

Abstract
Protein coats are supramolecular complexes that assemble on the cytosolic face of membranes to promote cargo sorting and transport carrier formation in the endomembrane system of eukaryotic cells. Several types of protein coat have been described, including COPI, COPII, AP-1, AP-2, AP-3, AP-4, AP-5 and retromer, which operate at different stages of the endomembrane system. Defects in these coats impair specific transport pathways, compromising the function and viability of the cells. In humans, mutations in subunits of these coats cause various congenital diseases that are collectively referred to as “coatopathies”. In this article, we review the fundamental properties of protein coats and the diseases that result from mutation of their constituent subunits.
Keywords: Protein trafficking, sorting signals, clathrin, coatomer, rare diseases, neurological disorders
INTRODUCTION
The endomembrane system of eukaryotic cells consists of an array of organelles including the endoplasmic reticulum (ER), ER-Golgi intermediate compartment (ERGIC), Golgi complex, trans-Golgi network (TGN), early endosomes, late endosomes, tubular endosomal network (TEN), endocytic recycling compartment, lysosomes, lysosome-related organelles (LROs) and different domains of the plasma membrane (Figure 1). Proteins and lipids (i.e., “cargos”) move through this system following various secretory, endocytic and intracellular transport routes. This movement is mediated by vesicular or tubular transport carriers that bud from a donor organelle and fuse with an acceptor organelle (reviewed by Bonifacino and Glick, 2004). While enabling the flow of cargo proteins through the system, the constituent organelles maintain their intrinsic composition of proteins and lipids, so as not to lose their integrity and function. Both the selectivity of cargo transport between organelles and the maintenance of organelle composition rely on mechanisms that ensure specific sorting of proteins and lipids at sites of transport carrier formation. A common mechanism at different stages of the endomembrane system involves “protein coats” (also referred to as “vesicle coats” or “membrane coats”) – supramolecular assemblies of proteins that associate with the cytosolic face of organelles, promoting both the formation of transport carriers and the incorporation of selected cargos into the carriers. Several types of protein coat have been described, including the “coatomer” complexes COPI (reviewed by Arakel and Schwappach, 2018) and COPII (reviewed by Miller and Schekman, 2013), the “adaptor protein” (AP)-based coats containing AP-1, AP-2, AP-3, AP-4 and AP-5 (reviewed by Robinson, 2015), and the “retromer” complex (reviewed by Seaman, 2012) (Figure 1 and Table 1). Studies using various model organisms have shown that all of these coats are essential for viability or normal physiology. In humans, mutations in subunits of these coats are the cause of various genetic disorders that we refer to as “coatopathies”. This article summarizes the most salient features of protein coats and discusses the coatopathies that result from genetic defects in some of their components.
Figure 1.

Schematic representation of the endomembrane system of a eukaryotic cell showing the location of protein coats discussed in this review. Arrows indicate the direction of sorting mediated by each coat. Some of these directions are well established, whereas others should be considered provisional. The plasma membrane is represented as having two distinct domains (1 and 2), as is the case in polarized cells. Although not represented here, AP-2 also plays a role in endocytosis from both plasma membrane domains. The term “tubular endosomal network” (TEN) is used to refer to a collection of tubules emanating from vacuolar endosomes.
Table 1.
Basic characteristics of protein coats
| Coat1 | Adaptor2,3 | Scaffold | Docking factors | Sorting signals4 | Localization5 | Functions6 |
|---|---|---|---|---|---|---|
| COPI | F-subcomplex: | B-subcomplex: | ARF1 | KKxx | ERGIC, Golgi | Retrograde transport from ERGIC and Golgi to ER |
| γ(1/2)-COP | α-COP | KxKxx | ||||
| β-COP | β’-COP | ØRxR | ||||
| δ-COP | ε-COP | FFXX[KR][KR] | ||||
| ζ(1/2)-COP | Ø[KR]xLx[KR] | |||||
| COPII | SEC23(A/B) | SEC13 | SAR1(A/B) | [DE]x[DE] | ERES | Export from ER to ERGIC and Golgi |
| SEC24(A/B/C/D) | SEC31(A/B) | ØØ | ||||
| [FY][FY] | ||||||
| ØxØxØ | ||||||
| LxxL[ME] | ||||||
| IxM | ||||||
| R[IL] | ||||||
| YNNSNPF | ||||||
| PPP | ||||||
| TM domains | ||||||
| AP-1 | AP-1 complex: | Clathrin: | ARF1, PtdIns(4)P | YxxØ | TGN/TEN | Bidirectional transport between TGN and endosomes; polarized sorting |
| γ(1/2)-adaptin | CHC17 (CHC22)7 | [DE]xxxL[LI] | ||||
| β1-adaptin | CLC (A/B) | Acidic clusters | ||||
| μ1(A/B) | Noncanonical | |||||
| σ1(A/B/C) | ||||||
| AP-2 | AP-2 complex: | Clathrin: | PtdIns(4,5)P | YxxØ | PM | Endocytosis |
| α(A/C)-adaptin | CHC17 (CHC22)7 | [DE]xxxL[LI] | ||||
| β2-adaptin | CLC (A/B) | Acidic clusters | ||||
| μ2 | Noncanonical | |||||
| σ2 | ||||||
| ARH | FxNPxY | PM | Endocytosis | |||
| AP-3 | AP-3 complex: | Clathrin? | ARF1, PtdIns(3)P | YxxØ | TEN | Transport from TEN to LRO, SV and DCV |
| δ-adaptin | VPS41? | [DE]xxxL[LI] | ||||
| β3(A/B)-adaptin | ||||||
| μ3(A/B) | ||||||
| σ3(A/B) | ||||||
| AP-4 | AP-4 complex: | Unknown | ARF1 | Yx[FYL][FL]E | TGN | Transport from TGN to PAS |
| ε-adaptin | Noncanonical | |||||
| β4-adaptin | ||||||
| μ4 | ||||||
| σ4 | ||||||
| AP-5 | AP-5 complex: | SPG118 | PtdIns(3)P | Unknown | LE | Retrieval from LE to Golgi |
| ζ-adaptin | SPG159 | |||||
| β5-adaptin | ||||||
| μ5 | ||||||
| σ5 | ||||||
| Retromer | SNX(3/12/27) | VPS26(A/B) | RAB7, PtdIns(3)P | Øx[LM] | TEN | Retrieval from endosomes to TGN and PM |
| VPS29 | NPxY | |||||
| VPS35 | [ST]xØ |
Each coat is named based on a unique key component. Only main coat components that are relevant to this review are listed.
The distinction between “adaptor” and “scaffold” is clear for some coats, such as COPII and those containing clathrin and either AP-1 or AP-2, but less clear for others.
Parentheses indicate alternative components, and those with numbers or letters separated by slashes indicate isoforms, such as γ1-COP and γ2-COP being two alternative isoforms of the γ-COP subunit.
Amino acids are represented in single letter code. Ø represents a bulky hydrophobic amino acid (L, I, V, M, F) and x any amino acid. Letters in brackets represent alternative amino acids at a given position.
Abbreviations: DCV, dense core vesicle; ER, endoplasmic reticulum; ERES, ER-Golgi exit site; ERGIC, ER-Golgi intermediate compartment; LE, late endosome; LRO, lysosome-related organelle; PAS, pre-autophagosomal structure; PM, plasma membrane; PtdIns, phosphatidylinositol; SV, synaptic vesicle; TEN, tubular endosomal network; TGN, trans-Golgi network;
Best-documented functions are listed.
The clathrin heavy chains (CHC) are indicated by the abbreviations of the human proteins (CHC17 and CHC22)
Also known as spatacsin.
Also known as spastizin.
GENERAL PROPERTIES OF PROTEIN COATS
The term “coat” stems from early transmission electron microscopy (EM) studies that described an electron-dense, proteinaceous deposit on the cytosolic face of some membrane buds and vesicles (Roth and Porter, 1964) (Figure 2). Later studies showed that these original coats contained the protein clathrin in combination with AP-1 or AP-2 as their main components (Pearse, 1975, Pearse and Robinson, 1984), and that they were structured as a polyhedral lattice encaging a membrane bud or vesicle (Crowther et al., 1976, Heuser, 1980, Fotin et al., 2004). Subsequent studies revealed the existence of additional, non-clathrin coats corresponding to COPI (Waters et al., 1991) and COPII (Barlowe et al., 1994), which by transmission EM also appeared as electron-dense deposits on a different set of buds and vesicles. Structural EM analyses, however, revealed that these coats had three-dimensional organizations that differed from that of clathrin coats (Zanetti et al., 2013, Dodonova et al., 2015, Bykov et al., 2017, Hutchings et al., 2018).
Figure 2.
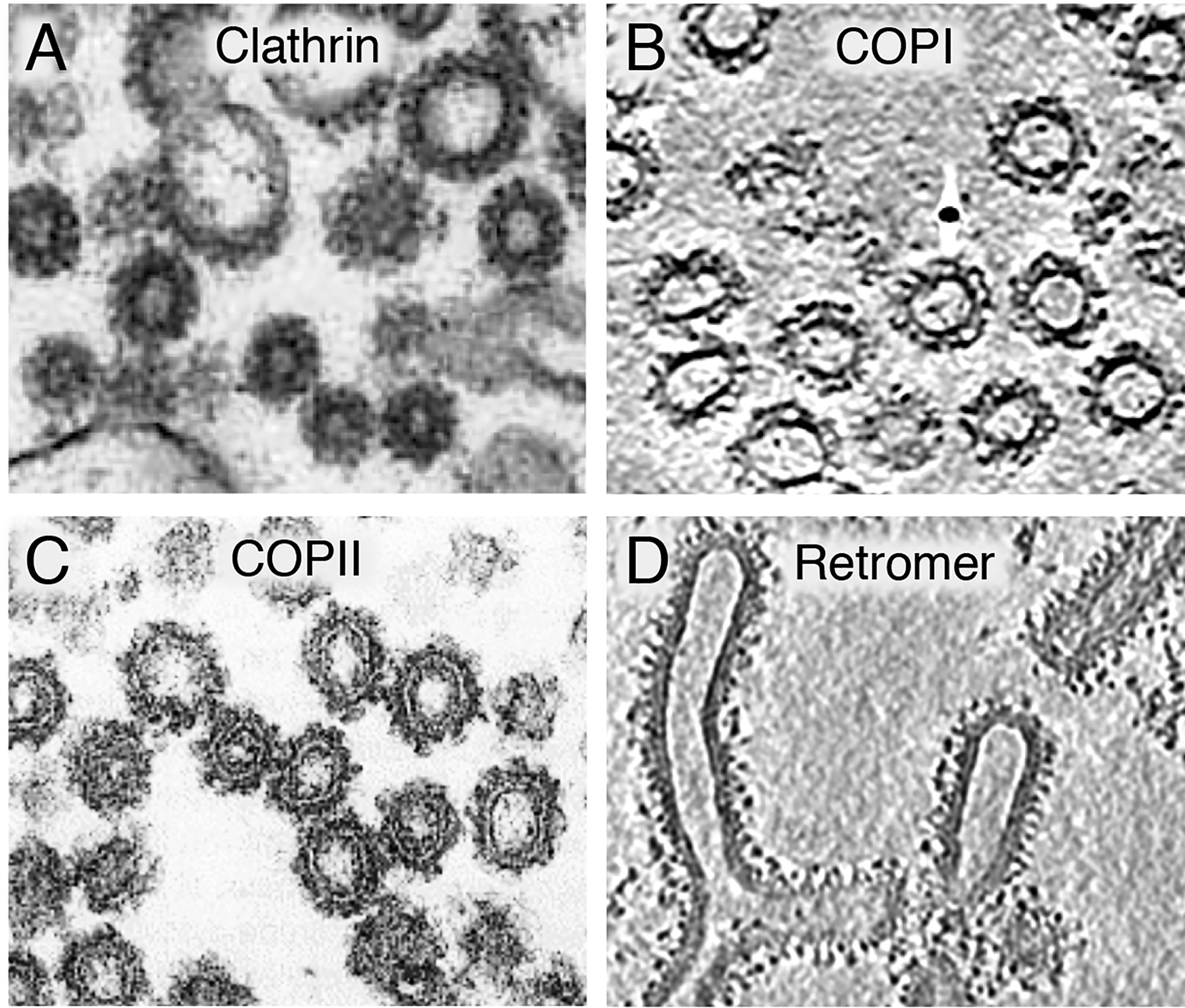
Electron micrographs of coated vesicles and tubules formed in vitro by assembly of purified coat components on liposomes or membrane fractions. (A) Thin-section electron microscopy of clathrin-coated vesicles formed from liposomes (Dannhauser and Ungewickell, 2012). (B) Section from a cryoelectron tomogram of COPI vesicles formed from giant unilamellar vesicles (Faini et al., 2012). (C) Thin-section electron microscopy of COPII vesicles formed from a membrane fraction (Barlowe et al., 1994). (D) Section from a cryoelectron tomogram of a yeast retromer complex assembled on liposome tubules (Kovtun et al., 2018). In all cases, notice the appearance of coats as electron-dense deposits on the surface of vesicles and tubules. Images reproduced with permission from the publishers.
Coats were initially proposed to be organized into (i) a membrane-proximal, “inner” or “adaptor” layer, and (ii) a membrane-distal, “outer” or “scaffold” layer, which sequentially assemble from cytosolic components onto the target membranes (Table 1). Biochemical and structural analyses borne out this organization and assembly mechanism for clathrin-containing and COPII coats. The inner layer of canonical clathrin coats comprises the heterotetrameric AP-1 or AP-2 complexes (Figures 3A and 4, and Table 1). The outer layer, on the other hand, consists of three-legged “triskelion” structures composed of three clathrin heavy chains (CHC) and three clathrin light chains (CLC) (CHC3-CLC3) (Ungewickell and Branton, 1981) (Figure 3A and Table 1), which assemble into the polyhedral lattice that is typical of clathrin coats (Fotin et al., 2004). Likewise, COPII comprises an inner layer of SEC23-SEC24 dimers and an outer layer of SEC132-SEC312 heterotetramers, which are sequentially recruited to form a flexible cage around vesicles (Zanetti et al., 2013, Hutchings et al., 2018) (Figure 3B and Table 1). In contrast, structural EM studies have shown that COPI does not have distinct inner and outer layers despite the homology of its F-subcomplex to AP-1 and AP-2, and of its B-subcomplex to clathrin (Dodonova et al., 2015, Bykov et al., 2017) (Figure 3C and Table 1). Instead, all seven COPI subunits are recruited en bloc, with all components lying in close proximity to the membrane. Other coats containing AP-3, AP-4 or AP-5 have not being visualized either in situ or in vitro, and their structural organization remains to be determined. The retromer complex was proposed to form a coat around tubules emanating from endosomes (Hierro et al., 2007, Lucas et al., 2016), a prediction that has been recently demonstrated by cryo-EM tomography of a fungal retromer complex (Kotvun et al., 2018) (Figure 2). Inner and outer layers can be discerned in the retromer coat, although its constituent subunits and overall organization are very different from those of other coats (Figure 3D and Table 1).
Figure 3.

Schematic representation of prototypic coats assembled on membrane buds. (A) AP-2. (B) COPII. (C) COPI. (D) Retromer. The coats represented in this figure are those for which there is both X-ray crystallographic and structural electron microscopy data. The schemes highlight some of the features of these coats but are not intended to be accurate representations of their three-dimensional structures. AP-2, COPII and retromer are shown as having membrane-proximal (inner/adaptor) and membrane-distal (outer/scaffold) layers. Despite the homology of COPI subunits to those of AP-2/clathrin, the F- and B-subcomplexes are not arranged as layers but parts of both subcomplexes are in close proximity to the membrane. The retromer complex represented in this figure corresponds to that containing sorting nexin 3 (SNX3), but there are other forms of retromer containing SNX27 and, in yeast, Vps5 and Vps17. For the complexes represented in this figure, cargo recognition is mainly mediated by AP-2, the B-subcomplex, SEC23-SEC24 and SNX3-VPS26, respectively, although additional modes of cargo recognition are possible. More details of the protein composition of AP complexes and COPI-F are shown in Figure 4.
Figure 4.

Schematic representation of AP complexes, COPI-F and ARH. The schemes represent the “unlocked” membrane-bound form of AP complexes and COPI-F. Subunit names are indicated. Homologous subunits are depicted in the same color.
