

Abstract
Reactions that activate carboxylates through acyl-adenylate intermediates are found throughout biology and include acyl- and aryl-CoA synthetases and tRNA synthetases. Here we describe the characterization of Aquifex aeolicus BioW, which represents a new protein fold within the superfamily of adenylating enzymes. Substrate-bound structures identified the enzyme active site and elucidated the mechanistic strategy for conjugating CoA to the seven-carbon α,ω-dicarboxylate pimelate, a biotin precursor. Proper position of reactive groups for the two half-reactions is achieved solely through movements of active site residues, as confirmed by site-directed mutational analysis. The ability of BioW to hydrolyze adenylates of noncognate substrates is reminiscent of pre-transfer proofreading observed in some tRNA synthetases, and we show that this activity can be abolished by mutation of a single residue. These studies illustrate how BioW can carry out three different biologically prevalent chemical reactions (adenylation, thioesterification, and proofreading) in the context of a new protein fold.
Biotin (vitamin B7 or coenzyme R) is a water-soluble essential cofactor in all domains of life, where it serves as the prosthetic group for numerous metabolic enzymes that catalyze carboxyltransfer reactions1. The chemical structure of biotin consists of a tetrahydroimidizalone ring fused with an organosulfur-containing tetrahydrothiophane ring that bears a valeric acid substituent2. Attachment of biotin to constituent enzymes occurs via an amide linkage between the carboxylate of the valeric acid moiety and the ε-amine of a specific Lys in biotin carrier protein, a small domain of roughly 80 residues3. The bicyclic rings of the resultant biotinylated protein extends outward, where the ureido ring N8 nitrogen can carry equivalents of CO2 (carboxybiotin) between the carboxylation and carboxyltransfer domains of biotin-dependent enzymes4.
Although mammals do not synthesize biotin de novo, intestinal bacteria produce biotin well in excess of the necessary daily requirements5. The unusual structure of biotin is derived from two precursors: alanine and a thioester of the C7 α,ω-dicarboxylic acid pimelate6 (1, Fig. 1). Genetic studies of Escherichia coli biotin auxotrophic mutants helped elucidate the structures of the intermediates in the late stages of the biosynthetic pathway7,8, and subsequent studies established that four enzymes catalyze the assembly of the bicyclic ring in an adenosine 5′-triphosphate (ATP)- and S-adenosylmethionine (SAM)-dependent manner9. However, the pathway for incorporation of pimelate into biotin has only recently been determined, and only for E. coli and Bacillus subtilis and closely related species10, revealing divergent biosynthetic routes to the pimeloyl-CoA intermediate (2).
Figure 1 |. Reaction catalyzed by BioW and representative homologs.
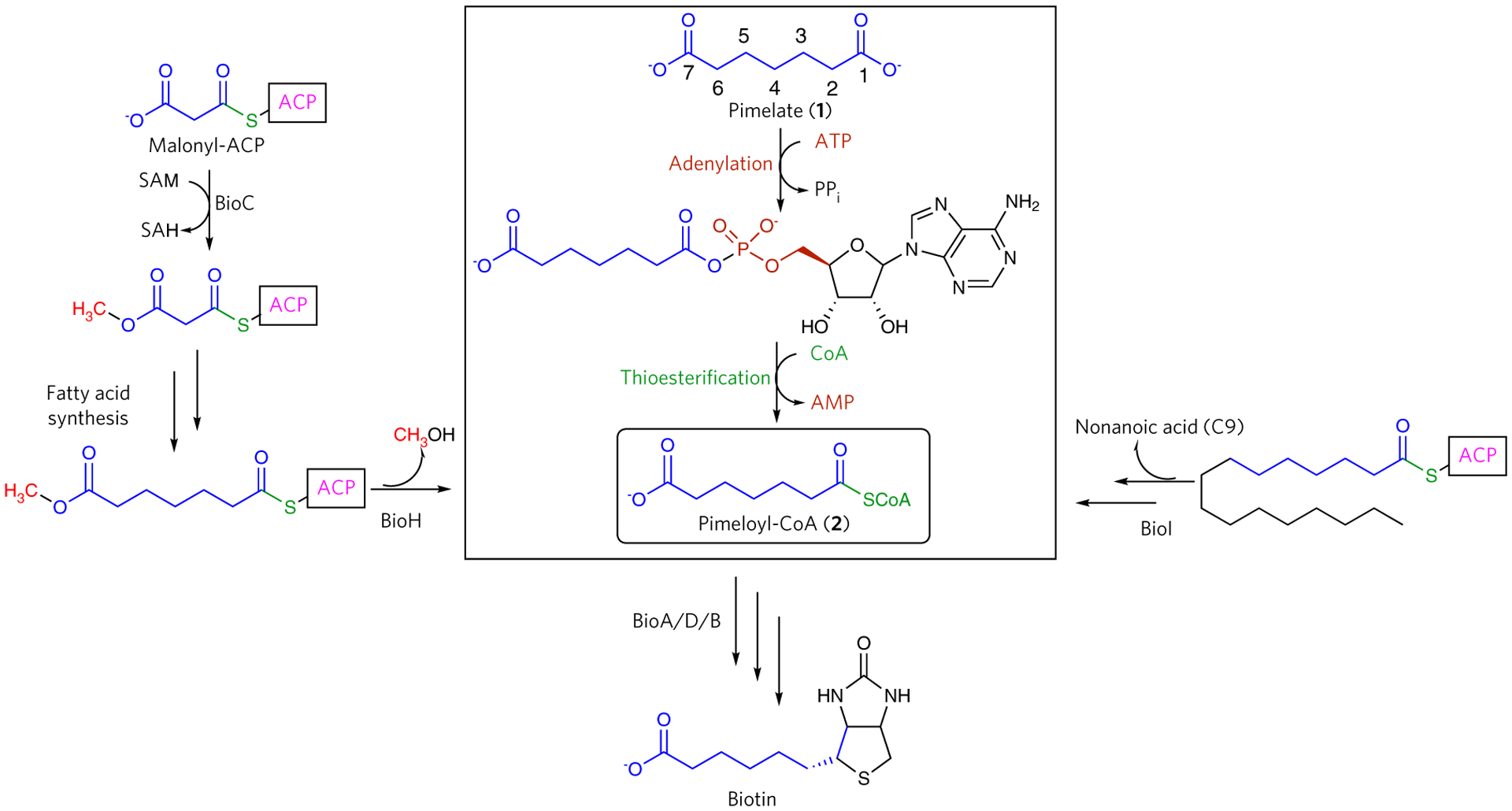
Convergent biosynthetic pathways for the incorporation of the pimelate dicarboxylate into biotin. The pathway highlighted in the rectangle illustrates the direct ligation of pimelic acid to pimeloyl-CoA via a two-step reaction catalyzed by BioW.
Isotopic labeling studies using E. coli cultures grown in differentially 13C -labeled acetate suggested that three acetate units are incorporated into the pimelate group of biotin11,12. The labeling patterns rule out a symmetric pimelate precursor and are consistent with a pimeloyl thioester as the likely intermediate. Genetic and biochemical studies show that two gene products essential for E. coli biotin biosynthesis (bioC and bioH) provide a route for the assembly of pimeloyl-acyl carrier protein (ACP) thioester by hijacking the fatty acid biosynthetic machinery13. Specifically, BioC is a SAM-dependent methyltransferase that converts the ω-carboxylate of malonyl-ACP into the corresponding methyl ester, providing a neutral charge mimic of fatty acyl chains that can be accommodated in the hydrophobic active sites of fatty acid synthetic enzymes. Following each round of condensation and reduction, BioH hydrolyzes the pimeloyl-ACP methyl ester and prevents further elongation14.
By contrast, B. subtilis and related strains (but not all bacilli) utilize two different orthogonal strategies for the production of pimeloyl thioesters (Fig. 1). In the first scheme, the cytochrome P450 enzyme BioI catalyzes the de novo synthesis of pimeloyl thioester from long-chain acyl-ACPs15–17. The ACP linkage fixes the long-chain acyl group into the active site along a bent tunnel, thus positioning the C7–C8 bond directly above the heme iron, where consecutive hydroxylations at C7 and C8 and subsequent oxidation of the vicinal diol produces the pimeloyl-ACP product18. A second route is the ATP-dependent synthesis of pimeloyl-CoA from the free α,ω-dicarboxylic acid pimelate by the acyl-CoA synthetase BioW19. The activity of BioW is strictly dependent on ATP, and the enzyme demonstrates specificity for pimelate13,15. This substrate specificity is strict, as the enzyme cannot utilize longer or shorter dicarboxylic acids or monocarboxylates of equivalent length (C6 or C7) to generate pimelate20.
