

Abstract
Viruses are known to perturb host cellular metabolism to enable their replication and spread. However, little is known about the interactions between Zika virus (ZIKV) infection and host metabolism. Using primary human retinal vascular endothelial (HRvEC) cells and an established human endothelial cell line (HUVEC), we investigated the role of AMPK, a master regulator of energy metabolism, in response to ZIKV challenge. ZIKV infection caused a time-dependent reduction in the active phosphorylated state of AMPK, and of its downstream target, ACC. Pharmacological activation of AMPK using AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), metformin, and a specific AMPKα activator (GSK621) attenuated ZIKV replication. This activity was reversed by an AMPK inhibitor (Compound C). Lentivirus-mediated knockdown of AMPK and the use of AMPKα−/− mouse embryonic fibroblasts (MEFs) provided further evidence that AMPK has an antiviral effect on ZIKV replication. Consistent with its antiviral effect, AMPK activation potentiated the expression of genes with antiviral properties (e.g., IFNs, OAS2, ISG15, MX1) and inhibited inflammatory mediators (e.g., TNFα, CCL5). Bio-energetic analysis showed that ZIKV infection evokes a glycolytic response, as evidenced by elevated ECAR (extracellular acidification rate) levels and increased expression of key glycolytic genes (GLUT1, HK2, TPI, MCT4); activation of AMPK by AICAR treatment reduced this response. Consistent with this, 2-deoxyglucose (2DG), an inhibitor of glycolysis, augmented AMPK activity and attenuated ZIKV replication. Thus, our study demonstrates that the anti-ZIKV effect of AMPK signaling in endothelial cells is mediated by reduction of viral-induced glycolysis and enhanced innate antiviral responses.
INTRODUCTION
Zika virus (ZIKV) is a mosquito-transmitted flavivirus, which has spread in recent years from Africa and Asia to the Caribbean, Latin America, and parts of the United States (1, 2). It has rapidly emerged as an important pathogen that can cause significant neurologic morbidity (3, 4). Recent ZIKV epidemics in Brazil (5, 6) and currently in India (7) have posed significant challenges, not only in the health care setting but also for the economies of affected countries, including the United States (8). While ZIKV infection during pregnancy has been primarily associated with microcephaly, a devastating birth defect, the full spectrum of ZIKV-associated disease, and the magnitude of risk remain undefined (9–13). Given the limited data and the lack of longitudinal studies, microcephaly may represent only one possible adverse outcome among the spectrum of conditions associated with congenital Zika syndrome (10). More recently, ocular ZIKV infections have been described in several clinical reports (14–19). Ocular pathology may thus be an underreported effect of the virus. ZIKV-induced ocular complications could lead to significant visual impairment in affected infants (2), with long-term social and economic implications.
Although, in the last two years, there has been considerable effort to understand the pathobiology of ZIKV infection (20–23), our understanding of ZIKV ocular pathogenesis is still in its infancy (24, 25). Moreover, there are no effective antiviral therapies or vaccines for the treatment or prevention of ZIKV infections. The ongoing threat posed by ZIKV warrants the identification of novel therapeutic targets for the development of antivirals that can be used to treat ZIKV infections, including ocular infections (26). To define pathogenic mechanisms of the ocular tissue injuries caused by ZIKV, our laboratory has been examining host-pathogen interactions between ZIKV and retinal cells, the primary target of ZIKV in the eye (26–28). We reported that ZIKV causes chorioretinal atrophy in experimental animal models and that cells lining the blood-retinal barrier (BRB) are permissive to ZIKV infection (27). Consistent with our findings, several independent studies also demonstrated the susceptibility of the BRB to ZIKV infection, including the retinal endothelium (25, 29, 30). These studies make it clear that ZIKV must overcome the BRB to gain access to the eye and cause ocular damage (1).
To better understand the molecular mechanisms of ZIKV infection in retinal cells, we recently performed a transcriptome analysis of ZIKV-infected RPE cells, which form the outer BRB (26). We also performed a meta-analysis of transcription profiles of ZIKV-infected cells and those infected with other flaviviruses such as dengue virus. Our study showed that in addition to the induction of genes involved in classical innate inflammatory and antiviral response, ZIKV perturbed genes involved in cellular metabolism, including lipid and ceramide metabolism in infected cells. These changes are consistent with the general concept that viruses have substantial metabolic requirements associated with the induction of robust cellular antiviral responses, replication of viral nucleic acids, and production of viral proteins and lipids for virion production. In addition, formation of virus replication factories requires an extensive proliferation of endoplasmic reticulum-associated membranes (26, 31). The biogenesis of this membrane-bounded compartment is dependent on cellular lipid and energy metabolism pathways. Because we had already demonstrated an important role for AMP-activated kinase (AMPK) in regulating retinal innate immunity during bacterial infection (32) and AMPK is well-established as a master regulator of energy and lipid metabolism, in this work, we investigated its role in ZIKV infection.