Coats are recruited to membranes through interactions with membrane lipids, particularly phosphoinositides, and/or small GTPases of the SAR/ARF or RAB families (Figure 3 and Table 1). Accordingly, coat recruitment also depends on phosphoinositide kinases and phosphatases, as well as SAR/ARF/RAB guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). The recruitment of coats to membranes is in some cases accompanied by conformational changes that enable interactions of the adaptors with scaffolds, regulators and cargos, as shown for AP-1 (Ren et al., 2013), AP-2 (Jackson et al., 2010, Kelly et al., 2014) and retromer (Gallon et al., 2014, Lucas et al., 2016). The coats interact with sorting signals present in the cytosolic tails of transmembrane cargos, leading to the selective concentration of cargos in coated areas of the membranes. In most cases, these signals are short, linear sequences that fit one of many consensus motifs (Table 1). Each coat recognizes a distinct, albeit in some cases overlapping, set of sorting signals. In addition to capturing cargo, coats induce membrane curvature, leading to the formation of coated buds, which eventually pinch off from the donor organelles as transport carriers. These carriers were originally thought to be coated vesicles of uniformly small size and spherical shape (60–150 nm diameter), as best exemplified by clathrin/AP-2-coated vesicles forming from the plasma membrane. More recent studies, however, have revealed a considerable diversity in the size and shape of transport carriers, including larger (>300 nm diameter) coated vesicles (Gorur et al., 2017), tubules (Arighi et al., 2004, Seaman, 2004), and pleiomorphic, tubular-vesicular structures having one or more coated domains (Presley et al., 1997, Polishchuk et al., 2006). This diversity extends to the fate of the coat, which can dissociate shortly after budding (Bykov et al., 2017), remain for longer periods on the carriers as they translocate through the cytoplasm (Polishchuk et al., 2006), or sort cargos into a membrane domain from which they are directly transferred to an adjacent organelle (Kurokawa et al., 2014, McCaughey et al., 2018).
The combined interactions with distinct phosphoinositides, small GTPases, cargos and other regulatory molecules determine the association of coats with different intracellular organelles (Figure 1 and Table 1). This allows the coats to participate in different transport pathways (Figure 1 and Table 1). The functional diversity of protein coats is further enhanced by the existence of subunit isoforms encoded by different genes (i.e., paralogs) (Table 1) or produced by alternative splicing (Jackson et al., 1987, Ball et al., 1995), which may endow the coats with different cargo recognition, architectural or regulatory properties.
EVOLUTION AND GENETICS OF PROTEIN COATS
Protein coats are found in all eukaryotes, and were already present in the last eukaryotic common ancestor 1–1.5 billion years ago (reviewed by Robinson, 2015, Rout and Field, 2017). COPI, AP-1, AP-2, AP-3, AP-4, AP-5 and another coat named TSET are all structurally related and thought to have evolved from a single ancestral complex (Robinson, 2015, Rout and Field, 2017) (Figure 4). The architecture of COPII is distinct but shares with the other coats structural elements such as β-propellers and α-solenoids, suggesting that they all evolved from an even more ancient “protocoatomer” complex (Rout and Field, 2017). The retromer complex is more divergent, although it also comprises α-solenoid and arrestin folds that are found in other coats (Shi et al., 2006, Hierro et al., 2007).
Throughout evolution, these coats have undergone adaptive changes and losses in some lineages. For example, humans and other vertebrates have COPI, COPII, AP-1 through AP-5, and retromer, but lack a full TSET complex (Robinson, 2015, Rout and Field, 2017). Furthermore, yeast, flies and nematodes have COPI, COPII, AP-1, AP-2, AP-3 and retromer, but lack AP-4, AP-5 and TSET (Robinson, 2015, Rout and Field, 2017). These latter organisms can thus be used as genetic model systems for the study of some but not all coats. Mice, on the other hand, have the same complement of coats as humans. In mice, biallelic ablation of genes encoding some of the COPII (Baines et al., 2013), AP-1 (Zizioli et al., 1999, (Meyer et al., 2000), AP-2 (Mitsunari et al., 2005) or retromer subunits (Lee et al., 1992) results in embryonic lethality, demonstrating that these coats are essential for viability. In contrast, mice homozygous for null mutations of genes for AP-3, AP-4 and AP-5 subunits are viable, but exhibit phenotypic abnormalities (Yang et al., 2000, Matsuda et al., 2008, Varga et al., 2015).
Mutations in subunits of all the coats have now been shown to be causes of various coatopathies (Tables 2–6). The inheritance of these conditions can be autosomal recessive, autosomal dominant or X-linked. For coats that are essential for viability, such as the COPI, COPII, AP-1 and AP-2 coats, the recessive or X-linked mutations usually involve complete absence or inactivation of a partially redundant subunit isoform, or partial loss of function due to decreased stability, assembly or function of a non-redundant subunit. In the case of the nonessential AP-3, AP-4 and AP-5 coats, recessive mutations are generally null. Finally, for all coats, autosomal-dominant mutations can cause disease through dominant-negative or constitutively-activating effects, or because of haploinsufficiency. In the following sections, we describe in more detail these coats and their corresponding coatopathies, beginning with those that function in the early part (i.e., ER and Golgi complex) and continuing with those that function in the late part (i.e., post-Golgi compartments) of the endomembrane system.
Table 2.
Diseases caused by mutations in COPI
| Coat | Component | Gene (locus) | Disease1 | OMIM2 | Inheritance | Mutation types | Key References |
|---|---|---|---|---|---|---|---|
| COPI | α-COP | COPA (1q23.2) | COPA syndrome (Autoimmune interstitial lung, joint, and kidney disease; AILJK) | 616414 | Autosomal dominant with incomplete penetrance | Missense | Watkin et al. 2015 |
| β’-COP | COPB2 (3q23) | Microcephaly 19, primary, autosomal recessive (MCPH19) | 617800 | Autosomal recessive | Missense | DiStasio et al., 2017 | |
| δ-COP | ARCN1 (11q23.3) | Short stature, rhizomelic, with microcephaly, micrognathia, and developmental delay (SRMMD) | 617164 | Autosomal recessive | Nonsense, Frameshift | Izumi et al., 2016 |
Disease acronyms or alternative names are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/).
Table 6.
Diseases caused by mutations in retromer
| Component | Gene (locus) | Disease1 | OMIM2 | Inheritance | Mutation types | Key references |
|---|---|---|---|---|---|---|
| VPS26A | VPS26A (10q22.1) | Atypical parkinsonism | None3 | Unknown | Missense, nonsense | Gustavsson et al., 2015 |
| VPS35 | VPS35 (16q11.2) | Parkinson disease-17 (PARK17) | 614203 | Autosomal dominant with incomplete penetrance | Missense | Vilarino-Guell et al., 2011, Zimprich et al., 2011 |
| SNX27 | SNX27 (1q21.3) | Infantile myoclonic epilepsy and neurodegeneration | None4 | Autosomal recessive | Frameshift | Damseh et al., 2015 |
Disease acronyms are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/).
No entry number has been assigned to the disease. The entry number for the gene is 605506.
No entry number has been assigned to the disease. The entry number for the gene is 611541.
COPI
COPI is a heteroheptameric complex that is mainly involved in the retrieval of escaped ER resident proteins and the recycling of transmembrane cargo receptors from the ERGIC and cis-Golgi cisternae to the ER, as well as in intra-Golgi retrograde transport (Arakel and Schwappach, 2018) (Figure 1 and Table 1). The subunits of COPI are named α-COP, β’-COP, ε-COP, β-COP, δ-COP, γ-COP and ζ-COP (Waters et al., 1991) (Table 1, and Figure 3C). In humans, there are two paralogs of the γ-COP and ζ-COP subunits named γ1-COP and γ2-COP, and ζ1-COP and ζ2-COP (Table 1). The α-COP, β’-COP, ε-COP subunits constitute the B-subcomplex, which is structurally related to clathrin, whereas β-COP, δ-COP, γ-COP and ζ-COP subunits constitute the F-subcomplex, which is homologous to AP complexes (Figures 3C and 4). The membrane recruitment of the COPI heteroheptamer is initiated by the GTP-bound form of ARF1, a small GTPase that associates with membranes via an N-myristoylated amphipathic α-helix (Serafini et al., 1991). Two ARF1 molecules bind one COPI complex through interactions with sites on the β-COP and γ-COP subunits (Yu et al., 2012). Three COPI complexes and their associated ARF1 molecules assemble into a curved “triad” that further interacts with other triads through multiple flexible domains (Bykov et al., 2017, Dodonova et al., 2015). The resulting coat is organized as a flexible network rather than a rigid cage (Dodonova et al., 2015, Bykov et al., 2017).
The best characterized cargos for COPI are ER-resident proteins that escape the ER and that must be retrieved from the ERGIC or the Golgi complex for maintenance of their steady-state distribution (Arakel and Schwappach, 2018). These cargos include ER transmembrane proteins having dibasic sorting motifs in their cytosolic tails (e.g., KKxx, KxKxx, ØRxR and variants thereof) (Table 1). KKxx and KxKxx signals directly bind to the N-terminal β-propeller “WD40” domains of α-COP and β’-COP, respectively (Cosson and Letourneur, 1994, Jackson et al., 2012, Ma and Goldberg, 2013), which, unlike the structurally homologous clathrin β-propeller domains, are positioned close to the membrane (Zerangue et al., 2001). Other ER resident proteins that escape the ER do not interact directly with COPI but do so through binding to transmembrane ER retrieval receptors, which in turn interact with COPI. These receptors include the KDEL receptor, which binds ER luminal proteins having C-terminal KDEL signals (Lewis and Pelham, 1990, Majoul et al., 1998), and RER1, which recognizes the transmembrane domain of ER transmembrane proteins (Sato et al., 2001). Both the KDEL receptor and RER1 lack canonical COPI-recognition signals, so it remains to be determined how they interact with COPI. Mutations in COPI or its cognate signals impair retrieval and result in leakage of ER proteins to more distal compartments of the secretory pathway. Additional COPI cargos are ER export receptors such as the CLN8 protein involved in delivery of newly-synthesized lysosomal enzyme precursors from the ER to the Golgi complex (di Ronza et al., 2018). CLN8 uses a COPII-interacting ØxØxØ motif for ER exit (see next section) and a KKxx signal for retrieval from the Golgi to the ER (di Ronza et al., 2018) (Table 1). Mutation of either of these motifs interrupts the cycling of CLN8 and impairs ER export of the lysosomal enzyme precursors (di Ronza et al., 2018).
A different type of COPI cargo is a subset of glycosyltransferases that are maintained in cis-Golgi cisternae by recycling from more distal compartments of the Golgi complex. This sorting is mediated by Ø[KR]xLx[KR] signals in the tails of the glycosyltransferases (Table 1), which interact with the δ-COP and ζ-COP subunits (Liu et al., 2018). In this case, mutation of the signals leads to missorting of the enzymes to lysosomes, where they are degraded (Liu et al., 2018), the same fate followed by RER1 in the absence of COPI (Sato et al., 2001). All of these findings underscore the critical role of COPI in maintaining the steady-state composition and function of early organelles of the secretory pathway.
Germline missense mutations in residues within the WD40 domains of α-COP and β’-COP have been implicated as the causes of two coatopathies with distinct clinical presentation and mode of inheritance (COPA syndrome and primary microcephaly-19), and nonsense or frameshift mutations in δ-COP have been reported to cause a third COPI coatopathy (Short stature, rhizomelic, with microcephaly, micrognathia, and developmental delay) (Table 2).
Monoallelic mutations in the gene encoding α-COP have been detected in multiple families with individuals suffering from a rare autoimmune syndrome affecting primarily the lungs (usually as interstitial lung disease with occasional cysts and/or haemorrhage) as well as the joints (inflammatory arthritis) and, less frequently, the kidneys (Watkin et al., 2015, Jensson et al., 2017, Noorelahi et al., 2018, Volpi et al., 2018; Taveira-DaSilva et al., 2018). Consistently with a disease mechanism involving autoimmunity and inflammation, the patients were reported to respond to immunosuppression (Watkin et al., 2015; Jensson et al., 2017, Noorelahi et al., 2018, Volpi et al., 2018, Taveira-DaSilva et al., 2018). The age of onset varies significantly, with some patients presenting clinically within their first year after birth and others presenting decades later. In fact, some members of the families included in the original description of the syndrome – now referred to as COPA syndrome – were found to carry pathological mutations and considered apparently normal at the time of analysis (Watkin et al., 2015). Accordingly, COPA syndrome is considered to be autosomal dominant with incomplete penetrance (Watkin et al., 2015) and variable expression (Taveira-DaSilva et al., 2018).
Remarkably, all of the missense mutations reported so far in unrelated cases of COPA syndrome fall within a relatively short stretch of amino acid residues, namely residues 230–241 of α-COP isoform 1, which is a 1233-residue protein. Given the mode of inheritance of the disease, and the unusual pattern of missense mutations in a protein region known to be involved in cargo recognition (Ma and Goldberg, 2013), it is tempting to speculate that the pathogenesis may involve a gain-of-function mechanism, such as unregulated interaction with potential cargo molecules. Using cell-based assays, Watkin and colleagues (Watkin et al., 2015) observed ER stress and altered autophagosomes upon α-COP knockdown or overexpression of the mutated protein, although the exact molecular mechanism that led to these organelle abnormalities remains to be defined.
DiStasio and colleagues (DiStasio et al., 2017) described a family with two siblings suffering from microcephaly, insufficient weight gain with normal height, cortical blindness and developmental delay. The two affected individuals, but not the parents or an apparently unaffected sibling, were found to share a large region of homozygosity, which spanned 16.8 Megabases of chromosome 3 and included a rare missense variant in the COPB2 gene encoding β’-COP (DiStasio et al., 2017). Identifying unrelated families with individuals carrying biallelic mutations in the same gene and a similar clinical presentation will be necessary to completely rule out the possibility that other genes within (or even outside) the above-mentioned region of homozygosity may have contributed to the disease, which is now referred to as primary microcephaly-19. Exactly the same missense variant found in the patients (p.R254C) was engineered in mice; while homozygous missense mutant mice appeared phenotypically normal, compound heterozygous carrying the p.R254C substitution over a null allele displayed morphological abnormalities similar to those of the patients, including small body and brain sizes as well as reduced cortical area (DiStasio et al., 2017). These observations led the authors to propose that the p.R254C variant acts as a hypomorphic allele and that the development of the human brain may be more sensitive to decreased COPI function than that of the mouse brain (DiStasio et al., 2017).
Monoallelic loss-of-function mutations in the gene encoding δ-COP (ARCN1) were found in four patients with a history of intrauterine growth retardation and suffering from short stature with disproportionally small proximal limbs (rhizomelia), microcephaly, micrognathia (small jaw), and developmental delay (Izumi et al., 2016). Consistent with the idea that this disease may arise from loss of only one functional copy of the ARCN1 gene, no loss-of-function alleles of this gene have been detected in large population sequencing projects, as compiled in the Genome Aggregation Database (Lek et al., 2016). Based on a family having an affected father and daughter sharing the same frameshift mutation in one copy of the δ-COP-encoding gene, the disorder was deemed to follow an autosomal dominant pattern of inheritance (Izumi et al., 2016). It was not clear whether the mutations found in other two unrelated patients were inherited or generated de novo.
Although the two coatopathies associated with mutations in the genes encoding β’-COP and δ-COP share some clinical features (reduced body weight, microcephalia and developmental delay) and are thought to stem from reduced COPI activity (due to hypomorphic alleles and haploinsufficiency, respectively), it should be noted that the clinical presentation of each syndrome includes manifestations not described as characteristic for the other. One possibility to consider is that the reported missense variant in the WD40 domain of β’-COP may affect recognition of only a subset of COPI cargos, while reduced expression of δ-COP could compromise all COPI-dependent activities. Another possibility is that the p.R254C variant in β’-COP may affect all COPI-dependent activities but represent a mild hypomorphic allele, a notion what would be consistent with the fact that mice homozygous for this variant displayed no obvious phenotype (DiStasio et al., 2017). Future work, and especially additional families carrying mutations in these genes, should help to clarify this point.
COPII
COPII is a two-layered coat associated with ER-exit sites (ERES), which mediates protein export from the ER towards the ERGIC and cis-Golgi cisternae (Miller and Schekman, 2013) (Figure 1). The core components of COPII are the small GTPase SAR1, the inner coat heterodimer SEC23-SEC24 and outer coat heterotetramer SEC132-SEC312 (Barlowe et al., 1994) (Figure 3B and Table 1). Most of the human COPII subunits exist as multiple paralogs (Table 1), which confer on COPII the ability to sort a wide variety of cargos. Assembly of COPII starts with GTP for GDP exchange on SAR1 by an ER-associated SAR1-GEF named SEC12 (Barlowe et al., 1994). Like ARF1-GTP, the activated SAR1-GTP attaches to the cytosolic leaflet of the ERES membrane through an amino-terminal myristoylated amphipathic α-helix, inducing membrane curvature and serving as a docking site for the SEC23-SEC24 dimer. The SEC132-SEC312 heterotetramer polymerizes on top of the SAR1-SEC23-SEC24 complex, resulting in the formation of a polyhedral lattice that gathers cargos at ERES while further contributing to membrane curvature (Zanetti et al., 2013, Hutchings et al., 2018). Another ER protein named SEC16, which in humans exists as SEC16A and SEC16B isoforms, contributes to the assembly and disassembly of COPII cages (Espenshade et al., 1995).
