

Abstract
Rationale:
Small molecule inhibitors of the acetyl-histone binding protein BRD4 have been shown to block cardiac fibrosis in pre-clinical models of heart failure (HF). However, since the inhibitors target BRD4 ubiquitously, it is unclear whether this chromatin reader protein functions in cell type-specific manner to control pathological myocardial fibrosis. Furthermore, the molecular mechanisms by which BRD4 stimulates the transcriptional program for cardiac fibrosis remain unknown.
Objective:
We sought to test the hypothesis that BRD4 functions in a cell-autonomous and signal-responsive manner to control activation of cardiac fibroblasts, which are the major extracellular matrix (ECM)-producing cells of the heart.
Methods and Results:
RNA-sequencing (RNA-seq), mass spectrometry and cell-based assays employing primary adult rat ventricular fibroblasts (ARVFs) demonstrated that BRD4 functions as an effector of TGF-β signaling to stimulate conversion of quiescent cardiac fibroblasts into Periostin (Postn)-positive cells that express high levels of ECM. These findings were confirmed in vivo through whole-transcriptome analysis of cardiac fibroblasts from mice subjected to transverse aortic constriction (TAC) and treated with the small molecule BRD4 inhibitor, JQ1. Chromatin immunoprecipitation-sequencing (ChIP-seq) revealed that BRD4 undergoes stimulus-dependent, genome-wide redistribution in cardiac fibroblasts, becoming enriched on a subset of enhancers and super-enhancers, and leading to RNA polymerase II activation and expression of downstream target genes. Employing the SERTA domain-containing protein 4 (Sertad4) locus as a prototype, we demonstrate that dynamic chromatin targeting of BRD4 is controlled, in part, by p38 mitogen-activated protein kinase (MAPK) and provide evidence of a critical function for Sertad4 in TGF-β-mediated cardiac fibroblast activation.
Conclusions:
These findings define BRD4 as a central regulator of the pro-fibrotic cardiac fibroblast phenotype, establish a p38-dependent signaling circuit for epigenetic reprogramming in HF, and uncover a novel role for Sertad4. The work provides a mechanistic foundation for the development of BRD4 inhibitors as targeted anti-fibrotic therapies for the heart.
Keywords: Fibrosis, Mechanisms, Myocardial Bilogy, Chromatin, Epigenetics, Fibroblast, Fibrosis, Heart Failure, Signaling
Graphical Abstract
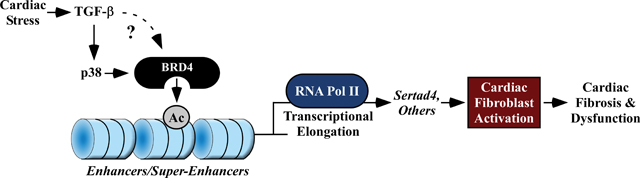
INTRODUCTION
Fibrosis is a stereotypical wound-healing response that is mounted after tissue injury or stress. A significant body of clinical data has demonstrated that myocardial fibrosis is strongly associated with adverse outcomes in several forms of human heart failure (HF), including HF with reduced ejection fraction (HFrEF), HF with preserved ejection fraction (HFpEF), and genetically driven cardiomyopathies 1, 2. While fibrotic responses may acutely serve to stabilize a focal area of myocardial damage, clinical and experimental studies support the contention that excessive, diffuse, or chronic activation of the fibrotic process can be deleterious to long-term cardiac function and patient survival. For example, interstitial fibrosis increases the passive stiffness of the myocardium, contributing to diastolic dysfunction 3, 4, and disrupts electrical conduction in the heart, causing arrhythmias and sudden cardiac death 5. Unfortunately, despite the well-accepted roles of fibrosis in cardiac dysfunction, no targeted anti-fibrotic drugs for the heart exist. As such, understanding the fundamental mechanisms driving cardiac fibrosis and discovering novel approaches to target this process are of significant scientific and therapeutic interest.
The adult heart contains a large number of resident cardiac fibroblasts, which are developmentally derived from the epicardium and play important roles in maintaining tissue architecture 6–8. In response to injury, resident tissue fibroblasts undergo a dramatic cell state transition to become activated fibroblasts 9, sometimes referred to as myofibroblasts, which are characterized by expression of the marker gene Periostin (Postn) 10, 11. This transition is associated with rewiring of the transcriptome to endow the activated fibroblasts with the capability to proliferate, migrate, secrete pro-inflammatory mediators and secrete fibrotic extracellular matrix (ECM). A causal role for activated fibroblasts in cardiac fibrosis was established employing mice in which a tamoxifen-inducible Cre cassette was used to selectively deplete Postn-positive fibroblasts using diphtheria toxin 11. In these mice, deletion of Postn-positive fibroblasts blunted cardiac fibrosis in response to angiotensin II infusion or myocardial infarction 11.
Cardiac fibroblast activation is driven, in part, by transforming growth factor-β (TGF-β), which signals through a heterodimeric cell surface receptor complex consisting of TGFβ receptor type I (TGFβRI) and II (TGFβRII) 12, 13. Canonical TGF-β signaling leads to phosphorylation and nuclear import of SMAD transcription factors, which bind to regulatory elements in a variety of pro-fibrotic genes. Deletion of TGF-β receptors or the SMAD3 family member in Postn-positive fibroblasts suppresses cardiac fibrosis in response to pressure overload in mice 14. TGF-β can also promote fibrosis by stimulating SMAD-independent, non-canonical pathways, such as those governed by mitogen-activated protein kinases (MAPKs). For example, cardiac fibroblasts lacking p38α are resistant to pro-fibrotic TGF-β signaling, and deletion of p38α in Postn-positive fibroblasts in vivo blunts cardiac fibrosis in response to myocardial infarction 15, 16. Transcriptional effectors of non-canonical TGF-β signaling in fibroblasts include nuclear factor of activated T cells (NFAT), serum response factor (SRF) and myocardin-related transcriptional cofactors (MRTFs) 17–21.
Recently, a family of epigenetic reader molecules called bromodomain and extraterminal (BET) acetyl-lysine binding proteins was shown to control pathological fibrosis 22. JQ1, a first-in-class, potent and specific inhibitor of BET bromodomains that functions by competitively displacing BET proteins from acetylated-histones 23, was found to block cardiac fibrosis in mice subjected to pressure overload or myocardial infarction 24–26. Bulk RNA-seq analysis of left ventricular tissue homogenates demonstrated the ability of JQ1 to block expression of fibroblasts activation markers, including Postn 25. However, the mechanism by which BET protein inhibition suppressed fibrotic remodeling in the heart remained undefined. Here, we show that JQ1 potently inhibits cardiac fibroblast activation, and that the BET family member, BRD4, functions as a downstream effector that couples pro-fibrotic TGF-β signaling to the epigenome in cardiac fibroblasts. The findings reveal a critical role for a chromatin reader protein in the epigenetic control of pathological cardiac fibrosis and HF.
