

Supplemental Digital Content is available in the text.
Keywords: alkaline phosphatase, arteries, calcification, lower extremity
Abstract
Objective:
The recessive disease arterial calcification due to deficiency of CD73 (ACDC) presents with extensive nonatherosclerotic medial layer calcification in lower extremity arteries. Lack of CD73 induces a concomitant increase in TNAP (tissue nonspecific alkaline phosphatase; ALPL), a key enzyme in ectopic mineralization. Our aim was to investigate how loss of CD73 activity leads to increased ALPL expression and calcification in CD73-deficient patients and assess whether this mechanism may apply to peripheral artery disease calcification.
Approach and Results:
We previously developed a patient-specific disease model using ACDC primary dermal fibroblasts that recapitulates the calcification phenotype in vitro. We found that lack of CD73-mediated adenosine signaling reduced cAMP production and resulted in increased activation of AKT. The AKT/mTOR (mammalian target of rapamycin) axis blocks autophagy and inducing autophagy prevented calcification; however, we did not observe autophagy defects in ACDC cells. In silico analysis identified a putative FOXO1 (forkhead box O1 protein) binding site in the human ALPL promoter. Exogenous AMP induced FOXO1 nuclear localization in ACDC but not in control cells, and this was prevented with a cAMP analogue or activation of A2a/2b adenosine receptors. Inhibiting FOXO1 reduced ALPL expression and TNAP activity and prevented calcification. Mutating the FOXO1 binding site reduced ALPL promoter activation. Importantly, we provide evidence that non-ACDC calcified femoropopliteal arteries exhibit decreased CD73 and increased FOXO1 levels compared with control arteries.
Conclusions:
These data show that lack of CD73-mediated cAMP signaling promotes expression of the human ALPL gene via a FOXO1-dependent mechanism. Decreased CD73 and increased FOXO1 was also observed in more common peripheral artery disease calcification.
Highlights.
In the absence of CD73 activity and downstream adenosine signaling, exogenous AMP induces phosphorylation of AKT, nuclear localization of FOXO1 (forkhead box O1 protein), and promotes expression and activity of ALPL/TNAP (tissue nonspecific alkaline phosphatase).
CD73 expression is decreased in a calcified femoropopliteal arteries compared with control samples
Within the same vessel, calcified areas exhibit increased FOXO1 and TNAP staining than in noncalcified areas.
See accompanying editorial on page 1614
Vascular calcification is an active process found in a variety of disease states. Luminal calcification is present in atherosclerotic plaques where it strongly correlates with an increased risk of coronary events.1 Medial layer calcification is a comorbidity and complication of prolonged diseases such as chronic kidney disease, diabetes mellitus, rheumatoid arthritis, and aging. In contrast to plaque calcification, medial calcification is often found along the elastic lamina and in areas surrounding smooth muscle cells.2 Currently, no therapy has been developed that is able to prevent, halt, or reverse ectopic calcification in the vasculature.3 To date, 2 genetic diseases have been identified in which mutations induce de novo calcifications in the medial layer of large elastic and smaller muscular arteries—generalized arterial calcification of infancy (OMIM # 208000) and arterial calcification due to deficiency of CD73 (ACDC, OMIM # 211800).4,5 Generalized arterial calcification of infancy and ACDC are rare autosomal recessive diseases caused by inactivating mutations in ENPP1 and NT5E (encoding for CD73 protein), respectively. ENPP1 and CD73 are extracellular enzymes that function as key components in the breakdown of extracellular nucleosides: ATP is metabolized to AMP and pyrophosphate by ENPP1, and CD73 converts AMP into adenosine and inorganic phosphate.6 The stresses thought to contribute to medial calcification, such as mechanical stress and inflammation, are the same stresses known to modulate ATP release and subsequent adenosine production.7–9
The CD73-deficient mouse does not recapitulate the vascular phenotypes seen in CD73-deficient patients with ACDC, thus this murine model is an ineffective tool to study the mechanisms driving the vascular phenotypes in ACDC pathogenesis and disease progression.10–12 We previously developed an in vivo disease model utilizing CD73-deficient induced pluripotent stem cells (iPSCs) and found accelerated calcification in CD73-deficient iPSC teratomas while control iPSC teratomas exhibited no such phenotype.13 CD73-deficient iPSC-derived mesenchymal stem cells more readily differentiated down the osteogenic lineage and increased expression and activity of TNAP (tissue nonspecific alkaline phosphatase; ALPL). While TNAP is a marker for stem cells, it is also a key enzyme in ectopic mineralization as it preferentially breaks down the endogenous mineralization inhibitor, pyrophosphate, into inorganic phosphate, a building block necessary for calcification.14 We found that reduced adenosine signaling because of CD73 mutations caused cells to upregulate ALPL gene expression, and that the TNAP enzyme can somewhat compensate for CD73 deficiency by hydrolyzing AMP to adenosine, but with 100-fold less efficiency.13 These observations confirm that CD73-mediated adenosine signaling is a critical component to vascular homeostasis.
While it is evident that TNAP is necessary and sufficient for ectopic mineralization, less clear are the factors inducing ectopic ALPL gene expression in nonbony tissue and cells. The objective of this study was to delineate the mechanism by which a lack of CD73-mediated adenosine signaling promotes calcification by upregulating expression of ALPL and activity of its protein product, TNAP.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. All patient samples were collected from individuals enrolled in studies that had been approved by the institutional review boards of the National Institutes of Health (control and ACDC cell lines) or procured from tissue donors after obtaining consent from next of kin according to protocols approved by the Nebraska Organ Recovery System (NORS).5,15 Tables 1 and 2 describe patient characteristics.
Table 1.
Baseline Characteristics of Patients for the Isolation of Control and ACDC Cell Lines
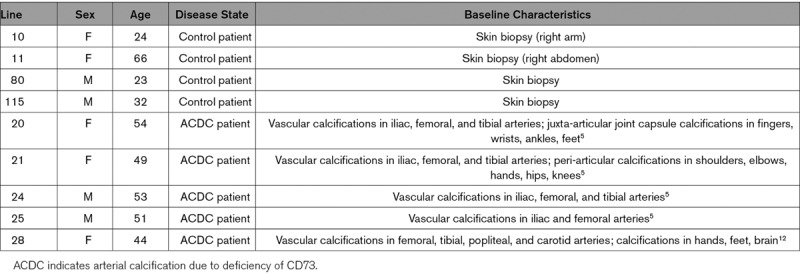
Table 2.
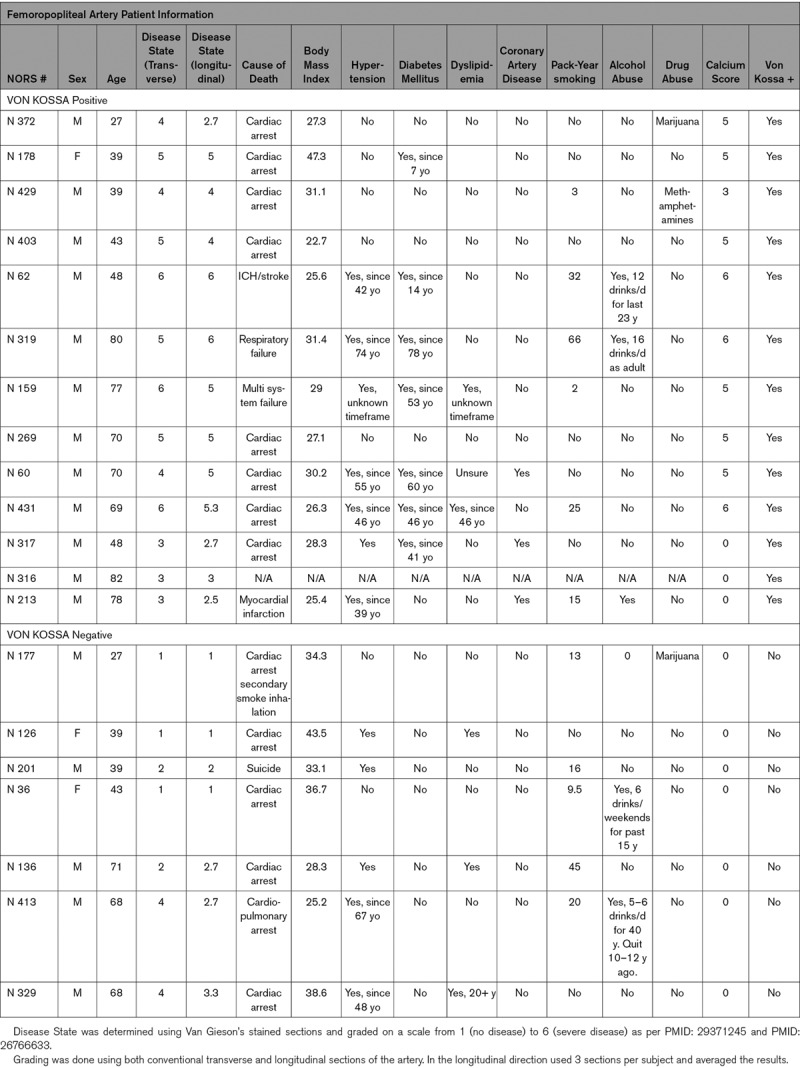
Patient Characteristics of Popliteal Artery Samples
Cell Culture
Four human Control (CT) and 5 human ACDC dermal fibroblast cultures were obtained from patient punch biopsy and grown in DMEM supplemented with 20% FBS and 1× penicillin-streptomycin (P/S). Primary fibroblasts were used between passages 5 and 16. Growth media was changed every 3 days and cells were split 1:2 when confluent. Post-expansion, all cells were plated with either 250 000 cells per 9.5 cm2, 125 000 cells per 4 cm2, or 75 000 cells per 1.7 cm2 with serum reduced to 10%. For cells undergoing osteogenic assay (also called in vitro disease model), no treatment media consisted of Gibco Minimum Essential Medium alpha+nucleosides supplemented with 10% FBS and 1× P/S while osteogenic treatment media consisted of no treatment media base with 10 mmol/L β glycerol phosphate, 50 µM ascorbic acid 2-phosphate, and 100 nM dexamethasone made fresh. Growth media was removed before the assay, and media was changed every 4 days throughout the time course.