Since COPII functions as a coat for virtually all proteins that exit the ER towards the ERGIC and Golgi complex, the mechanisms of cargo recognition by COPII are necessarily quite diverse. Transmembrane cargos can bind either directly to COPII or indirectly via transmembrane export receptors such as the cornichon, SCAP, iRhom and ERV26 proteins (reviewed by Barlowe and Helenius, 2016). Luminal proteins are topologically incapable of binding directly to COPII, so they are captured by other transmembrane export receptors such as the CLN8 protein mentioned in the previous section, as well as ERGIC53, ERV29 and p24 family members (Barlowe and Helenius, 2016). COPII transmembrane cargos and ER export receptors have cytosolic tails containing various sorting signals, including diacidic, dihydrophobic or diaromatic motifs (Table 1), most of which bind to different sites on SEC24 (Miller et al., 2003, Mossessova et al., 2003, Mancias and Goldberg, 2008). These cargos concentrate within small (60–80 nm) COPII-coated buds at ERES, which subsequently may pinch off as small COPII-coated vesicles. The vesicles are thought to uncoat prior to their fusion with the ERGIC or cis-Golgi cisternae, in part due to the activation of a SEC132-SEC312-stimulated GAP activity of SEC23 towards SAR1.
In addition to small cargos, COPII mediates the ER export of large luminal cargos such as procollagen fibrils with lengths of >300 nm (Stephens and Pepperkok, 2002). Loading of these fibrils into COPII carriers is mediated by a complex of two transmembrane proteins named TANGO1 (Saito et al., 2009) and cTAGE5 (Saito et al., 2011). Both of these proteins have multiple PPP motifs within proline-rich sequences that directly bind to SEC23 (Ma and Goldberg, 2016). These interactions enable packaging of procollagen into large buds or tubular carriers coated with a helical array of SEC23-SEC24 dimers and a rhomboidal lattice of SEC132-SEC312 heterotetramers (Zanetti et al., 2013). Like their smaller relatives, these large carriers may also shed their coat and fuse with the ERGIC or cis-Golgi. However, alternative scenarios have been proposed; these include maturation of the whole ERES into an ERGIC that detaches and moves towards the Golgi complex, and direct transfer of cargos from COPII-coated domains at the ERES to adjacent Golgi elements (Mironov et al., 2003, Kurokawa et al., 2014, McCaughey et al., 2018).
Clinically distinct coatopathies have been ascribed to mutations in each of the two human SEC23 paralogs and least one of the four human paralogs of the SEC24 paralogs (Table 3). Boyadjiev and colleagues (Boyadjiev et al., 2006) described six members of a large inbred family who displayed prominent and hyperpigmented forehead, broad nose with increased distance between the eyes, open or delayed closing of sutures in the skull, and congenital sutural cataracts. This disorder, named craniolenticulosutural dysplasia, was found to arise from a single non-conservative substitution in SEC23A, which in the six affected individuals was carried in homozygous form (Boyadjiev et al., 2006). Fibroblasts from the patients displayed normal levels of the mutant SEC23A protein but abnormally diffuse distribution of the SEC31 protein, as well as enlarged cisternae that contained ER markers and accumulated procollagen COL1A1 (Boyadjiev et al., 2006). Consistent with the idea that impaired function of COPII coats containing the SEC23A isoform lead to a form of skeletal dysplasia due to abnormal secretion of collagen or other components of the extracellular matrix, knockdown of the orthologous SEC23A in zebrafish resulted in skeletal malformation with cartilage hypoplasia (Boyadjiev et al., 2006).
Table 3.
Diseases caused by mutations in the COPII coat
| Coat | Component | Gene (locus) | Disease1 | OMIM2 | Inheritance | Mutation types | Key References |
|---|---|---|---|---|---|---|---|
| COPII | SEC23A | SEC23A (14q21.1) | Cranio-lenticulo-sutural dysplasia (CLSD; Boyadjiev-Jabs syndrome) | 607812 | Autosomal recessive | Missense | Boyadjiev et al., 2006 |
| SEC23B | SEC23B (20p11.23) | Congenital dyserythropoietic anemia type II (CDAII) | 224100 | Autosomal recessive | Missense, nonsense, frameshift | Schwarz et al., 2009; Bianchi et al., 2009 | |
| SEC24D | SEC24D (4q26) | Cole-Carpenter syndrome 2 (CLCRP2) | 616294 | Autosomal recessive | Missense, nonsense | Garbes et al., 2015 |
Disease acronyms or alternative names are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/)
Two research groups independently identified biallelic mutations (in homozygous or compound heterozygous forms) in the SEC23B gene as the cause of a form of congenital anemia characterized by defective generation of red blood cells from erythroblasts, which in samples from bone marrow can be seen as containing two or more nuclei (Schwarz et al., 2009, Bianchi et al., 2009). Given the wide variety of disease-causing mutations observed in patients from different families (Table 3), a mechanism involving at least partial loss of function of a COPII coat containing SEC23B has been invoked for this coatopathy, which is known as congenital dyserythropoietic anemia type II (Schwarz et al., 2009, Bianchi et al., 2009). Intriguingly, a monoallelic missense variation in the same gene (p.V594G) was implicated in an unrelated disorder, named Cowden syndrome, which is characterized by benign tumors known as hamartomas as well as an increased risk of developing certain types of cancer (Yehia et al., 2015). It should be noted, however, that this variant has recently been re-classified as a “variant of unknown significance” in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/RCV000210069/).
Although it would be reasonable to assume that the distinct coatopathies caused by mutations in SEC23A and SEC23B reflect functional specialization of COPII coats that contain either the SEC23A or SEC23B paralogs, Khoriaty and colleagues (Khoriaty et al., 2018) recently reported that SEC23A complemented SEC23B-null mice when expressed under control of the Sec23b promoter. The authors concluded that the two SEC23 paralogs are functionally interchangeable and that the two coatopathies may instead arise from relative differences in protein expression in disease-relevant cell types (e.g., not enough expression of SEC23A in erythroblasts to compensate for mutations in SEC23B). Consistent with this notion is the observation that mice deficient in SEC23B display apparently normal erythropoiesis (Khoriaty et al., 2014).
Yang et al. (2013) sequenced the SEC24B gene in 163 stillborn or miscarried fetuses with neural tube defects and detected monoallelic missense variants in four of them. However, each of these four missense variants has also been found in volunteers participating in large-scale sequencing projects (Lek et al., 2016), making it unlikely that these variants could lead to neural tube defects with high penetrance. Another case-control study suggested an increased number of potentially damaging mutations in the SEC24B gene among patients with mesial temporal lobe epilepsy with hippocampal sclerosis, relative to control individuals (Wong et al., 2018). For both diseases, ascertaining the potential role of SEC24B mutations will require future work.
Finally, compound heterozygous mutations in SEC24D were found in three cases of severe osteogenesis imperfecta from two families (Garbes et al., 2015). All three affected individuals displayed gross abnormalities in the ossification of the skull, and bone fractures prior to birth. Although some of the morphological features, as well as the cellular phenotype of enlarged ER cisternae, were reminiscent of craniolenticulosutural dysplasia caused by mutations in SEC23A, other clinical manifestations were unique to each of the two coatopathies (Garbes et al., 2015). The disease caused by SEC24D mutations is currently known as Cole-Carpenter syndrome type 2.
CLATHRIN
Clathrin functions as a common scaffold for various coats associated with the TGN, endosomes and the plasma membrane (reviewed by Kirchhausen et al., 2014). As mentioned above, the basic building block of the clathrin scaffold is a CHC3-CLC3 triskelion. There are two paralogous isoforms of both CHC (encoded by the CLTC and CLTCL1 genes) and CLC (encoded by the CLTA or CLTB genes) (Table 1). In humans, the CHC isoforms are referred to as CHC17 and CHC22 because of the location of their corresponding genes to chromosomes 17 and 22, respectively. CHC17 is the most abundantly and ubiquitously expressed in human tissues. Furthermore, CHC17 is conserved in all mammals, whereas the other paralog is not expressed in some mammals such as mice. Therefore, CHC17 is likely responsible for the general functions of clathrin, whereas CHC22 may have more specialized functions. In this latter regard, CHC22 has been reported not to interact with CLC and AP-2 (Liu et al., 2001), and not to be involved in clathrin-mediated endocytosis (Dannhauser et al., 2017) but in the formation of insulin-responsive GLUT4 compartments in muscle cells and adipocytes (Vassilopoulos et al., 2009). However, other studies have shown that CHC22 is a component of conventional clathrin-coated vesicles (Borner et al., 2006) and can substitute for CHC17 in clathrin-mediated endocytosis (Hood and Royle, 2009).
Structurally, each CHC consists of an N-terminal β-propeller domain with seven WD40 repeats (referred to as the “terminal domain”) harboring multiple binding sites for adaptor, accessory and regulatory proteins, followed by an α-helical solenoid domain that forms the backbone of the clathrin scaffold, a longer α-helix that mediates trimerization, and a short, unstructured sequence that mediates uncoating (reviewed by Kirchhausen et al., 2014). CLC binds to the C-terminal part of the α-helical solenoid domain and regulates assembly and disassembly of the clathrin scaffold. Triskelia assemble into polyhedral lattices featuring various combinations of hexagons and pentagons. Unlike the COPI B-subcomplex, the clathrin scaffold is a true outer coat that does not make direct contacts with the membrane bilayer. These contacts are instead mediated by a variety of adaptor proteins that connect (i.e., “adapt”) the clathrin terminal domain with membrane lipids and proteins, constituting the inner layer of the coats. Each of these adaptors exhibits a characteristic preference for phosphoinositides, ARF-family GTPases and cargos, which, together, specify clathrin recruitment and function at different locations in the endomembrane system. For example, TGN and/or endosomal clathrin coats contain the heterotetrameric AP-1 and/or monomeric GGA proteins, whereas plasma membrane clathrin coats contain AP-2 and ARH, among several other adaptors. Through coupling to these adaptors, clathrin is involved in multiple sorting processes, including selective transport from the TGN and/or endosomes, and endocytosis from the plasma membrane.
De novo, monoallelic mutations in the gene encoding CHC17 have been linked to global developmental delay combined with hypotonia and, in some of the patients, epilepsy (DeMari et al., 2016, Hamdan et al., 2017) (Table 4). At least one patient also presented with morphological abnormalities (DeMari et al., 2016). Although the disorder is currently listed in the Online Mendelian Inheritance in Man (OMIM) database as autosomal dominant mental retardation 56, it should be noted that only some of the patients displayed intellectual disability (Hamdan et al., 2017). Consistent with the notion that loss of function of a single copy of CHC17-encoding gene may cause disease, large-scale population sequencing projects (Lek et al., 2016) yielded very few variants predicted to represent loss-of-function alleles and less than one third of the number of missense variants expected by chance.
Table 4.
Diseases caused by mutations in clathrin, AP-1, AP-2 and AP-3
| Coat | Component | Gene (locus) | Disease1 | OMIM2 | Inheritance | Mutation types | Key references |
|---|---|---|---|---|---|---|---|
| Clathrin | CHC17 | CLTC (17q23.1) | Autosomal dominant mental retardation 56 (MRD56) | 617854 | De novo | Missense, frameshift, in-frame deletion | DeMari et al., 2016, Hamdan et al., 2017 |
| CHC22 | CLTCL1 (22q11.21) | Congenital insensitivity to pain and touch with cognitive delay | None3 | Autosomal recessive | Missense | Nahorski et al., 2015 | |
| AP-1 | σ1A | AP1S1 (7q22.1) | MEDNIK syndrome (Erythrokeratodermia variabilis 3, EKV3) | 609313 | Autosomal recessive | Frameshift | Montpetit et al., 2008 |
| σ1B | AP1S2 (Xp22.2) | Fried syndrome, Pettigrew syndrome (PGS), or X-linked syndromic mental retardation 5 (MRXS5) | 304340 | X-linked | Nonsense, frameshift, complete deletion, in-frame deletion | Tarpey et al., 2006, Saillour et al., 2007 | |
| σ1C | AP1S3 (2q36.1) | Susceptibility to pustular psoriasis 15 (PSOR15) | 616106 | Autosomal dominant with low penetrance | Missense | Setta-Kaffetzi et al., 2014 | |
| AP-2 | σ2 | AP2S1 (19q13.32) | Familial hypocalciuric hypercalcemia 3 (FHH3) | 600740 | Autosomal dominant | Missense | Nesbit et al., 2013 |
| ARH | LDLRAP1 (1p36.11) | Autosomal recessive hypercholesterolemia (ARH) | 603813 | Autosomal recessive | Missense, nonsense, frameshift | Garcia et al., 2001 | |
| AP-3 | β3A-adaptin | AP3B1 (5q14.1) | Hermansky-Pudlak syndrome 2 (HPS2) | 608233 | Autosomal recessive | Missense, nonsense, frameshift, in-frame deletion, in-frame insertion | Dell’Angelica et al., 1999b |
| β3B-adaptin | AP3B2 (15q25.2) | Early infantile epileptic encephalopathy 48 (EIEE48) | 617276 | Autosomal recessive | Nonsense, frameshift | Assoum et al., 2016 | |
| δ-adaptin | AP3D1 (19p13.3) | Hermansky-Pudlak syndrome 10 (HPS10) | 617050 | Autosomal recessive | Frameshift | Ammann et al., 2016 |
Disease acronyms are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/)
No entry number has been assigned to the disease. The entry number for the gene is 601273.
Nahorski and colleagues (Nahorski et al., 2015) studied a consanguineous family having two members suffering from developmental delay and inability to sense pain or soft touch (Table 4). The two affected individuals were found to share various regions of the genome in homozygous form (~1.5% of the total genome) and, within one of these regions, a missense variant in the gene encoding CHC22 (Nahorski et al., 2015). The same missense variant, p.E330K, has been observed at low frequency in apparently normal individuals (Lek et al., 2016) in heterozygous but not homozygous form. Additional, unrelated families carrying biallelic variants the CHC22-encoding gene and a similar clinical presentation will be necessary to ascertain the link between CHC22 and this disorder and to rule out the putative contribution of other gene variants within the genomic regions shared by the two patients. Although the CHC22-encoding gene maps to a region of human chromosome 22 that is often deleted in patients suffering from DiGeorge or velocardiofacial syndromes (Holmes et al., 1997), other genes within the region are considered relevant to the pathogenesis of these and related syndromes (Morrow et al., 2018).
AP-1
AP-1 is a component of clathrin coats associated with the TGN and endosomes (more specifically the TEN, Bonifacino and Rojas, 2006) (Figure 1 and Table 1). This complex is composed of four subunits named γ-adaptin, β1-adaptin, μ1 and σ1 (Table 1). The γ, μ1 and σ1 subunits occur as two or three paralogous isoforms named γ1- and γ2-adaptin, μ1A and μ1B, and σ1A, σ1B and σ1C. These isoforms are expressed in all mammalian tissues, with the exception of μ1B, which is specifically expressed in epithelial cells (Ohno et al., 1999). The AP-1 complex is structured as a large “core” domain comprising the N-terminal “trunk” portions of γ- and β1-adaptin plus the entire μ1 and σ1 subunits, with extended “hinge” and “ear” regions comprising the C-terminal portions of γ- and β1-adaptin. Both the sequence of the AP-1 subunits and the overall structure of the complex are homologous to those of the COPI F-subcomplex, AP-2, AP-3, AP-4 and AP-5 (Figure 4), a reflection of their common evolutionary origin (Robinson, 2015, Rout and Field, 2017). AP-1 is recruited to membranes by virtue of interactions with PtdIns(4)P and the GTP-bound form of ARF GTPases, particularly ARF1 (Wang et al., 2003, Ren et al., 2013), although the ARF-related protein 1 (ARFRP1) has also been shown to play a role in this process (Guo et al., 2013b). ARF1 binds to two sites near the N-termini of γ- and β1-adaptin that are equivalent to those on COPI, plus a third site on the central trunk region of γ-adaptin (Ren et al., 2013). ARF1 binding triggers a conformational change from a cytosolic, “locked” conformation (Heldwein et al., 2004) to a membrane-bound, “unlocked” conformation (Ren et al., 2013, Shen et al., 2015). In conjunction with certain cargos such as the Nef protein of HIV-1, ARF1 also contributes to the assembly of a hexagonal AP-1 lattice that serves as a template for the clathrin scaffold (Shen et al., 2015).