METHODS
The authors declare that all supporting data, analytical methods, and materials developed from this group within the article and its Online Data Supplement files are available. A detailed description of methods and materials is provided in the Online Data Supplement.
RESULTS
BRD4 inhibition blocks TGF-β-mediated cardiac fibroblast activation.
When placed in a collagen gel, activated fibroblasts exhibit the ability to contract due to expression of α-smooth muscle actin (α-SMA) 27, 28, and this process can be enhanced by stimulation with TGF-β 29. To begin to address the potential role of BRD4 in the control of cardiac fibroblast activation, primary adult rat ventricular fibroblast (ARVF) cultures were incorporated into collagen gels and stimulated with TGF-β in the absence or presence of JQ1. JQ1 significantly reduced basal and TGF-β-mediated collagen gel contraction in association with a profound reduction in α-SMA protein expression (Figure 1A-C). Dose-response analysis revealed that JQ1 suppressed α-SMA expression in ARVFs with a half-maximal inhibitory concentration (IC50) of 94 nM (Figure 1D).
Figure 1. JQ1 suppresses TGF-β-induced cardiac fibroblast activation.

A, ARVFs seeded on compressible collagen gel matrices were assayed for gel contraction following treatment with TGF-β1 (10ng/mL) and/or JQ1 (500nM) for 72 hours. B, Quantification of gel contraction images, reported as percentage contraction; (n=4 plates per condition). Data are presented as mean ± SEM. *P< 0.05 vs. TGF-β alone. Additional two-way ANOVA analysis at the 72hr time point indicated that the TGF-β Factor accounted for 78.04% of variation (P<0.0001), the JQ1 Factor 18.25% of variation (P<0.0001), and the interaction 1.877% of variation (P<0.05). C, ARVFs were treated with TGF-β1 in the absence or presence of JQ1 for 72 hours prior to fixation for indirect immunofluorescence detection of fibronectin (red) and α-SMA (green). Nuclei were stained with Hoechst 33342 (blue); scale bar = 100μm. D, ARVFs were treated with TGF-β1 in the presence of a concentration response of JQ1 (10μM to 5nM final concentration) for 72 hours prior to fixation for indirect immunofluorescence detection α-SMA. Each concentration was tested in duplicate wells and the data for α-SMA were normalized to the control wells of the plate to determine percent inhibition of the TGF-β1 stimulated α−SMA signal. The IC50 for inhibition of α-SMA was determined by non-linear regression of the normalized data. E and F, ARVFs were transfected with negative control siRNA (siCtrl) or siRNAs targeting BRD2, BRD3 or BRD4 and maintained in low serum medium for 48 hours. BET family member mRNA expression was determined by qRT-PCR (E) or immunoblotting (F); n=4 plates of cells per condition, *P < 0.05 vs siCtrl. α-tubulin served as a loading control. G, ARVFs were transfected with the indicated siRNAs and maintained in low serum medium or were treated with TGF-β1 for 48 hours prior to determination of α−SMA mRNA expression levels by qRT-PCR; n=4 plates of cells per condition, P<0.05 vs. unstimulated siControl. H, ARVFs were transfected with the indicated siRNAs and maintained in low serum medium or were treated with TGF-β1 for 48 hours prior to determination of BRD4 mRNA expression levels by qRT-PCR; n=4 plates of cells per condition, P<0.05 vs. unstimulated siControl. Additional two-way ANOVA showed the TGF-β Factor accounted for 3.35% of variation (P= 0.0117), the BRD4 Factor 93.24% of variation (P<0.0001), and the interaction <1% of variation (P<0.01). I, ARVFs were transfected with the indicated siRNAs and maintained in low serum medium or were treated with TGF-β1 for 48 hours prior to indirect immunofluorescence staining of α-SMA; nuclei are marked by Hoechst 33342 staining; scale bar = 10μm. J, Quantification of α-SMA staining; *P<0.05, ***P<0.0001. Additional Two-way ANOVA of all depicted groups showed the TGF-β Factor accounted for 51.08% of variation (P< 0.0001), the BET Factor 11.28 of variation (P<0.0001), and the interaction 13.7% of variation (P<0.0001). When two-way ANOVA was run individually (siCtrl vs siBET, one BET at a time), only the siBRD4 analysis showed that variance was attributed to BET targeting (16.4%, P<0.0001). All data are presented as mean +SEM. Statistical analysis was performed by unpaired t-test (E) or one-way ANOVA with Tukey’s post-hoc test (B, G, H, J) (J, siCtrl group not normally distributed, Kruskal-Wallis test P<0.05).
Resident cardiac fibroblasts undergo a burst of proliferation in response to stress signals 9. Consistent with this, treatment of ARVFs with TGF-β led to a dramatic increase in total cell number, which was normalized by JQ1 in a concentration-dependent manner (Online Figure IA and IB). In contrast, JQ1 had minimal effects on ARVF viability (Online Figure IC). These data suggest that, in addition to blocking cardiac fibroblast activation, JQ1 suppresses proliferation of this pro-fibrotic cell type.
Small interfering RNAs (siRNAs) were employed to address the roles of specific BET family members in the control of cardiac fibroblast activation. Quantitative PCR and immunoblotting confirmed selective knockdown of BRD2, BRD3 and BRD4 mRNA and protein expression in ARVFs (Figure 1E and 1F). Knockdown of BRD4, but not BRD2 or BRD3, suppressed TGF-β-mediated induction of α-SMA (Figure 1G), and, consistent with a role for BRD4 in agonist-induced ARVF activation, BRD4 mRNA expression was stimulated following TGF-β treatment (Figure 1H). Knockdown of BRD4 also blunted the formation of α-SMA-positive stress fibers in TGF-β-treated ARVFs; a modest reduction in α-SMA protein expression was also noted upon BRD2 knockdown (Figure 1I and 1J). These data suggest that BRD4 is the primary BET family member that regulates cardiac fibroblast activation
BRD4 inhibition blocks TGF-β-mediated mRNA and protein expression in cardiac fibroblasts.
