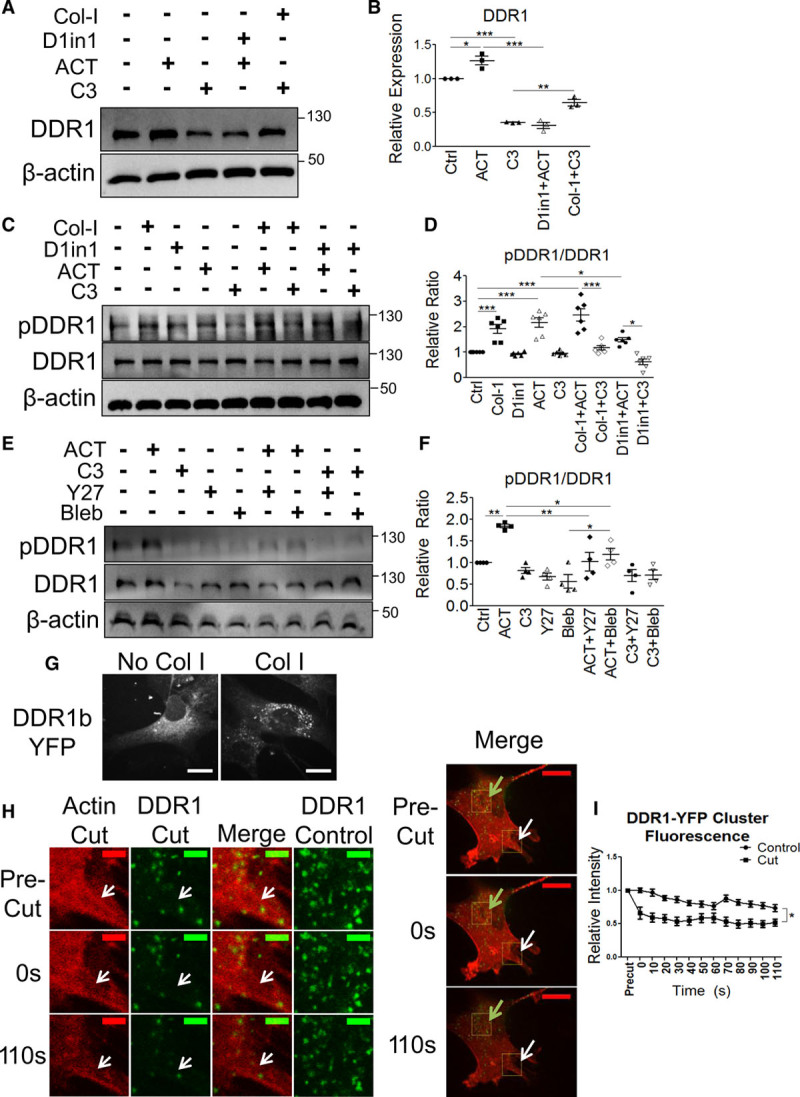
RhoA (ras homolog family member A) and actomyosin contractility mediate feedback regulation of DDR1 (discoidin domain receptor-1) expression, phosphorylation, and clustering. A, Serum-starved wild-type (WT) vascular smooth muscle cells (VSMCs) were treated with combinations of Col-I (collagen-I), D1in1 (DDR1-in-1), ACT (Rho activator II), or C3 (C3 exoenzyme) for 24 h, immunoblotted (n=3), and (B) DDR1 expression was quantified. C, Serum-starved WT VSMCs were treated for 3 h with Col-I or ACT or 4 h with D1in1 or C3, immunoblotted (n=6), and (D) pDDR1/DDR1 ratio was quantified. E, WT VSMCs were treated for 3 h with ACT or 4 h with C3, Y27 (Y-27632 [Rho-associated protein kinase inhibitor]), or blebbistatin (Bleb), immunoblotted (n=4), and F, pDDR1/DDR1 ratio was quantified. DDR1 expression or pDDR1/DDR1 ratios were quantified and expressed relative to control (Ctrl). G, WT VSMCs seeded on uncoated glass or Col-I -coated glass-bottomed dishes and transfected with DDR1b-YFP (yellow fluorescent protein-tagged full length DDR1b) plasmid were imaged at 600x magnification. Scale bar represents 50 μm. H, WT VSMCs expressing DDR1b-YFP and Actin-mApple were plated on Col-I. Stress fibers were cut with a UV laser then imaged for 2 min. Representative images for Actin-mApple and DDR1b-YFP at the cut site where the actin stress fiber was cut and DDR1b-YFP at a Ctrl site are shown. The merged images on the right show the cut site (white arrow) and the Ctrl site (green arrow). Scale bars represent 10 μm (left images) or 50 μm (right merged images). I, DDR1b-YFP cluster fluorescence intensity in the vicinity of the cut was quantified and compared to the Ctrl region. Means were derived from 17 clusters for Ctrl and 16 clusters for cut quantified from 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, bars or points represent means±SEM. D–F, Statistical analysis was performed by 1-way ANOVA or (I) 2-way ANOVA with Bonferroni post hoc test.