In experiments with exogenous AMP stimulation, cells were pretreated for 30 minutes with an adenosine deaminase inhibitor (EHNA 10 µM), then treated with vehicle or 100 µM exogenous AMP for 10 minutes. Forskolin and 3-isobutyl-1-methylxanthine (IBMX) were each used at a concentration of 10 µM with EHNA for pretreatment and with AMP for treatment. Cells were counted by staining with 0.4% Trypan Blue stain. Live/dead analysis was performed using the ReadyProbes Cell Viability Imaging Kit, Blue/Green (Invitrogen) and the Life Technologies Countess II FL according to manufacturer’s protocol.
All chemicals were dissolved in DMSO, which was administered in equal volume as vehicle control. Rapamycin (200 nM) was administered daily. D11 and L11 peptides were prepared according to manufacturer’s protocols (Novus Biologicals) and used at a concentration of 10 µM with treatment every 2 days. AS-1842856 was used at a concentration of 1 µM. 8-Br-cAMP was used at a concentration of 0.5 mmol/L. CGS-21680 was used at a concentration of 10 µM. BAY-60-6583 was used at a concentration of 10 µM. In 5-day experiments, drugs were administered daily, and in 21-day assays, cells were treated every other day.
In autophagy flux experiments, confluent cells were serum-starved for 24 to 48 hours in DMEM supplemented with 0.5% FBS. Cells were then treated with normal growth media or HBSS (GIBCO) with 10 nM bafilomycin (Sigma-Aldrich) or the same volume of DMSO as a vehicle control for 4 hours.
Transcriptional Analysis
RNA was extracted from cells using the RNeasy Mini Kit (Qiagen) and cDNA (cDNA) was prepared using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fischer) according to manufacturer’s protocols. Gene expression was quantified by real-time PCR reaction of 4 ng/µL cDNA, 1× SYBR Green (Thermo Fisher), 1 µM each forward and reverse primer and amplified using with the following parameters: 50°C for 2 minutes and then 95°C for 10 minutes followed by 40 cycles of 95°C for 20 seconds, 58°C for 20 seconds, 72°C for 1 minute on a CFX Connect Real-Time PCR System (Bio-Rad).
Western Blot Analysis
Cells were lysed in 1% CHAPS hydrate, 150 mmol/L sodium chloride, 25 mmol/L HEPES buffer supplemented with 1× protease and phosphatase inhibitor (Sigma-Aldrich). Cells were scraped into microcentrifuge tubes, vortexed for 10 minutes, freeze/thawed for 5 to 8 cycles, then centrifuged at 12 000×g for 10 minutes at 4°C. Protein lysate was combined with 1× Pierce LDS Sample Buffer, Non-Reducing (ThermoFisher Scientific) and 1× NuPAGE Sample Reducing Agent (Novex), brought to equal volume with CHAPS buffer, and denatured at 95°C for 15 minutes. Electrophoresis was performed on a 4% to 20% TGX stain-free polyacrylamide gel (Bio-Rad) in 1× Tris/Glycine/SDS Buffer (Bio-Rad). Proteins were transferred onto a 0.2 µm nitrocellulose membrane in 1× Trans-Blot Turbo Transfer Buffer (Bio-Rad) at 2.5 A and 25 V for 15 minutes using the Trans-Blot Turbo Transfer System (Bio-Rad). The membrane was blocked in Odyssey blocking buffer (PBS; Li-COR) and primary antibodies were incubated in Odyssey blocking buffer with 0.1% Tween 20. Membranes were washed in PBS with 0.1% Tween 20 before incubation with secondary antibodies. Membranes were imaged on an Odyssey CLx (LI-COR) and analyzed with Image Studio (Version 5.2, LI-COR) software. If blots were stripped, they were done so using 1× Restore Fluorescent Western Blot Stripping Buffer (Thermo Fisher) according to manufacturer’s protocols, washed with PBS, and placed directly into the next primary antibody solution.
Cyclic AMP Assay
Cells were serum starved and then pretreated with 100 µM IBMX for 30 minutes at 37°C before treatment with 30 µM AMP for 15 minutes. Cells were lysed in 0.1 M HCl and 0.5% Triton-X 100, and the Direct cAMP ELISA kit (Enzo) was used according to manufacturer’s instructions.
Alizarin Red Staining
Cells were washed with PBS and fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences) for 15 minutes at room temperature. Cells were then washed twice with deionized water and covered with 40 mmol/L Alizarin Red S (Sigma-Aldrich) at pH 4.1 to 4.3 for 20 minutes at room temperature. Unincorporated dye was removed, and cells were washed 2× to 3× with deionized water before images were acquired. After imaging, Alizarin Red S was extracted with 10% (v/v) acetic acid (Fisher Scientific) for 30 minutes, scraped into a microcentrifuge tube, vortexed, and then incubated at 85°C for 10 minutes. After chilling on ice for 5 minutes, the mixture was centrifuged at 20 000×g for 15 minutes at 4°C. Five hundred microliters of supernatant was transferred to a new tube and 10% (v/v) ammonium hydroxide (Fisher Scientific) was then added to the supernatant. Absorbance was read in triplicate at 405 nm using a 96-well plate spectrophotometer.
Tissue Nonspecific Alkaline Phosphatase Activity Staining
Cells were washed with 2× PBS and fixed with 4% PFA for 10 minutes at room temperature, then washed once with PBS and once with water. TNAP staining was produced using the SIGMAFAST BCIP/NBT tablet according to manufacturer’s instructions.
Immunofluorescence Imaging and Quantification
Cells were washed with PBS, then fixed with 4% PFA prepared in PBS 0.5% Triton-X 100 for 15 minutes. Cells were washed 3× with PBS, 0.5% Triton-X 100 for 5 minutes each, incubated at room temperature for 1 hour in blocking buffer (PBS, 0.5% Triton-X 100, 5% FBS), then incubated overnight at 4°C in primary antibody. Cells were washed with PBS, 0.1% TWEEN 20 3× for 5 minutes, then incubated with secondary antibody for 1 hour. Cells were washed 3× for 5 minutes each with PBS, 0.1% TWEEN 20, then once with PBS, and then stained for 30 minutes with AlexaFluor488 Phalloidin (Molecular Probes) for f-actin. Cells were washed a final time with PBS for 5 minutes, then mounted with Fluoroshield Mounting Medium with DAPI (Abcam). Slides were imaged within 24 hours of mounting.
To visualize p62 accumulation and localization, the Premo Autophagy Sensor RFP-p62 reagent (Life Technologies) was used according to manufacturer’s instructions, counter stained with AlexaFluor488 Phalloidin, and mounted with Fluoroshield Mounting Medium with DAPI (Abcam). Cells were imaged within 24 hours of mounting. In ImageJ, each image was split into blue, red, and green channels, and the pixel intensity of the red and blue channels was measured. RFP-p62 intensity of an image was normalized to DAPI intensity for that same image.