To further address the role of BRD4 in cardiac fibroblast activation, transcriptomic profiling by RNA sequencing (RNA-seq) was performed using RNA from ARVFs stimulated with TGF-β for 24 hours in the absence or presence of JQ1. Using a >two-fold expression change, minimum average expression of 5 FPKM, and a false discovery rate (FDR) of <0.05 as cutoffs, differential expression analysis showed that TGF-β increased expression of 174 genes and reduced expression of 236 genes which are depicted in heat map format (Figure 2A). Data from cells treated with TGF-β in the presence of JQ1 are also included in the heat map. Ingenuity Pathway Analysis (IPA) of TGF-β-induced transcripts showed a strong enrichment for fibrosis and inflammatory signaling (Figure 2B); the top five enriched pathways in TGF-β treated ARVFs in the absence or presence of JQ1 are shown. IPA also revealed that JQ1 profoundly suppressed expression of target genes that are known to be directly downstream of TGF-β signaling (Figure 2C and 2D). Additional enrichment analyses, which include data from ARVFs treated with JQ1 alone, reveal the complexities of TGF-β and JQ1 action, and demonstrate that not all TGF-β-inducible genes were inhibited by JQ1 (Online Figure II and Online Figure IV). Furthermore, subsets of transcripts were induced by JQ1, either in the absence or presence of TGF-β treatment, suggesting that BRD4 may also function as a transcriptional repressor in cardiac fibroblasts. In this regard, a truncated isoform of BRD4, which remains sensitive to JQ1, has previously been shown to function as a transcriptional co-repressor 30.
Figure 2. JQ1 globally suppresses pro-fibrotic gene expression in cardiac fibroblasts.

ARVFs were treated with TGF-β1 in the absence or presence of JQ1 or vehicle control (DMSO) for 24 hours prior to extraction of RNA for RNA-seq analysis. A, Heat map of genes significantly altered by TGF-β1. Each column represents data from a distinct plate of cells. Color indicates row-normalized expression from +2 (red) to −2 (blue). B, Ingenuity pathway analysis (IPA) was used to determine canonical pathways significantly altered by TGF-β stimulation relative to vehicle control or versus TGF-β + JQ1 treatment. The top five affected pathways for each analysis are reported; three redundant cholesterol biosynthesis pathways were removed from the lower table. C, IPA analysis also indicated TGF-β as the strongest upstream effector molecule in the dataset. Induced genes that led to this determination are displayed (red indicates increased expression following TGF-β treatment). D, Comparison of expression of these same genes in TGF-β-treated versus TGF-β + JQ1 treated cells revealed that JQ1 blocked induction of the majority of TGF-β downstream target genes (blue indicates decreased expression following JQ1 treatment, grey indicates no significant change). Connecting lines in C and D represent IPA prediction of direct TGF-β effects on gene expression in the TGF-β vs unstimulated analysis (C); orange = predicted and measured activation, grey = predicted effect without predicted directionality, and yellow = predicted effect opposite of measured effect in the dataset. Shape key:  =cytokine,
=cytokine,  =enzyme,
=enzyme,  =G-protein couple receptor,
=G-protein couple receptor, =kinase,
=kinase, =peptidase,
=peptidase,  =transcription regulator,
=transcription regulator,  =transporter,
=transporter,  =transmembrane receptor,
=transmembrane receptor,  =other.
=other.
Postn was among the most strongly downregulated transcripts in JQ1 treated ARVFs, further supporting a role for BRD4 in the control of cardiac fibroblast activation (Figure 2C and 2D, black arrows). Quantitative mass spectrometry confirmed that the BET inhibitor blocked TGF-β-induced expression of Periostin protein (Figure 3A). Furthermore, principal component analysis of relative protein abundance in whole-cell ARVF homogenates indicated clear segregations in the constellation of proteins expressed in unstimulated ARVFs compared to those in cells treated with TGF-β in the absence or presence of JQ1 (Figure 3B). Differential expression analysis revealed that TGF-β treatment significantly induced the expression of 64 proteins (adjusted P-value < 0.05). As depicted in the heat map, JQ1 blocked induction of a subset, but not all, TGF-β-induced proteins (Figure 3C; Periostin is indicated with a black arrow).
Figure 3. JQ1 suppresses TGF-β-induced expression of protein markers of cardiac fibroblast activation.

ARVFs were treated with DMSO vehicle or TGF-β in the absence or presence of JQ1 for 48 hours. Total protein was subjected to LC-MS analysis, as described in the Methods section. A, Label-free quantification of Periostin protein expression. Box: interquartile range; whiskers: min to max, with n=3 plates of cells per condition; Normalized Spectral Abundance Factor (NSAF). B, Principal component analysis of relative protein abundance revealed clear segregation of each treatment group. Each box represents data from an independent plate of ARVFs. C, Heat map of standardized protein values depicts proteins that were increased in expression upon TGF-β stimulation, and impact of JQ1 treatment on this induction (Benjamini & Hochberg adjusted P-value <0.1). Color indicates row-normalized expression from +2 (red) to −2 (blue).
BRD4 inhibition blocks TAC-induced changes in cardiac fibroblast gene expression in vivo.
To determine whether BRD4 inhibition also suppresses cardiac fibroblast activation in the heart in vivo, mice were subjected to transverse aortic constriction (TAC) and treated with JQ1 or vehicle control by intraperitoneal injection every other day for two weeks (Figure 4A). At the completion of the study, cardiac fibroblasts were rapidly isolated from the hearts of the mice as well as sham controls, and RNA was prepared from the cells immediately following isolation; gravimetric analysis of the hearts prior to cell/RNA isolation confirmed that the TAC procedure induced cardiac hypertrophy, and that JQ1 blocked the hypertrophic response (Online Figure III). Transcriptomic profiling of the cardiac fibroblasts was performed by RNA-seq. Using a >two-fold expression change and a FDR <0.05 as cutoffs, 64 transcripts were found to be significantly upregulated in cardiac fibroblasts following TAC and significantly inhibited by JQ1 (Figure 4B). Consistent with the cell culture data, TAC-induced Postn mRNA expression was dramatically repressed in fibroblasts from JQ1-treated mice (Figure 4B, arrow). Additionally, IPA analysis revealed a strong enrichment for fibrosis and inflammatory signaling in cardiac fibroblasts post-TAC (Figure 4C); the top five enriched pathways in cardiac fibroblasts from mice subjected to pressure overload in the absence or presence of JQ1 are shown. The impact of TAC and JQ1 on expression of fibrosis- and inflammation-associated mRNA transcripts is depicted (Figure 4D). Together, the findings suggest that BRD4 regulates cardiac fibroblast activation in response to pressure overload in vivo.
Figure 4. JQ1 suppresses pressure overload-induced pro-fibrotic gene expression in cardiac fibroblasts in vivo.