The BioW-catalyzed reaction has been shown to proceed through the formation of stoichiometric quantities of an acyl-adenylate intermediate20, a common feature amongst acyl-CoA ligases. However, the primary sequence of the enzyme does not reveal any of the sequence motifs that are common amongst the different clades of acyl-CoA ligases21,22. Adenylation chemistry is prevalent throughout biology, and a recent classification of adenylating enzymes superfamily defines three classes: class I, which includes adenylation domains from nonribosomal peptide synthetases (NRPS) and acyl- and aryl-CoA ligases; class II, which encompasses the aminoacyl-tRNA synthetases (aaRSs); and class III, which includes the siderophore synthetases22. Biochemical analysis of reaction intermediates demonstrated that recombinant B. subtilis BioW can hydrolyze noncognate substrates20, an activity that is similar to pre-transfer proofreading observed in aaRSs that define class II members of the superfamily.
Here we report several crystal structures of a functionally active BioW from Aquifex aeolicus. These structures reveal a previously uncharacterized fold for an acyl-CoA ligase. The enzyme demonstrates a strict specificity for pimelic acid and has the ability to proofread and hydrolyze noncognate acyl-adenylates. Biochemical characterization of structure-based variants identifies the residues that establish both the strict specificity for the correct length dicarboxylic acid, as well as dictate the proofreading of noncognate acyl-adenylates.
RESULTS
Identification and characterization of BioW homologs
As primary sequence analysis failed to identify obvious structural homologs, we used the Enzyme Similarity Tool from the Enzyme Function Initiative (EFI-EST)23 to analyze the relationship among different sequences in the BioW superfamily. An alignment score corresponding to a cutoff of at least 60% identity in sequence prouced a total of 2,857 edges in the similarity network. We cloned representative BioW homologs from different genera and screened each for crystallization. Crystals could be readily obtained for A. aeolicus (hereafter called AaBioW) and Bacillus amyloliquefaciens (hereafter called BaBioW). We determined the kinetic parameters for AaBioW by measuring the rate of AMP production using anaytical HPLC (Fig. 2a,b). As A. aeolicus is a thermophilic organism, all of the kinetic experiments were carried out at 323 K, a suitable temperature below the natural growth environment of the organism (358 K) in which formation of nonenzymatic side products was negligible. The Km of AaBioW for pimelate is 10.7 ± 0.96 μM with a kcat value of 7.45 ± 0.023 × 10−1 s−1. The Km value is similar to that obtained for the well-characterized BioW from B. subtilis, the kinetic parameters for which were determined using TLC analyses20. By contrast, the kcat value for AaBioW is nearly four orders of magnitude greater than that of the Bacillus enzyme (kcat = 8.4 ± 1.3 × 10−5 s−1). Attempts to determine the kinetic parameters for BaBioW were hampered by a propensity of the enzyme to precipitate and by the loss of activity upon prolonged incubation.
Figure 2 |. Biochemical activity of AaBioW.
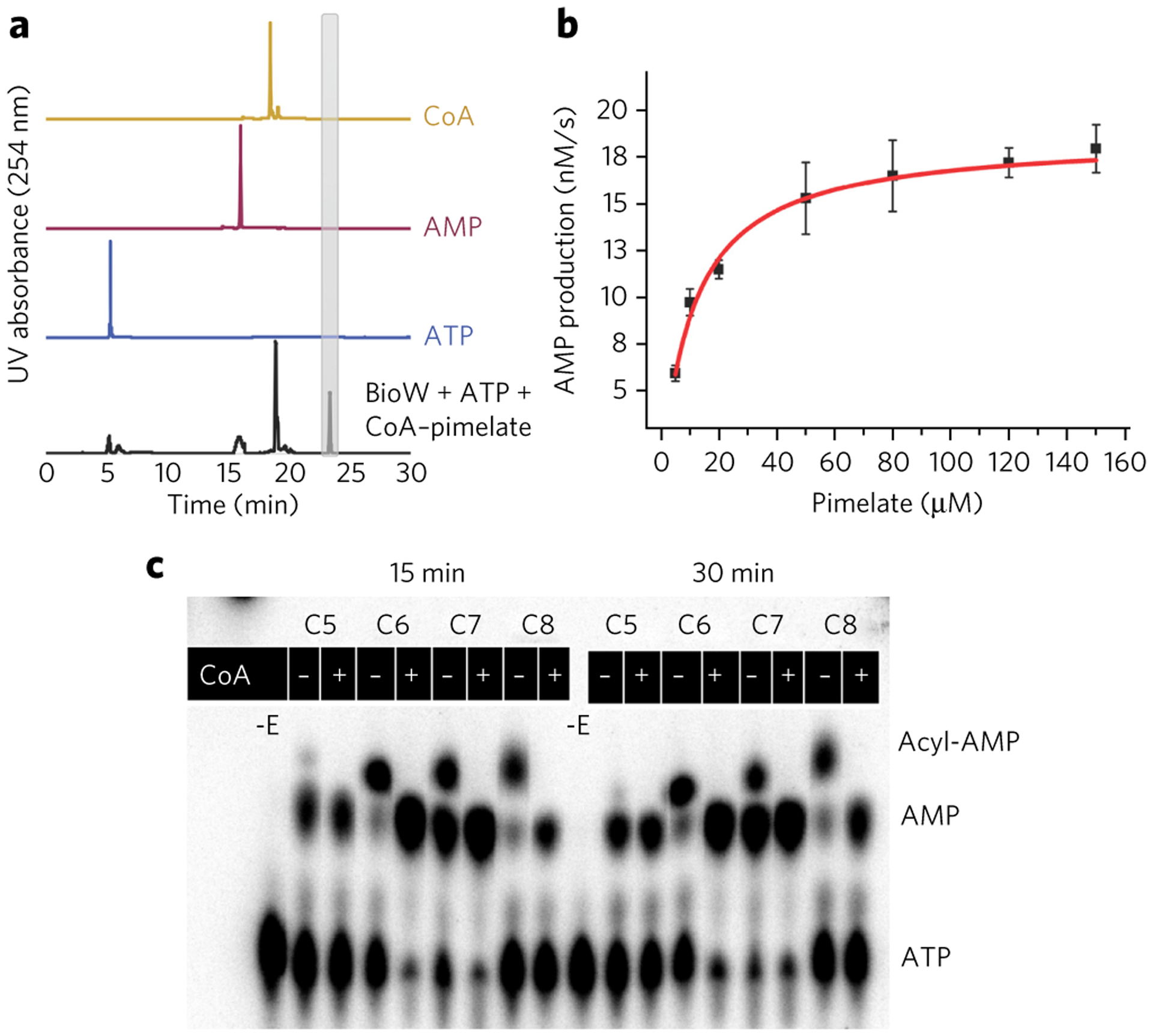
(a) Formation of pimeloyl-CoA by AaBioW analyzed by HPLC analysis (black trace). The elution profiles for the isolated standards are shown in the lines above. (b) Michaelis–Menten curve obtained by measuring the AMP production over varying concentrations of pimelate with fixed concentrations of ATP (0.4 mM) and CoA (0.3 mM). Error bars represent mean ± s.d. Measurements were conducted in triplicate. (c) The proofreading activity of AaBioW was monitored using a range of dicarboxylic acids, including the cognate (C7) pimelate. –E, reactions that lacked enzyme.
The overall ligase reaction catalyzed by BioW consists of two half-reactions, namely, ATP-dependent adenylation of pimelate followed by acyl transfer to CoA. We sought to characterize the interaction between AaBioW and each of the two substrates (ATP and CoA) using isothermal titration calorimetry (Supplementary Results, Supplementary Fig. 1). Experiments were conducted in duplicate, and the results are the means of both experiments. Each of the ligands bound to the enzyme with a stoichiometry of one and with respective dissociation constant (Kd) values of 13 ± 3 μM for ATP and 14 ± 2.5 μM for CoA. The calorimetric data support the biochemical results and demonstrate direct binding of each ligand by the enzyme.
All known adenylate-forming enzymes that have been characterized are metal dependent, with a preference for Mg2+ (ref. 22). Typically, the metal ion coordinates to the phosphate groups of ATP, acts to stabilize charges that develop in the transition state, and then facilitates release of the pyrophosphate product. We tested the Mg2+ requirement for AaBioW by measuring total acyl-CoA production in end-point experiments at various concentrations of the metal ion (Supplementary Fig. 2). In reactions carried out with 1 mM EDTA in the absence of Mg2+, no consumption of ATP can be observed. The production of both AMP and the cognate pimeloyl-CoA varied in a linear fashion with increasing concentrations of exogenous Mg2+ and saturated above stoichiometric concentrations of the metal. These data demonstrate that, like B. subtilis BioW20, the adenyl transfer reaction catalyzed by AaBioW is Mg2+ dependent.