Next, we assessed the effect of Col-I stimulation and RhoA on DDR1 phosphorylation. Treating WT VSMCs with Col-I or ACT alone increased pDDR1 (phosphorylated DDR1) levels to a similar extent, whereas C3 treatment alone did not affect pDDR1 levels compared to control (Figure 5C and 5D). Treatment with both Col-I and ACT had a mild additive effect on DDR1 phosphorylation compared with either treatment alone (Figure 5C and 5D). Treatment with D1in1 and ACT attenuated ACT-induced increase in pDDR1, and C3 inhibitor treatment attenuated the Col-I–mediated increase in pDDR1 (Figure 5C and 5D). Taken together, this data suggests that DDR1 activation is dependent upon Col-I stimulation and RhoA activity.
Finally, since RhoA can promote actomyosin contractility, we sought to determine whether cell contractility is important for DDR1 activation. Contractility is stimulated by ROCK (Rho-associated protein kinase) and is mediated by NMMIIA activity on actin fibers. Y27 (Y-27632 [Rho-associated protein kinase inhibitor]) was used to inhibit ROCK, and blebbistatin was used to inhibit NMMIIA downstream of RhoA. Treatment with either Y27 or blebbistatin inhibited ACT-mediated increases in pDDR1, suggesting that feedback regulation of DDR1 activity occurs at the level of actomyosin contractility (Figure 5E and 5F). Concomitant treatment with C3 and Y27 or blebbistatin did not further decrease pDDR1 compared to C3 alone. Thus, positive feedback regulation of DDR1 activity occurs through a linear pathway with cell contraction downstream of RhoA (Figure III in the Data Supplement).
DDR1 activation requires DDR1 clustering followed by receptor phosphorylation,24,25 so we also measured receptor clustering. WT VSMCs were transfected with DDR1b-YFP, and clusters were visualized using confocal microscopy. DDR1b-YFP clusters formed on Col-I coated glass, but not on uncoated glass (Figure 5G). To determine whether DDR1 clustering was influenced by actomyosin contractility, Actin-mApple labeled stress fibers were cut by a UV laser and DDR1 clusters were imaged by time-lapse confocal microscopy. After cutting a stress fiber, cluster fluorescence intensity in the cut region was significantly reduced compared with clusters in the control region (Figure 5H and 5I).
Because RhoA affects actin polymerization, we sought to determine whether DDR1 activation was dependent upon actin polymerization or actin stabilization. We used Jas (jasplakinolide), an actin stabilizing molecule that promotes actin polymerization, and LatA (latrunculin A), an actin-depolymerizing agent which stabilizes monomeric G-actin.26,27 Concomitant treatment of Jas or LatA with ACT attenuated RhoA-mediated increases in pDDR1 (Figure IV in the Data Supplement). Also, both Jas and LatA when added in conjunction with C3 further reduced DDR1 phosphorylation compared with either treatment alone. We conclude that it is the loss of dynamically remodeling stress fibers that suppress DDR1 phosphorylation.
RhoA Activity and Actomyosin Contractility Regulate Runx2 Nuclear Localization and Expression and VSMC Calcification
We examined whether RhoA and actomyosin contractility regulate Runx2 activity and VSMC calcification. First, the effect of RhoA on Runx2 nuclear localization was assessed. WT VSMCs were cultured in calcifying media for 2 days supplemented with ACT or C3 before immunostaining for Runx2. ACT treatment resulted in a statistically significant increase in nuclear to cytoplasmic ratio of Runx2 compared to control cells, whereas C3 did not significantly affect the ratio compared to control cells (Figure 6A and 6B). We think that Runx2 activation is a surrogate marker for calcification, and thus calcification would likely be promoted by RhoA activity. Next, WT VSMCs were cultured for 2 days in calcifying media supplemented with either ACT, C3, or blebbistatin and immunoblotted for pDDR1, DDR1, and Runx2 (Figure 6C). Both pDDR1 and DDR1 was increased with ACT and reduced with C3 or blebbistatin treatment compared with control. Runx2 expression was significantly increased with ACT and significantly decreased with C3 or blebbistatin compared with control (Figure 6C and 6D). Finally, WT or knockout VSMCs were cultured for 12 days with or without blebbistatin and stained with Alizarin Red to determine whether actomyosin contractility was required for VSMC calcification. WT VSMCs had greater calcification than knockout VSMCs, whereas blebbistatin treatment attenuated WT VSMC calcification to a similar level as knockout VSMCs (Figure 6E and 6F).