A, Schematic representation of the experiment. B, Heat map of genes significantly upregulated by transverse aortic constriction (TAC) and suppressed by JQ1. Color indicates row-normalized expression from +2 (red) to −2 (blue). C, IPA was used to determine which gene expression pathways were significantly altered in cardiac fibroblasts isolated from mice subjected to TAC isolated versus Sham controls, or in cardiac fibroblasts from TAC +JQ1 treated mice versus TAC alone. The top five affected pathways for each analysis are shown. D, The diagram depicts genes from the IPA ‘hepatic fibrosis pathway’ that were upregulated in cardiac fibroblasts from mice subjected to TAC. E, Comparison of expression of these same genes in TAC versus TAC + JQ1-treated mice revealed that JQ1 blocked induction of the majority of the target genes in this pathway (D and E, blue indicates decreased expression, red indicates increased expression, and gray indicates no change; shape key:  =cytokine,
=cytokine,  =enzyme,
=enzyme,  =kinase,
=kinase,  =transmembrane receptor,
=transmembrane receptor,  =other).
=other).
Gene set enrichment analysis revealed congruence in TGF-β-induced/repressed mRNAs (RNA-Seq) and proteins (mass spectrometry) in ARVFs (Online Figure IVA and IVB), as well as similarities in TGF-β- and TAC-induced gene expression in ARVFs and resident cardiac fibroblasts, respectively (Online Figure IVC). However, transcripts that were suppressed following TGF-β treatment of ARVFs did not show significant enrichment with mRNAs that were inhibited in resident cardiac fibroblasts following TAC (Online Figure IVD). JQ1 targeted largely overlapping gene sets in TGF-β-treated ARVFs and in fibroblasts from the TAC overloaded hearts (Online Figure IVG). IPA analysis was performed on the transcripts found to be enriched in the analyzed data sets, and the top affected pathways are included for each analysis (Online Figure IV). Induced genes that led to significant enrichment scores between the data sets were often associated with metabolic processes, including amino acid synthesis, degradation, or modification. Enrichment scores from each analysis are provided in Online Table V.
BRD4 binding to cardiac fibroblast enhancers is dynamically regulated by TGF-β.
To address the mechanism by which BRD4 promotes cardiac fibroblast activation, ARVFs were subjected to whole-genome chromatin immunoprecipitation-sequencing (ChIP-seq) to map gene regulatory elements bound by BRD4. ARVFs were treated with vehicle control or stimulated with TGF-β for 24 hours prior to preparation of sheared chromatin (Figure 5A). ChIP was performed separately with BRD4- and RNA Pol II-specific antibodies, and associated cardiac fibroblast DNA was subject to deep sequencing. Aggregate analysis of the two groups revealed well-defined peaks of BRD4 binding to 7,813 gene enhancers, with enhancers defined as being at least 2,500 base pairs (bp) from the transcription start site (TSS) of the associated gene (Figure 5B). Prominent BRD4 binding was also detected at 4,069 gene promoters, defined as TSSs +/− 1000 bp (Online Figure V).
Figure 5. Chromatin targeting of BRD4 in cardiac fibroblasts correlates with RNA pol II elongation and downstream target gene expression.

A, Schematic representation of the experiment. B, Heat map of BRD4- bound enhancers in TGF-β and unstimulated ARVFs covering 10kb upstream and downstream of the enhancer summit, where enhancers are grouped by increased, decreased or unchanged BRD4 binding in TGF-β stimulated ARVFs compared to unstimulated cells; mean reads per million mapped reads per base pair (RPM/bp). C, Top – a histogram of cumulative BRD4 binding to enhancer/promoters of 4,674 active genes ranked by cumulative BRD4 enhancer abundance in response to TGF-β treatment. Bottom - a second histogram depicts the relative mRNA expression of genes proximal to the BRD4 bound enhancers/promoters. The downstream target gene mRNAs were clustered into bins of 100, ranked left-to-right based on cumulative BRD4 abundance at the enhancer/promoter (above); the data are presented as means ±SEM. D, RNA Pol II binding to gene bodies (top) and promoters (bottom) of the corresponding genes is also shown as a histogram. RNA Pol II-bound genes were clustered into bins of 100, ranked left-to-right based on log2 fold-change of cumulative BRD4 abundance at the enhancer/promoter (above); the data are presented as means ±SEM. E and F, RNA Pol II rpm/bp plotted for 400 genes where TGF-β treatment led to increased BRD4 binding (E), or decreased BRD4 binding (F). G and H, BRD4 rpm/bp plotted for 400 genes where TGF-β treatment led to increased BRD4 binding (G), or decreased BRD4 binding to proximal enhancers (H). The insets magnify gene bodies, highlighting RNA Pol II elongation behavior and BRD4 at intragenic enhancers.
To assess whether genomic targeting of BRD4 in cardiac fibroblasts is under signal-dependent control, we evaluated binding of the protein to 4,674 genes (enhancers and promoters combined), which are defined as active genes based on the presence of RNA Pol II in the TSS +/− 1000 bp region, and an fpkm >= 10. Three general patterns of BRD4 dynamics were observed: (i) increased BRD4 binding upon TGF-β stimulation, (ii) loss of BRD4 binding following TGF-β stimulation, and (iii) constitutive BRD4 binding that is unchanged by TGF-β treatment (Figure 5C, top panel); the genes where BRD4 association with regulatory elements was most dramatically increased and decreased are highlighted in red and green, respectively. BRD4 abundance correlated with downstream target gene expression (Figure 5C, bottom panel).
Comparison of BRD4 and RNA Pol II ChIP-seq signals suggested that association of BRD4 with regulatory elements for the 4,674 genes is coupled to productive transcription elongation, since RNA Pol II abundance within the bodies of the genes correlated with the degree of BRD4 binding (Figure 5D, top panel); the amount of RNA Pol II within the gene body only increased in cases where TGF-β stimulation enhanced BRD4 binding. Paradoxically, TGF-β treatment led to decreased RNA Pol II binding to promoter elements for all of the 4,674 genes examined, although the degree of the diminution paralleled the extent of BRD4 binding (Figure 5D, bottom panel).
RNA Pol II dynamics were further scrutinized by focusing on the top 400 genes where BRD4 binding was most dramatically increased or decreased in response to TGF-β. ChIP-seq showed that TGF-β stimulation enhanced gene-body enrichment of RNA Pol II when BRD4 was gained at enhancers/promoters, but decreased gene-body enrichment of RNA Pol II when the presence of BRD4 was reduced at regulatory sites upon cellular stimulation (Figure 5E and 5F). These findings corroborate a critical role for BRD4 in TGF-β-coupled pause release of RNA poll II at upregulated genes in cardiac fibroblasts.
TGF-β-mediated recruitment of BRD4 to Sertad4 enhancers and super-enhancers.