In addition to catalyzing the synthesis of pimeloyl-CoA, B. subtilis BioW is demonstrated to catalyze the hydrolysis of noncognate substrates (proofreading)20. We tested the proofreading activity of AaBioW against a panel of α,ω-dicarboxylate substrates including glutarate (C5, 3), adipate (C6, 4), and suberate (C8, 5) using TLC (Fig. 2c). In the absence of CoA, adenylated intermediates can be observed for the cognate C7, as well as for the noncognate C6 and C8 substrates. However, while addition of CoA to the pimeloyl (C7)-adenylate produced the corresponding pimeloyl-CoA, its addition to the noncognate C6 and C8 adenylates simply resulted in hydrolysis to yield AMP and the free acids (Fig. 2c and Supplementary Fig. 3). By contrast, reactions carried out using glutarate in the absence of CoA resulted only in the accumulation of AMP, suggesting that AaBioW can catalyze the cleavage of glutaryl-adenylate, as was previously established for B. subtilis BioW. Likewise, increase in the pH of the reaction buffer resulted in the expected hydrolysis of the cognate pimeloyl-adenylate, but had minimal effect on the hydrolysis of the noncognate glutaryl-adenylate (Supplementary Fig. 3). These data are consistent with the prior observations for B. subtilis BioW20 that AMP production from the corresponding noncognate adenylates is a function of proofreading by AaBioW and not the result of nonenzymatic solvent hydrolysis.
Crystal structures of BioW
We determined the crystal structures of BaBioW and AaBioW at 3.1 Å and 2.5 Å resolutions, respectively. Relevant data collection and refinement statistics are reported in Supplementary Table 2. The structures of the two homologs are architecturally similar, and the overall fold consists of an N-terminal domain of roughly 55 residues that contain three anti-parallel β-strands with a single intervening α-helix and a C-terminal domain of roughly 200 residues that constitutes a α/β domain (Fig. 3a). A DALI search24 against the Protein Data Bank25 fails to identify any structural homologs of the entire polypeptide. Consequently, the overall topology and structure of BioW represents a new protein fold with no resemblance to those of known adenylating enzymes Supplementary Fig. 4).
Figure 3 |. Crystal structures of AaBioW and ligand complexes.
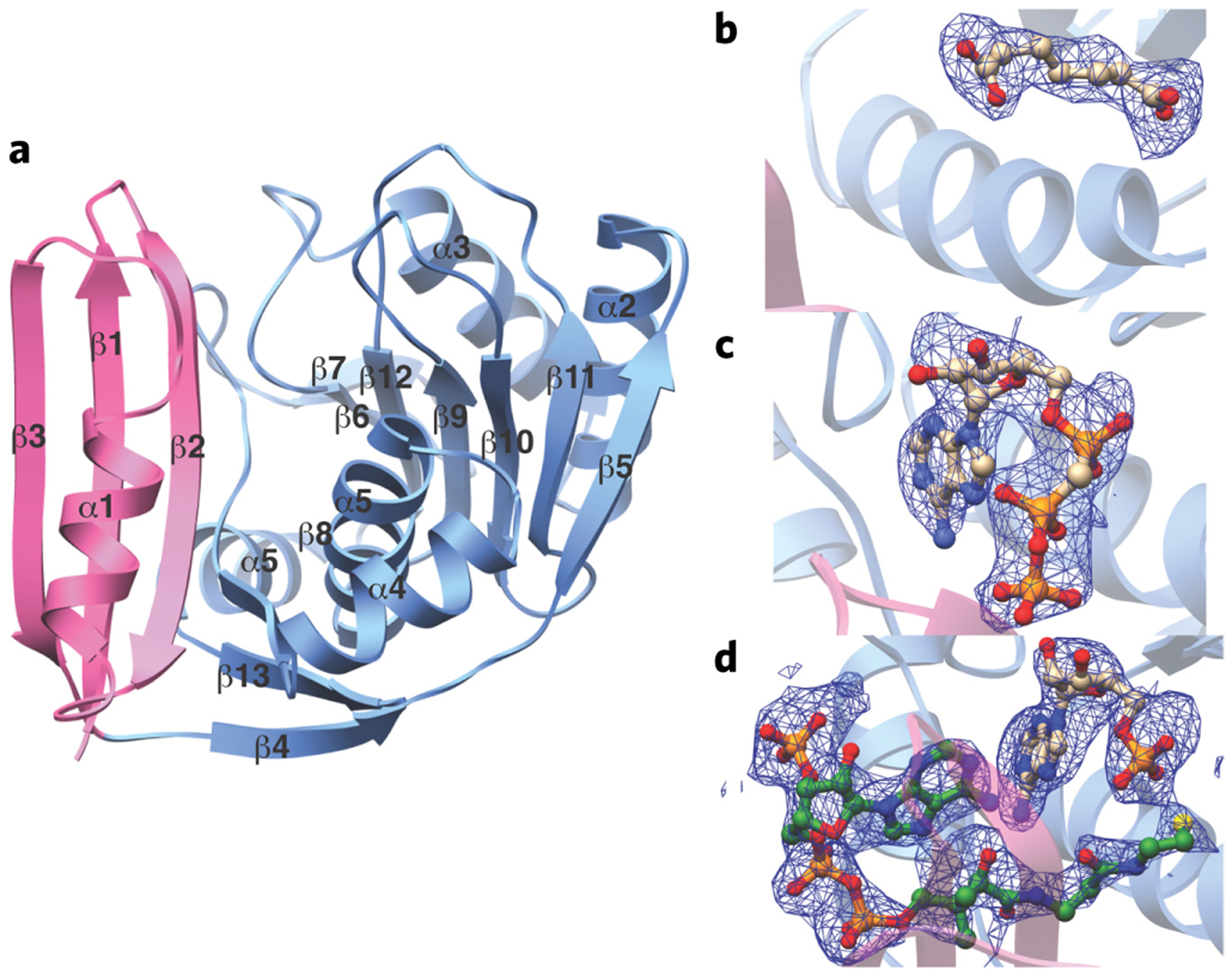
(a) Ribbon diagram of AaBioW illustrating the orientation of the PII domain (in pink) and the Rossmann fold domain (in light blue). Secondary structural elements are demarcated. (b–d) Simulated annealing difference Fourier maps (Fo – Fc) of AaBioW complexes contoured to 2.5 σ (blue) showing the bound pimelate (b), AMP–CPP–Mg2+ (c), and AMP–CoA (d). The coordinates for ligand were omitted before map calculations. The coordinates of the complexes are superimposed and the ligands are shown in ball-and-stick representation.
Structure-based similarity searches using the isolated domains of the protein revealed that the N terminus resembles the PII proteins that modulate bacterial signaling pathways via interactions with a wide-range of protein targets26 (50–60 Cα aligned with Z-scores around 5.0) (Supplementary Fig. 5a,b). The physiological role of PII proteins involves regulation of cellular nitrogen levels. The homotrimeric PII proteins are modulated by binding to ligand effectors, most notably nucleotides such as ATP and ADP27. More distant topological relationships can also be discerned with the B domains of ATP-grasp enzymes, such as l-amino acid ligases (43 Cα aligned with a Z-score of 3.2). The B domain is positioned away from the main body of ATP-grasp enzymes and undergoes conformational movements to encapsulate the active site upon binding of the nucleotide28.
The BioW C-terminal domain resembles a modified Rossmann fold, a structural motif that is commonly found in nucleotide-binding proteins29. However, the connectivity of the secondary structural elements of this domain in BioW is markedly different from that of the alternating β–α–β motif found in canonical Rossmann fold enzymes (Supplementary Fig. 4) such as protein kinases and class II aaRSs30,31. The closest structural homolog of this domain is the amino-terminal nucleotide-binding domain of the di-adenylate cyclase DisA (PDB Code 3C23; Z-score of 4.1; r.m.s. deviation of 3.7 Å over 102 aligned Cα atoms) (Supplementary Fig. 5c)32. This enzyme is a homotetramer that orients bound ATP from each protomer in an antiparallel fashion to facilitate the synthesis of bis-(3′,5′)-cyclic dimeric adenosine monophosphate (c-di-AMP), and this activity is coordinated with the binding of a branched DNA substrate by the C-terminal domain. The location of the bound nucleotide in AaBioW is considerably different from that found in DisA (Supplementary Fig. 5a,c), as expected, given their different topologies and overall reactions catalyzed by each enzyme. A more distant structural conservation is observed with (3′,5′)-cyclic phosphodiesterases (PDB Code 3DBA; Z-score of 3.0; r.m.s. deviation of 4.0 Å over 88 aligned Cα atoms)33, for which the nucleotide-binding site is entirely different from that in BioW. There are also some structural similarities with the ‘little finger’/polymerase-associated domain (LF/PAD) unique to the Y-family of translesion DNA polymerases. The LF/PAD domain is flexible and interacts with the DNA duplex upstream from a mismatch to help guide the template strand into the polymerase active site34.