The unlocking of AP-1 exposes binding sites for at least two types of sorting signal fitting the consensus motifs YXXØ (i.e., tyrosine-based signals) and [DE]xxxL[LI] (i.e., dileucine-based signals) (Table 1). Whereas YXXØ signals bind to a site on the C-terminal domain of μ1 (Ohno et al., 1995, Mattera et al., 2014), [DE]xxxL[LI] signals bind to a site on σ1, with some contribution from γ-adaptin (Janvier et al., 2003, Mattera et al., 2011). The μ1 subunit also binds acidic clusters (Navarro Negredo et al., 2017) and non-canonical sequences (Guo et al., 2013a). The μ1A and μ1B isoforms (Guo et al., 2013a), as well as σ1A, σ1B and σ1C isoforms, bind distinct but overlapping sets of signals, broadening the repertoire of signal recognition by AP-1 (Mattera et al., 2011).
In general, AP-1 functions in bidirectional transport between the TGN and endosomes (Hirst et al., 2012) (Figure 1 and Table 1). However, in polarized cells such as epithelial cells and neurons, AP-1 plays a more specific role in sorting to the basolateral (Folsch et al., 1999, Gravotta et al., 2012) and somatodendritic domains (Farías et al., 2012) of the plasma membrane, respectively. This latter function does not mean that AP-1 is generally required for transport to the plasma membrane (Chen et al., 2017, but reflects a function of AP-1 in keeping proteins away from the apical domain of epithelial cells and the axonal domain of neurons (Bonifacino, 2014). As mentioned above, homozygous knockout of the genes encoding γ1-adaptin and μ1A in mouse caused early embryonic lethality (Meyer et al., 2000, Zizioli et al., 1999). Homozygous knockout of the epithelially-expressed gene encoding μ1B, on the other hand, resulted in a mouse with altered polarity and hyperproliferation of intestinal epithelial cells (Hase et al., 2013).
Consistent with the specific role of AP-1 in polarized sorting in neurons and epithelial cells, mutations in each of the three σ1 subunit isoforms have been found to be the cause of a different neuroepithelial disorder (Table 4). Biallelic mutations in the AP1S1 (σ1A) gene cause an autosomal-recessive, multisystem disorder characterized by mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma (known by the acronym MEDNIK) (Montpetit et al., 2008, Martinelli et al., 2013) (Table 4). Before the identification of the σ1A mutations and further clinical characterization, this disease had been referred to as erythrokeratodermia variabilis-3 based on the hyperkeratosis and reddening of the skin found in patients from the Kamouraska region of Québec (Saba et al., 2005). In addition, MEDNIK patients feature copper metabolism defects, including hypocupremia, hypoceruloplasminemia and liver copper overload, suggestive of missorting of the copper transporters ATP7A and ATP7B (Martinelli et al., 2013). The AP1S1 mutations described to date include a splice site mutation and single exonic G insertion, both of which result in a frameshift and consequent production of a truncated, inactive protein (Montpetit et al., 2008, Martinelli et al., 2013, Incecik et al., 2018).
Biallelic mutations in the AP1S2 (σ1B) gene cause a severe neurodevelopmental disorder known by several names, including Fried syndrome, Pettigrew syndrome and X-linked syndromic mental retardation type 59 (Tarpey et al., 2006, Saillour et al., 2007) (Table 4). In addition to intellectual disability, patients exhibit facial dysmorphism, involuntary movements, progressive spasticity and seizures. Medical imaging of these patients showed hydrocephalus, basal ganglia calcifications, and cerebellar hypoplasia with enlargement of the fourth ventricle and posterior fossa (i.e., Dandy-Walker malformation). A variety of AP1S2 mutations have been identified in Fried/Pettigrew patients, including nonsense, frameshift, in-frame deletion and complete deletion (Tarpey et al., 2006, Saillour et al., 2007), which lead to the production of truncated or internally deleted proteins, or to the total absence of the protein.
Whole-genome sequencing of patients with pustular psoriasis, an inflammatory skin condition, identified two monoallelic missense variants in AP1S3 (p.R33W and p.F4C) that were 4–5-fold more frequent in the patients than in the general population (Setta-Kaffetzi et al., 2014) (Table 4). It should be noted that both of these variants are found in the general population with allele frequencies of 0.7–0.8%, and that healthy individuals who are homozygous for each of these variants can be found in large-scale sequencing projects (Lek et al., 2016). Accordingly, the effects of these variants must have low penetrance. It is for this reason that this form of the disease is referred to as “susceptibility to pustular psoriasis 15” (Table 4). A heterozygous patient with variants of both AP1S3 and IL36RN, another gene implicated in heterozygous pustular psoriasis, had more severe symptoms, suggesting that the disease involves complex genetic interactions (Mahil et al., 2016). The p.F4C variant was shown to be conformationally unstable, whereas the p.R33W variant failed to incorporate into the AP-1 complex, suggesting that both represented loss-of-function substitutions (Mahil et al., 2016). Cells expressing these variants displayed enhanced Toll-like receptor 2/6 signaling, impaired autophagy, NF-κB activation and increased IL-36α expression in skin keratinocytes, all of which likely contribute to the skin inflammation that underlies this disorder (Setta-Kaffetzi et al., 2014, Mahil et al., 2016).
Since all the mutations in σ1 isoforms described to date cause loss of function, the corresponding diseases must result from the absence of a particular variant of the AP-1 complex in particular cells and tissues. Although all three σ1 isoforms are ubiquitously expressed, their abundance ratios are likely variable among different cell types. Thus, loss of one isoform in a cell type in which the isoform is normally abundant could lead to low overall levels of AP-1 in those cells. Alternatively, each σ1 isoform could participate in recognition of a distinct subset of dileucine-containing cargos. Both of these factors could explain the different phenotypes resulting from loss of each σ1 isoform. Nevertheless, there must also be a certain level of functional redundancy among σ1 isoforms for the affected neonates to be viable, in contrast to the early embryonic lethality of mice lacking the γ1-adaptin and μ1A subunits (Meyer et al., 2000, Zizioli et al., 1999).
AP-2
AP-2 is a component of clathrin coats associated with the plasma membrane (Figures 1, 3 and 4, and Table 1). It comprises four ubiquitously-expressed subunits named α-adaptin, β2-adaptin, μ2 and σ2. α-adaptin exists as two paralogous isoforms named αA- and αC-adaptin. Additional diversification is achieved by alternative splicing of the αC transcript in the brain (Ball et al., 1995). There are no paralogs of the other AP-2 subunits, although β1-adaptin can be incorporated to some extent into the AP-2 complex in the absence of β2-adaptin (Li et al., 2010). AP-2 is recruited to the inner face of the plasma membrane mainly through interaction with PtdIns(4,5)P2 via binding sites near the N-terminus of α-adaptin and on the C-terminal domain of μ2 (Collins et al., 2002). A role for ARF-family members in AP-2 function has also been proposed (Moravec et al., 2012), but this is less well established than for COPI and AP-1. Binding to plasma membrane PtdIns(4,5)P2 triggers a conformational change from a locked to an unlocked form of AP-2 (Jackson et al., 2010). This conformational change also allows AP-2 to bind YxxØ and [DE]xxxL[LI] signals similar to those recognized by AP-1, through interaction with the μ2 and σ2-α-adaptin subunits, respectively (Ohno et al., 1995, Chaudhuri et al., 2007, Doray et al., 2007, Owen and Evans, 1998, Kelly et al., 2008). The μ2 subunit also binds acidic clusters (Singh et al., 2018). At the plasma membrane, AP-2 establishes a network of interactions with numerous accessory proteins (e.g., FCHO1/2, EPS15), as well as with clathrin, leading to the formation of clathrin-coated pits, and then clathrin-coated vesicles, which deliver endocytic cargos to endosomes.
The function of AP-2 in endocytosis is critical for viability, as demonstrated by the early embryonic lethality of mice with homozygous ablation of the μ2-encoding gene (Mitsunari et al., 2005). In contrast, mice with homozygous disruption of the β2-encoding gene die perinatally and exhibit a cleft palate (Li et al., 2010); this latter phenotype is likely due to defective endocytosis of signaling receptors involved in palate development. The differential requirement of these AP-2 subunits for embryonic development is due to the fact that μ2 cannot be substituted by any other μ subunit, whereas β2 can be partially replaced by β1 (Li et al., 2010).
To date, only one Mendelian coatopathy associated to AP-2 has been described (Table 4). Nesbit and colleagues (Nesbit et al., 2013) studied several unrelated cases of familial hypocalciuric hypercalcemia (FHH), which is characterized by increased levels of calcium in blood combined with decreased levels of calcium in urine, and among those that were negative for mutations in the CASR gene (associated with the majority of FHH cases) observed a high rate (~20%) of monoallelic missense variants in the AP2S1 gene encoding the σ2 subunit of AP-2. All of the missense variants identified in the initial studies (Nesbit et al., 2013, Hannan et al., 2015) involved the same amino-acid residue in σ2, namely R15, which is evolutionary conserved and known to be directly involved in the recognition of [DE]xxxL[LI] signals (Kelly et al., 2008). More recently, monoallelic missense variants involving other residues of σ2, which instead of interacting with cargo participate in assembly with α-adaptin, have been found in some individuals with mild hypercalcemia (Gorvin et al., 2018). Based on the genetic basis of three known types of FHH and functional studies, a pathogenic mechanism involving impaired calcium sensing seems likely. Indeed, FHH1 is caused by mutations in the CASR gene, which encodes a calcium-sensing G-protein-coupled receptor, FHH2 is caused by mutations in the GNA11 gene encoding the G-protein subunit α11, and FHH3 is the type caused by mutations in AP2S1 (reviewed by Lee and Shoback, 2018). All of these defects likely cause decreased intracellular signaling in response to extracellular calcium levels, which in parathyroid and kidney cells result in a higher threshold for extracellular calcium to suppress secretion of parathyroid hormone and to stimulate calcium excretion into urine, respectively (reviewed by Lee and Shoback, 2018).
ARH
ARH (an acronym for autosomal recessive hypercholesterolemia) is a monomeric clathrin adaptor associated with plasma membrane AP-2/clathrin-coated pits (He et al., 2002. Mishra et al., 2002) (Figures 1 and 4, and Table 1). This protein comprises an N-terminal phosphotyrosine-binding domain that interacts with both PtdIns(4,5)P2 and unphosphorylated FxNPxY sorting signals, and an unstructured C-terminal domain having a motif for binding to the clathrin terminal domain (Mishra et al., 2002). FxNPxY signals are present in various endocytic receptors, particularly members of the low-density lipoprotein (LDL) receptor (LDLR) family (Chen et al., 1990). Recognition of this motif by ARH promotes clathrin-mediated endocytosis of the receptors and their ligands from the plasma membrane into endosomes.
Prior to the identification of the ARH protein as a component of coats containing clathrin and AP-2 at the plasma membrane (He et al., 2002. Mishra et al., 2002), biallelic mutations in its encoding gene, now known as LDLRAP1, were shown to cause a rare form of autosomal recessive hypercholesterolemia (Garcia et al., 2001) (Table 4). Unlike the most common form of familial hypercholesterolemia (FH), caused by mutations in the LDLR gene (encoding the main receptor for LDL in hepatocytes and other cell types) and exhibiting a co-dominant mode of inheritance (i.e., heterozygous individuals display clinical symptoms, which are more severe in homozygous patients), the form of the disease caused by mutations in LDLRAP1 develops only when both the paternal and maternal copies of the gene are affected (reviewed by Fellin et al., 2015). Yet, the two types of hypercholesterolemia are mechanistically linked, as both arise from defective internalization of cholesterol-rich LDL into hepatocytes, in one case (FH) due to reduced expression and/or function of the LDLR and in the other (ARH) due to failure to incorporate LDLR (with bound LDL) into clathrin-coated pits for endocytosis (Sirinian et al., 2005, Garuti et al., 2005).
AP-3
AP-3 is another member of the heterotetrameric AP complex family (Dell’Angelica et al., 1997, Simpson et al., 1997), which in mammals is found in association with buds on endosomal tubules that are part of the TEN (Dell’Angelica et al., 1998, Peden et al., 2004) (Figure 1). In keeping with the nomenclature of AP complexes, its four subunits are named δ-adaptin, β3-adaptin, μ3 and σ3 (Figure 4 and Table 1). The β3, μ3 and σ3 subunits exist as A and B paralogous isoforms. Whereas β3A, μ3A, σ3A and σ3B are expressed in all tissues, β3B and μ3B are mainly expressed in the brain. Like COPI and AP-1, AP-3 is recruited to endosomal tubules by the GTP form of ARF GTPases, most probably ARF1 (Ooi et al., 1998). PtdIns(3)P additionally contributes to AP-3 recruitment (Baust et al., 2008). AP-3 also recognizes YxxØ and [DE]xxxL[LI] signals through the μ3 and σ3-δ subunits, respectively (Ohno et al., 1996, Janvier et al., 2003). The YxxØ binding site on μ3 (Mardones et al., 2013) is at a similar location than those on μ1 (Jia et al., 2014) and μ2 (Owen and Evans, 1998). It remains to be determined how AP-3 recognizes [DE]xxxL[LI] signals and whether the AP-3 core also undergoes conformational activation.
Mammalian AP-3 interacts with the terminal domain of clathrin via the same motif found in AP-1 and AP-2 (Dell’Angelica et al., 1998). Furthermore, immunoelectron microscopy analyses showed that about half of AP-3-coated buds also contain clathrin (Dell’Angelica et al., 1998, Peden et al., 2004, Theos et al., 2005). However, at least some functions of AP-3 are independent of clathrin (Peden et al., 2002, Zlatic et al., 2013). In addition, AP-3 was shown to interact with the HOPS subunit VPS41, which, like clathrin, has β-propeller and α-solenoid domains, and can assemble into lattice-like structures in vitro (Asensio et al., 2013). Further work is needed to ascertain the physiological significance of the interactions of AP-3 with clathrin and VPS41.
In fibroblasts, AP-3 enhances the transport of lysosomal membrane proteins from endosomes to lysosomes and decreases their recycling to the plasma membrane (Le Borgne et al., 1998, Dell’Angelica et al., 1999b, Peden et al., 2004). However, AP-3 has a more critical function in specialized cell types having LROs. For example, in melanocytes, AP-3 mediates sorting of the transmembrane enzyme tyrosinase to melanosomes via a [DE]xxxL[LI] signal (Theos et al., 2005). Accordingly, homozygous mutations of the δ- and β3A-adaptin subunits in mouse cause a pigmentation defect (Kantheti et al., 1998, Feng et al., 1999). In addition, mice with homozygous mutations in δ-adaptin have neurological and behavioral defects (Kantheti et al., 1998), which is consistent with additional roles of AP-3 in the biogenesis of synaptic vesicles (SVs) in neurons (Newell-Litwa et al., 2009) and dense core vesicles (DCVs) in neuroendocrine cells (Sirkis et al., 2013).
Biallelic loss-of-function mutations in the genes encoding β3A- and β3B-adaptin result in two coatopathies with non-overlapping clinical features (Hermansky-Pudlak syndrome type 2 [HPS2] and early infantile epileptic encephalopathy-48 [EIEE48], respectively) and those in the gene encoding the unique δ-adaptin result in a third coatopathy (Hermansky-Pudlak syndrome type 10 [HPS10]) characterized by a combination of the clinical manifestations of the other two (Table 4).