To further interrogate signal-dependent regulation of BRD4 genomic localization in cardiac fibroblasts, subsequent analyses focused on enhancers and super-enhancers (SEs), which are long-range gene regulatory elements that have been defined in cancer and immune cells based on abundant BRD4 binding above a threshold level found at typical enhancers 31–33. Dynamic, signal-regulated targeting of BRD4 in cardiac fibroblasts is exemplified by the Sertad4 locus, where the abundance of the chromatin reader was dramatically increased at the promoter region and six distinct proximal enhancers (E1 – E6) following TGF-β stimulation of ARVFs (Figure 6A, top panel). TGF-β treatment led to enhanced binding of BRD4 to each of these sites, with profound enrichment occurring at E1 and E2 (Figure 6A, bottom panel). Based on the level of BRD4 association, both E1 and E2 were categorized as SEs following stimulation with the pro-fibrotic agonist. Consistent with a regulatory role for BRD4, TGF-β-induced expression of Sertad4 was blocked by JQ1 (Figure 6B).
Figure 6. TGF-β stimulates binding of BRD4 to enhancers and super-enhancers associated with the Sertad4 gene.

A, Top - gene track of the Sertad4 locus showing BRD4 binding to the promoter and six distinct proximal enhancers (E) in unstimulated ARVFs. Bottom – upon TGF-β stimulation for 24 hours, BRD4 binding to all of these sites was increased, reaching a threshold for definition of E1 and E2 as super-enhancers (SE). B, ARVFs were treated with TGF-β for 48 hours in the absence or presence of JQ1 or DMSO vehicle, and Sertad4 mRNA expression was determined by qRT-PCR. Data are presented as mean ± SEM; n=3 plates of cells per condition, P<0.05 vs. vehicle. C, Hockey-stick plots of BRD4-enriched enhancers in unstimulated ARVFs and ARVFs stimulated with TGF-β for 24 hours. SEs, based on a threshold level of BRD4 binding, are boxed. The abundance of BRD4 at E/SE1 and E/SE2 of the Sertad4 locus is indicated. D, Schematic representation of the ChIP-PCR experiment. E - K, BRD4-binding to the indicated Sertad4 gene regulatory elements was significantly increased by TGF-β stimulation, and these increases were blocked by JQ1. Data are presented as mean ± SEM; n=4 plates of cells per condition, P<0.05 vs. (−) TGF-β/(−) JQ1. For B and E - K, statistical analysis was performed by one-way ANOVA with Tukey’s post-hoc test (G, JQ1 group not normally distributed, Kruskal-Wallis test, P<0.05).
In total, 147 and 116 BRD4-enriched SEs were detected in unstimulated and TGF-β stimulated cardiac fibroblasts, respectively (Figure 6C). The ranking of E1 and E2, commensurate with BRD4 abundance before and after TGF-β stimulation, is indicated (Figure 6C, yellow and green circles). Follow-up ChIP-PCR experiments with independent preparations of ARVFs confirmed that TGF-β stimulates loading of BRD4 onto the Sertad4 promoter and its six distal enhancers (Figure 6D – 6K). In all cases, signal-dependent accumulation of BRD4 on the regulatory region was blocked by JQ1, establishing that targeting of BRD4 to these sites is dependent on its bromodomains.
By comparison, while BRD4 was detected at enhancer and promoter elements for the canonical fibroblast activation genes encoding α-SMA and periostin, the level of this BET family member at these sites was only increased upon stimulation with TGF-β at the periostin enhancers (Online Figure VI). JQ1 blocks expression of both α-SMA and Postn (above). Thus, the data suggest that inducible recruitment of BRD4 to Postn regulatory elements is involved in stimulation of this gene, while BRD4 loading is necessary but not sufficient for agonist-dependent expression of α-SMA. Sertad4 was chosen for follow-up investigation given the magnitude of agonist-dependent loading of BRD4 on its enhancers, and because there is a paucity of information regarding the role of SERTAD4 in fibroblast activation.
p38 MAP kinase regulates TGF-β-mediated recruitment of BRD4 to Sertad4 enhancers and super-enhancers.
The Sertad4 locus was employed as a prototype to begin to define the signaling pathways that couple TGF-β receptor activation to chromatin targeting of BRD4 in cardiac fibroblasts. Initially, a panel of pharmacological inhibitors was employed to address whether known effectors of cardiac fibroblast activation regulate agonist-dependent expression of Sertad4 (Figure 7A). As shown in Figure 7B, inhibition of p38 mitogen-activated protein kinase, but not extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) or the calcineurin phosphatase, suppressed TGF-β-induced Sertad4 mRNA expression in ARVFs. Inhibition of p38 activity also blunted TGF-β−mediated induction of α-SMA, consistent with prior studies illustrating a role for this kinase in cardiac fibroblast activation (Figure 7C). Subsequently, ChIP-PCR was used to address the hypothesis that p38 kinase activity contributes to signal-dependent chromatin targeting of BRD4 (Figure 7D). Consistent with the mRNA expression data, pan-p38 inhibition with SB203580 led to significant decreases in binding of BRD4 to the promoter region of the Sertad4 gene, as well as each of the six enhancers/super-enhancers defined above (Figure 7E). These data suggest that p38 signaling is involved in controlling BRD4 loading on gene regulatory elements in cardiac fibroblasts.
Figure 7. p38 inhibition suppresses recruitment of BRD4 to enhancers and super-enhancers associated with the Sertad4 gene.

A, Schematic representation of mRNA expression experiment. B, Inhibition of p38 (SB203580), but not ERK (PD98059), JNK (SP600125) and calcineurin (CN; cyclosporin A), blunted TGF-β-induced Sertad4 mRNA expression in ARVFs. C, SB203580 also suppressed TGF-β-induced expression of α-SMA. Data for B and C are presented as mean +SEM; n=3–6 plates of cells per condition. *P < 0.05 vs unstimulated, #P < 0.05 vs TGF-β alone by one-way ANOVA with Tukey’s post-hoc test (B p38i and C TGF-β/p38i groups not normally distributed, Kruskal-Wallis/Mann-Whitney tests P<0.05). D, Schematic representation of the ChIP-PCR experiment. E, BRD4-binding to the Sertad4 gene regulatory elements was significantly reduced in TGF-β stimulated ARVFs treated with the p38 inhibitor. Values were normalized to input levels are presented as mean +SEM; n=7 per condition. *P < 0.05 vs TGF-β alone by unpaired t-test (E, E3, p38i group not normally distributed, Mann-Whitney test p<0.05).
Sertad4 regulates TGF-β-mediated induction of markers of cardiac fibroblast activation.