Figure 6.
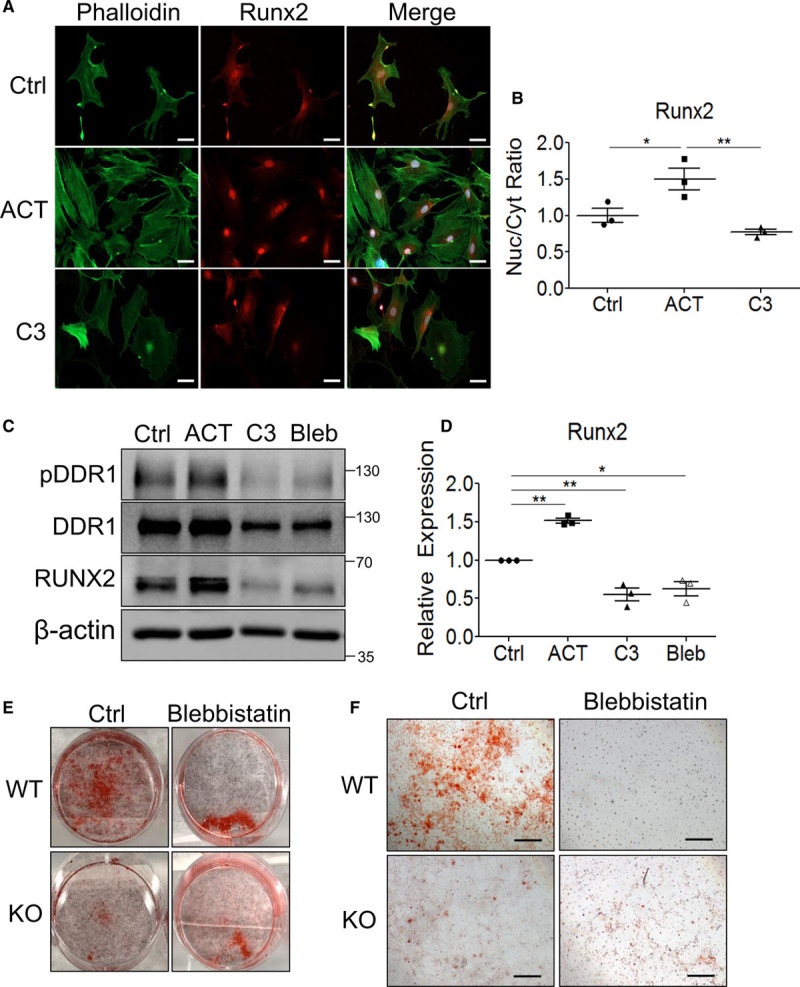
RhoA (ras homolog family member A) and actomyosin contractility promote Runx2 (Runt-related transcription factor 2) nuclear (Nuc) localization and expression as well as vascular smooth muscle cells (VSMC) calcification. A, Wild-type (WT) VSMCs cultured in calcifying media for 2 days with ACT (Rho activator II) or C3 (C3 exoenzyme) were stained with AlexaFluor488 phalloidin and Runx2 primary antibody with AlexaFluor568 anti-rabbit secondary antibody and imaged at ×200 magnification by confocal microscopy (n=3). Scale bar represents 50 μm. B, Nuc to cytoplasmic (Cyt) ratio was calculated with 10 to 30 cells per treatment group for each replicate. Ratios are expressed relative to control (Ctrl). C, WT VSMCs cultured in calcifying media for 2 days with ACT or C3 were immunoblotted (n=3). D, Runx2 expression was quantified by densitometry and normalized to β-actin. E, WT and knockout (KO) VSMCs were cultured in calcifying media for 12 days with or without blebbistatin before staining with Alizarin Red (n=3). F, Higher magnification images were taken from the wells shown in E. Scale bar represents 500 μm. B and D, *P<0.05, **P<0.01, bars represent means±SEM. Statistical analysis was performed by 1-way ANOVA with Bonferroni post hoc test. DDR1 indicates discoidin domain receptor-1.