Hermansky-Pudlak syndrome (HPS) defines a group of at least ten Mendelian diseases that share the common clinical manifestations of oculocutaneous albinism (hypopigmentation of the skin, hair and eyes) and bleeding diathesis (prolonged bleeding) (Huizing et al., 2008). These manifestations arise from defects in two LROs, melanosomes and platelet dense granules, respectively. Additional clinical features are characteristic of specific types of HPS, which are defined by the identity of the mutated gene. HPS2, in particular, is caused by biallelic mutations in the AP3B1 gene encoding the ubiquitously expressed β3A-adaptin (Dell’Angelica et al., 1999b, Shotelersuk et al., 2000), and represents the first coatopathy described in the literature. The albinism observed in HPS2 patients is, at least in part, due to defective sorting of the melanogenic enzyme tyrosinase from the TEN to maturing melanosomes (Huizing et al., 2001, Theos et al., 2005). The bleeding diathesis, on the other hand, has been ascribed to missorting of SLC35D3 (Meng et al., 2012) and/or multidrug resistance protein 4 (Schaletzki et al., 2017) to platelet dense bodies. Recently, blood endothelial cells isolated from a patient suffering from HPS2 were reported to display impaired exocytosis of another LRO, Weibel-Palade bodies (Karampini et al., 2019), which could additionally contribute to bleeding diathesis. Besides albinism and bleeding diathesis, HPS2 patients typically present with innate immunodeficiency, which is thought to arise from defects in immune cell types containing LROs, namely, neutrophils, cytotoxic T cells and natural killer cells (Fontana et al., 2006, Clark et al., 2003, Lorenzi et al., 2013). Other LROs, such as lamellar bodies in alveolar type 2 cells, may be affected as well (Korogi et al., 2019).
As mentioned above, β3B is expressed almost exclusively in brain. In fact, this protein was originally identified as a neuron-specific antigen recognized by antibodies from a patient with autoimmune cerebellar degeneration (Newman et al., 1995). Two decades later, biallelic mutations in the encoding gene, AP3B1, were reported to cause EIEE48, which is characterized by global developmental delay, hypotonia, poor visual contact, and, in most cases, seizures and microcephaly (Assoum et al., 2016). On the other hand, no pigmentation or hematological abnormalities were noted (Assoum et al., 2016). The neurological presentation of β3B deficiency may result from alterations in SV and DCV biogenesis, in which AP-3 has also been implicated (Newell-Litwa et al., 2009, Sirkis et al., 2013), or from a more general role of AP-3 containing β3B in brain development.
Finally, biallelic mutations in the AP3D1 gene encoding δ-adaptin were found in four patients from two unrelated families displaying a complex syndrome referred to as HPS10 (Ammann et al., 2016, Mohammed et al., 2018), which can be described as a combination of symptoms in HPS2 and EIEE48, plus some dysmorphic features. Like HPS2 patients, those suffering from HPS10 display oculocutaneous albinism, abnormal platelet function and innate immunodeficiency associated to both neutropenia and impaired degranulation of natural killer and cytotoxic T cells (Ammann et al., 2016, Mohammed et al., 2018). In addition, and like patients suffering from EIEE48, HPS10 patients presented with global developmental delay, microcephaly, hypotonia, seizures and lack of ocular fixation (Ammann et al., 2016, Mohammed et al., 2018). HPS10 patients also displayed abnormalities in ear and jaw morphology as well as the distance between eyes (Ammann et al., 2016, Mohammed et al., 2018), probably reflecting an involvement of AP-3 in processes other than LRO, SV and DCV biogenesis (Le Borgne et al., 1998, Dell’Angelica et al., 1999b, Peden et al., 2004).
AP-4
Unlike AP-1, AP-2 and AP-3, AP-4 does not interact with clathrin and is thus thought to be part of a non-clathrin coat (Dell’Angelica et al., 1999a, Hirst et al., 1999) (Figure 4 and Table 1). It is currently unknown if AP-4 has a scaffold like the other coats or uses a different mechanism to perform its functions in cargo sorting and transport carrier formation. Microscopic analyses have shown that AP-4 is primarily associated with the TGN (Dell’Angelica et al., 1999a, Hirst et al., 1999) by virtue of interactions with ARF1 (Boehm et al., 2001) (Figure 1 and Table 1). Its subunits are named ε-adaptin, β4-adaptin, μ4 and σ4, are ubiquitously expressed and have no isoforms. The μ4 subunit binds tyrosine-based signals fitting the consensus motif YX[FYL][FL]E (Burgos et al., 2010), found in the cytosolic tails of members of the amyloid precursor protein (APP) family (i.e., APP, APLP1 and APLP2) (Burgos et al., 2010) and the ATG9 autophagy protein family (i.e., ATG9A, ATG9B) (Mattera et al., 2017). Although YX[FYL][FL]E sequences represent a subset of the YXXØ motif, they bind to a site on μ4 that is on the opposite face from the YXXØ-binding site on μ1, μ2 and μ3 (Burgos et al., 2010). The μ4 subunit also binds non-canonical sequences containing critical phenylalanine and/or tyrosine residues found in the cytosolic tails of the δ2 glutamate receptor (Yap et al., 2003) and the transmembrane AMPA receptor regulatory proteins (TARPs) (Matsuda et al., 2008); the structural mechanism for the recognition of these sequences is unknown. The “serine incorporator” proteins SERINC1 and SERINC3 have also been identified as AP-4 cargos, but it’s also unknown how they are recognized by AP-4.
Insights into the function of AP-4 have been obtained by mutation of its subunit genes in the mouse. Biallelic ablation of the genes encoding ε-adaptin (De Pace et al., 2018) and β4-adaptin (Matsuda et al., 2008) resulted in mice that were viable and fertile, but exhibited motor deficits and behavioral abnormalities. Moreover, histological analyses of the mutant mouse brains revealed a thinner corpus callosum and widespread swellings along the axons of various neuronal types (Matsuda et al., 2008, De Pace et al., 2018). These findings indicated that AP-4 is particularly important for the function of the central nervous system, probably due to a role in sorting specific neuronal cargos. Indeed, the normally somatodendritic δ2 and AMPA glutamate receptors were found to accumulate in axonal autophagosomes (Matsuda et al., 2008), and the ATG9A protein, which normally cycles between the TGN and pre-autophagosomal structures, was found to be retained at the TGN in both neuronal and non-neuronal cells (Mattera et al., 2017, De Pace et al., 2018, Davies et al., 2018). This latter defect impairs the maturation of pre-autophagosomal structures (Mattera et al., 2017, De Pace et al., 2018, Davies et al., 2018) and hinders the ability of neurons to dispose of axonal aggregates (De Pace et al., 2018). These studies thus demonstrated that AP-4 thus a role in export of specific cargos out of the TGN (Figure 1).
Defects in the genes encoding each of the four AP-4 subunits in humans have been shown to be the cause of an autosomal-recessive, complicated form of hereditary spastic paraplegia (HSP) referred to as “AP-4-deficiency syndrome” (Verkerk et al., 2009, Abou Jamra et al., 2011, Moreno-De-Luca et al., 2011, Bauer et al., 2012) (Table 5). The genetic defects include 5’-deletion, nonsense, missense, frameshift and splice-site mutations that either prevent expression of the proteins or result in expression of abnormal proteins. As a consequence of this primary defect, the remaining subunits are degraded to varying extents. Like other forms of HSP, AP-4-deficiency syndrome features progressive weakness and stiffness of the legs (i.e., paraplegia) due to degeneration of upper motor neurons running from the brain through the spinal cord. However, AP-4-deficient patients exhibit additional symptoms, including developmental delay, intellectual disability and seizures. The condition of the patients worsens with age, with some of them losing use of their arms in addition to their legs (i.e., tetraplegia) and requiring a wheelchair. Anatomical findings in this disease include a thin corpus callosum and white matter loss. The similarities of these motor and behavioral deficits to those of mice with mutations in AP-4 genes suggest a common pathogenetic mechanism involving defective sorting of AP-4 cargos and consequent cellular defects. Indeed, skin fibroblasts from AP-4-deficient patients also display accumulation of ATG9A at the TGN (Davies et al., 2018, De Pace et al., 2018). These findings in the patients and mutant mice makes it likely that defective autophagy underlies at least some of the motor neuron dysfunction characteristic of this disorder.
Table 5.
Diseases caused by mutations in AP-4 and AP-5 coats
| Coat | Component | Gene (locus) | Disease1 | OMIM2 | Inheritance | Mutation types | Key references |
|---|---|---|---|---|---|---|---|
| AP-4 | β4-adaptin | AP4B1 (1p13.2) | Hereditary spastic paraplegia 47 (SPG47) | 614066 | Autosomal recessive | Nonsense, missense, frameshift | Abou Jamra et al., 2011, Bauer et al., 2012 |
| μ4 | AP4M1 (7q22.1) | Hereditary spastic paraplegia 50 (SPG50) | 612936 | Autosomal recessive | Nonsense, missense, frameshift | Verkerk et al., 2009 | |
| ε-adaptin | AP4E1 (15q21.2) | Hereditary spastic paraplegia 51 (SPG51) | 613744 | Autosomal recessive | Nonsense, frameshift, 5’ deletion | Moreno-De-Luca et al., 2011, Abou Jamra et al., 2011 | |
| Familial persistent stuttering 1 (STUT1) | 184450 | Autosomal dominant | Missense, nonsense, frameshift | Raza et al., 2015 | |||
| σ4 | AP4S1 (14q12) | Hereditary spastic paraplegia 52 (SPG52) | 614067 | Autosomal recessive | Nonsense, frameshift | Abou Jamra et al., 2011 | |
| AP-5 | ζ-adaptin | AP5Z1 (7p22.1) | Hereditary spastic paraplegia 48 (SPG48) | 613647 | Autosomal recessive | Nonsense, missense, frameshift | Slabicki et al., 2010 |
| SPG113 | SPG11 (15q21.1) | Hereditary spastic paraplegia 11 (SPG11) | 604360 | Autosomal recessive | Nonsense, frameshift | Stevanin et al., 2007 | |
| Juvenile amyotrophic lateral sclerosis 5 (ALS5) | 602099 | Autosomal recessive | Nonsense, frameshift | Orlacchio et al., 2010 | |||
| Charcot-Marie-Tooth disease type 2X (CMT2X) | 616668 | Autosomal recessive | Nonsense, missense, frameshift | Montecchiani et al., 2016 | |||
| SPG154 | ZFYVE26 (14q24.1) | Hereditary spastic paraplegia 15 (SPG15) | 270700 | Autosomal recessive | Nonsense, frameshift | Hanein et al., 2008 |
Disease acronyms are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/).
Also known as spatacsin.
Also known as spastizin.
Monoallelic missense variants of the AP4E1 (ε-adaptin) gene were also linked to familial persistent stuttering, a volitional speech disorder (Raza et al., 2015). Most of the variant proteins were stable and assembled into the AP-4 complex (Raza et al., 2015), so the pathogenetic mechanism remains unclear. It is possible that these variations nonetheless impair some activity of AP-4, perhaps causing dysfunction in a subset of motor neurons that control speech initiation.
AP-5
AP-5 is part of another non-clathrin coat that associates with late endosomes (Hirst et al., 2011, Hirst et al., 2013). Like other AP complexes, it is composed of four subunits named ζ-adaptin, β5-adaptin, μ5 and σ5 (Figure 4 and Table 1). However, these subunits form a tightly assembled heterohexamer with two other proteins known as SPG11 (also named spatacsin) and SPG15 (also named spastizin) (Slabicki et al., 2010, Hirst et al., 2013) (Table 1). All subunits of this complex are ubiquitously expressed in mammalian tissues and have no paralogs. SPG11 is predicted to have N-terminal β-propeller and C-terminal α-solenoid domains like CHC and the α and β’ subunits of COPI, indicating that it likely functions as a scaffold. SPG15, on the other hand, has a PtdIns(3)P-interacting FYVE domain that may target this coat to late endosomes. The FYVE domain is flanked by α-solenoid structures that may further contribute to the assembly of the scaffold. SPG15 was shown to interact with the transmembrane cation-independent mannose 6-phosphate receptor (CI-MPR) involved in sorting of acid hydrolases to lysosomes (Hirst et al., 2013), and knockout of AP-5 subunits impaired retrieval of the CI-MPR from endosomes to the TGN (Hirst et al., 2013). This defect was exacerbated by knockdown of retromer subunits (see next section), suggesting that these two coats mediate parallel pathways of CI-MPR retrieval to the TGN (Hirst et al., 2013) (Figure 1). In addition, SPG15 was shown to interact with sortilin, another transmembrane protein that cycles between endosomes and the TGN, and that may serve as an intracellular sorting receptor for retrieval of Golgi proteins (Hirst et al., 2013).
As is the case for AP-4, orthologs of the four subunits of AP-5 are not present in Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster. However, orthologs of SPG11 and SPG15 are found in Drosophila melanogaster, suggesting that some functions of these proteins are independent of AP-5. Mice with homozygous knockout of the genes encoding SPG11 (Varga et al., 2015, Branchu et al., 2017) or SPG15 (Khundadze et al., 2013) exhibit progressive spastic paraplegia with loss of cortical neurons, Purkinje cells and upper and lower motor neurons. In accordance with a role of AP-5-SPG11-SPG15 in protein recycling from late endosomes to the TGN, cellular findings in these mutant mice include aberrant lysosomes and autophagosomes, defects that may contribute to neuronal loss.
Missense and frameshift mutations in human ζ-adaptin were identified as the cause of another complicated form of HSP named SPG48 (Slabicki et al., 2010) (Table 5). In addition to the characteristic spasticity of the legs, these patients exhibit mild intellectual disability and parkinsonism (Slabicki et al., 2010). The onset of the disease ranges from childhood to late adulthood. As in AP-4 deficiency syndrome, SPG48 patients exhibit thinning of the corpus callosum and white matter loss. Despite the similarities of these disorders, cells with knockout of ζ-adaptin exhibit normal distribution of ATG9A (Mattera et al., 2017), suggesting a different pathomechanism. Consistent with their role as a putative scaffold for the AP-5 coat, nonsense or frameshift mutations in the genes encoding SPG11 or SPG15 were found to cause a disorder that was similar, albeit more severe, than SPG48 (Stevanin et al., 2007, Hanein et al., 2008) (Table 5). Nonsense or frameshift mutations in SPG11 were also identified in patients with juvenile amyotrophic lateral sclerosis 5 (Orlacchio et al., 2010) or Charcot-Marie-Tooth disease type 2X (Montecchiani et al., 2016), reflecting the fact that these diseases fall within a spectrum of related motor neuron disorders. Like in the mutant mice, neurons and skin fibroblasts from patients with mutations in ζ-adaptin, SPG11 or SPG15 exhibit enlarged endolysosomal organelles containing multilamellar structures and other inclusion materials similar to those found in lysosomal storage disorders (Renvoisé et al., 2014, Hirst et al., 2015, Denora et al., 2016). It is thus likely that the inability of mannose 6-phosphate receptors to recycle from endosomes to the TGN in AP-5-deficient patients impairs sorting of lysosomal enzyme precursors to lysosomes, resulting in the accumulation of undegraded materials in lysosomes, with consequent harm to motor neuron function and viability.
RETROMER
Although also considered a coat, the retromer complex is structurally distinct from COPI, COPII and AP coats (Seaman, 2012). The mammalian retromer comprises a VPS26-VPS29-VPS35 heterotrimer that associates with various sorting nexin (SNX) proteins (Figure 3 and Table 1). VPS35 is structured as a long α-solenoid onto which VPS26 and VPS29 assemble at the N- and C-termini, respectively (Lucas et al., 2016). Mammalian VPS26 exists as two paralogous isoforms named VPS26A and VPS26B, whereas VPS29 and VPS35 are unique proteins. Of the more than 30 SNX proteins encoded in the human genome, two have been shown to physically associate with the mammalian retromer: SNX3 and SNX27 (Strochlic et al., 2007, Temkin et al., 2011). These SNX proteins comprise a PX domain that binds phosphoinositides, a defining characteristic of all members of the SNX family. SNX27 has additional FERM and PDZ domains. Both the VPS and SNX components of the retromer coat play multiple roles. The VPS heterotrimer serves as a scaffold for cross-linking of the SNX proteins (Kovtun et al., 2018), as well as for binding regulatory proteins such as the WASH actin nucleating complex (Gomez and Billadeau, 2009) and the TBC1D5 RAB7 GAP (Seaman et al., 2009). In addition, the VPS heterotrimer participates in the recognition of cargo signals fitting a ØX[LM] motif (Seaman, 2007) through a site at the interface of VPS26 and SNX3 (Lucas et al., 2016). The SNX proteins, on the other hand, mediate the recruitment of retromer to membranes through their phosphoinositide-binding activity, while also contributing to recognition of some cargos (Lucas et al., 2016). In the case of SNX27, the FERM and PDZ domains recognize cargos having NPxY and PDZ-ligand [ST]xØ motifs, respectively (Ghai et al., 2013, Temkin et al., 2011). The small GTPase RAB7 additionally contributes to the recruitment of retromer to endosomes (Rojas et al., 2008, Seaman et al., 2009).