MitoSOX red reagent (Molecular Probes) was diluted to 5 µM in media and added to cells in a 4-well chamber slide which were incubated in the dark for 10 minutes at 37°C. Slides were prepared as above with the addition of AlexaFluor488 Phalloidin (Molecular Probes) and then mounted with Fluoroshield Mounting Medium with DAPI and imaged within 24 hours.
To quantify immunofluorescent stains, ImageJ was used to split each image into red, green, and blue channels, and the pixel intensity of the red and blue (DAPI) channels was measured. The measured intensity of a given stain was normalized to DAPI intensity of that same image.
PCR Mutagenesis, Transfection, and Luciferase Assays
The pEZX-LvPG04 backbone plasmid contains the 1353 base pairs upstream of the ATG start site of the human ALPL gene (NM_000478) and this promoter sequence drives expression of Gaussia luciferase (Genecopoeia). QuickChange II XL Site-Directed Mutagenesis Kit (Agilent) was used to substitute the putative FOXO1 (forkhead box O1 protein) recognition site TGTTG with TATTA using forward primer: 5’-TCTGTCTCTGTGTCTGTTAATATATCTGG CTTTCTCTGGGTC-3’ and reverse primer: 5’-GACCCAGAGAAAGCCAGATATATTAACAG ACACAGAGACAGA-3’ according to manufacturer’s protocol. Mutant plasmids were selected and confirmed by Sanger sequencing (Genewiz) and transfected into cells using the Lipofectamine LTX Reagent with PLUS Reagent (Invitrogen) according to manufacturer’s protocols. Luciferase was quantified using the Secrete-Pair Dual Luminescence Assay Kit (Genecopoeia) according to manufacturer’s protocols and normalized to SEAP secretion.
Von Kossa Staining
Slides with adhered paraffin histological sections were warmed to 65°C for 1 hour and then deparaffinized through xylene and graded alcohol baths before being placed in distilled water. The slides were then stained using the Von Kossa Method of Calcium Kit (Polysciences, 24633-1) according to manufacturer’s instructions.
Immunofluorescent Staining on Paraffin Sections
Slides were warmed for 1 hour at 65°C and deparaffinized by xylene and graded alcohol baths, rehydrated in ddH2O, and heated for 20 minutes in antigen unmasking solution (Vector Labs). Slides were cooled to room temperature, then washed with PBS for 5 minutes. The tissues were incubated in blocking buffer (PBS, 3% fish skin gelatin, 10% horse serum) for 1 hour, then incubated in primary antibody overnight. Tissues were washed 3× for 5 minutes each in wash buffer (PBS, 3% fish skin gelatin, 0.1% TWEEN 20), then for 5 minutes in PBS. Tissues were incubated with secondary antibody covered from light for 1 hour, then washed with wash buffer 3× for 5 minutes each. Tissues were mounted with Fluoroshield Mounting Medium with DAPI (Abcam) and imaged within 24 hours.
Statistics
Statistical analysis was performed with GraphPad Prism 8 (GraphPad Software, Inc) and data shown are mean±SD. Statistical analysis used, exact n values, biological replicates, and P values are stated within each figure legend. A P value equal to or less than 0.05 was considered significant.
Results
Excess AMP Induces Aberrant Signaling in CD73-Deficient Cells
Inactivating mutations in CD73 render the enzyme incapable of converting extracellular AMP into adenosine and inorganic phosphate, thus CD73-deficient cells are exposed to an excess of AMP resulting in a concomitant reduction in adenosine and downstream adenosine receptor signaling (Figure 1A). Adenosine receptors are G-protein coupled receptors, which were initially classified by their ability to pair with the Gαi (A1, A3) and Gαs (A2a, A2b) G-protein subunits to inhibit or induce, respectively, adenylyl cyclase production of cAMP.16 We found no differences in expression of adenosine receptor genes between control and ACDC cells (Figure 1B). A cAMP ELISA assay showed exogenous AMP induced increased production of cAMP in control, but not CD73-deficient fibroblasts, indicative of adenosine primarily signaling via the A2a or A2b adenosine receptors (Figure 1C). As a consequence of elevated cAMP levels, control cells showed activation of VASP (vasodilator-stimulated phosphoprotein; Figure 1D). In contrast, phosphorylation of VASP was not observed in CD73-deficient ACDC cells treated with exogenous AMP; instead, these cells exhibited increased levels of phosphorylated AKT at T308 and S473 (Figure 1D). To determine if elevation of cAMP could prevent phosphorylation of AKT and therefore rescue the absence of CD73 activity, ACDC cells were pretreated with the adenylyl cyclase agonist forskolin and the phosphodiesterase inhibitor IBMX. Direct activation of adenylyl cyclase induced phosphorylation of VASP and importantly, attenuated phosphorylation of AKT at T308 and S473 in ACDC cells (Figure 1E). These data suggest that lack of CD73/adenosine receptor-mediated production of cAMP contributes to aberrant AKT activation.
Figure 1.
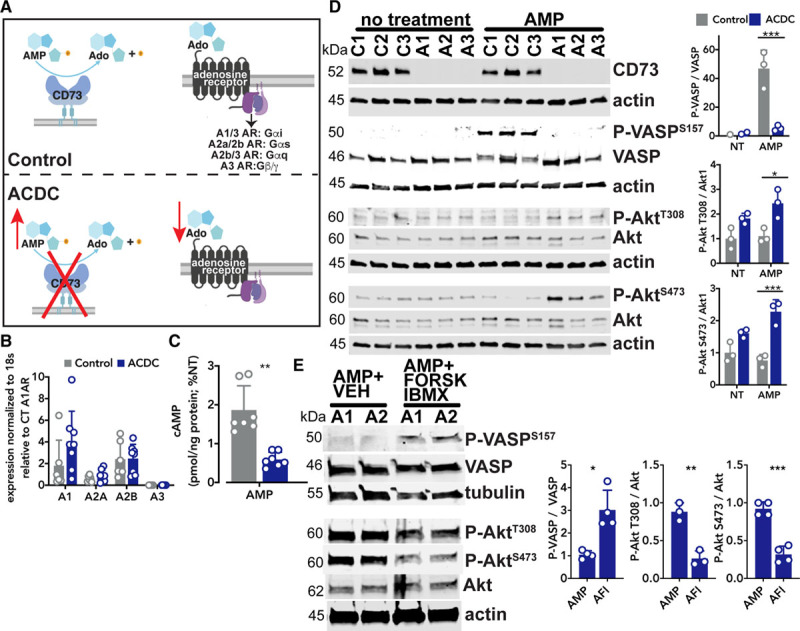
AMP induces aberrant signaling in arterial calcification due to deficiency of CD73 (ACDC) cells. A, Schematic of CD73 and downstream adenosine receptor signaling. B, Baseline measurement of adenosine receptor gene expression. Results representative of n=6 to 8 per group using 4 control and 3 ACDC patient cell lines. Statistical analysis performed using unpaired t test with Welch correction. C, Quantification of cAMP levels in control and ACDC cells in response to exogenous AMP. Cells were pretreated with 0.5 mmol/L IBMX for 30 min then treated with 30 µM AMP for 15 min. Results representative of n=7 per group using 3 control and 4 ACDC patient cell lines. **P=0.0013 using unpaired t test with Welch correction. D, Control and ACDC fibroblasts were given exogenous 100 µM AMP for 10 min and total cell lysates were analyzed by Western blotting for VASP and AKT phosphorylation. Results representative of n=3 per group using 3 control patient and 3 ACDC patient cell lines. For P-VASP, ***P=0.0003, P-AKT T308 *P=0.0155, P-AKT S473 ***P=0.0005. Two-way ANOVA with Tukey multiple comparisons test was used for statistical analysis. E, ACDC fibroblasts were treated with 100 µM exogenous AMP or pretreated with 10 µM forskolin and 10 µM IBMX for 30 min before treatment with 100 µM exogenous AMP, 10 µM forskolin, and 10 µM IBMX (AFI) for 10 min. Total cell lysates were analyzed by Western blotting for VASP and AKT phosphorylation. Results representative of n=3 to 4 per group using 2 ACDC patient cell lines. For P-VASP *P=0.0183, P-AKT T308 **P=0.0025, P-AKT S473 ***P=0.0002. One-way ANOVA with Tukey multiple comparisons test was used for statistical analysis.