Discussion
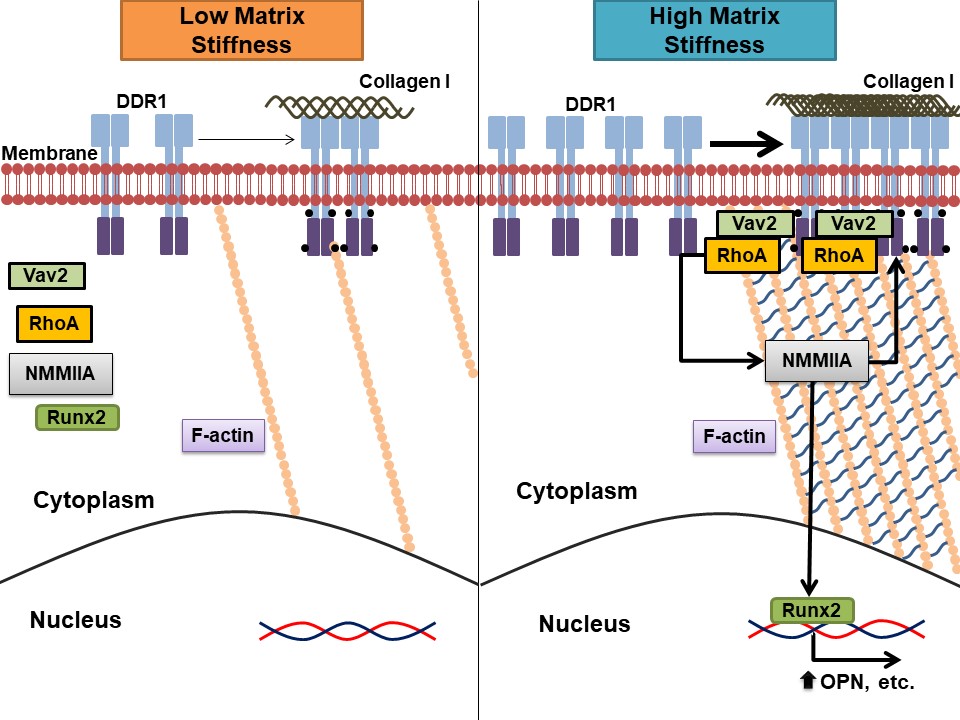
Our work makes an important contribution showing matrix stiffness as a driver of VSMC calcification and identifies DDR1 as a critical mechanosensor in this process. We have shown previously that DDR1 deficiency attenuated calcification and inhibited VSMC osteochondrocytic transdifferentiation both in vitro in primary VSMCs and in vivo in Ldlr−/− mice fed a high-fat diabetogenic diet.5 In this study, we show that increased substrate stiffness, over the range seen from fibro-fatty to mineralized atherosclerotic plaques,28–30 promotes VSMC osteochondrocytic transdifferentiation and calcification. RhoA and actomyosin contractility regulates Runx2 expression and nuclear localization and in vitro calcification. By contrast, the phenotypic shift and calcification are inhibited in knockout VSMCs. The underlying mechanism involves a DDR1-Vav2–mediated increase in RhoA activity and stress fiber formation. We show that DDR1 activity and expression are increased with increased substrate stiffness. The mechanism involves positive feedback between DDR1 and actomyosin contractility, with cytoskeletal tension necessary for DDR1 clustering and activation.
VSMC transdifferentiation to an osteochondrocytic phenotype is critical to the pathogenesis of vascular calcification.31 Runx2 is a master regulator of a pro-osteogenic transcriptional network and Runx2 in VSMCs is a major driver of both intimal and medial calcification.3,32 Here, we show that DDR1 and increased substrate stiffness promote nuclear localization of Runx2 in VSMCs in calcifying media. Furthermore, increased stiffness promoted osteochondrocytic marker expression only in WT VSMCs, suggesting that stiffness-dependent VSMC transdifferentiation requires DDR1.
The pathogenesis of vascular calcification is characterized as a sequence of initiation and propagation events. Extracellular vesicle deposition forms foci for initiation and propagation.33 Stiffness mediated enhancement of VSMC transdifferentiation and subsequent calcification offers a complementary explanation for this progression. Regional heterogeneity of matrix stiffness in the vessel may result in foci above the threshold stiffness for osteochondrocytic transdifferentiation. Previous modeling of the heterogeneity of human atherosclerotic plaque stiffness showed a range of stiffnesses as low as 9 kPa and as high as 100 to 200 kPa.30 Initial VSMC transdifferentiation at stiffer matrix foci could lead to mineralization, stiffening the matrix, and triggering another wave of transdifferentiation. Subsequent waves of stiffening and VSMC transdifferentiation likely contribute to the propagation phase of vascular calcification.