Characterization of the substrate-binding sites in AaBioW
Individual co-crystal structures of AaBioW with substrate pimelate (2.45 Å), the nonhydrolyzable analog AMP–CPP–Mg2+–pimelate (2.55 Å), and AMP–CoA (2.25 Å) allowed elucidation of the active sites of the enzyme (Fig. 3b–d). The AaBioW–AMP–oA and AMP-CPP-Mg2+–pimelate structures show unambiguous density corresponding to the nucleotide in the C-terminal Rossmann fold domain that is flanked on the opposite side by the N-terminal PII-like domain. In both structures, the adenine moiety is bracketed between two Arg residues (Arg135 and Arg215) mainly through van der Waals contacts rather than the more common cation–π stacking interaction (Figs. 3c,d and 4a). These residues, along with a third Arg113, and two aliphatic residues (Gly114 and Leu166) delineate the binding pocket for the nucleobase. There are minimal hydrogen bonding interactions with the enzyme, and such interactions are only through main chain atoms, namely between N6 and N1 of adenine and the backbone amide and carbonyl oxygen, respectively, of Thr136. Similarly, the O2 atom of the ribose is within hydrogen bonding distance of the backbone carbonyls of Met112 and Gly114, and the O3′ atom is engaged by the side chain of Asp183. The α-phosphate oxygen is engaged via interactions with Ser182, Asp183, and Arg215, with the side chain Arg159 located ~3.4 Å away. In the AMP–CPP–Mg2+–pimelate structure, one of the oxygen atoms on the β-phosphate is within hydrogen bonding distance to Asp183, and the oxygen atoms of the γ phosphate are proximal to, but beyond hydrogen bonding distance from, His16, Arg39, and Arg135. A single Mg2+ ion positioned at the β-phosphate of the nucleotide is additionally coordinated by Asp183 and is located ~3.3 Å away from the α-phosphate. Notably, the nucleotide-binding site lacks all of the canonical features of representative adenyltransferases, such as stacking interactions that stabilize the nucleobase and carboxylate residues that are poised to facilitate metal-assisted pyrophosphate hydrolysis. Lastly, although some ATP-grasp enzymes contain a multidomain architecture consisting of an A domain that bears a Rossmann fold and a B domain that superficially resembles a PII-like fold28, the architecture of BioW is distinct, as is the nature of the nucleotide-binding site.
Figure 4 |. Structure-based mutational analysis of the AaBioW active site.
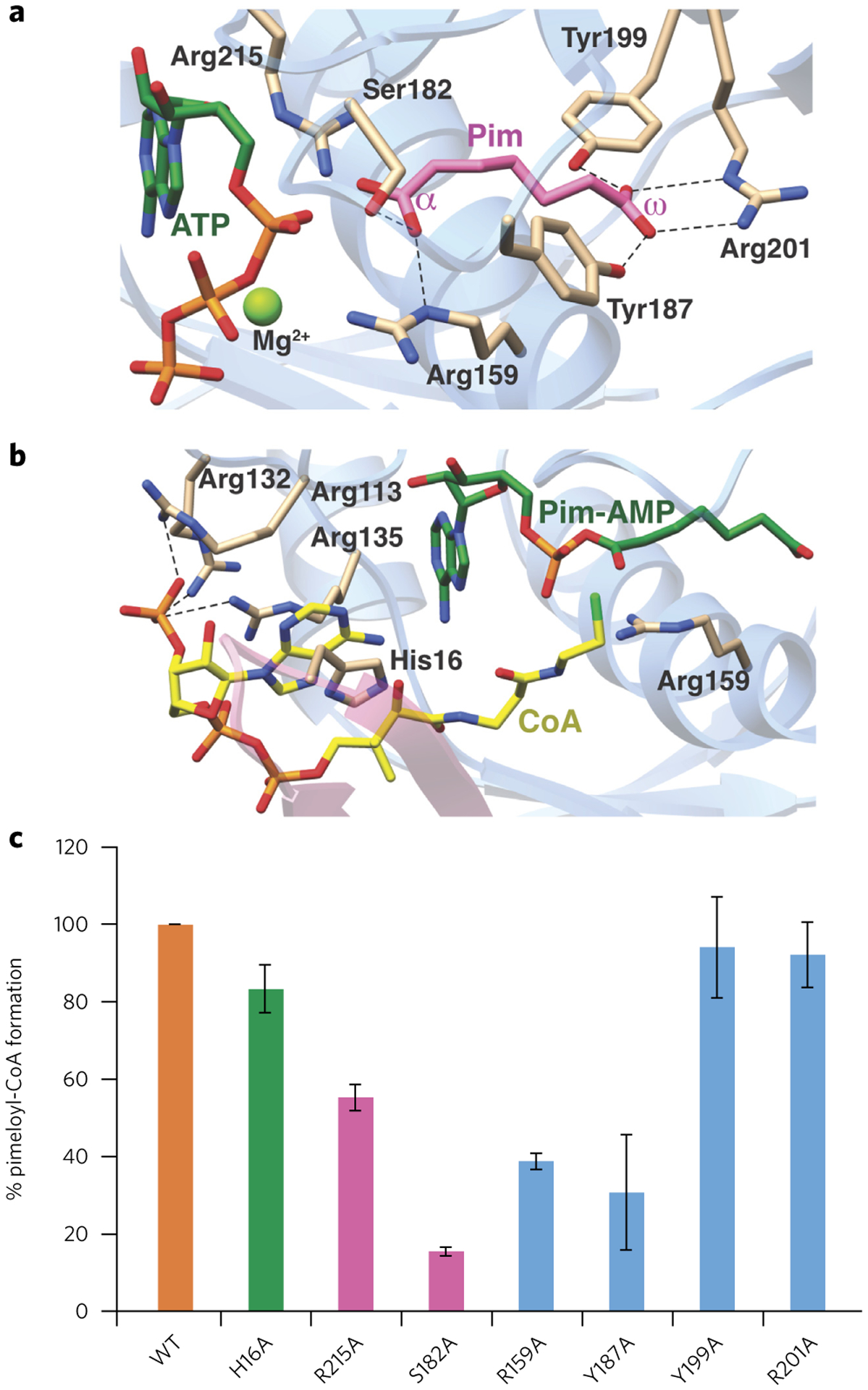
(a,b) Model of the AaBioW active site structure during the adenylation (a) and thioester (b) formation steps, generated by superimposing the crystal structures of relevant ligand-bond complexes. Green sticks, nucleotide; purple, pimelate (Pim); yellow, CoA. Active site residues that may play a role in catalysis are shown as tan sticks. (c) Biochemical activities of site-specific variants of active site residues in AaBioW are identified in a and b. The efficiency of each variant is measured as the amount of pimeloyl-CoA formed relative to the wild-type enzyme. Experiments in c were conducted in triplicate. Error bars represent mean ± s.d.
The binding site for pimelate could be readily discerned in electron density maps derived from the AaBioW–pimelate and AMP–CPP–Mg2+–pimelate co-crystal structures. Bifurcated density, characteristic of the α- and ω-carboxylates, situates the bound pimelate at a location adjacent to the nucleotide-binding site, where we designate the α-carboxylate as that closest to the phosphate of AMP (Fig. 3b,c). Both of the substrate carboxylates are engaged through numerous interactions with polar residues, and a hydrophobic environment that is established by Ala163, Tyr187, and Tyr191 encapsulate the methylene carbons of the substrate. The α-carboxylate is poised to interact with the side chains of Arg159, Ser182, and Arg215, whereas the side chains of Tyr187, Tyr199, and Arg201 engage the ω-carboxylate (Fig. 4b). Most notably, the orientation of the side chain of Arg159 precisely matches the length of the pimelate and may serve as a ‘ruler’ to set the length of the correct substrate. The orientation of Arg159 also sequesters the ligand away from bulk solvent.