Inducing Autophagy Reduces Calcification but Autophagy Flux Is Not Altered in CD73-Deficient Cells
We previously observed that CD73-deficient iPSCs and iPSC-derived mesenchymal stem cells exhibit an accelerated osteogenic differentiation that was inhibited in vitro and in vivo with rapamycin.13 AKT is an upstream regulator of many signaling pathways, including mTOR (mammalian target of rapamycin). Activation of mTOR by AKT decreases autophagy, and decreased autophagy induces stem cells to differentiate into osteoblasts as well as promotes the calcification of smooth muscle cells.17,18 We observed that the mTOR inhibitor rapamycin reduced ALPL gene expression and TNAP activity and prevented calcification of CD73-deficient cells (Figure 2A through 2C). As rapamycin can affect many signaling pathways, autophagy was directly stimulated with a cell-penetrating peptide of Beclin-1 (D11), which mirrored rapamycin’s anti-calcification effects (Figure 2D), illustrating that inducing autophagy alone is sufficient to prevent calcification.19
Figure 2.
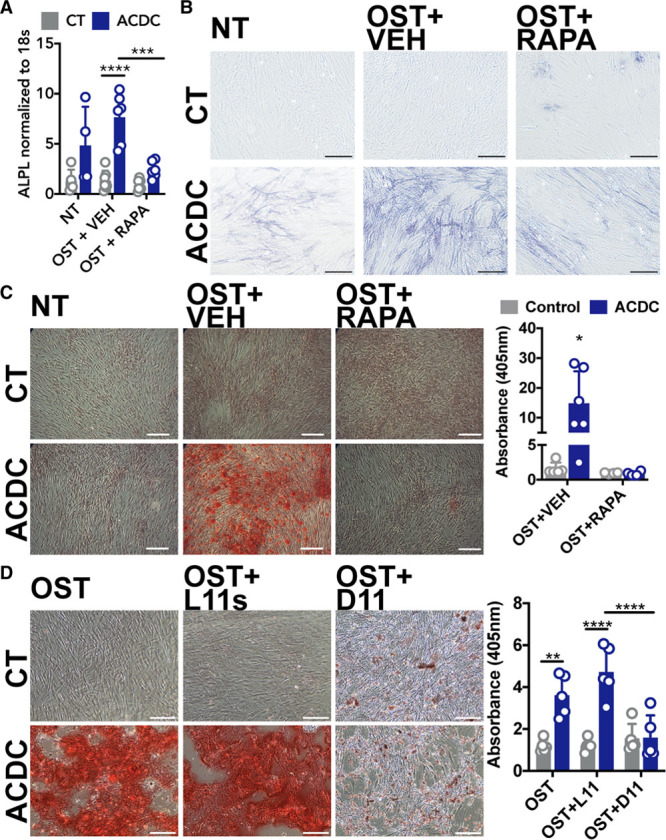
Inducing autophagy reduces calcification in CD73-deficient cells. A, ALPL gene expression. Results representative of n=4 to 7 per group using 3 control and 3 arterial calcification due to deficiency of CD73 (ACDC) patient cell lines. ***P=0.003 ****P<0.0001. B, Staining for TNAP activity in control and ACDC fibroblasts cultured in osteogenic media and supplemented with 200 nM rapamycin for 5 d. Results representative of n=4 to 7 per group using 3 control and 3 ACDC patient cell lines. C, Alizarin Red S stain was used to image and quantify calcification content in control and ACDC fibroblasts cultured under osteogenic conditions for 21 d and supplemented with 200 nM rapamycin. Results representative of n=6 per group using 2 control and 2 ACDC patient cell lines. *P=0.037. D, Alizarin Red S staining of control and ACDC cells treated with autophagy-inducing peptide Tat-Beclin 1 D11. Results representative of n=5 per group using 4 control and 3 ACDC patient cell lines. **P=0.0043 ****P<0.0001. Scale bar represents 200 µm. Two-way ANOVA with Tukey multiple comparisons test was used for all statistical analyses in this figure.
We next wanted to determine whether there were alterations in autophagy in CD73-deficient cells at baseline and under osteogenic conditions. Neither overnight treatment of exogenous AMP nor serum starvation altered the levels of the autophagosome cargo protein p62, or the ratio of LC3-II/I, the autophagosome biogenesis protein (Figure IA and IB in the Data Supplement), and autophagy flux experiments show no differences between control and CD73-deficient cells (Figure IC in the Data Supplement). Further, no differences were observed in the localization or intensity of a p62-RFP conjugate protein transfected into cells treated under osteogenic conditions (Figure ID in the Data Supplement), and long-term treatment under osteogenic stimulation with or without rapamycin did not result in differences in p62 or LC3-II/I ratios in ACDC cells (Figure IE in the Data Supplement). Importantly, ACDC cells show increased LC3-II/I ratios upon treatment with D11 which illustrates their ability for autophagosome biogenesis. We found that exogenous AMP did not alter cell proliferation of control and ACDC cells under baseline or exogenous AMP stimulation for 21 days (Figure IIA in the Data Supplement). Comparison of the percentage of live/dead cells showed exogenous AMP increased the number of live control cells compared with untreated control cells, and this increase was also seen in untreated ACDC cells at day 6; however, these differences were not seen at the day 15 and 21 timepoints (Figure IIA in the Data Supplement). Exogenous AMP also did not induce ER stress response and did not activate AMPK (AMP-activated protein kinase; Figure IIB and IIC in the Data Supplement), and the levels of mitochondrial oxidative stress at baseline and after 21 days of exogenous AMP showed no differences (Figure ID in the Data Supplement). From these data, we conclude that proliferation, apoptosis, or mitochondrial oxidative stress are not significant contributors to the calcification seen in CD73-deficient cells. In summary, while treatment with rapamycin or the Beclin-1 peptide can prevent calcification, we do not observe alterations in autophagy processes in CD73-deficient cells.
Exogenous AMP Promotes FOXO1 Nuclear Localization in CD73-Deficient Cells
Increased TNAP activity is one of the main features observed in CD73-deficient fibroblasts when these cells are stimulated with osteogenic medium as well as in ACDC patient tissue.13 As TNAP is a key enzyme in ectopic mineralization, we sought to determine which transcription factors may contribute to activation of ALPL gene expression in the absence of CD73 activity. Using the TRANSFAC database, we screened 1350 bp upstream of the transcriptional start site of the human ALPL gene (NM_ 000478) and identified 227 sites with both a core score and matrix score of 1, indicative of a high likelihood of being a functional binding site (online Table I).20 Of interest was a site for FOXO1, located 445 base pairs upstream of the transcriptional start site (Figure 3A). Treatment with exogenous AMP promoted the localization of FOXO1 to the nucleus in ACDC but not control cells. This localization was prevented when cells were pretreated with the cAMP analogue 8-Br-cAMP or by activation of the A2a and A2b adenosine receptors, which activate adenylyl cyclase (Figure 3B). FOXO1 can be regulated by a variety of kinases, and proteasome-dependent degradation of FOXO1 is thought to be dependent on AKT phosphorylation.21,22 While exogenous AMP activates AKT in CD73-deficient cells, we could not detect significant differences in the levels of FOXO1 phosphorylation between control and ACDC whole cell lysates after 30 minutes of stimulation using whole-cell lysate (Figure III in the Data Supplement). Together, these data suggest that in the absence of CD73-generated adenosine signaling via the A2a and A2b receptor, exogenous AMP promotes the nuclear localization of FOXO1.
Figure 3.