The retromer coat associates with a tubular compartment emanating from vacuolar endosomes (Arighi et al., 2004, Seaman, 2004), which is part of the TEN (Bonifacino and Rojas, 2006) (Figure 1). At this location, the retromer promotes recycling of a large number of transmembrane proteins to the TGN and the plasma membrane (Steinberg et al., 2013). Knockout or knockdown of retromer subunit genes in cultured cells often results in missorting of cargos to lysosomes (Steinberg et al., 2013). Disruption of retromer genes in mouse causes early embryonic lethality, demonstrating that the function of retromer is critical for embryonic development (Lee et al., 1992).
A monoallelic missense mutation in VPS35 (p.D620N) was linked to both familial and sporadic forms of late-onset Parkinson’s Disease (PD) (Table 6), a neurodegenerative disorder characterized by resting tremor, muscle rigidity, slowed movement, postural instability, cognitive decline and dementia (Vilarino-Guell et al., 2011, Zimprich et al., 2011). An underlying cause of these symptoms is the loss of nigrostriatal dopaminergic neurons, which normally control movement, behavior and cognition. The p.D620N mutation occurs worldwide, albeit with relatively low frequency (~0.1% of all PD patients). Additional missense variants of VPS35 have been identified, but their linkage to PD remains to be established. The p.D620N mutation does not prevent assembly of the retromer complex nor its association with endosomes, but decreases the affinity of retromer for the WASH complex and its associated proteins (McGough et al., 2014, Zavodszky et al., 2014). This likely leads to impaired F-actin polymerization on retromer-containing endosomal domains and consequent missorting of retromer cargos such as the CI-MPR (McGough et al., 2014, Follett et al., 2014). Other processes that are altered by the p.D620N mutation include AMPA receptor trafficking (Munsie et al., 2015), maintenance of monoamine transporter levels (Cataldi et al., 2018) and autophagy (Zavodszky et al., 2014), although it is unclear if these alterations are direct or indirect effects of retromer dysfunction. Another intriguing defect in p.D620N mutant cells is an enhanced phosphorylation of Rab8A, Rab10 and Rab12 by the LRRK2 kinase, another protein that is mutated in cases of familial PD (Mir et al., 2018). Finally, the p.D620N mutation also causes mitochondrial dysfunction (Wang et al., 2016), a common cause of other forms of familial PD (Pickrell and Youle, 2015).
Two missense (p.K93E, p.M112V) and one nonsense variant (p.K297X) in VPS26A were identified in three out of 635 sequenced patients with atypical parkinsonism but not in control individuals (Gustavsson et al., 2015) (Table 6). Atypical parkinsonism refers to a variety of disorders that share some but not all features of PD. Functional analyses showed that these VPS26A variants assembled into the retromer complex and localized to endosomes (McMillan et al., 2016). They also interacted with all tested retromer partners, including WASH, to the same extent as normal VPS26A. The exception was p.K297X, which exhibited decreased interaction with SNX27 (McMillan et al., 2016). As a consequence, cells expressing the p.K297X variant displayed missorting of a well-established retromer and SNX27 cargo, the glucose transporter GLUT1, which instead of recycling to the plasma membrane was delivered to lysosomes (McMillan et al., 2016). Therefore, the p.K297X variant is likely pathogenic, whereas the significance of the other variants for atypical parkinsonism remains to be determined.
Finally, a biallelic frameshift mutation in the retromer-associated SNX27 was identified in four members of a consanguineous family with infantile myoclonic (i.e., “muscle-jerk) epilepsy and neurodegeneration (Damseh et al., 2015). SNX27 comprises, in N- to C-terminal order, PDZ, PX and FERM domains. The mutation results in the deletion of most of the PX domain and the FERM domain. The absence of the phosphoinositide-binding PX domain likely renders the mutant protein unable to associate with endosomes. The functional effects of this mutation, however, have not been investigated. It is noteworthy that mutations in VPS35/VPS26A and SNX27 are associated with different neurological disorders. The fact that VPS35/VPS26A mutations are monoallelic whereas the SNX27 mutation is biallelic may explain the greater severity of the SNX27 deficiency. Furthermore, SNX27 might have additional functions that are independent of retromer.
ARF1 AND SAR1B
As mentioned in previous sections, the small GTPase ARF1 mediates the recruitment of COPI, AP-1, AP-3 and AP-4 to membranes. Ge et al. (2016) reported three unrelated patients carrying de novo, monoallelic missense variants in ARF1 and presenting with severe brain malformation (Table 7). In two of the patients this malformation was characterized as periventricular heterotopia (altered distribution of subsets of neurons near ventricles caused by impaired cell migration during fetal brain development), and in the third one it was described as cerebral underdevelopment with delayed myelination. Interestingly, biallelic mutations in ARF1GEF2, which encodes a GEF for ARF1, also result in periventricular heterotopia, though combined with microcephaly (Sheen et al., 2004, Banne et al., 2013) (Table 7). These diseases could result from defective function of any or all of the coats regulated by ARF1.
Table 7.
Diseases caused by mutations in docking, regulatory and accessory coat proteins
| Protein | Coats | Gene (locus) | Function | Disease1 | OMIM2 | Inheritance | Key references |
|---|---|---|---|---|---|---|---|
| AAGAB | AP-1 | AAGAB (15q23) | AP-1 and AP-2 assembly | Punctate palmoplantar keratoderma type IA (PPKP1A) | 148600 | Autosomal dominant | Giehl et al., 2012, Pohler et al., 2012 |
| AP-2 | |||||||
| ARF1 | COPI | ARF1 (1q42.13) | Docking factor | Periventricular nodular heterotopia 8 (PVNH8) | 618185 | De novo | Ge et al., 2016 |
| AP-1 | |||||||
| AP-3 | |||||||
| AP-4 | |||||||
| ARFGEF23 | AP-1 | ARFGEF2 (20q13.13) | ARF1 GEF | Periventricular heterotopia with microcephaly (ARPHM; West syndrome) | 608097 | Autosomal recessive | Banne et al., 2013, Sheen et al., 2004 |
| Auxilin | AP-1 | DNAJC6 (1p31.3) | Clathrin uncoating | Parkinson disease 19A (PARK19A) and 19B (PARK18B) | 615528 | Autosomal recessive | Edvardson et al., 2012, Elsayed et al., 2016 |
| AP-2 | |||||||
| Dynamin 1 | AP-2 | DNM1 (9q34.11) | Coated vesicle scission | Early infantile epileptic encephalopathy 31 (EIEE31) | 616346 | De novo | EuroEPINOMICS-RES et al., 2017, |
| Dynamin 2 | AP-2 | DNM2 (19p13.2) | Coated vesicle scission | Centronuclear myopathy 1 (CNM1) | 160150 | Autosomal dominant | Bitoun et al., 2005 |
| Charcot-Marie-Tooth disease, axonal type 2M (CMT2M) and dominant intermediate B (CMTDIB) | 606482 | Autosomal dominant | Züchner et al., 2005; Fabrizi et al., 2007 | ||||
| Lethal congenital contracture syndrome 5 (LCCS5) | 615368 | Autosomal recessive | Koutsopoulos et al., 2013 | ||||
| NECAP1 | AP-1 | NECAP1 (12p13.31) | AP-1 and AP-2 vesicle formation | Early infantile epileptic encephalopathy 21 (EIEE21) | 615833 | Autosomal recessive | Alazami et al., 2014 |
| AP-2 | |||||||
| OCRL | AP-1 | OCRL (Xq26.1) | Inositol polyphosphate 5-phosphatase | Dent disease 2 | 300555 | X-linked | Hoopes et al., 2005 |
| AP-2 | |||||||
| Oculocerebrorenal syndrome of Lowe | 309000 | X-linked | Attree et al., 1992 | ||||
| RUSC24 | AP-4 | RUSC2 (9p13.3) | Movement of AP-4 vesicles | Autosomal recessive mental retardation-61 (MRT61) | 617773 | Autosomal recessive | Alwadei et al., 2016 |
| SAR1B | COPII | SAR1B (5q31.1) | Docking factor | Chylomicron retention disease (CMRD; Anderson disease) | 246700 | Autosomal recessive | Jones et al., 2003 |
| Synaptojanin 1 | AP-2 | SYNJ1 (21q22.11) | Polyphosphoinositide phosphatase | Early infantile epileptic encephalopathy 53 (EIEE53) | 617389 | Autosomal recessive | Hardies et al., 2016 |
| Parkinson disease 20, early-onset (PARK20) | 615530 | Autosomal recessive | Krebs et al., 2013, Quadri et al., 2013 |
Disease acronyms or alternative names are given in parentheses.
Online Mendelian Inheritance in Man (https://www.omim.org/).
Also known as BIG2.
Also known as Iporin.
Unlike ARF1, the small GTPase SAR1 is exclusively involved in membrane recruitment of the COPII coat. Two paralogs of SAR1, encoded by the SAR1A and SAR1B genes, exist in humans. Biallelic loss-of-function mutations in SAR1B have been found in several unrelated patients suffering from failure to thrive due to malabsorption of dietary lipids and fat-soluble vitamins. This phenotype likely results from defective secretion of chylomicrons from enterocytes into the lymphatic system. Among the alternative names given to this disorder, chylomicron retention disease appears to be the most descriptive (Table 7). However, some of the patients with biallelic mutations in SAR1B, which is expressed in many cell types besides enterocytes, also displayed muscular abnormalities (Silvain et al., 2008). These findings suggest that SAR1B plays a role in selective export of chylomicrons and other from the ER.
CONCLUSIONS AND PROSPECTS
In addition to the diseases that have been described in the previous sections, the definition of coatopathies can be extended to diseases caused by mutations of other regulatory and accessory proteins (Table 7). Most of these proteins are not structural components of coats, but transiently associate with them to regulate coat assembly and disassembly, vesicle budding and interactions with the cytoskeleton. Importantly, there are clear overlaps among the diseases caused by mutations in core coat components and regulatory/accessory proteins (Table 2–7). It is also noteworthy that, although most coatopathies are multi-system disorders, >80% affect the central nervous system. This is likely due to the complex organization of neurons and glial cells, having very long, differentiated processes that make them particularly vulnerable to defects in intracellular transport.
Because coatopathies are congenital disorders that affect fundamental components of the cellular machinery, treatment of these diseases is particularly challenging. An obvious approach would be to correct the genetic defect by gene therapy, as it is currently being done for other diseases (Ginn et al., 2018). However, this approach has yet to be tried for coatopathies. One problem with coatopathies is how to deliver the therapeutic agent to all the affected cells, since most coat proteins are ubiquitously expressed. As a first approximation it would be sensible to target the most critically affected organs or those that are most accessible to the available delivery methods. Another challenge is that most coatopathies have a developmental component that is already manifested at birth. Nevertheless, some coatopathies also have a subsequent degenerative phase that progresses over time. Gene therapy could thus be used both to ameliorate an existing problem or to prevent further deterioration of the patients.
While gene therapy remains the best hope for correction of the primary defect, there is also a need for more effective symptomatic treatments. A variety of pharmacologic, surgical and rehabilitation approaches are currently being used to treat specific symptoms and thus improve the patient’s quality of life. A better understanding of the biology of protein coats and the pathogenesis of these disorders could be instrumental in the development of new interventions. Because the main role of protein coats is to sort cargos, the identification of the whole repertoire of cargos for each coat could be particularly helpful. Indeed, many of these cargos are receptors or transporters for which specific drugs may already exist or could be developed. An example are copper metabolism defects in the AP-1 coatopathy MEDNIK syndrome (Montpetit et al., 2008), which are likely caused by missorting of the copper transporters ATP7A and/or ATP7B (Martinelli et al., 2013). Because these defects are similar to those of Wilson disease, caused by mutation in ATP7B, MEDNIK patients were treated with a well-established therapeutic agent for Wilson disease, zinc acetate, with considerable improvement of their clinical condition (Martinelli et al., 2013).
Finally, another approach that should be considered is pharmacologically inducing the expression of a paralogous isoform in cells in which an isoform is mutated, as suggested by Khoriaty and colleagues for treatment of congenital dyserythropoietic anemia type-II caused by SEC23B deficiency (Khoriaty et al., 2018). This approach would be analogous to the treatment of β-thalassemia or sickle-cell disease caused by genetic defects in genes encoding the α- or β-globin chains of hemoglobin A, through induction of expression of the paralogous γ-globin chain of fetal hemoglobin (Wienert et al., 2018). We expect that further research into the properties, functions and regulation of protein coats will aid in the development of new therapeutic approaches for other coatopathies.