The structure of AaBioW in complex with AMP–CoA establishes the location of the CoA binding site (Fig. 3d). The ligand is positioned almost exclusively within the N-terminal PII-like domain, an unexpected finding given that CoA binding is not a function that has been previously attributed to PII-like domains. The adenosine 3′-phosphate is accommodated by minor rearrangements of side chains. The CoA adenine is engaged via π-stacking interactions with the side chain of His16, and the 3′-phosphate is positioned to interact with the side chains of Arg113, Arg132, and Arg135 (Fig. 4b). Side chain rotations of all of these residues are necessary in order to accommodate the binding of CoA (Supplementary Fig. 6). Numerous basic residues including Arg7, Arg9, and Lys49 engage the CoA diphosphate. The phosphopantetheine moiety runs along a groove between the N- and C-terminal domains, and the thiol is positioned in proximity to the nucleotide- and pimelate-binding regions of the active site. All of the residues involved in interactions with nucleotide, CoA, and pimelate are conserved across BioW sequences (Supplementary Fig. 7).
Mutational analysis of the function of active site residues
A superposition of all of the ligand-bound structures yields plausible models for Michaelis complexes for the two half-reactions (adenylation and thioester formation) (Fig. 4a,b). These models suggest that each of the substrates is bound in the enzyme active site with the optimal orientation to facilitate catalysis. In the adenylation complex model, the α-phosphate of ATP is oriented roughly 3.6 Å away from the α-carboxylate of pimelate, and minor movement of either substrate would be necessary to facilitate the attack of the weakly nucleophilic carboxylate on the weakly electrophilic phosphate (Fig. 4a). The β- and γ-phosphates of ATP are positioned in the region between the PII and Rossmann fold domains to allow displacement to bulk solvent upon adenyl transfer. The function of residues that interact with ATP and pimelate were probed through site-specific variants (Fig. 4c; Supplementary Fig. 8). End-point analyses demonstrate that the Y199A and R201A mutations have little effect on product formation, consistent with the role for these residues in simply anchoring the ω-carboxylate of the substrate pimelate. By contrast, the R159A and Y187A variants demonstrate a notable reduction in turnover, which is in line with the function of these residues in forming the exterior wall of the pimelate-binding cavity. Lastly, both Ser182 and Arg215 are situated immediately adjacent to the location of the incipient phosphoester bond, where they are poised to stabilize the intermediate formed during the in-line attack of the pimelate oxygen on the α-phosphorus atom of ATP in the adenylation reaction. Consequently, the S182A and R215A each demonstrate a substantial reduction in product formation.
The hypothetical model for the pre-catalytic thioester-forming complex shows a paucity of residues that could participate in the chemistry, consistent with the energetic favorability of this half-reaction (Fig. 4b). In the model, the thiol–sulfur atom is positioned at a distance suitable to favor nucleophilic attack on the carbon atom of the adenylate anhydride. Although His16 forms a π-stacking interaction with the adenine, the H16A variant displays only a modest 20% loss in activity relative to the wild type, reflecting the importance of these other interacting residues in stabilizing CoA binding. A slight 2.5 Å movement of Arg159 in the thioester-forming model positions this residue to stabilize the negative charge that would develop on the oxygen atom of the α-carboxylate upon attack by the thiol. Similar movements of amino acid side chains occur throughout the CoA binding site, notably at Arg7, Arg113, Arg132, and Arg135 (Supplementary Fig. 6). These changes serve to both avoid steric clashes upon ligand binding and facilitate favorable interactions with polar groups in CoA. In the adenylation conformation, formation of the adenylate would presumably be protected from solvent hydrolysis by these Arg residues, which would then be displaced upon CoA binding. An interesting discovery from the mutational analysis is the identification of an R132A variant that demonstrates far more robust activity than the wild-type enzyme. Inspection of the product distribution of this variant demonstrates a notable increase in production of both pimeloyl-CoA and AMP relative to the wild-type AaBioW (Supplementary Fig. 9). In the AMP–CoA crystal structure, Arg132 engages the adenosine 3′-phosphate of CoA.
Crystal structures of AaBioW with bound substrate reveal that the enzyme engages both carboxylates of the substrate through numerous interactions, using the side chain of Arg159 to establish the distance between the two carboxylates in the cognate pimelate substrate. Hence, it seemed plausible that single-residue variants of AaBioW at residues that engage either of the two carboxylates in the substrate might utilize either monocarboxylate or dicarboxylate substrates of varying length. To test this hypothesis, we generated Ala mutants at Arg159 and Arg215 (which engage the α-carboxylate) and at Arg201 (which engages the ω-carboxylate) and tested these variants against a panel of mono- and dicarboxylate substrates (Supplementary Figs. 10,11). However, none of these variants demonstrated any activity against any substrate other than pimelate. Given that adipic acid (4) does not occur naturally, it is not surprising that BioW does not show ligase activity for the molecule. However, both glutaric acid (3), and suberic acid (5) occur naturally and are byproducts of amino acid metabolism and oxidation of castor oil, respectively. The lack of activity against these other dicarboxylates suggests that AaBioW has been adapted to utilize only the C7 dicarboxylate for CoA ligation. This is an important property, because homologs having shortened or extended valerate moieties cannot replace biotin35.
A gatekeeper residue involved in proofreading
An unusual property of B. subtilis BioW is the ability of this enzyme to catalyze hydrolytic cleavage of acyl-adenylates other than the cognate pimeloyl (C7)-adenylate20, as we have also demonstrated with AaBioW for the noncognate glutaryl (C5)-adenylate. A comparison of the AaBioW co-crystal structures provides some hints regarding residues that may facilitate this proofreading activity (Supplementary Fig. 6). The movement of Arg159, which serves to position this residue to assist in thioester formation, is reminiscent of the domain movement in acetyl-CoA synthetase that positions a catalytic lysine important for adenylation away from the active site to facilitate thioester formation36. A surface rendering of the thioester formation state illustrates the position of Arg159 directly above the α-phosphate, consistent with a role for this residue in protecting the cognate pimeloyl-adenylate (Supplementary Fig. 12). Conceivably, misalignment of the Arg159 side chain upon formation of complexes with noncognate adenylates could facilitate hydrolytic cleavage, perhaps through electronic effects. To decipher any roles that this residue may play in proofreading, we carried out HPLC analysis of R159A AaBioW using cognate pimelate, as well as non-cognate di- and mono-acids as substrates. The R159A variant demonstrated the ability to generate the CoA thioester using pimelate as a substrate, albeit at lower concentrations than that of the wild-type enzyme (Fig. 5a). However, while the variant enzyme can generate the corresponding adenylates using any of the noncognate mono- or di-acid substrates, the acyl-adenylates are no longer hydrolyzed or converted into the corresponding thioesters in the presence of CoA (Fig. 5b–e). Hence, R159A AaBioW lacks the ability to proofread, which strongly suggests a role for Arg159 in this activity.
Figure 5 |. Proofreading activity of the wild-type and R159A AaBioWs.