Activation of RhoA mediates actin polymerization and stress fiber formation, and actomyosin contractility, allowing for the transmission of mechanical forces into the cell. We have shown previously that DDR1 deficiency disrupts both microtubule and actin cytoskeletons.5,34 In the current study, we focused on the actin cytoskeleton given the prominent role of actomyosin contractility in mechanosensing and the previous reports that DDR1 associates with NMMIIA.15 Stress fiber formation and RhoA activity increased with matrix stiffness in WT VSMCs, while these responses were attenuated in knockout VSMCs suggesting that DDR1 was important in stiffness-dependent RhoA activation. This was mediated via the RhoGEF, Vav2. Vav2 phosphorylation was enhanced by increased stiffness, which was prevented in knockout VSMCs. Col-I stimulation also increased the association of DDR1 with Vav2. Furthermore, Col-I stimulation promoted stress fiber formation, which was blocked by Vav2 siRNA. A limitation of our study is that Vav2 also regulates the Rho GTPases Rac1 (ras-related C3 botulinum toxin substrate 1) and Cdc42 (cell division control protein 42 homolog),21,35 and previous studies have implicated these GTPases in vascular calcification,36 so it remains to be determined if they are downstream of DDR1-Vav2 signaling in calcifying VSMCs. Nonetheless, our data suggest that DDR1 mediates stress fiber formation through Vav2. Increased matrix stiffness can promote RhoA activity which was found here to promote DDR1 activation and expression, which can then feedforward to further enhance RhoA activity.
An unexpected finding was that pharmacological activation of RhoA could activate DDR1 phosphorylation independent of Col-I, while inhibition of RhoA with C3 exoenzyme prevented DDR1 phosphorylation. This suggested that DDR1 activation was downstream of RhoA. This was confirmed by experiments showing that ROCK and NMMIIA inhibition prevented DDR1 phosphorylation and that laser ablation of stress fibers inhibited DDR1 clustering, which showed that DDR1 activation requires actomyosin contractility. The cycle between Col-I and actomyosin contractility in the regulation of DDR1 activation may be a mechanism for short-term responses to matrix stiffening.
Previous studies have suggested that fibrillar collagens potentiate DDR1 activation by clustering receptors along fibrils, allowing for lateral phosphorylation of DDR1 dimers.24 However, soluble (nonfibrillar) Col-I is also a potent activator of DDR1. DDR1 has been shown to promote fibril formation in soluble Col-I solutions, but DDR1 clustering occurs well before fibril formation.37 Thus, these studies suggest that binding to fibrils is not the primary driver of DDR1 clustering. Rather, DDR1 clustering and activation are likely initiated via another collagen receptor and mediated by actomyosin contractility. Our studies support the notion that force generation is required for DDR1 clustering and activation, and collagen fibers may reinforce this response.
We have also discovered that DDR1 protein expression is dependent upon the interplay between DDR1 and RhoA. We found that ACT treatment alone increased DDR1 expression in WT VSMCs, which was reduced by concomitant treatment with DDR1-in-1. Conversely, DDR1 expression decreased with C3 exoenzyme treatment but was rescued in part by concomitant treatment with soluble Col-I. This data was also supported by the reduction in DDR1 expression following Vav2 knockdown. In our experiments, it is unknown whether the increase in DDR1 protein expression was due to transcription or turnover. In fibroblasts, prolonged stimulation with Col-I increased DDR1 mRNA expression, suggesting an increase in transcription.23 The regulation of DDR1 protein expression by substrate stiffness and RhoA may be a mechanism for cells to acclimate to matrix stiffening over the long term.
Finally, in response to DDR1 signaling from a stiffening matrix, we found that RhoA and actomyosin contractility regulate Runx2 activity and expression and VSMC calcification. It is interesting to note that increased stiffness and contractility increase TGF (transforming growth factor)-β1 and BMP-2 release from the ECM (extracellular matrix),38,39 and also promote Smad (small mothers against decapentaplegic) and Wnt (wingless-related integration site) signaling.40,41 These pathways also promote Runx2 activation during calcification.36,42–44 We show that BMP-2 expression in WT VSMCs increases with increased stiffness, but is attenuated in knockout VSMCs. We have yet to determine if Smads are activated. However, DDRs promote BMP-Smad signaling in other organisms, such as Ciona robusta.45 Our previous work showed reduced pGSK3β in DDR1 knockout VSMCs, which implicates cross-talk with canonical Wnt signaling.5 Also, noncanonical Wnt signaling is associated with DDR1 activation in breast cancer.46 Further studies are needed to define the interplay between BMP, Wnt, and DDR1 signaling in VSMC mechanotransduction and calcification. Breaking the cycle of DDR1 and RhoA activation may be a potential therapeutic target for stiffness-mediated vascular calcification.