ACKNOWLEDGMENTS
We thank the following colleagues for helpful discussions and advice: Matthew Farrer, Lauren Jackson. Work in the authors’ laboratories is supported by the Intramural Program of NICHD (ZIA HD001607) (JSB) and NIH grant R01GM112942 (ECD).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abou Jamra R, Philippe O, Raas-Rothschild A, Eck SH, Graf E, et al. 2011. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 88: 788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Hijazi H, Kentab AY, Alkuraya FS. 2014. NECAP1 loss of function leads to a severe infantile epileptic encephalopathy. J Med Genet. 51: 224–8. [DOI] [PubMed] [Google Scholar]
- Alwadei AH, Benini R, Mahmoud A, Alasmari A, Kamsteeg EJ, et al. 2016. Loss-of-function mutation in RUSC2 causes intellectual disability and secondary microcephaly. Dev Med Child Neurol. 58: 1317–22. [DOI] [PubMed] [Google Scholar]
- Ammann S, Schulz A, Krägeloh-Mann I, Dieckmann NM, Niethammer K, et al. 2016. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 127: 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakel EC, Schwappach B 2018. Formation of COPI-coated vesicles at a glance. J Cell Sci. 131: [DOI] [PubMed] [Google Scholar]
- Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. 2004. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. 165: 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio CS, Sirkis DW, Maas JW, Egami K, To TL, et al. 2013. Self-assembly of VPS41 promotes sorting required for biogenesis of the regulated secretory pathway. Dev Cell. 27: 425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoum M, Philippe C, Isidor B, Perrin L, Makrythanasis P, et al. 2016. Autosomal-Recessive Mutations in AP3B2, Adaptor-Related Protein Complex 3 Beta 2 Subunit, Cause an Early-Onset Epileptic Encephalopathy with Optic Atrophy. Am J Hum Genet. 99: 1368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, et al. 1992. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 358: 239–42. [DOI] [PubMed] [Google Scholar]
- Baines AC, Adams EJ, Zhang B, Ginsburg D 2013. Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS One. 8: e61114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball CL, Hunt SP, Robinson MS. 1995. Expression and localization of alpha-adaptin isoforms. J Cell Sci. 108: 2865–75. [DOI] [PubMed] [Google Scholar]
- Banne E, Atawneh O, Henneke M, Brockmann K, Gärtner J, et al. 2013. West syndrome, microcephaly, grey matter heterotopia and hypoplasia of corpus callosum due to a novel ARFGEF2 mutation. J Med Genet. 50: 772–5. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Helenius A 2016. Cargo capture and bulk flow in the early secretory pathway. Annu Rev Cell Dev Biol. 32: 197–222. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, et al. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77: 895–907. [DOI] [PubMed] [Google Scholar]
- Bauer P, Leshinsky-Silver E, Blumkin L, Schlipf N, Schroder C, et al. 2012. Mutation in the AP4B1 gene cause hereditary spastic paraplegia type 47 (SPG47). Neurogenetics. 13: 73–6. [DOI] [PubMed] [Google Scholar]
- Baust T, Anitei M, Czupalla C, Parshyna I, Bourel L, et al. 2008. Protein networks supporting AP-3 function in targeting lysosomal membrane proteins. Mol Biol Cell. 19: 1942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, et al. 2009. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 30: 1292–8. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Maugenre S, Jeannet PY, Lacène E, Ferrer X, et al. 2005. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 37: 1207–9. [DOI] [PubMed] [Google Scholar]
- Boehm M, Aguilar RC, Bonifacino JS. 2001. Functional and physical interactions of the adaptor protein complex AP-4 with ADP-ribosylation factors (ARFs). EMBO J. 20: 6265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS. 2014. Adaptor proteins involved in polarized sorting. J Cell Biol. 204: 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. 2004. The mechanisms of vesicle budding and fusion. Cell. 116: 153–66. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R 2006. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 7: 568–79. [DOI] [PubMed] [Google Scholar]
- Borner GH, Harbour M, Hester S, Lilley KS, Robinson MS. 2006. Comparative proteomics of clathrin-coated vesicles. J Cell Biol. 175: 571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, et al. 2006. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 38: 1192–7. [DOI] [PubMed] [Google Scholar]
- Branchu J, Boutry M, Sourd L, Depp M, Leone C, et al. 2017. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol Dis. 102: 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos PV, Mardones GA, Rojas AL, daSilva LL, Prabhu Y, et al. 2010. Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18: 425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykov YS, Schaffer M, Dodonova SO, Albert S, Plitzko JM, et al. 2017. The structure of the COPI coat determined within the cell. Elife. 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldi S, Follett J, Fox JD, Tatarnikov I, Kadgien C, et al. 2018. Altered dopamine release and monoamine transporters in Vps35 p.D620N knock-in mice. NPJ Parkinsons Dis. 4: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. 2007. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 81: 3877–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS. 1990. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low-density lipoprotein receptor. J Biol Chem. 265: 3116–23. [PubMed] [Google Scholar]
- Chen Y, Gershlick DC, Park SY, Bonifacino JS. 2017. Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers. J Cell Biol. 216: 4141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, et al. 2003. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol. 4: 1111–20. [DOI] [PubMed] [Google Scholar]
- Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 109: 523–35. [DOI] [PubMed] [Google Scholar]
- Cosson P, Letourneur F 1994. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 263: 1629–31. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Finch JT, Pearse BM. 1976. On the structure of coated vesicles. J Mol Biol. 103: 785–98. [DOI] [PubMed] [Google Scholar]
- Damseh N, Danson CM, Al-Ashhab M, Abu-Libdeh B, Gallon M, et al. 2015. A defect in the retromer accessory protein, SNX27, manifests by infantile myoclonic epilepsy and neurodegeneration. Neurogenetics. 16: 215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannhauser PN, Camus SM, Sakamoto K, Sadacca LA, Torres JA, et al. 2017. CHC22 and CHC17 clathrins have distinct biochemical properties and display differential regulation and function. J Biol Chem. 292: 20834–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannhauser PN, Ungewickell EJ. 2012. Reconstitution of clathrin-coated bud and vesicle formation with minimal components. Nat Cell Biol. 14: 634–9. [DOI] [PubMed] [Google Scholar]
- Davies AK, Itzhak DN, Edgar JR, Archuleta TL, Hirst J, et al. 2018. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun. 9: 3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pace R, Skirzewski M, Damme M, Mattera R, Mercurio J, et al. 2018. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 14: e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. 1998. Association of the AP-3 adaptor complex with clathrin. Science. 280: 431–4. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Mullins C, Bonifacino JS. 1999a. AP-4, a novel protein complex related to clathrin adaptors. J Biol Chem. 274: 7278–85. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, et al. 1997. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 16: 917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. 1999b. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 3: 11–21. [DOI] [PubMed] [Google Scholar]
- DeMari J, Mroske C, Tang S, Nimeh J, Miller R, et al. CLTC as a clinically novel gene associated with multiple malformations and developmental delay. Am J Med Genet A. 170A: 958–66. [DOI] [PubMed] [Google Scholar]
- Denora PS, Smets K, Zolfanelli F, Ceuterick-de Groote C, Casali C, et al. 2016. Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions. Brain. 139: 1723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Ronza A, Bajaj L, Sharma J, Sanagasetti D, Lotfi P, et al. 2018. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat Cell Biol. 20: 1370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStasio A, Driver A, Sund K, Donlin M, Muraleedharan RM, et al. 2017. Copb2 is essential for embryogenesis and hypomorphic mutations cause human microcephaly. Hum Mol Genet. 26: 4836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodonova SO, Diestelkoetter-Bachert P, von Appen A, Hagen WJ, Beck R, et al. 2015. A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science. 349: 195–8. [DOI] [PubMed] [Google Scholar]
- Doray B, Lee I, Knisely J, Bu G, Kornfeld S 2007. The gamma/sigma1 and alpha/sigma2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Mol Biol Cell. 18: 1887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, et al. 2012. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 7: e36458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed LE, Drouet V, Usenko T, Mohammed IN, Hamed AA, et al. 2016. A Novel Nonsense Mutation in DNAJC6 Expands the Phenotype of Autosomal-Recessive Juvenile-Onset Parkinson’s Disease. Ann Neurol. 79: 335–7. [DOI] [PubMed] [Google Scholar]
- Espenshade P, Gimeno RE, Holzmacher E, Teung P, Kaiser CA. 1995. Yeast SEC16 gene encodes a multidomain vesicle coat protein that interacts with Sec23p. J Cell Biol. 131: 311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES, Consortium. 2017. De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. Am J Hum Genet. 100: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizi GM, Ferrarini M, Cavallaro T, Cabrini I, Cerini R, et al. 2007. Two novel mutations in dynamin-2 cause axonal Charcot-Marie-Tooth disease. Neurology. 69: 291–5. [DOI] [PubMed] [Google Scholar]
- Faini M, Prinz S, Beck R, Schorb M, Riches JD, et al. 2012. The structures of COPI-coated vesicles reveal alternate coatomer conformations and interactions. Science. 336: 1451–4. [DOI] [PubMed] [Google Scholar]
- Farías GG, Cuitino L, Guo X, Ren X, Jarnik M, et al. 2012. Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron. 75: 810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin R, Arca M, Zuliani G, Calandra S, Bertolini S 2015. The history of Autosomal Recessive Hypercholesterolemia (ARH). From clinical observations to gene identification. Gene. 555: 23–32. [DOI] [PubMed] [Google Scholar]
- Feng L, Seymour AB, Jiang S, To A, Peden AA, et al. 1999. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet. 8: 323–30. [DOI] [PubMed] [Google Scholar]
- Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, et al. 2014. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic. 15: 230–44. [DOI] [PubMed] [Google Scholar]
- Folsch H, Ohno H, Bonifacino JS, Mellman I 1999. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell. 99: 189–98. [DOI] [PubMed] [Google Scholar]
- Fontana S, Parolini S, Vermi W, Booth S, Gallo F, et al. 2006. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. 107: 4857–64. [DOI] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, et al. 2004. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 432: 573–9. [DOI] [PubMed] [Google Scholar]
- Gallon M, Clairfeuille T, Steinberg F, Mas C, Ghai R, et al. 2014. A unique PDZ domain and arrestin-like fold interaction reveals mechanistic details of endocytic recycling by SNX27-retromer. Proc Natl Acad Sci U S A. 111: E3604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, et al. 2015. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet. 96: 432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, et al. 2001. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 292: 1394–8. [DOI] [PubMed] [Google Scholar]
- Garuti R, Jones C, Li WP, Michaely P, Herz J, et al. 2005. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem. 280: 40996–1004. [DOI] [PubMed] [Google Scholar]
- Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, et al. 2016. Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. NPJ Genom Med. 1: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai R, Bugarcic A, Liu H, Norwood SJ, Skeldal S, et al. 2013. Structural basis for endosomal trafficking of diverse transmembrane cargos by PX-FERM proteins. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl KA, Eckstein GN, Pasternack SM, Praetzel-Wunder S, Ruzicka T, et al. 2012. Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type Buschke-Fischer-Brauer. Am J Hum Genet. 91: 754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. 2018. Gene therapy clinical trials worldwide to 2017: An update. J Gene Med 20: e3015. [DOI] [PubMed] [Google Scholar]
- Gomez TS, Billadeau DD. 2009. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. 17: 699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorur A, Yuan L, Kenny SJ, Baba S, Xu K, et al. 2017. COPII-coated membranes function as transport carriers of intracellular procollagen I. J Cell Biol. 216: 1745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvin CM, Metpally R, Stokes VJ, Hannan FM, Krishnamurthy SB, et al. 2018. Large-scale exome datasets reveal a new class of adaptor-related protein complex 2 sigma subunit (AP2σ) mutations, located at the interface with the AP2 alpha subunit, that impair calcium-sensing receptor signalling. Hum Mol Genet. 27: 901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravotta D, Carvajal-Gonzalez JM, Mattera R, Deborde S, Banfelder JR, et al. 2012. The Clathrin Adaptor AP-1A Mediates Basolateral Polarity. Dev Cell. 22: 811–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Mattera R, Ren X, Chen Y, Retamal C, et al. 2013a. The Adaptor Protein-1 μ1B Subunit Expands the Repertoire of Basolateral Sorting Signal Recognition in Epithelial Cells. Dev. Cell 27: 353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zanetti G, Schekman R 2013b. A novel GTP-binding protein-adaptor protein complex responsible for export of Vangl2 from the trans Golgi network. elife. 2: e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson EK, Guella I, Trinh J, Szu-Tu C, Rajput A, et al. 2015. Genetic variability of the retromer cargo recognition complex in parkinsonism. Mov Disord. 30: 580–4. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, et al. 2017. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet. 101: 664–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, et al. 2008. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 82: 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan FM, Howles SA, Rogers A, Cranston T, Gorvin CM, et al. 2015. Adaptor protein-2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype-phenotype correlations, codon bias and dominant-negative effects. Hum Mol Genet. 24: 5079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies K, Cai Y, Jardel C, Jansen AC, Cao M, et al. 2016. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain. 139: 2420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase K, Nakatsu F, Ohmae M, Sugihara K, Shioda N, et al. 2013. AP-1B-Mediated Protein Sorting Regulates Polarity and Proliferation of Intestinal Epithelial Cells in Mice. Gastroenterology. [DOI] [PubMed] [Google Scholar]
- He G, Gupta S, Yi M, Michaely P, Hobbs HH, et al. 2002. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J Biol Chem. 277: 44044–9. [DOI] [PubMed] [Google Scholar]
- Heldwein EE, Macia E, Wang J, Yin HL, Kirchhausen T, et al. 2004. Crystal structure of the clathrin adaptor protein 1 core. Proc Natl Acad Sci U S A. 101: 14108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J 1980. Three-dimensional visualization of coated vesicle formation in fibroblasts. J Cell Biol. 84: 560–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierro A, Rojas AL, Rojas R, Murthy N, Effantin G, et al. 2007. Functional architecture of the retromer cargo-recognition complex. Nature. 449: 1063–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MN, et al. 2011. The fifth adaptor protein complex. PLoS Biol. 9: e1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Borner GH, Antrobus R, Peden AA, Hodson NA, et al. 2012. Distinct and Overlapping Roles for AP-1 and GGAs Revealed by the “Knocksideways” System. Curr Biol. 22: 1711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Borner GH, Edgar J, Hein MY, Mann M, et al. 2013. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 24: 2558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Bright NA, Rous B, Robinson MS. 1999. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 10: 2787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Edgar JR, Esteves T, Darios F, Madeo M, et al. 2015. Loss of AP-5 results in accumulation of aberrant endolysosomes: defining a new type of lysosomal storage disease. Hum Mol Genet. 24: 4984–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes SE, Riazi MA, Gong W, McDermid HE, Sellinger BT, et al. 1997. Disruption of the clathrin heavy chain-like gene (CLTCL) associated with features of DGS/VCFS: a balanced (21;22)(p12;q11) translocation. Hum Mol Genet. 6: 357–67. [DOI] [PubMed] [Google Scholar]
- Hood FE, Royle SJ. 2009. Functional equivalence of the clathrin heavy chains CHC17 and CHC22 in endocytosis and mitosis. J Cell Sci. 122: 2185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes RR, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, et al. 2005. Dent Disease with mutations in OCRL1. Am J Hum Genet. 76: 260–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. 2008. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 9: 359–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Sarangarajan R, Strovel E, Zhao Y, Gahl WA, et al. 2001. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell. 12: 2075–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchings J, Stancheva V, Miller EA, Zanetti G 2018. Subtomogram averaging of COPII assemblies reveals how coat organization dictates membrane shape. Nat Commun. 9: 4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incecik F, Bisgin A, Yılmaz M 2018. MEDNIK syndrome with a frame shift causing mutation in AP1S1 gene and literature review of the clinical features. Metab Brain Dis. 33: 2065–8. [DOI] [PubMed] [Google Scholar]
- Izumi K, Brett M, Nishi E, Drunat S, Tan ES, et al. 2016. ARCN1 Mutations Cause a Recognizable Craniofacial Syndrome Due to COPI-Mediated Transport Defects. Am J Hum Genet. 99: 451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AP, Seow HF, Holmes N, Drickamer K, Parham P 1987. Clathrin light chains contain brain-specific insertion sequences and a region of homology with intermediate filaments. Nature. 326: 154–9. [DOI] [PubMed] [Google Scholar]
- Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, et al. 2010. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. 141: 1220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LP, Lewis M, Kent HM, Edeling MA, Evans PR, et al. 2012. Molecular basis for recognition of dilysine trafficking motifs by COPI. Dev Cell. 23: 1255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janvier K, Kato Y, Boehm M, Rose JR, Martina JA, et al. 2003. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 gamma-sigma1 and AP-3 delta-sigma3 hemicomplexes. J Cell Biol. 163: 1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensson BO, Hansdottir S, Arnadottir GA, Sulem G, Kristjansson RP, et al. 2017. COPA syndrome in an Icelandic family caused by a recurrent missense mutation in COPA. BMC Med Genet. 18: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, et al. 2014. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. Elife. 3: e02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, et al. 2003. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 34: 29–31. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, et al. 1998. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 21: 111–22. [DOI] [PubMed] [Google Scholar]
- Karampini E, Schillemans M, Hofman M, van Alphen F, de Boer M, et al. 2019. Defective AP-3-dependent VAMP8 trafficking impairs Weibel-Palade body exocytosis in Hermansky-Pudlak Syndrome type 2 blood outgrowth endothelial cells. Haematologica. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BT, Graham SC, Liska N, Dannhauser PN, Höning S, et al. 2014. Clathrin adaptors. AP2 controls clathrin polymerization with a membrane-activated switch. Science. 345: 459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BT, McCoy AJ, Späte K, Miller SE, Evans PR, et al. 2008. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. 456: 976–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoriaty R, Hesketh GG, Bernard A, Weyand AC, Mellacheruvu D, et al. 2018. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc Natl Acad Sci U S A. 115: E7748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoriaty R, Vasievich MP, Jones M, Everett L, Chase J, et al. 2014. Absence of a red blood cell phenotype in mice with hematopoietic deficiency of SEC23B. Mol Cell Biol. 34: 3721–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, et al. 2013. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet. 9: e1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T, Owen D, Harrison SC. 2014. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol 6: a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogi Y, Gotoh S, Ikeo S, Yamamoto Y, Sone N, et al. 2019. In Vitro Disease Modeling of Hermansky-Pudlak Syndrome Type 2 Using Human Induced Pluripotent Stem Cell-Derived Alveolar Organoids. Stem Cell Reports. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsopoulos OS, Kretz C, Weller CM, Roux A, Mojzisova H, et al. 2013. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet. 21: 637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun O, Leneva N, Bykov YS, Ariotti N, Teasdale RD, et al. 2018. Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature. 561: 561–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, et al. 2013. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 34: 1200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Okamoto M, Nakano A 2014. Contact of cis-Golgi with ER exit sites executes cargo capture and delivery from the ER. Nat Commun. 5: 3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R, Alconada A, Bauer U, Hoflack B 1998. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J Biol Chem. 273: 29451–61. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Radice G, Perkins CP, Costantini F 1992. Identification and characterization of a novel, evolutionarily conserved gene disrupted by the murine H beta 58 embryonic lethal transgene insertion. Development. 115: 277–88. [DOI] [PubMed] [Google Scholar]