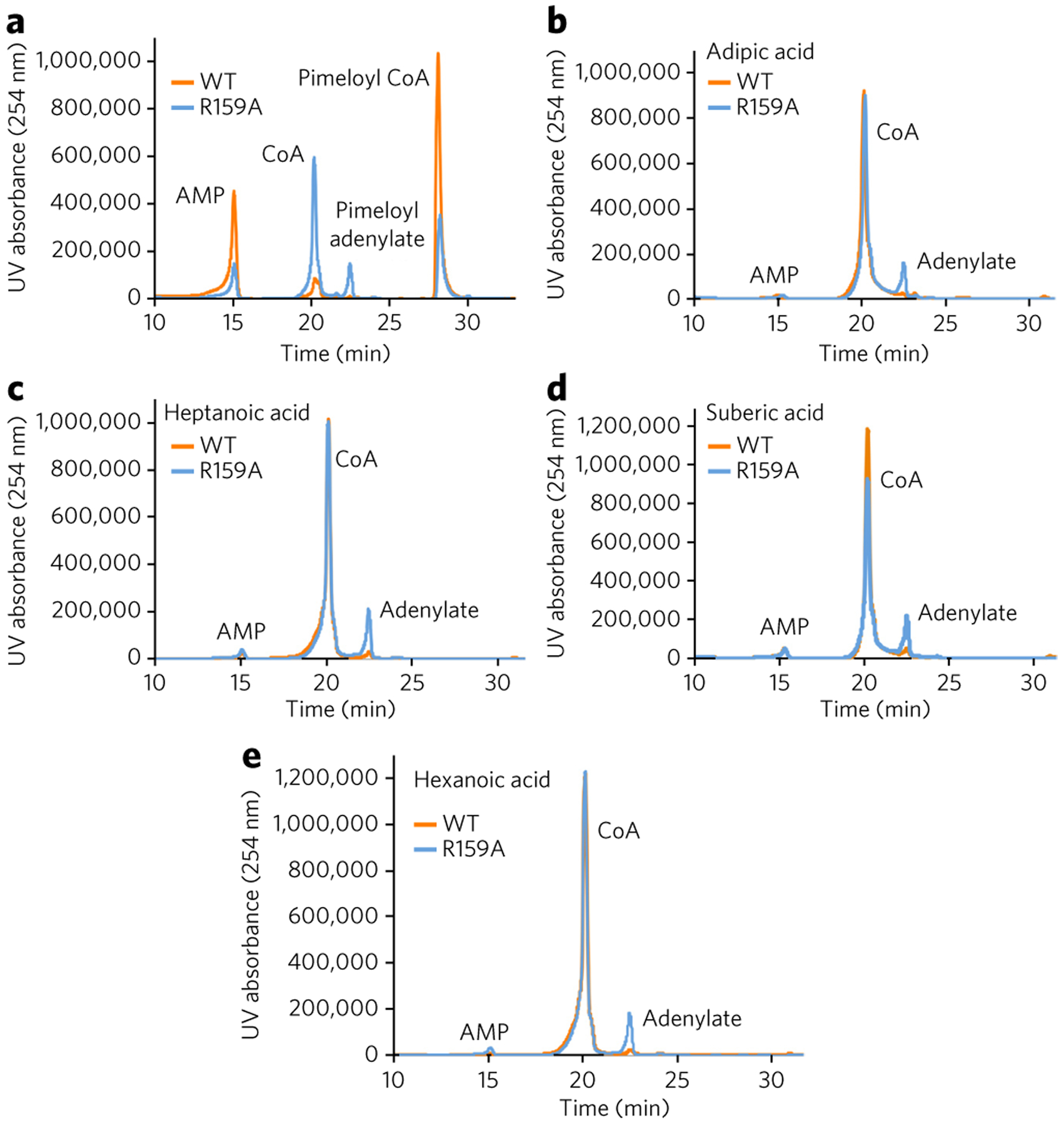
(a) Although the R159A variant can no longer proofread, the enzyme still retains ligase activity and can catalyze the formation of pimeloyl-CoA. (b–e) HPLC traces (absorbance at 254 nm for the AaBioW R159A mutant with different mono- and di-acid substrates: adipic acid (4; b), heptanoic acid (6; c), suberic acid (5; d), and hexanoic acid (7; e). Experiments were conducted in triplicate; data shown are representative of one to three measurements.
DISCUSSION
Our biochemical and structural analyses of BioW expands upon the growing superfamily of adenylating enzymes22. Although structurally distinct, BioW employs catalytic strategies that are congruous with those of other adenylating enzymes, including the use of a two-step reaction scheme to activate the substrate by coupling it to AMP, after which the adenylate undergoes attack by a nucleophile. All known adenylation enzymes contain one or more basic residues located adjacent to the α-phosphate of ATP; class I enzymes contain an invariant Lys (Lys517 in gramicidin synthetase S37; Lys529 in firefly luciferase38) located in motif A10 within this subfamily. A highly conserved Thr (Thr190 in gramicidin synthetase S) is also located near the α-phosphate in class I members. Similarly, both class II and III enzymes employ a conserved Arg. This Arg is found in motif 2 of tRNA synthetases (Arg262 in lysine tRNA synthetase)39 in class II members and the conserved Arg305 in the class III NRPS-independent siderophore synthetase AcsD40. Along with the catalytically requisite divalent metal ion, these positively charged residues both enhance the electrophilicity of the α-phosphorus and track the accumulation of negative charge through the pentavalent transition state to the pyrophosphate product. In AaBioW, Arg215 is poised to play an equivalent role in catalysis, and Ser182 likely serves a role equivalent to the conserved Thr in class I members. We show that single-residue mutations of these residues compromise, but do not eliminate, catalytic activity, and this may reflect the overlapping roles that these two residues, along with the Mg2+ ion, play in catalysis.
A superposition of the various co-crystal structures illustrates that many of the active site Arg residues move in the thioester-forming state (Supplementary Fig. 6), and this movement is necessary for binding of CoA. Side chain movement may explain, in part, the observed greater activity of the Arg132→Ala variant, as Arg132 buttresses the position of Arg113 and Arg135. Movement of the latter two residues could only occur after Arg132 is shifted out of the way, and consequently, the Arg132→Ala variant may provide a more expedient means of accommodating the necessary shifts in Arg113 and Arg135. Although Arg132 engages the adenine 3′-phosphate of CoA, multiple other residues are also involved in interactions with the ligand. Biotin synthetic enzymes are generally poor catalysts9 given the low physiological demand from biotin, and, consequently, there would be no evolutionary pressure to produce a more efficient BioW. Hence, given the role in ligand binding, Arg132 is preserved amongst BioW enzymes, as mutations that may confer an increase in catalytic efficiency do not yield a biosynthetic competitive advantage.
These data provide some insights into a possible evolutionary role of this catalyst with respect to class I enzymes that carry out the adenylation of fatty acid substrates. Massive domain movements occur during the transition from the adenylation to the transesterification half reactions in the class I catalysts36. By contrast, the various co-crystal structures of AaBioW show no change in the overall structure of the enzyme throughout the presumed catalytic cycle other than movements of the side chains of various Arg residues for optimal contact with the respective substrates. It has been proposed that the domain alteration strategy used by class I adenylating enzymes was necessary, as simple fatty acid substrates lack any additional functional groups that could be used to properly position the substrate during the adenylation reaction21. In such enzymes, substrate positioning during adenylation occurs through the carboxylate, and domain movement would be necessary to allow access of the CoA thiol to the adenylate during the thioesterification step. By contrast, as the substrates for class II and class III adenylating enzymes contain additional functional groups (i.e., polar side chains and/or the α-amine of amino acids for class II members such as aaRSs or additional carboxylates for class III NRPS-independent siderophore synthetases). Positioning of the reactive carboxylate for adenylation can be achieved through interactions with these other groups, eliminating the need for a domain alteration strategy. The co-crystal structures of AaBioW presented here reinforce this hypothesis, as the ω-carboxylate provides an additional chemical handle for substrate positioning.
Biochemical studies of AaBioW recapitulate the unexpected observation that the enzyme can hydrolyze adenylates of noncognate dicarboxylates, and this proofreading activity was previously observed for B. subtilis BioW20. As proofreading of the misactivated dicarboxylate occurs after the formation of the adenylate, the activity is analogous to the pre-transfer proofreading activities observed in aaRSs. Our studies demonstrate that this activity does not require a second active site, as is the case with several aaRSs, where proofreading activity occurs in a region of the polypeptide distinct from where adenylation is catalyzed41. However, proofreading activities are also observed in aaRSs that lack obvious proofreading domains, including MetRS42, LysRS-II43, SerRS44, and yeast mitochondrial ThrRS45, each of which use unique strategies to avoid misactivation. The proofreading activity of BioW is most comparable to that observed in SerRS44 and mitochondrial ThrRS46, wherein the enzyme active site can directly catalyze the hydrolysis of misactivated aminoacyl-adenylate. In the case of BioW, pH stability experiments demonstrate that the noncognate adenylates are not as easily hydrolyzed as cognate adenylates (Supplementary Fig. 3), and such differences may facilitate the proofreading activity of AaBioW. Analysis of the R159A variant is consistent with at least a partial role for this residue in proofreading, perhaps through electronic or substrate orientation effects.
Prior sequence-based searches against various databases failed to identify any recognizable motif in B. subtilis BioW20, which is surprising considering that the enzyme uses two cofactors (CoA and ATP) for which there is extensive literature on structure-based classifications of binding motifs47. In light of the small size of the protein, it is surprising that multiple activities, including the capability of proofreading misadenylated substrates, can be encoded within the BioW fold. Nature has achieved this feat through judicious use of the flexibility of multiple Arg residues to provide the necessary interactions to facilitate productive catalysis. The strategy is successful only because the chemical makeup of the substrate, which provides unconstrained flexibility, along with dual carboxylate functional groups as chemical handles offer a straightforward means to achieve an otherwise elaborate catalytic scheme.
ONLINE METHODS
Protein expression and purification.
Oligonucleotides were purchased from Integrated DNA Technologies Inc. (Coralville, IA). Molecular biology reagents were purchased from New England BioLabs (Ipswich, MA), Thermo Fisher Scientific (Waltham, MA), or Gold Biotechnology Inc. (St. Louis, MO). Chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The bioW genes of A. aeolicus and B. amyloliquefaciens were individually amplified from genomic DNA using primers based in the published gene sequence (Supplementary Table 1). The genes were each cloned into pET28a resulting in N-terminal His6 tag proteins. All mutants were cloned into pET-His6 Sumo TEV LIC cloning vector (2S-T). All sequences were verified using dideoxy sequencing by ACGT Inc. (Wheeling, IL).