Acknowledgments
We acknowledge Alan Lam for protocols to prepare silicon substrates and Guangpei Hou for isolating mouse vascular smooth muscle cells.
Sources of Funding
This research was funded by a research grant MOP133592 from the Canadian Institutes of Health Research to Michelle P. Bendeck. D. Ngai was funded by a doctoral award from the Canadian Institutes of Health Research.
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- ACT
- Rho activator II
- BMP-2
- bone-morphogenetic protein-2
- C3
- C3 exoenzyme
- Col-I
- collagen-I
- D1in1
- DDR1-in-1
- DDR
- discoidin domain receptor
- ECM
- extracellular matrix
- Jas
- jasplakinolide
- LatA
- latrunculin A
- NMMIIA
- nonmuscle myosin IIA
- OPN
- osteopontin
- RhoA
- ras homolog family member A
- RhoGEF
- rho guanine nucleotide exchange factor
- ROCK
- Rho-associated Protein Kinase
- Runx2
- Runt-related transcription factor 2
- Sox9
- SRY (sex-determining region Y)-box 9
- TGF
- transforming growth factor
- VSMC
- vascular smooth muscle cell
- Y27
- Y-27632 (rho-associated protein kinase inhibitor)
For Sources of Funding and Disclosures, see page 1775.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314697.
References
- 1.Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5:185–197. doi: 10.2147/vhrm.s4822. doi: 10.2147/vhrm.s4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ngai D, Lino M, Bendeck MP. Cell-matrix interactions and matricrine signaling in the pathogenesis of vascular calcification. Front Cardiovasc Med. 2018;5:174. doi: 10.3389/fcvm.2018.00174. doi: 10.3389/fcvm.2018.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552. doi: 10.1161/CIRCRESAHA.112.267237. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad PJ, Trcka D, Xue S, Franco C, Speer MY, Giachelli CM, Bendeck MP. Discoidin domain receptor-1 deficiency attenuates atherosclerotic calcification and smooth muscle cell-mediated mineralization. Am J Pathol. 2009;175:2686–2696. doi: 10.2353/ajpath.2009.080734. doi: 10.2353/ajpath.2009.080734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lino M, Wan MH, Rocca AS, Ngai D, Shobeiri N, Hou G, Ge C, Franceschi RT, Bendeck MP. Diabetic vascular calcification mediated by the collagen receptor discoidin domain receptor 1 via the phosphoinositide 3-kinase/akt/runt-related transcription factor 2 signaling axis. Arterioscler Thromb Vasc Biol. 2018;38:1878–1889. doi: 10.1161/ATVBAHA.118.311238. doi: 10.1161/ATVBAHA.118.311238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackey RH, Venkitachalam L, Sutton-Tyrrell K. Calcifications, arterial stiffness and atherosclerosis. Adv Cardiol. 2007;44:234–244. doi: 10.1159/000096744. doi: 10.1159/000096744. [DOI] [PubMed] [Google Scholar]
- 7.Sims TJ, Rasmussen LM, Oxlund H, Bailey AJ. The role of glycation cross-links in diabetic vascular stiffening. Diabetologia. 1996;39:946–951. doi: 10.1007/BF00403914. doi: 10.1007/BF00403914. [DOI] [PubMed] [Google Scholar]
- 8.Lan TH, Huang XQ, Tan HM. Vascular fibrosis in atherosclerosis. Cardiovasc Pathol. 2013;22:401–407. doi: 10.1016/j.carpath.2013.01.003. doi: 10.1016/j.carpath.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Stabley JN, Towler DA. Arterial calcification in diabetes mellitus: preclinical models and translational implications. Arterioscler Thromb Vasc Biol. 2017;37:205–217. doi: 10.1161/ATVBAHA.116.306258. doi: 10.1161/ATVBAHA.116.306258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yahagi K, Kolodgie FD, Lutter C, Mori H, Romero ME, Finn AV, Virmani R. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2017;37:191–204. doi: 10.1161/ATVBAHA.116.306256. doi: 10.1161/ATVBAHA.116.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 12.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 13.Coelho NM, McCulloch CA. Mechanical signaling through the discoidin domain receptor 1 plays a central role in tissue fibrosis. Cell Adh Migr. 2018;12:348–362. doi: 10.1080/19336918.2018.1448353. doi: 10.1080/19336918.2018.1448353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51:7420–7432. doi: 10.1021/bi300758e. doi: 10.1021/bi300758e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coelho NM, Arora PD, van Putten S, Boo S, Petrovic P, Lin AX, Hinz B, McCulloch CA. Discoidin domain receptor 1 mediates myosin-dependent collagen contraction. Cell Rep. 2017;18:1774–1790. doi: 10.1016/j.celrep.2017.01.061. doi: 10.1016/j.celrep.2017.01.061. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, Ashcraft K, Jahid MJ, April C, Ghajar CM, Ruan J, Wang H, Foster M, Hughes DC, Ramirez AG, et al. Regulation of adipose oestrogen output by mechanical stress. Nat Commun. 2013;4:1821. doi: 10.1038/ncomms2794. doi: 10.1038/ncomms2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Azreq MA, Kadiri M, Boisvert M, Pagé N, Tessier PA, Aoudjit F. Discoidin domain receptor 1 promotes Th17 cell migration by activating the RhoA/ROCK/MAPK/ERK signaling pathway. Oncotarget. 2016;7:44975–44990. doi: 10.18632/oncotarget.10455. doi: 10.18632/oncotarget.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou G, Vogel W, Bendeck MP. The discoidin domain receptor tyrosine kinase DDR1 in arterial wound repair. J Clin Invest. 2001;107:727–735. doi: 10.1172/JCI10720. doi: 10.1172/JCI10720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calve S, Simon HG. Biochemical and mechanical environment cooperatively regulate skeletal muscle regeneration. FASEB J. 2012;26:2538–2545. doi: 10.1096/fj.11-200162. doi: 10.1096/fj.11-200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Gonzalez R, Zallen JA. Oscillatory behaviors and hierarchical assembly of contractile structures in intercalating cells. Phys Biol. 2011;8:045005. doi: 10.1088/1478-3975/8/4/045005. doi: 10.1088/1478-3975/8/4/045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, Burridge K, Der CJ. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–10149. doi: 10.1074/jbc.275.14.10141. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- 22.Lemeer S, Bluwstein A, Wu Z, Leberfinger J, Müller K, Kramer K, Kuster B. Phosphotyrosine mediated protein interactions of the discoidin domain receptor 1. J Proteomics. 2012;75:3465–3477. doi: 10.1016/j.jprot.2011.10.007. doi: 10.1016/j.jprot.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz PA, Jarai G. Collagen I induces discoidin domain receptor (DDR) 1 expression through DDR2 and a JAK2-ERK1/2-mediated mechanism in primary human lung fibroblasts. J Biol Chem. 2011;286:12912–12923. doi: 10.1074/jbc.M110.143693. doi: 10.1074/jbc.M110.143693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Juskaite V, Corcoran DS, Leitinger B. Collagen induces activation of DDR1 through lateral dimer association and phosphorylation between dimers. Elife. 2017;6:e25716. doi: 10.7554/eLife.25716. doi: 10.7554/eLife.25716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corcoran DS, Juskaite V, Xu Y, Görlitz F, Alexandrov Y, Dunsby C, French PMW, Leitinger B. DDR1 autophosphorylation is a result of aggregation into dense clusters. Sci Rep. 2019;9:17104. doi: 10.1038/s41598-019-53176-4. doi: 10.1038/s41598-019-53176-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 27.Yarmola EG, Somasundaram T, Boring TA, Spector I, Bubb MR. Actin-latrunculin A structure and function. Differential modulation of actin-binding protein function by latrunculin A. J Biol Chem. 2000;275:28120–28127. doi: 10.1074/jbc.M004253200. doi: 10.1074/jbc.M004253200. [DOI] [PubMed] [Google Scholar]
- 28.Sicard D, Fredenburgh LE, Tschumperlin DJ. Measured pulmonary arterial tissue stiffness is highly sensitive to AFM indenter dimensions. J Mech Behav Biomed Mater. 2017;74:118–127. doi: 10.1016/j.jmbbm.2017.05.039. doi: 10.1016/j.jmbbm.2017.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong KK, Thavornpattanapong P, Cheung SC, Sun Z, Tu J. Effect of calcification on the mechanical stability of plaque based on a three-dimensional carotid bifurcation model. BMC Cardiovasc Disord. 2012;12:7. doi: 10.1186/1471-2261-12-7. doi: 10.1186/1471-2261-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akyildiz AC, Speelman L, van Velzen B, Stevens RRF, van der Steen AFW, Huberts W, Gijsen FJH. Intima heterogeneity in stress assessment of atherosclerotic plaques. Interface Focus. 2018;8:20170008. doi: 10.1098/rsfs.2017.0008. doi: 10.1098/rsfs.2017.