- Lee JY, Shoback DM. 2018. Familial hypocalciuric hypercalcemia and related disorders. Best Pract Res Clin Endocrinol Metab. 32: 609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E,. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR. 1990. A human homologue of the yeast HDEL receptor. Nature. 348: 162–3. [DOI] [PubMed] [Google Scholar]
- Li W, Puertollano-Moro R, Bonifacino J, Overbeek P, Everett E 2010. Disruption of the murine Ap2b1 gene causes nonsyndromic cleft palate. Cleft Palate Craniofac. J 47: 566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Doray B, Kornfeld S 2018. Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc Natl Acad Sci U S A. 115: 8984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SH, Towler MC, Chen E, Chen CY, Song W, et al. 2001. A novel clathrin homolog that co-distributes with cytoskeletal components functions in the trans-Golgi network. EMBO J. 20: 272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi L, Tabellini G, Vermi W, Moratto D, Porta F,. 2013. Occurrence of nodular lymphocyte-predominant hodgkin lymphoma in hermansky-pudlak type 2 syndrome is associated to natural killer and natural killer T cell defects. PLoS One. 8: e80131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, et al. 2016. Structural Mechanism for Cargo Recognition by the Retromer Complex. Cell. 167: 1623–1635.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Goldberg J 2013. Rules for the recognition of dilysine retrieval motifs by coatomer. EMBO J. 32: 926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Goldberg J 2016. TANGO1/cTAGE5 receptor as a polyvalent template for assembly of large COPII coats. Proc Natl Acad Sci U S A. 113: 10061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahil SK, Twelves S, Farkas K, Setta-Kaffetzi N, Burden AD, et al. 2016. AP1S3 Mutations Cause Skin Autoinflammation by Disrupting Keratinocyte Autophagy and Up-Regulating IL-36 Production. J Invest Dermatol. 136: 2251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoul I, Sohn K, Wieland FT, Pepperkok R, Pizza M, et al. 1998. KDEL receptor (Erd2p)-mediated retrograde transport of the cholera toxin A subunit from the Golgi involves COPI, p23, and the COOH terminus of Erd2p. J Cell Biol. 143: 601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Goldberg J 2008. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 27: 2918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardones GA, Burgos PV, Lin Y, Kloer DP, Magadan JG, et al. 2013. Structural Basis for the Recognition of Tyrosine-based Sorting Signals by the μ3A Subunit of the AP-3 Adaptor Complex. J. Biol. Chem 288: 9563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli D, Travaglini L, Drouin CA, Ceballos-Picot I, Rizza T, et al. 2013. MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain. 136: 872–81. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Miura E, Matsuda K, Kakegawa W, Kohda K, et al. 2008. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron. 57: 730–45. [DOI] [PubMed] [Google Scholar]
- Mattera R, Boehm M, Chaudhuri R, Prabhu Y, Bonifacino JS. 2011. Conservation and diversification of dileucine signal recognition by adaptor protein (AP) complex variants. J Biol Chem. 286: 2022–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Farias GG, Mardones GA, Bonifacino JS. 2014. Co-assembly of viral envelope glycoproteins regulates their polarized sorting in neurons. PLoS Pathog. 10: e1004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Park SY, De Pace R, Guardia CM, Bonifacino JS. 2017. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc Natl Acad Sci U S A. 114: E10697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaughey J, Stevenson NL, Cross S, Stephens DJ. 2018. ER-to-Golgi trafficking of procollagen in the absence of large carriers. J Cell Biol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough IJ, Steinberg F, Jia D, Barbuti PA, McMillan KJ, et al. 2014. Retromer Binding to FAM21 and the WASH Complex Is Perturbed by the Parkinson Disease-Linked VPS35(D620N) Mutation. Curr Biol. 24: 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan KJ, Gallon M, Jellett AP, Clairfeuille T, Tilley FC, et al. 2016. Atypical parkinsonism-associated retromer mutant alters endosomal sorting of specific cargo proteins. J Cell Biol. 214: 389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng R, Wang Y, Yao Y, Zhang Z, Harper DC, et al. 2012. SLC35D3 delivery from megakaryocyte early endosomes is required for platelet dense granule biogenesis and is differentially defective in Hermansky-Pudlak syndrome models. Blood. 120: 404–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, et al. 2000. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19: 2193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, et al. 2003. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 114: 497–509. [DOI] [PubMed] [Google Scholar]
- Miller EA, Schekman R 2013. COPII - a flexible vesicle formation system. Curr Opin Cell Biol. 25: 420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, et al. 2018. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J. 475: 1861–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Beznoussenko GV, Trucco A, Lupetti P, Smith JD, et al. 2003. ER-to-Golgi carriers arise through direct en bloc protrusion and multistage maturation of specialized ER exit domains. Dev Cell. 5: 583–94. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Watkins SC, Traub LM. 2002. The autosomal recessive hypercholesterolemia (ARH) protein interfaces directly with the clathrin-coat machinery. Proc Natl Acad Sci U S A. 99: 16099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunari T, Nakatsu F, Shioda N, Love PE, Grinberg A, et al. 2005. Clathrin adaptor AP-2 is essential for early embryonal development. Mol Cell Biol. 25: 9318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed M, Al-Hashmi N, Al-Rashdi S, Al-Sukaiti N, Al-Adawi K, et al. 2018. Biallelic mutations in AP3D1 cause Hermansky-Pudlak syndrome type 10 associated with immunodeficiency and seizure disorder. Eur J Med Genet. In press. [DOI] [PubMed] [Google Scholar]
- Montecchiani C, Pedace L, Lo Giudice T, Casella A, Mearini M, et al. 2016. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain. 139: 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit A, Cote S, Brustein E, Drouin CA, Lapointe L, et al. 2008. Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. PLoS Genet. 4: e1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moravec R, Conger KK, D’Souza R, Allison AB, Casanova JE. 2012. BRAG2/GEP100/IQSec1 interacts with clathrin and regulates α5β1 integrin endocytosis through activation of ADP ribosylation factor 5 (Arf5). J Biol Chem. 287: 31138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca A, Helmers SL, Mao H, Burns TG, Melton AM, et al. 2011. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet. 48: 141–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ. 2018. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A. 176: 2070–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Bickford LC, Goldberg J 2003. SNARE selectivity of the COPII coat. Cell. 114: 483–95. [DOI] [PubMed] [Google Scholar]
- Munsie LN, Milnerwood AJ, Seibler P, Beccano-Kelly DA, Tatarnikov I, et al. 2015. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum Mol Genet. 24: 1691–703. [DOI] [PubMed] [Google Scholar]
- Nahorski MS, Al-Gazali L, Hertecant J, Owen DJ, Borner GH, et al. 2015. A novel disorder reveals clathrin heavy chain-22 is essential for human pain and touch development. Brain. 138: 2147–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro Negredo P, Edgar JR, Wrobel AG, Zaccai NR, Antrobus R, et al. 2017. Contribution of the clathrin adaptor AP-1 subunit μ1 to acidic cluster protein sorting. J Cell Biol. 216: 2927–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, et al. 2013. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 45: 93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Salazar G, Smith Y, Faundez V 2009. Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell. 20: 1441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman LS, McKeever MO, Okano HJ, Darnell RB. 1995. Beta-NAP, a cerebellar degeneration antigen, is a neuron-specific vesicle coat protein. Cell. 82: 773–83. [DOI] [PubMed] [Google Scholar]
- Noorelahi R, Perez G, Otero HJ. 2018. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr Radiol. 48: 279–82. [DOI] [PubMed] [Google Scholar]
- Ohno H, Fournier MC, Poy G, Bonifacino JS. 1996. Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J Biol Chem. 271: 29009–15. [DOI] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, et al. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 269: 1872–5. [DOI] [PubMed] [Google Scholar]
- Ohno H, Tomemori T, Nakatsu F, Okazaki Y, Aguilar RC, et al. 1999. Mu1B, a novel adaptor medium chain expressed in polarized epithelial cells. FEBS Lett. 449: 215–20. [DOI] [PubMed] [Google Scholar]
- Ooi CE, Dell’Angelica EC, Bonifacino JS. 1998. ADP-Ribosylation factor 1 (ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J Cell Biol. 142: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, et al. 2010. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. 133: 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DJ, Evans PR. 1998. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science. 282: 1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse BM. 1975. Coated vesicles from pig brain: purification and biochemical characterization. J Mol Biol. 97: 93–8. [DOI] [PubMed] [Google Scholar]
- Pearse BM, Robinson MS. 1984. Purification and properties of 100-kd proteins from coated vesicles and their reconstitution with clathrin. EMBO J. 3: 1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, et al. 2004. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 164: 1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Rudge RE, Lui WW, Robinson MS. 2002. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 156: 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 85: 257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohler E, Mamai O, Hirst J, Zamiri M, Horn H, et al. 2012. Haploinsufficiency for AAGAB causes clinically heterogeneous forms of punctate palmoplantar keratoderma. Nat Genet. 44: 1272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk RS, San Pietro E, Di Pentima A, Tete S, Bonifacino JS. 2006. Ultrastructure of long-range transport carriers moving from the trans Golgi network to peripheral endosomes. Traffic. 7: 1092–103. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, et al. 1997. ER-to-Golgi transport visualized in living cells. Nature. 389: 81–5. [DOI] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, et al. 2013. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 34: 1208–15. [DOI] [PubMed] [Google Scholar]
- Raza MH, Mattera R, Morell R, Sainz E, Rahn R, et al. 2015. Association between Rare Variants in AP4E1, a Component of Intracellular Trafficking, and Persistent Stuttering. Am J Hum Genet. 97: 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Farias GG, Canagarajah BJ, Bonifacino JS, Hurley JH. 2013. Structural Basis for Recruitment and Activation of the AP-1 Clathrin Adaptor Complex by Arf1. Cell. 152: 755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoisé B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, et al. 2014. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol. 1: 379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. 2015. Forty Years of Clathrin-coated Vesicles. Traffic. 16: 1210–38. [DOI] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, et al. 2008. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J Cell Biol. 183: 513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TF, Porter KR. 1964. Yolk protein uptake in the oocyte of the mosquito Aedes aegypti. J Cell Biol. 20: 313–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Field MC. 2017. The Evolution of Organellar Coat Complexes and Organization of the Eukaryotic Cell. Annu Rev Biochem. 86: 637–57. [DOI] [PubMed] [Google Scholar]
- Saba TG, Montpetit A, Verner A, Rioux P, Hudson TJ, et al. 2005. An atypical form of erythrokeratodermia variabilis maps to chromosome 7q22. Hum Genet. 116: 167–71. [DOI] [PubMed] [Google Scholar]
- Saillour Y, Zanni G, Des Portes V, Heron D, Guibaud L, et al. 2007. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J Med Genet. 44: 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Chen M, Bard F, Chen S, Zhou H, et al. 2009. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 136: 891–902. [DOI] [PubMed] [Google Scholar]
- Saito K, Yamashiro K, Ichikawa Y, Erlmann P, Kontani K, et al. 2011. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell. 22: 2301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sato M, Nakano A 2001. Rer1p, a retrieval receptor for endoplasmic reticulum membrane proteins, is dynamically localized to the Golgi apparatus by coatomer. J Cell Biol. 152: 935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaletzki Y, Kromrey ML, Bröderdorf S, Hammer E, Grube M, et al. 2017. Several adaptor proteins promote intracellular localisation of the transporter MRP4/ABCC4 in platelets and haematopoietic cells. Thromb Haemost. 117: 105–15. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, et al. 2009. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 41: 936–40. [DOI] [PubMed] [Google Scholar]
- Seaman MN. 2004. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 165: 111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. 2007. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci. 120: 2378–89. [DOI] [PubMed] [Google Scholar]
- Seaman MN. 2012. The retromer complex - endosomal protein recycling and beyond. J Cell Sci. 125: 4693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, Harbour ME, Tattersall D, Read E, Bright N 2009. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J Cell Sci. 122: 2371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Orci L, Amherdt M, Brunner M, Kahn RA, et al. 1991. ADP-ribosylation factor is a subunit of the coat of Golgi-derived COP-coated vesicles: a novel role for a GTP-binding protein. Cell. 67: 239–53. [DOI] [PubMed] [Google Scholar]
- Setta-Kaffetzi N, Simpson MA, Navarini AA, Patel VM, Lu HC, et al. 2014. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum Genet. 94: 790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, et al. 2004. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 36: 69–76. [DOI] [PubMed] [Google Scholar]
- Shen QT, Ren X, Zhang R, Lee IH, Hurley JH. 2015. HIV-1 Nef hijacks clathrin coats by stabilizing AP-1:Arf1 polygons. Science. 350: aac5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Rojas R, Bonifacino JS, Hurley JH. 2006. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat Struct Mol Biol. 13: 540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shotelersuk V, Dell’Angelica EC, Hartnell L, Bonifacino JS, Gahl WA. 2000. A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med. 108: 423–7. [DOI] [PubMed] [Google Scholar]
- Silvain M, Bligny D, Aparicio T, Laforêt P, Grodet A, et al. 2008. Anderson’s disease (chylomicron retention disease): a new mutation in the SARA2 gene associated with muscular and cardiac abnormalities. Clin Genet. 74: 546–52. [DOI] [PubMed] [Google Scholar]
- Simpson F, Peden AA, Christopoulou L, Robinson MS. 1997. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 137: 835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Stoneham C, Lim C, Jia X, Guenaga J, et al. 2018. Phosphoserine acidic cluster motifs bind distinct basic regions on the μ subunits of clathrin adaptor protein complexes. J Biol Chem. 293: 15678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirinian MI, Belleudi F, Campagna F, Ceridono M, Garofalo T, et al. 2005. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein (LDL) binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem. 280: 38416–23. [DOI] [PubMed] [Google Scholar]
- Sirkis DW, Edwards RH, Asensio CS. 2013. Widespread dysregulation of peptide hormone release in mice lacking adaptor protein AP-3. PLoS Genet. 9: e1003812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, et al. 2010. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 8: e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, et al. 2013. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 15: 461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DJ, Pepperkok R 2002. Imaging of procollagen transport reveals COPI-dependent cargo sorting during ER-to-Golgi transport in mammalian cells. J Cell Sci. 115: 1149–60. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, et al. 2007. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 39: 366–72. [DOI] [PubMed] [Google Scholar]
- Strochlic TI, Setty TG, Sitaram A, Burd CG. 2007. Grd19/Snx3p functions as a cargo-specific adapter for retromer-dependent endocytic recycling. J Cell Biol. 177: 115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Stevens C, Teague J, Edkins S, O’Meara S, et al. 2006. Mutations in the gene encoding the Sigma 2 subunit of the adaptor protein 1 complex, AP1S2, cause X-linked mental retardation. Am J Hum Genet. 79: 1119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveira-DaSilva AM, Markello TC, Kleiner DE, Jones AM, Groden C, et al. 2018. Expanding the phenotype of COPA syndrome: a kindred with typical and atypical features. J Med Genet. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin P, Lauffer B, Jäger S, Cimermancic P, Krogan NJ, et al. 2011. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 13: 715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Tenza D, Martina JA, Hurbain I, Peden AA, et al. 2005. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 16: 5356–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E, Branton D 1981. Assembly units of clathrin coats. Nature. 289: 420–2. [DOI] [PubMed] [Google Scholar]
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, et al. 2015. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 11: e1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos S, Esk C, Hoshino S, Funke BH, Chen CY, et al. 2009. A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science. 324: 1192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Schot R, Dumee B, Schellekens K, Swagemakers S, et al. 2009. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 85: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, et al. 2011. VPS35 mutations in Parkinson disease. Am J Hum Genet. 89: 162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, et al. 2018. Type I interferon pathway activation in COPA syndrome. Clin Immunol. 187: 33–6. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, et al. 2016. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med. 22: 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, et al. 2003. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 114: 299–310. [DOI] [PubMed] [Google Scholar]
- Waters MG, Serafini T, Rothman JE. 1991. ‘Coatomer’: a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature. 349: 248–51. [DOI] [PubMed] [Google Scholar]
- Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, et al. 2015. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 47: 654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienert B, Martyn GE, Funnell APW, Quinlan KGR, Crossley M 2018. Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies. Trends Genet. 34: 927–40. [DOI] [PubMed] [Google Scholar]
- Wong JKL, Gui H, Kwok M, Ng PW, Lui CHT, et al. 2018. Rare variants and de novo variants in mesial temporal lobe epilepsy with hippocampal sclerosis. Neurol Genet. 4: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Li C, Ward DM, Kaplan J, Mansour SL. 2000. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci. 113: 4077–86. [DOI] [PubMed] [Google Scholar]
- Yap CC, Murate M, Kishigami S, Muto Y, Kishida H, et al. 2003. Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol Cell Neurosci. 24: 283–95. [DOI] [PubMed] [Google Scholar]
- Yehia L, Niazi F, Ni Y, Ngeow J, Sankunny M, et al. 2015. Germline Heterozygous Variants in SEC23B Are Associated with Cowden Syndrome and Enriched in Apparently Sporadic Thyroid Cancer. Am J Hum Genet. 97: 661–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Breitman M, Goldberg J 2012. A structure-based mechanism for Arf1-dependent recruitment of coatomer to membranes. Cell. 148: 530–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Prinz S, Daum S, Meister A, Schekman R, et al. 2013. The structure of the COPII transport-vesicle coat assembled on membranes. Elife. 2: e00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, et al. 2014. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 5: 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, J et al. 2001. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci U S A. 98: 2431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, et al. 2011. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 89: 168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizioli D, Meyer C, Guhde G, Saftig P, von Figura K, et al. 1999. Early embryonic death of mice deficient in gamma-adaptin. J Biol Chem. 274: 5385–90. [DOI] [PubMed] [Google Scholar]
- Zlatic SA, Grossniklaus EJ, Ryder PV, Salazar G, Mattheyses AL, et al. 2013. Chemical-genetic disruption of clathrin function spares adaptor complex 3-dependent endosome vesicle biogenesis. Mol Biol Cell. 24: 2378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, et al. 2005. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 37: 289–94. [DOI] [PubMed] [Google Scholar]