Each of the clones was transformed into E. coli Rosetta2 (DE3) cells and plated on LB agar plates supplemented with chloramphenicol (0.025 mg/ml) and either kanamycin (0.05 mg/ml) or ampicillin (0.1 mg/ml). A single colony was used to inoculate 6 ml of LB broth supplemented with the appropriate antibiotics and grown for 12–15 h at 37 °C. The starter culture was used to inoculate 2 L of Luria–Bertani broth supplemented with both kanamycin and chloramphenicol for expression of wild-type proteins, and ampicillin and chloramphenicol for mutant proteins. Cultures were grown at 37 °C until the OD600 reached 0.6–0.8. Protein expression was induced by addition of IPTG (isopropyl β-d-thiogalactopyranoside) to a final concentration of 0.3 mM. The cells were further incubated with shaking at 18 °C for 18 h. Cells were harvested by centrifugation, resuspended in 20 mM Tris–HCl (pH 8.0), 500 mM NaCl, and 10% glycerol buffer, and lysed by sonication. The lysate was centrifuged and the supernatant was loaded into a 5 ml His-Trap nickel column. The column was washed with buffer containing 20 mM Tris–HCl (pH 8.0), 1 M NaCl, and 30 mM imidazole. An increasing imidazole concentration gradient was used to elute the protein. The protein was incubated overnight at 4 °C with thrombin or TEV protease to remove His×6 tag or Sumo, respectively. Size-exclusion chromatography (HiLoad 16/60 Superdex 200) was used to further purify the protein using a 20 mM Na-HEPES (pH 7.5) buffer with 100 mM KCl for A. aeolicus wild type and mutants or 500 mM KCl for B. amyloliquefaciens wild type and mutants. Selenomethionine-labeled (SeMet) protein was produced by repression of methionine biosynthesis48, and was purified as described above.
Kinetic analysis of wild-type AaBioW.
Kinetic parameters were obtained by measuring AMP production (peak area at 254 nm) during the reaction progress. The 60 μl reactions contained 20 mM Na-HEPES (pH 7.0), 100 mM NaCl, 2 mM MgCl2, 0.2 mM DTT, 0.3 mM CoA, 0.4 mM ATP and 27 nM BioW with varying concentrations of pimelate (5 μM to 150 μM). The reactions were run at 50 °C for 15, 30, 45 and 60 s, and were started by the addition of pimelate. Reactions were stopped by adding methanol to a final concentration of 50% and flash freezing in liquid nitrogen. Once thawed, precipitated protein was removed by centrifuging for 10 min and 100 μl of the reaction mixture was then injected into a HPLC (Shimadzu prominence LC 20A) equipped with a C18 analytical column. Solvent A was 50 mM ammonium acetate pH 5 and solvent B was 100% methanol. The elution method consisted of 0% B for 5 min, then a 35 min gradient from 0% to 70% B, followed by a 70% to 100% B gradient in 5 min, finishing with 100% B for 5 min for 10 min at a flow rate of 1 ml/min at room temperature. The area under the AMP peak was converted to concentration using an AMP calibration curve generated with known concentration of standard, using the same column and instrument. Calibration curve and reaction rates were obtained using Microsoft Excel. Rates were plotted against substrate concentration using Origin and fitted to the Michaelis–Menten equation. Rates were plotted as an average of two independent experiments.
End-point analysis of BioW mutants.
Reactions contained 20 mM Na-HEPES (pH 7.0), 100 mM NaCl, 2 mM MgCl2, 0.2 mM DTT, 1 mM CoA, 1 mM ATP, 5 μM protein, and 1 mM substrate. The reactions were incubated for 18 h at 37 °C and stopped by addition of methanol to a final concentration of 50%. Precipitated protein was removed by filtration using 10,000 Da molecular weight cutoff Amicon centrifugal filters before loading the reactions to a C18 analytical column connected to a Shimadzu HPLC. CoA-substrates and adenylated substrates were detected by measuring the absorbance at 254 nm. The percent product formation reported is the average of two independent experiments.
Isothermal titration calorimetry (ITC).
Calorimetric analysis was conducted at 25 °C using a VP-ITC microcalorimeter (Microcal Inc.). For binding experiments, 30 μM AaBioW in a buffer composed of 20 mM HEPES (pH 7.0), 100 mM KCl, and 900 μM MgCl2 was maintained in the cell. Either ATP or CoA (concentration of 900 μM) was dissolved into the buffer solution that was identical to that used for protein dialysis. The ligand was injected into the cell containing AaBioW in 28 consecutive injections (6 μl for ATP and 8 μl for CoA) at 420 s intervals. Reference cell power was set at 2.3 μcal/s and stirring speed was 310 r.p.m. Nonlinear regression with a single-site fitting model (MicroCal Origin) was applied for data analysis, and the thermodynamic parameters were calculated using the Gibbs free energy equation (ΔG = ΔH – TΔS) and the relationship ΔG = −RTlnKa. Results are means ± s.e.m. of duplicate experiments.
Sequence similarity network.
The sequence similarity network was created using the Enzyme Function Initiative Enzyme Similarity Tool, EFI-EST (http://efi.igb.illinois.edu/efi-est/index.php). The AaBioW sequence was used to generate the data set containing related sequences obtained from UniProtKB using an E-value of 1e−10. The initial data analysis was done with an alignment score of 25 (ref. 23). The final network was created with an alignment score of 83 and visualized with Cytoscape version 3.3.0.
Thin layer Chromatographic Assays.
All TLC experiments were conducted as described previously20 using an ATP concentration of 25 μM. The proofreading activity of AaBioW were tested at 50 °C with an enzyme concentration 0.15 mg/ml for 15 or 30 min. Experiments with the R159A mutant were run for 30 min with an enzyme concentration of 0.2 mg/ml. The reactions were spotted onto cellulose TLC plates followed by development and autoradiography5.
Crystallization of AaBioW and ligand complexes.
Initial crystallization conditions were determined by the sparse matrix sampling method using commercial screens. AaBioW and complexes with bound ligands were crystallized using various compositions of polyethylene glycols (average weight of either 3,350 or 4,000 Da) using the hanging-drop method. Briefly, following the size-exclusion chromatographic step, purified protein samples were concentrated to 8–10 mg/ml using 10,000 Da molecular weight cutoff Amicon centrifugal filters. Crystals grew within 5–7 d at 9 °C, and were transiently soaked in precipitant solution supplemented with 30% glycerol before vitrification by direct immersion in liquid nitrogen. Ligand bound crystals were grown using protein samples that were incubated with 2–5 mM of the appropriate ligand for 30 min before crystallization.
X-ray crystallographic data collection and structure determination.
Initial crystallographic phases were determined solved by single wavelength anomalous diffraction using anomalous scattering from selenium-substituted AaBioW–pimelate. A 21-fold redundant data set was collected at the selenium absorption edge to a limiting resolution of 2.45 Å (overall Rmerge = 0.09, I/σ (I) = 1.8 in the highest resolution shell) using a Mar 300 CCD detector (LS-CAT, Sector 21 ID-D, Advanced Photon Source, Argonne, IL, USA). Data were indexed and scaled using HKL2000 (ref. 49). Selenium sites were identified using HySS50, and subsequently refined in SHARP51 to yield an initial figure of merit of 0.31 at 2.5 Å resolution. The resultant electron density map was of exceptional quality and permitted automatically tracing of nearly the entire main chain using Parrot and Buccaneer52. The remainder of the model was fitted using COOT53 and further improved by rounds of refinement with REFMAC5 (ref. 54) and manual building until convergence. Cross-validation, using 5% of the data for the calculation of the free R factor55, was used throughout the model-building process in order to monitor building bias.
Crystallographic data for unliganded AaBioW, and other ligand-bound complexes were collected at LS-CAT using either a Mar 225 (Sector 21 ID-F) or Mar 300 (Sector 21 ID-G) detector. Data were indexed and scaled using either HKL2000 (ref. 49) or XDS56. Phases for these structures were calculated by molecular replacement50 using the final refined coordinates of the SeMet AaBioW–pimelate (without ligand or solvents) as a search probe. Each of the structures was built, refined and validated using the procedures detailed above. Ligands were built in COOT, and water molecules were added using the ARP/wARP57 solvent building software of the CCP4 suite. For each of the structures, the stereochemistry of the model was monitored throughout the course of refinement using Procheck58 and MolProbity59 for quality assurance.