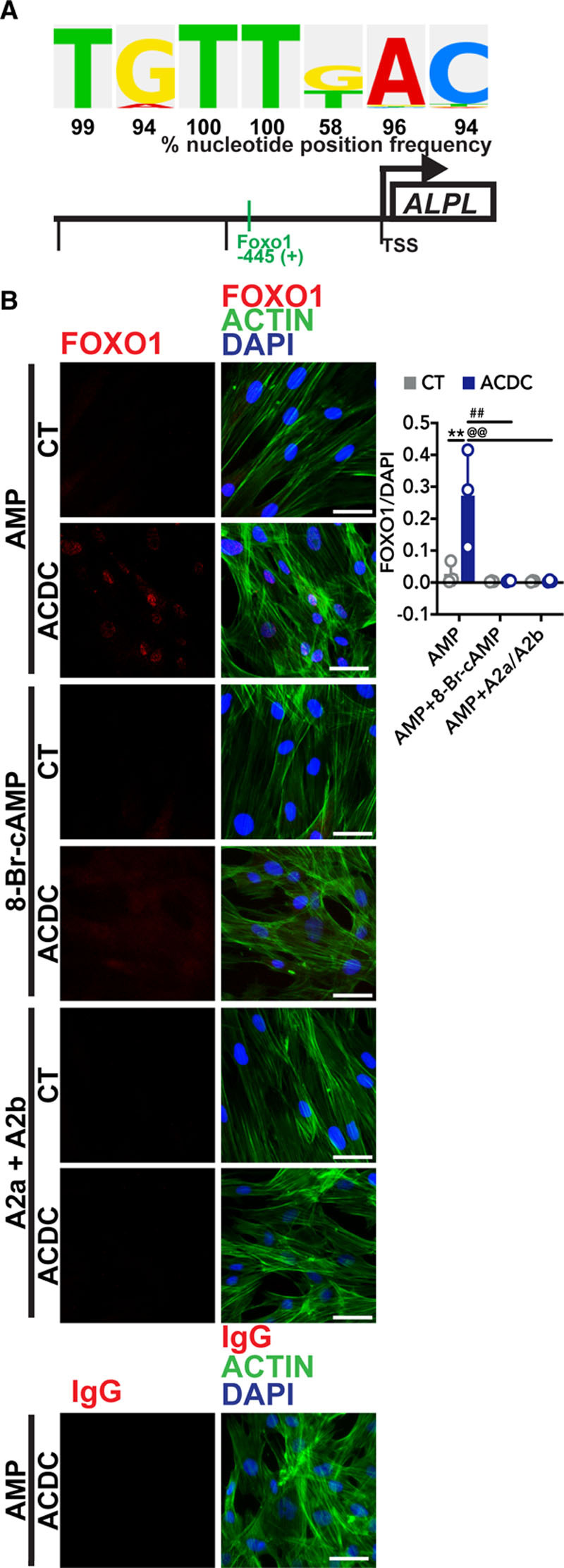
AMP induces FOXO1 (forkhead box O1 protein) nuclear localization. A, The putative FOXO1-binding site in the human ALPL promoter shown with the percentage that the sequence located 445 upstream of the transcriptional start site (TSS) on the human ALPL promoter is found in experimentally proven FOXO1-binding sites according to the TRANSFAC database. B, Immunofluorescent staining was used to detect FOXO1 localization in response to exogenous AMP. Control and arterial calcification due to deficiency of CD73 (ACDC) fibroblasts were pretreated with vehicle, 0.5 mmol/L 8-Br-cAMP, or 10 µM each of CGS-21680 and BAY 60-6583 (A2a and A2b agonists, respectively) for 30 min, then 100 µM AMP was added to each group for 2 h. Cells were stained with anti-FOXO1 antibody and an IgG control, and pixel intensity was quantified and normalized to DAPI. **P=0.0054, ##P=0.0027, @@P=0.0027 using 2-way ANOVA with Tukey multiple comparisons test for statistical analysis. Results representative of 5 replicates of 3 control patient cell lines and 3 ACDC patient cell lines per group. Scale bar represents 50 µm.
FOXO1 Inhibition Reduces ALPL Expression, TNAP Activity, and Calcification
CD73-deficient ACDC cells increase ALPL gene expression when cultured under osteogenic conditions, and treatment with the FOXO1 inhibitor AS-1842856 (1 µmol/L) reduced ALPL transcription (Figure 4A). Similarly, AS-1842856 treatment decreased TNAP enzymatic activity in CD73-deficient ACDC cells cultured under osteogenic conditions, and further prevented cells from calcifying in the 21-day osteogenic assay (Figure 4B and 4C) illustrating that FOXO1 function is required for calcification in the absence of CD73.
Figure 4.
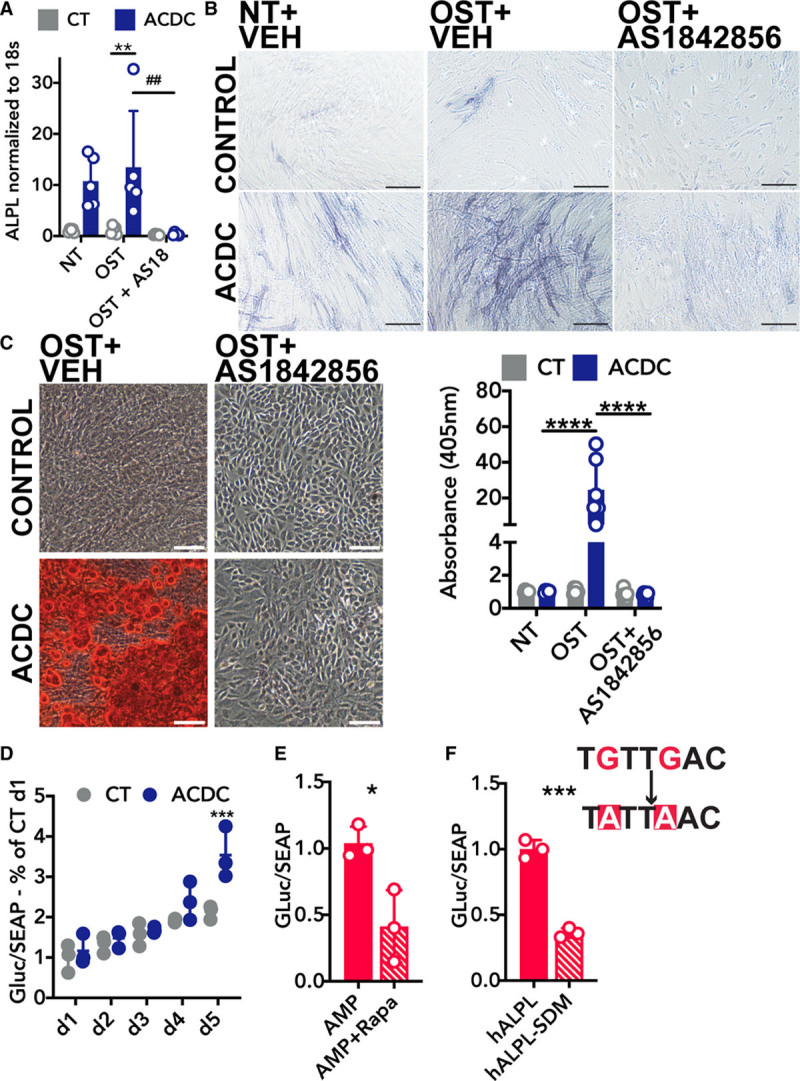
FOXO1 (forkhead box O1 protein) inhibitor prevents calcification via reducing ALPL promoter activation. A, RT-qPCR analysis of ALPL mRNA isolated from cells in osteogenic conditions receiving 1 µM AS-184856 for 5 d. Results representative of n=5 per group using 3 control and 3 arterial calcification due to deficiency of CD73 (ACDC) patient cell lines. **P=0.0077, ##P=0.0039. Two-way ANOVA with Tukey multiple comparison test was used for statistical analysis. B, TNAP activity staining in cells cultured for 5 d in osteogenic media supplemented daily with the FOXO1 inhibitor AS-1842856 (1 µM) or vehicle control. Results representative of n=5 per group using 4 control and 3 ACDC patient cell lines. Scale bar represents 200 µm. C, Alizarin Red S stain in cells cultured under osteogenic conditions for 21 d and supplemented 1 µM AS-1842856. Results representative of n=6 per group using 4 control and 3 ACDC patient cell lines. ****P>0.0001 by 2-way ANOVA with Tukey multiple comparison test. Scale bar represents 200 µm. D, Luciferase assay of plasmid DNA containing the Gaussia luciferase under control of the human ALPL promoter (hALPL). Cells were transfected with 1 µg of plasmid and cultured for 5 d under osteogenic stimulation. Results representative of n=3 per group using 3 control and 3 ACDC patient cell lines. ***P=0.0011 by 2-way ANOVA with Sidak multiple comparisons test. E, Luciferase assay of hALPL plasmid in cells treated with 100 µmol/L AMP with and without pretreatment of 200 nM rapamycin. Results representative of n=3 using 1 ACDC patient cell line. *P=0.0406. F, Luciferase assay of hALPL and Luciferase assay of hALPL with site-directed mutagenesis (SDM) of the putative FOXO1-binding site. Results representative of n=3 using 1 ACDC patient cell line. ***P=0.0008. Unpaired t test with Welch correction was used for E and F.
Plasmid DNA harboring the Gaussia luciferase gene under the control of the human ALPL gene promoter (hALPL) was used to assess the role of the putative FOXO1 binding site located 445 base pairs upstream from the transcriptional start site. Transfection of hALPL into control and ACDC cells shows promoter activity is increased at day 5 in CD73-deficient ACDC cells (Figure 4D). In ACDC cells transfected with hALPL, pretreatment with 200 nM rapamycin reduced AMP-induced hALPL activity compared with the vehicle control, mirroring results in Figure 2 (Figure 4E). Site-directed PCR mutagenesis was performed to change the putative FOXO1 binding site from TGTTGAC to TATTAAC. This mutation reduced promoter activity 63% in response to AMP treatment (Figure 4F), indicating that this site is operative in the expression of human ALPL.