Sertad4 has not previously been implicated in the regulation of cardiac fibroblast activation. Thus, to address the functional relevance of BRD4-mediated control of this factor, RNA interference was used to knock down expression of Sertad4 in ARVFs. Two independent lentivirus-encoded shRNAs efficiently repressed basal and TGF-β-induced expression of Sertad4 (Figure 8A). Furthermore, Sertad4 knockdown markedly reduced TGF-β stimulated expression of α-SMA and Postn in the cardiac fibroblasts (Figure 8B and 8C), suggesting that SERTAD4 protein contributes to cardiac fibroblast activation.
Figure 8. Sertad4 knockdown represses cardiac fibroblast activation.

ARVFs were infected with lentiviruses encoding control short-hairpin RNA (shRNA; shCtrl) or two independent shRNAs targeting Sertad4 (shSertad4 [#1] and [#2]). The cells were subsequently treated with TGF-β or DMSO control for 48 hours, and qRT-PCR analysis of Sertad4 (A), α-SMA (B) and periostin (C) mRNA expression was performed. Data are presented as mean +SEM; n=4 plates of cells per condition. *P < 0.05 vs shControl by one-way ANOVA with Tukey’s post-hoc test (A, shSertad4(#1)/TGF-β group not normally distributed, Kruskal-Wallis/Mann-Whitney tests P<0.05). To assess relative effects on α-SMA expression, an additional two-way ANOVA analysis was performed. The TGF-β Factor accounted for 57.27% of variation (P<0.0001), and the Sertad4 Factor accounted for 21.69% of variation (P<0.0001), while the interaction between TGF-β and Sertad4 accounted for 9.21% of variation (P<0.01). For periostin expression, the TGF-β Factor accounted for 34.95% of variation (P<0.0001), the Sertad4 Factor for 33.06% of variation (P<0.0001), and the interaction for 15.01% of variation (P<0.01). (D) A model for BRD4-mediated regulation of cardiac fibroblast activation. Cardiac stress signals, including TGF-β, stimulate p38 and likely other pathways to target BRD4 to gene regulatory elements, resulting in RNA Pol II elongation and expression of downstream targets, including Sertad4, which promote fibroblast activation.
DISCUSSION
Epigenetic mechanisms that regulate cardiac fibrosis remain poorly understood. Here, we demonstrate that the BRD4 chromatin reader protein is crucially involved in promoting cardiac fibroblast activation. BRD4 binding to discrete gene enhancers in fibroblasts can be altered by TGF-β receptor signaling in a p38 kinase-dependent manner, providing a circuit for coupling extracellular cues to the cardiac epigenome to drive pro-fibrotic gene expression (Figure 7D).
Our prior RNA-seq analyses demonstrated the ability of JQ1 to block expression of canonical markers of fibroblast activation in the LV in mouse TAC and MI models 25. However, interpretation of these data was complicated by the use of bulk RNA from whole LV tissue homogenates. In the current study, we demonstrate that JQ1 profoundly inhibits expression of activation markers and ECM proteins in cultured cardiac fibroblasts treated with TGF-β, and in cardiac fibroblasts isolated from LVs of animals subjected to TAC. JQ1 also suppressed the contractile activity of cardiac fibroblasts in association with repression of expression of β-SMA. These data suggest that BRD4 functions in a fibroblast-autonomous manner to promote fibrotic remodeling of the heart.
We performed parallel ChIP-seq and RNA-seq to address, mechanistically, how BRD4 regulates fibroblast activation. Remarkably, to our knowledge, the resulting data represent the first genome-wide evaluation of epigenetic regulatory events in cardiac fibroblasts. The findings revealed that BRD4 is positioned at thousands of discrete enhancers and promoters throughout the cardiac fibroblast genome. In response to TGF-β treatment, the amount of BRD4 increases at a subset of these sites, while in other cases it decreases or is unaffected by the pro-fibrotic signal. This signal-dependent regulation of BRD4 is analogous to what we previously observed in cardiomyocytes treated with a hypertrophic agonist 34, although the genomic localization of BRD4 in fibroblasts versus myocytes is clearly distinct. In the current study, assessment of genes proximal to the BRD4 peaks demonstrated a correlation between BRD4 abundance at enhancers/promoters, RNA Pol II level within the gene body, and mRNA expression in cardiac fibroblasts. This relationship is consistent with the ability of BRD4 to stimulate gene expression by promoting transcriptional elongation and release of paused RNA Pol II 35–37. It is interesting to note that the number of BRD4-enriched enhancers in unstimulated cardiac fibroblasts actually decreased following TGF-β stimulation, suggesting that BRD4 activity needs to be repressed at certain genes in order for fibroblasts to become activated and secrete ECM. The data also imply that BRD4 serves important functions in quiescent cardiac fibroblasts. While we cannot rule out the possibility that disrupting BRD4 with JQ1 may elicit undesirable effects in unstressed fibroblasts, it is noteworthy that the BET inhibitor did not significantly impact ARVF viability.
The mechanisms governing BRD4 recruitment to and dissociation from specific genomic loci in cardiac fibroblasts remain unclear. In other cellular contexts, coordinately deployed transcription factors recruit BRD4 to active cis-regulatory elements in a manner that is dependent on the underlying acetyl-histone landscape of chromatin 38. We posit that signal-dependent alterations in transcription factor occupancy and histone acetyltransferase/deacetylase activity govern genome-wide redistribution of BRD4 to drive cardiac fibroblast activation.
ChIP-PCR analysis demonstrated that treatment of cardiac fibroblasts with the p38 inhibitor, SB203580, blunted TGF-β-mediated recruitment of BRD4 to enhancers proximal to the gene encoding Sertad4, resulting in reduced Sertad4 mRNA expression. p38 kinase has long been recognized as an effector of pathological cardiac remodeling 39, and a recent report demonstrated that p38α within cardiac fibroblasts mediates pro-fibrotic TGF-β signaling in the heart 15, 16. Deletion of p38α (Mapk14 gene) in cultured fibroblasts blocked differentiation of the cells into α-SMA-positive fibroblasts in response to TGF-β, angiotensin II (Ang II) or cyclic stretching. This block could be overcome by ectopic overexpression of the canonical transient receptor potential 6 (TRPC6) channel, a constitutively active form of the calcineurin phosphatase, or the serum response factor (SRF) transcription factor, suggesting that p38α lies upstream of these proteins, all of which have been demonstrated to regulate myofibroblast differentiation 17. Our data define BRD4 as another downstream effector of pro-fibrotic p38 signaling. The mechanism by which p38 controls BRD4, and the extent to which this kinase regulates the genome-wide distribution of BRD4 in cardiac fibroblasts and other cell types, remain to be determined.