AMPK is a heterotrimeric protein complex that serves as the primary cellular energy sensor (33). The AMPK complex consists of a catalytic alpha subunit and regulatory beta and gamma subunits. Activation of the complex is triggered by binding of AMP or ADP to the gamma subunit, leading to increased phosphorylation of the alpha subunit at threonine-172 (33–35). The important role of AMPK activity in the survival of stressed cells has been studied in the context of type II diabetes, obesity, metabolic syndrome, and cancer (36, 37). AMPK controls a diverse array of cellular functions, including cellular metabolism and innate immune signaling; its activity is frequently altered during infection (32, 33, 38). In prior studies, AMPK was found to have both pro- and antiviral properties (39–41) in cells infected with ZIKV and other related flaviviruses. However, AMPK-mediated regulation of innate antiviral responses and modulation of glycolysis during ZIKV infection has not been studied.
Here, we provide multiple lines of evidence (pharmacological activation/inhibition, shRNA knockdown, and AMPK deficient MEFs) demonstrating that activation of AMPK inhibits ZIKV infection of endothelial cells by two distinct mechanisms: potentiating innate antiviral responses and inhibition of virus-induced glycolysis. We have thus identified new potential targets for the development of anti-ZIKV therapeutic modalities.
MATERIALS AND METHODS
Cells and virus strains.
Human umbilical cord vein endothelial cells (HUVEC, ATCC CRL-1730) and primary human retinal vascular endothelial cells [HRvEC, Cell Systems, ACBRI 181, possessing endothelial morphology and cell markers (vWF and CD34)] (42, 43) were maintained in F12K media (Invitrogen) supplemented with 10% Fetal bovine serum (FBS), 1% penicillin-streptomycin (PS) solution (Invitrogen), endothelial cell growth supplement (ECGS) (Corning, 6 μg/ml) and heparin (Sigma, 0.1 mg/ml). Vero cells were grown in Dulbecco’s minimal essential medium (DMEM, Invitrogen) supplemented with 10% FBS and 1% PS solution. Cells were grown in at 37°C in 5% CO2 with humidity. Zika virus strain PRVABC59 (NR-50240) was originally isolated from human blood in Puerto Rico in December 2015 and was obtained through BEI Resources, National Institute of Allergy and Infectious Diseases (NIAID), NIH (26).
Cell viability assay.
The HRvEC cells were treated with the drugs at different concentrations and incubated for 48h followed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay. Briefly, the MTT reagent (0.5mg/mL), in the cell culture medium was added onto the cells for 4hours followed by lysis of the cells using 20% SDS (Sodium dodecyl sulfate) in 50% DMF (Dimethylformamide) solution. The readings were collected at 570nm and the values were plotted with reference to the untreated control as cell viability (%).
Viral attachment and entry assay.
Viral attachment protocol was adapted from He et al., 2018 (44) wherein HRvEC were pre-incubated at 4°C for attachment assay and 37°C for entry assay with the drugs at specified concentrations for an hour followed by ZIKV infected at MOI 1. After adsorption, the cells were gently washed with 1X PBS followed by viral RNA isolation from whole cells using viral RNA isolation kit (Qiagen, Germany). ZIKV RNA copy number was determined using RT-qPCR.
Direct inactivation of ZIKV stock by the drugs.
ZIKV stocks were diluted to 106 PFU/mL in serum-free media and the drugs were added at a final concentration as used in the study. The virus and the drug mixtures were incubated at 37°C for 2 hours and the mixture was used for quantification of virus yield by plaque assay on Vero cell monolayer.
Virus infection and Plaque assay.
The HRvEC/HUVEC cells were seeded at 70–80% confluence and incubated at 37°C in a humidified CO2 incubator. Cells were washed with 1X PBS and infected with ZIKV at a multiplicity of infection (MOI) of 1 or mock-infected in serum-free media. The cells were incubated with the virus for 2h with intermittent shaking every 15 min. The culture medium was removed and supplemented with F12K media with ECGS, 2% FBS (Fetal bovine serum) and 1% PS solution for the desired period of incubation time. The cell culture supernatant was harvested at different time points for the virus titer estimation by plaque assay.
For plaque assay, Vero cells were seeded at confluence in 6-well tissue culture plates and washed once with 1X PBS after adherence. Serial dilutions of culture supernatants were prepared in serum-free DMEM media and added onto the cells for 2 h. The virus was removed, and an equal mixture of DMEM and Noble agar (1:1) was added onto the monolayer and allowed to solidify. A nutrient mix of BSA (Bovine serum albumin) (50 mg/ml), MgCl2 (1 M), oxaloacetic acid (400 mM) with 1% PS solution along with DMEM was added onto the overlay and incubated for 5 days (37°C in a CO2 incubator with humidity). The monolayer was fixed with 10% trichloroacetic acid (TCA) for 15 min followed by careful removal of the solution and the overlay media. The cells were stained using crystal violet solution for 15 min and washed with distilled water to visualize the plaques. The plaques were counted, and the viral titer was expressed as log10 PFU/ml. The experiment was conducted in triplicates and represented with statistical analysis.
Antibodies and drugs.