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600. doi: 10.1093/cvr/cvy010. doi: 10.1093/cvr/cvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin ME, Chen T, Leaf EM, Speer MY, Giachelli CM. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am J Pathol. 2015;185:1958–1969. doi: 10.1016/j.ajpath.2015.03.020. doi: 10.1016/j.ajpath.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med. 2018;5:36. doi: 10.3389/fcvm.2018.00036. doi: 10.3389/fcvm.2018.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu S, Bala S, Bendeck MP. Discoidin domain receptor 1 deficiency in vascular smooth muscle cells leads to mislocalisation of N-cadherin contacts. Biol Open. 2019;8:bio041913. doi: 10.1242/bio.041913. doi: 10.1242/bio.041913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabbiano S, Menacho-Márquez M, Sevilla MA, Albarrán-Juárez J, Zheng Y, Offermanns S, Montero MJ, Bustelo XR. Genetic dissection of the vav2-rac1 signaling axis in vascular smooth muscle cells. Mol Cell Biol. 2014;34:4404–4419. doi: 10.1128/MCB.01066-14. doi: 10.1128/MCB.01066-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng SL, Ramachandran B, Behrmann A, Shao JS, Mead M, Smith C, Krchma K, Bello Arredondo Y, Kovacs A, Kapoor K, et al. Vascular smooth muscle LRP6 limits arteriosclerotic calcification in diabetic LDLR−/− mice by restraining noncanonical Wnt signals. Circ Res. 2015;117:142–156. doi: 10.1161/CIRCRESAHA.117.306712. doi: 10.1161/CIRCRESAHA.117.306712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeung DA, Shanker N, Sohail A, Weiss BA, Wang C, Wellmerling J, Das S, Ganju RK, Miller JLC, Herr AB, et al. Clustering, spatial distribution, and phosphorylation of discoidin domain receptors 1 and 2 in response to soluble collagen I. J Mol Biol. 2019;431:368–390. doi: 10.1016/j.jmb.2018.11.015. doi: 10.1016/j.jmb.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wells RG, Discher DE. Matrix elasticity, cytoskeletal tension, and TGF-beta: the insoluble and soluble meet. Sci Signal. 2008;1:pe13. doi: 10.1126/stke.110pe13. doi: 10.1126/stke.110pe13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilde F, Fourel L, Guillot R, Pignot-Paintrand I, Okada T, Fitzpatrick V, Boudou T, Albiges-Rizo C, Picart C. Stiffness-dependent cellular internalization of matrix-bound BMP-2 and its relation to Smad and non-Smad signaling. Acta Biomater. 2016;46:55–67. doi: 10.1016/j.actbio.2016.09.014. doi: 10.1016/j.actbio.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lv H, Li L, Sun M, Zhang Y, Chen L, Rong Y, Li Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res Ther. 2015;6:103. doi: 10.1186/s13287-015-0083-4. doi: 10.1186/s13287-015-0083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du J, Zu Y, Li J, Du S, Xu Y, Zhang L, Jiang L, Wang Z, Chien S, Yang C. Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Sci Rep. 2016;6:20395. doi: 10.1038/srep20395. doi: 10.1038/srep20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Yang HY, Giachelli CM. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis. 2008;199:271–277. doi: 10.1016/j.atherosclerosis.2007.11.031. doi: 10.1016/j.atherosclerosis.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280:33132–33140. doi: 10.1074/jbc.M500608200. doi: 10.1074/jbc.M500608200. [DOI] [PubMed] [Google Scholar]
- 44.Cheng SL, Behrmann A, Shao JS, Ramachandran B, Krchma K, Bello Arredondo Y, Kovacs A, Mead M, Maxson R, Towler DA. Targeted reduction of vascular Msx1 and Msx2 mitigates arteriosclerotic calcification and aortic stiffness in LDLR-deficient mice fed diabetogenic diets. Diabetes. 2014;63:4326–4337. doi: 10.2337/db14-0326. doi: 10.2337/db14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernadskaya YY, Brahmbhatt S, Gline SE, Wang W, Christiaen L. Discoidin-domain receptor coordinates cell-matrix adhesion and collective polarity in migratory cardiopharyngeal progenitors. Nat Commun. 2019;10:57. doi: 10.1038/s41467-018-07976-3. doi: 10.1038/s41467-018-07976-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jönsson M, Andersson T. Repression of Wnt-5a impairs DDR1 phosphorylation and modifies adhesion and migration of mammary cells. J Cell Sci. 2001;114(Pt 11):2043–2053. doi: 10.1242/jcs.114.11.2043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.