SERTAD4 is a member of the SERTAD family of proteins 40, 41, which are also known as Trip-Br or SEI proteins 42. SERTAD proteins are characterized by the presence of a conserved SERTA (SEI, RBT1, TARA) domain 40, 42, 43. Members of this family have been shown to regulate cell cycle progression by virtue of their ability to interact with CDK/cyclin complexes 44, and also by functioning as transcriptional co-factors to positively or negatively control E2F-dependent transcription 43. The biological function of SERTAD4, which is a nuclear protein that appears to lack intrinsic transcriptional activity 45, is largely unknown. However, the related protein, SERTAD1, was found to act as a transcriptional co-activator of SMAD1 to stimulate BMP target gene expression 46. We demonstrate that TGF-β stimulates Sertad4 expression, and that knockdown of this factor blunts TGF-β-induced expression of activation markers in cardiac fibroblasts. It is intriguing to speculate that, analogous to the BMP/SERTAD1 axis, SERTAD4 protein functions within a positive feedback circuit to promote TGF-β-dependent gene expression via SMADs, resulting in pathological cardiac fibrosis. This validation of SERTAD4 function highlights the ability of our epigenomic discovery approach to identify new regulators of cardiac fibroblast activation.
The BET family consists of BRD2, BRD3, BRD4 and BRDT, all of which contain tandem bromodomains. JQ1, which is a specific pan-inhibitor of all BET family members, has shape complementarity to the acetyl-lysine binding pocket within the bromodomains of these proteins 23. JQ1 competitively and reversibly displaces BETs from acetyl-histones on active enhancers, thereby disrupting downstream signaling events to RNA Pol II. We previously demonstrated that JQ1 treatment reduces interstitial fibrosis in mouse TAC and MI models 24–26. Recently, the ability of JQ1 to block cardiac fibrosis in the TAC model was confirmed by an independent group, and was associated with reduced endothelial to mesenchymal transition 47. JQ1 has also been shown to blunt fibrosis in other organ systems, including lung 48–50, kidney 51, liver 52, pancreas 53, and skin explants from patients with systemic sclerosis 54. Collectively, the data underscore the therapeutic potential of BET inhibitors for the treatment of diverse fibrotic diseases. Nonetheless, there is reasonable concern about potential toxicities of pan-BET inhibitors 55, which is highlighted by to adverse effects such as thrombocytopenia and nausea observed in clinical trials evaluating bromodomain inhibitors in cancer clinical trials 56.
BRD4 is a key pro-fibrotic BET family member. The data presented here enhance our understanding of the molecular mechanisms by which BRD4 controls pathogenic gene expression in fibroblasts. Together with forthcoming findings, this knowledge should yield a cumulative framework that guides the development of more selective BRD4 inhibitors, as opposed to pan-BET inhibitors, with the potential to provide a therapeutic window that is suitable for the treatment of chronic fibrotic diseases such as HF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
TGF-β induces cardiac fibroblast activation via signaling through canonical Smad-mediated and non-canonical pathways, including p38 MAPK.
Though beneficial for scar formation post-myocardial infarction, unconstrained cardiac fibroblast activation leads to interstitial fibrosis that impairs cardiac function and contribute to the progression of heart failure.
What New Information Does This Article Contribute?
TGF-β acts in part via p38 to redistribute the transcriptional co-activator, BRD4, to new locations in the cardiac fibroblast genome, which activates pro-fibrotic gene expression while concomitantly silencing genes associated with cell fate plasticity/pluripotency.
BRD4-dependent Sertad4 expression is indispensable for robust cardiac fibroblast activation in response to TGF-β stimulation.
Genome-, transcriptome- and proteome-wide expression datasets specific to cardiac fibroblasts generated using ChIP-seq (BRD4, RNA Pol II), RNA-seq (in vivo, cell culture), and quantitative mass spectrometry, respectively, provide a rich resource for data mining.
While multiple cardiomyocyte-focused studies have been conducted to investigate the role of BET proteins in heart failure, there is a paucity of information pertaining to the roles these acetyl-lysine binding proteins play in non-muscle cardiac cells. Here, we identify BRD4 as a crucial mediator of the fibroblast activation in the heart. When BRD4 is redistributed in the genome in response to pro-fibrotic TGF-β stimulation, RNA Pol II activity and gene expression is locally activated. Overlapping BRD4 ChIP-seq and RNA-seq experiments identified Sertad4 as a gene that is strongly activated by BRD4-bound enhancers in response to TGF-β stimulation. When Sertad4 was targeted with shRNAs, fibroblast activation marker expression was significantly blunted. This work uncovers a novel role for Sertad4, and provides a mechanistic foundation for the development of BRD4 inhibitors as targeted anti-fibrotic therapies for the heart.
ACKNOWLEDGEMENTS
We thank Charles Danan (Baylor College of Medicine) for RNA-seq library preparation.
SOURCES OF FUNDING
This work was supported by the NIH (HL116848, HL147558 and DK119594 to T.A.M,. and HL127240 to C.Y.L., S.M.H and T.A.M) and the American Heart Association (16SFRN31400013 to T.A.M). M.S.S. was funded by K01AG056848, 5T32HL007822, and F32HL126354. R.A.B. received funding from the Canadian Institutes of Health Research (FRN-216927), and M.B.F. was supported by an American Heart Association fellowship (19POST34380603). M.P.Y.L. was supported by the NIH (HL127302 and HL141278). M.A. was funded by a Swiss National Science Foundation Postdoctoral Fellowship.
Nonstandard Abbreviations and Acronyms:
- α-SMA
α-Smooth muscle actin
- ARVFs
Adult rat ventricular fibroblasts
- ChIP
Chromatin immunoprecipitation
- ECM
Extracellular matrix
- ERK
Extracellular signal-regulated kinase
- FDR
False discovery rate
- HF
Heart failure
- HFpEF
Heart failure with preserved ejection fraction
- HFrEF
Heart failure with reduced ejection fraction
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen-activated protein kinase
- MRTFs
Myocardin-related transcriptional cofactors
- NFAT
Nuclear factor of activated T cells
- Postn
Periostin
- SE
Super-enhancer
- Sertad4
SERTA domain-containing protein 4
- SRF
Serum response factor
- TAC
Transverse aortic constriction
- TGF-β
Transforming growth factor-β
- TSS
Transcription start site
Footnotes
DISCLOSURES
S.M.H. is an executive and shareholder of Amgen, Inc. and is a shareholder of Tenaya Therapeutics. The other authors declare no competing financial interests.