CD73 Is Decreased and FOXO1 Is Increased in Calcified Femoropopliteal Arteries
Patients with CD73 deficiency exhibit extensive calcification and vessel tortuosity in their lower extremity arteries. While CD73 deficiency is a rare disease with only a handful of individuals identified, one overarching goal of studying the mechanisms driving ACDC pathogenesis is to uncover novel pathways that may be operative in more common forms of medial artery calcification.5,23–28 To that end, paraffin embedded femoropopliteal arteries from 20 age-matched donors exhibiting calcification and no calcification were obtained,15 and Von Kossa stain used to confirm calcification state. Staining for CD73, FOXO1, and TNAP was performed on adjacent sections (Figure 5). We found calcified arteries exhibited dramatically lower CD73 staining than control arteries. We also found that the levels of FOXO1 staining are significantly higher in the calcified areas of Von Kossa-positive calcified vessels than the control vessels. Importantly, when comparing noncalcified regions to calcified regions in Von Kossa-positive vessels, we found that FOXO1 staining was substantially decreased in noncalcified areas. While the differences between TNAP staining in control versus calcified vessels did not reach statistical significance, similar to FOXO1 staining, TNAP staining was higher in the calcified regions of Von Kossa-positive vessels compared with noncalcified regions from Von Kossa-positive vessels. These data suggest that a CD73/FOXO1/TNAP axis may be operative in more common forms of nonatherosclerotic medial layer vascular calcification.
Figure 5.
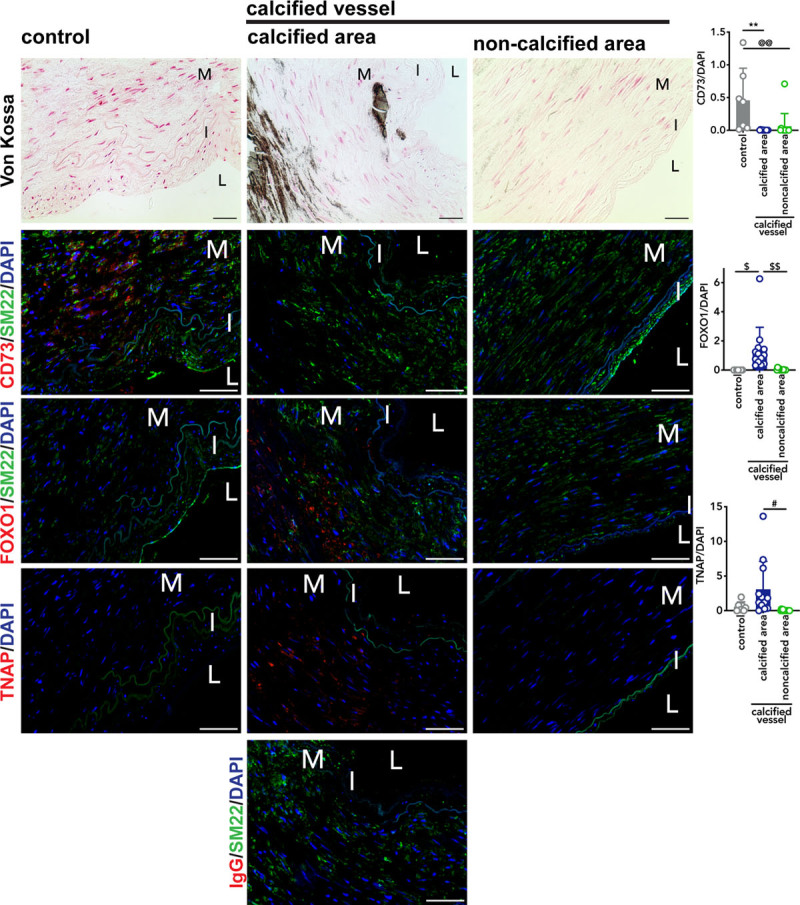
CD73 levels reduced in nonarterial calcification due to deficiency of CD73 (ACDC) calcified femoropopliteal artery exhibiting higher levels of FOXO1 (forkhead box O1 protein). Sections of femoropopliteal arteries from 20 different patients were stained with Von Kossa stain to detect calcification. Of the 20 patients, no calcification was detected in 7 vessels and the remaining 13 show calcification. Serial sections were stained with CD73, FOXO1, and TNAP antibody, and an IgG control to match concentration of TNAP at 20 µg/mL. Pixel quantification was done using ImageJ and normalized to DAPI. **P=0.0023, @@P=0.0067, $P=0.0151, $$P=0.0043, #P=0.0133. M=tunica media. I=tunica intima. L=lumen. Von Kossa scale bar represents 100 µm, fluorescent images scale bar represents 50 µm.
Discussion
The initial discovery that inactivating mutations in the gene encoding for CD73 protein result in extensive nonatherosclerotic lower extremity medial layer calcification identified a novel pathway involved in vascular calcification. Several CD73 knockout mouse models are available and while these mice exhibit calcification in the juxta-articular spaces in their joints, similar to that seen in patients with ACDC, there is no evidence of calcification in any of the vascular beds.10,29–31 This underscores that vascular mineralization potential is very different in mice compared with humans and justifies our use of primary human cells in an in vitro disease model to study the mechanism driving ACDC pathogenesis.11 The cell source for this in vitro disease model is dermal fibroblasts, which is a limiting factor of this model. However, the use of fibroblasts in disease modeling has been well established,32 and our published studies illustrate that this system is sufficient to recapitulate this calcification phenotype and therefore suitable to define the underlying mechanisms promoting the ectopic calcification phenotype seen in patients with ACDC.
Two sites on AKT are considered to be rate-limiting for the propagation of downstream signaling; phosphorylation of AKT at T308 is mediated via PDK1, and S473 via mTORC2.33 We have now shown that in the absence of CD73 activity, exogenous AMP induces phosphorylation of AKT at these sites, and this phosphorylation is mitigated when adenylyl cyclase is directly activated to produce cAMP (Figure 1). Our findings tie in nicely with previous biochemical studies which illustrated that cAMP inhibits AKT phosphorylation by blocking the localization of PDK1 to the cell membrane and inhibiting the lipid kinase activity of PI3K, and blocks insulin and amino acid starvation-induced mTORC1 activation.34,35 Additionally, a lack of cAMP production due to inactivating mutations in GNAS, the gene encoding for adenylyl cyclase, was found to contribute in the pathogenesis of another ectopic calcification disorder, progressive osseous heteroplasia. This coupled with our results suggests that reduced intracellular cAMP production may be shared in various forms dystrophic vascular calcification.36 What remains to be determined is the mechanism through which exogenous AMP induces AKT phosphorylation. One report suggests that AMP is able to signal through binding the A1 adenosine receptor. Experiments were conducted with physiological concentrations of exogenous AMP; however in this reporter, system cells were transfected with plasmid DNA to overexpress the A1 adenosine receptor as well as the Gαi G-protein subunit.37 The A1 adenosine receptor density on the cell surface as well as the internal Gαi were under constitutively active promoters and thus not present at physiological levels; thus evidence of a true AMP receptor in human cells or tissues is lacking.
To date, hundreds of AKT substrates have been identified and we became interested in the role of CD73 in regulating autophagy, as AKT activation can promote mTORC1-mediated inhibition of autophagy. While upregulated under stressed conditions such as nutrient deprivation, autophagy is active even at baseline and is necessary to ensure proper homeostasis. One study modeling chronic kidney disease–induced vascular calcification identified a link between the activation of autophagy and a means to prevent calcification.18 We hypothesized that increased AMP-induced AKT activation may lead to reduced levels of autophagy which would promote calcification. Although AKT was phosphorylated in CD73-deficient cells, we found no defect in autophagy flux.