Data availability.
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under the following accession codes: Native AaBioW (5TV5), SeMet AaBioW with pimelate (5TV6), AaBioW with AMP–CPP–Mg2+–pimelate (5TV8), and AaBioW with CoA–AMP (5TVA). All other data that support the findings of this study are contained within the published article (and its supplementary information files) or are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank the Mining Microbial Genomes theme at the Carl R. Woese Institute for Genomic Biology for access to the LC-MS equipment. We also thank K. Brister and the staff at the Life Sciences Collaborative Access Team (Sector 21) at the Argonne National Labs for facilitating data collection. This work was supported by NIH grant AI15650 (to J.C.).
Footnotes
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.McMahon RJ Biotin in metabolism and molecular biology. Annu. Rev. Nutr 22, 221–239 (2002). [DOI] [PubMed] [Google Scholar]
- 2.DeTitta GT, Edmonds JW, Stallings W & Donohue J Molecular structure of biotin. Results of two independent crystal structure investigations. J. Am. Chem. Soc 98, 1920–1926 (1976). [DOI] [PubMed] [Google Scholar]
- 3.Chapman-Smith A & Cronan JE Jr. Molecular biology of biotin attachment to proteins. J. Nutr 129 (Suppl. 1), 477S–484S (1999). [DOI] [PubMed] [Google Scholar]
- 4.Tong L Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci 70, 863–891 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Streit WR & Entcheva P Biotin in microbes, the genes involved in its biosynthesis, its biochemical role and perspectives for biotechnological production. Appl. Microbiol. Biotechnol 61, 21–31 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Eisenberg MA & Star C Synthesis of 7-oxo-8-aminopelargonic acid, a biotin vitamer, in cell-free extracts of Escherichia coli biotin auxotrophs. J. Bacteriol 96, 1291–1297 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenberg MA & Krell K Dethiobiotin synthesis from 7,8-diaminolargonic acid in cell-free extracts of a biotin auxotroph of Escherichia coli K-12. J. Biol. Chem 244, 5503–5509 (1969). [PubMed] [Google Scholar]
- 8.Eisenberg MA & Krell K Synthesis of desthiobiotin from 7,8-diaminopelargonic acid in biotin auxotrophs of Escherichia coli K-12. J. Bacteriol 98, 1227–1231 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S & Cronan JE Closing in on complete pathways of biotin biosynthesis. Mol. Biosyst 7, 1811–1821 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Cronan JE & Lin S Synthesis of the α,ω-dicarboxylic acid precursor of biotin by the canonical fatty acid biosynthetic pathway. Curr. Opin. Chem. Biol 15, 407–413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ifuku O et al. Origin of carbon atoms of biotin. 13C-NMR studies on biotin biosynthesis in Escherichia coli. Eur. J. Biochem 220, 585–591 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Sanyal I, Lee S-L & Flint DH Biosynthesis of pimeloyl-CoA, a biotin precursor in Escherichia coli, follows a modified fatty acid synthesis pathway: 13C-labeling studies. J. Am. Chem. Soc 116, 2637–2638 (1994). [Google Scholar]
- 13.Lin S, Hanson RE & Cronan JE Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat. Chem. Biol 6, 682–688 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal V, Lin S, Lukk T, Nair SK & Cronan JE Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc. Natl. Acad. Sci. USA 109, 17406–17411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bower S et al. Cloning, sequencing, and characterization of the Bacillus subtilis biotin biosynthetic operon. J. Bacteriol 178, 4122–4130 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cryle MJ & De Voss JJ Carbon-carbon bond cleavage by cytochrome p450(BioI)(CYP107H1). Chem. Commun. (Camb.) 1, 86–87 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Stok JE & De Voss J Expression, purification, and characterization of BioI: a carbon-carbon bond cleaving cytochrome P450 involved in biotin biosynthesis in Bacillus subtilis. Arch. Biochem. Biophys 384, 351–360 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Cryle MJ & Schlichting I Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc. Natl. Acad. Sci. USA 105, 15696–15701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ploux O, Soularue P, Marquet A, Gloeckler R & Lemoine Y Investigation of the first step of biotin biosynthesis in Bacillus sphaericus. Purification and characterization of the pimeloyl-CoA synthase, and uptake of pimelate. Biochem. J 287, 685–690 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manandhar M & Cronan JE Proofreading of noncognate acyl adenylates by an acyl-coenzyme a ligase. Chem. Biol 20, 1441–1446 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulick AM Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol 4, 811–827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmelz S & Naismith JH Adenylate-forming enzymes. Curr. Opin. Struct. Biol 19, 666–671 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerlt JA et al. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm L & Park J DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Berman HM et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osanai T & Tanaka K Keeping in touch with PII: PII-interacting proteins in unicellular cyanobacteria. Plant Cell Physiol. 48, 908–914 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Forchhammer K & Lüddecke J Sensory properties of the PII signalling protein family. FEBS J. 283, 425–437 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Fawaz MV, Topper ME & Firestine SM The ATP-grasp enzymes. Bioorg. Chem 39, 185–191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossmann MG & Argos P The taxonomy of binding sites in proteins. Mol. Cell. Biochem 21, 161–182 (1978). [DOI] [PubMed] [Google Scholar]
- 30.Cheek S, Zhang H & Grishin NV Sequence and structure classification of kinases. J. Mol. Biol 320, 855–881 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Moras D Structural and functional relationships between aminoacyl-tRNA synthetases. Trends Biochem. Sci 17, 159–164 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Witte G, Hartung S, Büttner K & Hopfner KP Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Martinez SE, Heikaus CC, Klevit RE & Beavo JA The structure of the GAF A domain from phosphodiesterase 6C reveals determinants of cGMP binding, a conserved binding surface, and a large cGMP-dependent conformational change. J. Biol. Chem 283, 25913–25919 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prakash S, Johnson RE & Prakash L Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem 74, 317–353 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Balcher MR & Lichstein HC Growth promotion and antibiotin effect of homobiotin and norbiotin. J. Bacteriol 58, 579–583 (1949). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gulick AM, Starai VJ, Horswill AR, Homick KM & Escalante-Semerena JC The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Conti E, Stachelhaus T, Marahiel MA & Brick P Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Branchini BR, Murtiashaw MH, Magyar RA & Anderson SM The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase. Biochemistry 39, 5433–5440 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Onesti S, Miller AD & Brick P The crystal structure of the lysyl-tRNA synthetase (LysU) from Escherichia coli. Structure 3, 163–176 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Schmelz S et al. AcsD catalyzes enantioselective citrate desymmetrization in siderophore biosynthesis. Nat. Chem. Biol 5, 174–182 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yadavalli SS & Ibba M Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv. Protein Chem. Struct. Biol 86, 1–43 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Fersht AR & Dingwall C An editing mechanism for the methionyl-tRNA synthetase in the selection of amino acids in protein synthesis. Biochemistry 18, 1250–1256 (1979). [DOI] [PubMed] [Google Scholar]
- 43.Jakubowski H Misacylation of tRNALys with noncognate amino acids by lysyl-tRNA synthetase. Biochemistry 38, 8088–8093 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Gruic-Sovulj I, Rokov-Plavec J & Weygand-Durasevic I Hydrolysis of non-cognate aminoacyl-adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett. 581, 5110–5114 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Ling J, Peterson KM, Simonovic I, Söll D & Simonovic M The mechanism of pre-transfer editing in yeast mitochondrial threonyl-tRNA synthetase. J. Biol. Chem 287, 28518–28525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minajigi A & Francklyn CS Aminoacyl transfer rate dictates choice of editing pathway in threonyl-tRNA synthetase. J. Biol. Chem 285, 23810–23817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinjo AR & Nakamura H Comprehensive structural classification of ligand-binding motifs in proteins. Structure 17, 234–246 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Walden H Selenium incorporation using recombinant techniques. Acta Crystallogr. D Biol. Crystallogr 66, 352–357 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Borek D, Majewski W & Minor W Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 (2003). [DOI] [PubMed] [Google Scholar]
- 50.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vonrhein C, Blanc E, Roversi P & Bricogne G Automated structure solution with autoSHARP. Methods Mol. Biol 364, 215–230 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Cowtan K The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA & Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Brünger AT Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 66, 133–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langer G, Cohen SX, Lamzin VS & Perrakis A Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R & Thornton JM AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under the following accession codes: Native AaBioW (5TV5), SeMet AaBioW with pimelate (5TV6), AaBioW with AMP–CPP–Mg2+–pimelate (5TV8), and AaBioW with CoA–AMP (5TVA). All other data that support the findings of this study are contained within the published article (and its supplementary information files) or are available from the corresponding author upon reasonable request.