REFERENCES
- 1.Schuetze KB, McKinsey TA and Long CS. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol. 2014;70:100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travers JG, Kamal FA, Robbins J, Yutzey KE and Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M and Martinez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–7. [DOI] [PubMed] [Google Scholar]
- 4.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ and Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francis Stuart SD, De Jesus NM, Lindsey ML and Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol. 2016;91:114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore-Morris T, Cattaneo P, Puceat M and Evans SM. Origins of cardiac fibroblasts. J Mol Cell Cardiol. 2016;91:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tallquist MD. Cardiac fibroblasts: from origin to injury. Curr Opin Physiol. 2018;1:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tallquist MD and Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z and Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol. 2018;114:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD and Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S and Wettschureck N. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res. 2016;118:1906–17. [DOI] [PubMed] [Google Scholar]
- 12.Bujak M and Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis J and Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J and Molkentin JD. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K and Davis J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stratton MS, Koch KA and McKinsey TA. p38alpha: A Profibrotic Signaling Nexus. Circulation. 2017;136:562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis J, Burr AR, Davis GF, Birnbaumer L and Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lighthouse JK and Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol. 2016;91:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5:794–804. [DOI] [PubMed] [Google Scholar]
- 20.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K and Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN and Small EM. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013;110:16850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stratton MS, Haldar SM and McKinsey TA. BRD4 inhibition for the treatment of pathological organ fibrosis. F1000Res. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S and Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE and Haldar SM. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA and Haldar SM. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, Bradner JE and McKinsey TA. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol. 2013;63:175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arora PD and McCulloch CA. Dependence of collagen remodelling on alpha-smooth muscle actin expression by fibroblasts. J Cell Physiol. 1994;159:161–75. [DOI] [PubMed] [Google Scholar]
- 28.Bell E, Ivarsson B and Merrill C. Production of a tissue-like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro. Proc Natl Acad Sci U S A. 1979;76:1274–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montesano R and Orci L. Transforming growth factor beta stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proc Natl Acad Sci U S A. 1988;85:4894–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou MM and Ott M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol Cell. 2017;67:1001–1012 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, Yang T, Wang H, Luscinskas FW, Croce K, Bradner JE and Plutzky J. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA and Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI and Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, Ambardekar AV, Sucharov CC, Bradner JE, Haldar SM and McKinsey TA. Signal-Dependent Recruitment of BRD4 to Cardiomyocyte Super-Enhancers Is Suppressed by a MicroRNA. Cell Rep. 2016;16:1366–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bisgrove DA, Mahmoudi T, Henklein P and Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104:13690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN and Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–34. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K and Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–45. [DOI] [PubMed] [Google Scholar]
- 38.Roe JS, Mercan F, Rivera K, Pappin DJ and Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arabacilar P and Marber M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front Pharmacol. 2015;6:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennetts JS, Fowles LF, Berkman JL, van Bueren KL, Richman JM, Simpson F and Wicking C. Evolutionary conservation and murine embryonic expression of the gene encoding the SERTA domain-containing protein CDCA4 (HEPP). Gene. 2006;374:153–65. [DOI] [PubMed] [Google Scholar]
- 41.Calgaro S, Boube M, Cribbs DL and Bourbon HM. The Drosophila gene taranis encodes a novel trithorax group member potentially linked to the cell cycle regulatory apparatus. Genetics. 2002;160:547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe-Fukunaga R, Iida S, Shimizu Y, Nagata S and Fukunaga R. SEI family of nuclear factors regulates p53-dependent transcriptional activation. Genes Cells. 2005;10:851–60. [DOI] [PubMed] [Google Scholar]
- 43.Hsu SI, Yang CM, Sim KG, Hentschel DM, O’Leary E and Bonventre JV. TRIP-Br: a novel family of PHD zinc finger- and bromodomain-interacting proteins that regulate the transcriptional activity of E2F-1/DP-1. EMBO J. 2001;20:2273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimoto M, Nakamura T, Ohtani N, Hampson L, Hampson IN, Shimamoto A, Furuichi Y, Okumura K, Niwa S, Taya Y and Hara E. Regulation of CDK4 activity by a novel CDK4-binding protein, p34(SEI-1). Genes Dev. 1999;13:3027–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusano S, Shiimura Y and Eizuru Y. I-mfa domain proteins specifically interact with SERTA domain proteins and repress their transactivating functions. Biochimie. 2011;93:1555–64. [DOI] [PubMed] [Google Scholar]
- 46.Peng Y, Zhao S, Song L, Wang M and Jiao K. Sertad1 encodes a novel transcriptional co-activator of SMAD1 in mouse embryonic hearts. Biochem Biophys Res Commun. 2013;441:751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song S, Liu L, Yu Y, Zhang R, Li Y, Cao W, Xiao Y, Fang G, Li Z, Wang X, Wang Q, Zhao X, Chen L, Wang Y and Wang Q. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-beta-induced endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell Cardiol. 2018;127:83–96. [DOI] [PubMed] [Google Scholar]
- 48.Tang X, Peng R, Phillips JE, Deguzman J, Ren Y, Apparsundaram S, Luo Q, Bauer CM, Fuentes ME, DeMartino JA, Tyagi G, Garrido R, Hogaboam CM, Denton CP, Holmes AM, Kitson C, Stevenson CS and Budd DC. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am J Pathol. 2013;183:470–9. [DOI] [PubMed] [Google Scholar]
- 49.Tang X, Peng R, Ren Y, Apparsundaram S, Deguzman J, Bauer CM, Hoffman AF, Hamilton S, Liang Z, Zeng H, Fuentes ME, Demartino JA, Kitson C, Stevenson CS and Budd DC. BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol Pharmacol. 2013;83:283–93. [DOI] [PubMed] [Google Scholar]
- 50.Tian B, Zhao Y, Sun H, Zhang Y, Yang J and Brasier AR. BRD4 mediates NF-kappaB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1183–L1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou B, Mu J, Gong Y, Lu C, Zhao Y, He T and Qin Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-beta-mediated Nox4 expression. Redox Biol. 2017;11:390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M, He M, Liddle C, Downes M and Evans RM. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci U S A. 2015;112:15713–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar K, DeCant BT, Grippo PJ, Hwang RF, Bentrem DJ, Ebine K and Munshi HG. BET inhibitors block pancreatic stellate cell collagen I production and attenuate fibrosis in vivo. JCI Insight. 2017;2:e88032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin JY, Beckett JD, Bagirzadeh R, Creamer TJ, Shah AA, McMahan Z, Paik JJ, Sampedro MM, MacFarlane EG, Beer MA, Warren D, Wigley FM and Dietz HC. Epigenetic activation and memory at a TGFB2 enhancer in systemic sclerosis. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrieu G, Belkina AC and Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016;19:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doroshow DB, Eder JP and LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28:1776–1787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.