Moving away from defects in autophagy, we conducted an in silico screen of the human ALPL promoter and focused on a putative FOXO1 transcription factor binding site located 445 base pairs upstream of the ALPL transcriptional start site. Forkhead box transcription factors are an evolutionarily conserved family that in mammals regulate a wide array of cellular processes, including proliferation, apoptosis, metabolism, and oxidative stress resistance.21 Their necessity in development was illustrated first in Drosophila, and FoxO1 knockout mice die around embryonic day 11 primarily due to defects in vascular development in the embryo and yolk sac.38,39 While necessary in development, in disease states FOXO transcription factors can contribute to disease pathology. For example, increased FOXO1 signaling in liver and pancreatic cells induces diabetes mellitus via impairing insulin signaling.40 There have been various and opposing reports of these transcription factors in osteogenesis and ectopic calcification. In vitro and in vivo genetic knockdown shows that FOXO1 helps drive the differentiation of mesenchymal stem cells into osteoblasts, and bone-specific knockout murine models provide evidence that FOXO1 is necessary for osteoblast function and bone homeostasis.41,42 In opposition to these studies in bone, in mice harboring a smooth muscle cell-specific deletion of Pten gene (phosphatase and tensin homolog), loss of PTEN promoted calcification by stabilizing RUNX2 protein levels, which are normally targeted for ubiquitination and proteasome mediated degradation via FOXO1 and FOXO3.43 PTEN is a phosphatase that acts on AKT to inhibit AKT signaling, thus these mice exhibit constitutively active AKT in their smooth muscle cells. In our human system, we observe elevated AKT activation in response to exogenous AMP.
Although RUNX2 is a regulator of genes necessary for the osteogenic transition of stem and vascular smooth muscle cells,44–47 there is evidence for RUNX2-independent upregulation of ALPL in response to some osteogenic stimuli. Specifically, TNF-α and IL-1β decreased RUNX2 expression in human mesenchymal stem cells but increased expression of ALPL, and this increase of ALPL and subsequent TNAP activity is sufficient to induce calcification.48 The necessity of functional TNAP is clear in both humans and mice, where genetic mutation or deletion of the ALPL gene causes hypophosphatasia, also known as rickets, and one study found that FOXO1 binds and activates murine Alpl promoter, which supports our finding on the role of FOXO1 in activating ALPL promoter activity in human cells.49–51 Our data show that in the absence of CD73 activity, AMP induces FOXO1 nuclear localization which stimulates ALPL promoter activation; nuclear localization of FOXO1 is prevented when cAMP levels are increased.
Last, our data suggest that mechanisms driving ACDC pathogenesis may contribute to calcification in non-ACDC peripheral arteries. Calcification in the lower extremity arteries is relatively common and can present alongside or independent of atherosclerosis. Differences in calcification properties and patterns have been observed in the vascular beds. Carotid artery calcification is most commonly found in the necrotic core of atherosclerotic plaques, while lower-extremity calcification is often plaque-independent and localizes in the medial layer along the elastic fibers.15,52 Carotid, femoral, and femoropopliteal arteries exhibit microcalcification, sheet calcification, and nodular calcification; however, the lower extremity vessels exhibited true bone formation (osteoid metaplasia), indicative of a cell phenotypic switching from a vascular to an osteogenic-type cell.53 Supporting this, lower-extremity arteries exhibited elevated expression of bone development genes, and relevant to our findings, ENPP1, the enzyme directly upstream of CD73 that converts extracellular ATP to AMP, was found to be drastically downregulated in lower limb arteries, suggesting that its absence may contribute to decreases in the available adenosine.
The femoropopliteal artery of mobile individuals experiences mechanical stretches and stresses during limb flexion which may induce cells to release ATP.54 ATP in the extracellular space is rapidly converted to adenosine when CD73 is present, and this adenosine helps protect cells from this stress, hence the notion that adenosine is a retaliatory metabolite.55 Calcified femoropopliteal arteries exhibit medial remodeling and are stiffer in both longitudinal and circumferential directions.15 A recent study classified 431 femoropopliteal artery samples into 7 groups that histologically exhibit varying degrees of calcification and found that stiffness precedes evidence of extensive calcification.15 Mechanical stretch can induce the expression of CD73 on lung epithelial cells and the adenosine generated is protective against acute lung injury.56 We found robust staining for CD73 in control femoropopliteal arteries; however, CD73 was nearly absent in calcified arteries. We found that calcified femoropopliteal arteries exhibit lower levels of CD73. Future studies will explore whether stiffness decreases CD73 expression and whether aberrant CD73/adenosine receptor signaling precedes calcification in non-ACDC contexts.
From these studies and our observations, we postulate that CD73-mediated adenosine signaling may protect the femoropopliteal artery during mechanical stretch; thus, a reduction of CD73 would prevent this protection. What is clear from our data is that the study of this rare monogenetic disease is able to inform us about potential novel mechanisms operative in more common forms of peripheral medial layer vascular calcification.
Acknowledgments
We would like to thank Jason Dobbins for insightful discussion and critical reading of this manuscript. We would also like to acknowledge Live On Nebraska for their help and support, and thank tissue donors and their families for making this study possible. Some figures created with Biorender.com.
Sources of Funding
C.S.H. is supported by the National Heart, Lung, and Blood Institute K22 HL117917, the Pittsburgh Heart, Lung, and Blood Vascular Medicine Institute, and the University of Pittsburgh School of Medicine Division of Cardiology. A.K. and J.N.M. were funded in part by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Numbers HL125736 and HL147128.
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- ACDC
- arterial calcification due to deficiency of CD73
- AMPK
- AMP-activated protein kinase
- FOXO1
- forkhead box O1 protein
- iPSC
- induced pluripotent stem cell
- mTOR
- mammalian target of rapamycin
- TNAP
- tissue nonspecific alkaline phosphatase
- VASP
- vasodilator-stimulated phosphoprotein
These authors contributed equally to this article.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.119.313765.
For Sources of Funding and Disclosures, see page 1692.
References
- 1.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 2.Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, St Hilaire C, Shanahan C. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35:1515–1525. doi: 10.1093/eurheartj/ehu163. doi: 10.1093/eurheartj/ehu163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.St Hilaire C, Liberman M, Miller J Early Career Committee. Bidirectional translation in cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2016;36:e19–e24. doi: 10.1161/ATVBAHA.115.307056. doi: 10.1161/ATVBAHA.115.307056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 5.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. doi: 10.1056/NEJMoa0912923. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. 2012;95:269–280. doi: 10.1093/cvr/cvs187. doi: 10.1093/cvr/cvs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto CM, Prendergast B. Aortic-valve stenosis–from patients at risk to severe valve obstruction. N Engl J Med. 2014;371:744–756. doi: 10.1056/NEJMra1313875. doi: 10.1056/NEJMra1313875. [DOI] [PubMed] [Google Scholar]
- 8.Sauer H, Hescheler J, Wartenberg M. Mechanical strain-induced Ca(2+) waves are propagated via ATP release and purinergic receptor activation. Am J Physiol Cell Physiol. 2000;279:C295–C307. doi: 10.1152/ajpcell.2000.279.2.C295. doi: 10.1152/ajpcell.2000.279.2.C295. [DOI] [PubMed] [Google Scholar]
- 9.Murata N, Ito S, Furuya K, Takahara N, Naruse K, Aso H, Kondo M, Sokabe M, Hasegawa Y. Ca2+ influx and ATP release mediated by mechanical stretch in human lung fibroblasts. Biochem Biophys Res Commun. 2014;453:101–105. doi: 10.1016/j.bbrc.2014.09.063. doi: 10.1016/j.bbrc.2014.09.063. [DOI] [PubMed] [Google Scholar]
- 10.Li Q, Price TP, Sundberg JP, Uitto J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: similarities to patients with NT5E mutations. Cell Cycle. 2014;13:2609–2615. doi: 10.4161/15384101.2014.943567. doi: 10.4161/15384101.2014.943567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joolharzadeh P, St Hilaire C. CD73 (Cluster of Differentiation 73) and the differences between mice and humans. Arterioscler Thromb Vasc Biol. 2019;39:339–348. doi: 10.1161/ATVBAHA.118.311579. doi: 10.1161/ATVBAHA.118.311579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011;103:44–50. doi: 10.1016/j.ymgme.2011.01.018. doi: 10.1016/j.ymgme.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin H, St Hilaire C, Huang Y, Yang D, Dmitrieva NI, Negro A, Schwartzbeck R, Liu Y, Yu Z, Walts A, et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci Signal. 2016;9:ra121. doi: 10.1126/scisignal.aaf9109. doi: 10.1126/scisignal.aaf9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamenskiy A, Poulson W, Sim S, Reilly A, Luo J, MacTaggart J. Prevalence of calcification in human femoropopliteal arteries and its association with demographics, risk factors, and arterial stiffness. Arterioscler Thromb Vasc Biol. 2018;38:e48–e57. doi: 10.1161/ATVBAHA.117.310490. doi: 10.1161/ATVBAHA.117.310490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.St Hilaire C, Yang D, Schreiber BM, Ravid K. B-Myb regulates the A(2B) adenosine receptor in vascular smooth muscle cells. J Cell Biochem. 2008;103:1962–1974. doi: 10.1002/jcb.21586. doi: 10.1002/jcb.21586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliver L, Hue E, Priault M, Vallette FM. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012;21:2779–2788. doi: 10.1089/scd.2012.0124. doi: 10.1089/scd.2012.0124. [DOI] [PubMed] [Google Scholar]
- 18.Dai XY, Zhao MM, Cai Y, Guan QC, Zhao Y, Guan Y, Kong W, Zhu WG, Xu MJ, Wang X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013;83:1042–1051. doi: 10.1038/ki.2012.482. doi: 10.1038/ki.2012.482. [DOI] [PubMed] [Google Scholar]
- 19.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–D110. doi: 10.1093/nar/gkj143. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 22.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120(Pt 15):2479–2487. doi: 10.1242/jcs.001222. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 23.de Nijs T, Albuisson J, Ockeloen CW, Legrand A, Jeunemaitre X, Schultze Kool LJ, Riksen NP. Isolated arterial calcifications of the lower extremities: A clue for NT5E mutation. Int J Cardiol. 2016;212:248–250. doi: 10.1016/j.ijcard.2016.03.068. doi: 10.1016/j.ijcard.2016.03.068. [DOI] [PubMed] [Google Scholar]
- 24.Ichikawa N, Taniguchi A, Kaneko H, Kawamoto M, Sekita C, Nakajima A, Yamanaka H. Arterial Calcification Due to Deficiency of CD73 (ACDC) as one of rheumatic diseases associated with periarticular calcification. J Clin Rheumatol. 2015;21:216–220. doi: 10.1097/RHU.0000000000000245. doi: 10.1097/RHU.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 25.Kochan Z, Karbowska J, Gogga P, Kutryb-Zajac B, Slominska EM, Smolenski RT. Polymorphism in exon 6 of the human NT5E gene is associated with aortic valve calcification. Nucleosides Nucleotides Nucleic Acids. 2016;35:726–731. doi: 10.1080/15257770.2016.1180393. doi: 10.1080/15257770.2016.1180393. [DOI] [PubMed] [Google Scholar]
- 26.Rothe H, Brandenburg V, Haun M, Kollerits B, Kronenberg F, Ketteler M, Wanner C. Ecto-5’ -Nucleotidase CD73 (NT5E), vitamin D receptor and FGF23 gene polymorphisms may play a role in the development of calcific uremic arteriolopathy in dialysis patients - Data from the German Calciphylaxis Registry. PLoS One. 2017;12:e0172407. doi: 10.1371/journal.pone.0172407. doi: 10.1371/journal.pone.0172407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshioka K, Kuroda S, Takahashi K, Sasano T, Furukawa T, Matsumura A. Calcification of joints and arteries with novel NT5E mutations with involvement of upper extremity arteries. Vasc Med. 2017;22:541–543. doi: 10.1177/1358863X17724263. doi: 10.1177/1358863X17724263. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Calcification of joints and arteries: second report with novel NT5E mutations and expansion of the phenotype. J Hum Genet. 2015;60:561–564. doi: 10.1038/jhg.2015.85. doi: 10.1038/jhg.2015.85. [DOI] [PubMed] [Google Scholar]
- 29.Koszalka P, Ozüyaman B, Huo Y, Zernecke A, Flögel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sévigny J, et al. Targeted disruption of cd73/ecto-5’-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821. doi: 10.1161/01.RES.0000144796.82787.6f. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- 30.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5’-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5’-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown MS, Goldstein JL. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell. 1976;9(4 PT 2):663–674. doi: 10.1016/0092-8674(76)90130-6. doi: 10.1016/0092-8674(76)90130-6. [DOI] [PubMed] [Google Scholar]
- 33.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S, Jee K, Kim D, Koh H, Chung J. Cyclic AMP inhibits Akt activity by blocking the membrane localization of PDK1. J Biol Chem. 2001;276:12864–12870. doi: 10.1074/jbc.M001492200. doi: 10.1074/jbc.M001492200. [DOI] [PubMed] [Google Scholar]
- 35.Xie J, Ponuwei GA, Moore CE, Willars GB, Tee AR, Herbert TP. cAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell Signal. 2011;23:1927–1935. doi: 10.1016/j.cellsig.2011.06.025. doi: 10.1016/j.cellsig.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adegbite NS, Xu M, Kaplan FS, Shore EM, Pignolo RJ. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am J Med Genet A. 2008;146A:1788–1796. doi: 10.1002/ajmg.a.32346. doi: 10.1002/ajmg.a.32346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rittiner JE, Korboukh I, Hull-Ryde EA, Jin J, Janzen WP, Frye SV, Zylka MJ. AMP is an adenosine A1 receptor agonist. J Biol Chem. 2012;287:5301–5309. doi: 10.1074/jbc.M111.291666. doi: 10.1074/jbc.M111.291666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Nakayama K, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 39.Weigel D, Jürgens G, Küttner F, Seifert E, Jäckle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645–658. doi: 10.1016/0092-8674(89)90133-5. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- 40.Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–253. doi: 10.1038/ng890. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 41.Teixeira CC, Liu Y, Thant LM, Pang J, Palmer G, Alikhani M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J Biol Chem. 2010;285:31055–31065. doi: 10.1074/jbc.M109.079962. doi: 10.1074/jbc.M109.079962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, Depinho RA, Kousteni S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010;11:147–160. doi: 10.1016/j.cmet.2010.01.001. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng L, Huang L, Sun Y, Heath JM, Wu H, Chen Y. Inhibition of FOXO1/3 promotes vascular calcification. Arterioscler Thromb Vasc Biol. 2015;35:175–183. doi: 10.1161/ATVBAHA.114.304786. doi: 10.1161/ATVBAHA.114.304786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol. 2000;20:8783–8792. doi: 10.1128/mcb.20.23.8783-8792.2000. doi: 10.1128/mcb.20.23.8783-8792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, et al. Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1387–1396. doi: 10.1161/ATVBAHA.110.222547. doi: 10.1161/ATVBAHA.110.222547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Zheng B, Zhou PP, Zhang RN, He M, Yang Z, Wen JK. Vascular calcification is coupled with phenotypic conversion of vascular smooth muscle cells through Klf5-mediated transactivation of the Runx2 promoter. Biosci Rep. 2014;34:e00148. doi: 10.1042/BSR20140103. doi: 10.1042/BSR20140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin ME, Chen T, Leaf EM, Speer MY, Giachelli CM. Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. Am J Pathol. 2015;185:1958–1969. doi: 10.1016/j.ajpath.2015.03.020. doi: 10.1016/j.ajpath.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian JC, Hardouin P, Magne D. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. 2009;84:499–504. doi: 10.1016/j.lfs.2009.01.013. doi: 10.1016/j.lfs.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millán JL, MacGregor GR, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015–2026. doi: 10.1359/jbmr.1999.14.12.2015. doi: 10.1359/jbmr.1999.14.12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss MJ, Cole DE, Ray K, Whyte MP, Lafferty MA, Mulivor RA, Harris H. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc Natl Acad Sci USA. 1988;85:7666–7669. doi: 10.1073/pnas.85.20.7666. doi: 10.1073/pnas.85.20.7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatta M, Daitoku H, Matsuzaki H, Deyama Y, Yoshimura Y, Suzuki K, Matsumoto A, Fukamizu A. Regulation of alkaline phosphatase promoter activity by forkhead transcription factor FKHR. Int J Mol Med. 2002;9:147–152. [PubMed] [Google Scholar]
- 52.Yahagi K, Kolodgie FD, Otsuka F, Finn AV, Davis HR, Joner M, Virmani R. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol. 2016;13:79–98. doi: 10.1038/nrcardio.2015.164. doi: 10.1038/nrcardio.2015.164. [DOI] [PubMed] [Google Scholar]
- 53.Steenman M, Espitia O, Maurel B, Guyomarch B, Heymann MF, Pistorius MA, Ory B, Heymann D, Houlgatte R, Gouëffic Y, et al. Identification of genomic differences among peripheral arterial beds in atherosclerotic and healthy arteries. Sci Rep. 2018;8:3940. doi: 10.1038/s41598-018-22292-y. doi: 10.1038/s41598-018-22292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mikolajewicz N, Mohammed A, Morris M, Komarova SV. Mechanically stimulated ATP release from mammalian cells: systematic review and meta-analysis. J Cell Sci. 2018;131:jcs223354. doi: 10.1242/jcs.223354. [DOI] [PubMed] [Google Scholar]
- 55.Newby AC. Adenosine and the concept of ‘retaliatory metabolites’. Trends Biochem Sci. 1984;9:42–44. [Google Scholar]
- 56.Eckle T, Füllbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.