The antibodies used in the study were: anti-phospho (Thr172)-AMPKα, anti-phospho acetyl-CoA carboxylase (Ser79) (ACC), anti-AMPKα, anti-HK 2 (Cell Signaling Technology, Beverly, MA), anti-flavivirus 4G2 IgG antibody (EMD Millipore) anti-ZIKV NS3 (Genetax), anti-GLUT1 (Santa Cruz Biotechnologies), at dilutions of 1:1000, and anti-β-actin (Sigma Aldrich) at a dilution of 1:5000. The secondary antibodies used were goat anti-rabbit/mice-IgG-HRP (Bio-Rad, Hercules, CA), rabbit anti-mouse Alexa 594 (Invitrogen). The drugs used in the study were AICAR (1 mM) (Toronto Research Chemicals, Inc. (ON, Canada), Compound C (10 μM) (Toronto Research Chemicals, Inc. (ON, Canada), GSK621 (10 μM) (Sigma-Aldrich), 2-DG (1 mM) (Sigma-Aldrich) and Metformin (20 mM) (Ranbaxy Laboratories (Princeton, NJ). For AMPK inhibition and activation studies, cells were pre-treated with respective drugs for an hour prior to virus challenge, removed during adsorption and then added again in fresh media and kept for the entire duration of the experiment.
Immunoblotting.
For immunoblotting analysis, cells were lysed in RIPA lysis buffer (Thermo Scientific), and the protein concentrations were estimated using a BCA assay kit (Thermo Scientific) as per manufacturer’s instructions. Denatured proteins were resolved on 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred onto 0.45 μm nitrocellulose membranes (Bio-Rad, Hercules, CA). The membrane was blocked with 5% non-fat skim milk followed by a wash with 1X TBST (Tris-glycine buffer with Tween 20). Blots were incubated with the primary antibody in 3% bovine serum albumin overnight at 4°C with shaking. Membranes were washed thrice with TBST followed by incubation with the secondary antibody at room temperature. The membrane was washed thrice with 1X TBST and the protein bands were visualized using Supersignal West Femto chemiluminescent substrate (Thermo Scientific, Rockford, IL). Relative intensities of protein bands were quantified using ImageJ software (NIH, USA).
Immunofluorescence assay.
Cells were cultured in four-well chamber slides (Fisher Scientific) and infected with ZIKV at MOI 1, then fixed with 4% paraformaldehyde in 1X PBS overnight at 4°C. The cells were washed thrice with PBS and then permeabilized and blocked using 1% BSA, 0.4% Triton X-100 in 1X PBS for 1 hour at room temperature. The cells were then incubated with mouse anti-Flavivirus 4G2 IgG (1:100) in the dilution buffer in a humidified chamber overnight at 4⁰C followed by three washes with 1X PBS. The cells were then incubated with anti-mouse Alexa Fluor 594 (Invitrogen) for 1 hour at room temperature in a humidified chamber. The cells were washed with 1X PBS thrice and mounted in Vectashield anti-fade mounting medium (Vector Laboratories). The cells were visualized using the Eclipse 90i fluorescence microscope (Nikon).
Bioenergetics analysis.
A Seahorse Bioscience XFe 96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) was used to monitor the extracellular acidification rate (ECAR). Briefly, 1×105 cells/well were plated in poly-lysine-coated 96-well culture plates. Growth medium was removed, the wells were washed two times, and 175 μl of bicarbonate-free glycolysis stress test (GST) media with glutamine (pre-warmed at 37°C) was added to each well. The cells were then incubated at 37°C for 1h. ECAR was measured by adding glucose (Glu) (10 μM) (injection 1), oligomycin (OM) (2 μM) (injection 2) and 2-deoxyglucose (2-DG) (100 μM) (injection 3) to the indicated final concentrations using the included ports on the XFe 96 cartridges. The data obtained were normalized with cell number counts, as described previously (45). Basal glycolysis and glycolytic capacity were calculated in mock-infected control, AICAR-treated, ZIKV-infected, and ZIKV-infected / AICAR-treated groups. Basal glycolysis was calculated as ECARGlu - ECAR2DG when glucose (Glu) was injected in port 1. ECAROM - ECAR2DG indicates the maximal glycolysis attained by cells when mitochondrial respiration is inhibited with oligomycin (inhibitor of ATPase).
RNA isolation and quantitative reverse transcriptase PCR (qRT-PCR).
Total RNA was extracted from the cells infected and treated with the drugs as specified using Trizol (Invitrogen) reagent as per the manufacturer’s instructions. The cDNA was synthesized using 1 μg of RNA using Maxima First Strand cDNA Synthesis kit (Thermo Scientific) according to the manufacturer’s instructions. qRT-PCR was performed using gene-specific primers as described previously (27) in a StepOnePlus Real-time PCR system (Applied Biosystems). Quantification of gene expression was determined via the comparative ΔΔCT method and expressed as relative fold-changes.
Enzyme-linked immunosorbent assay (ELISA).
The HRvEC and HUVEC cells were pretreated with AICAR for 1h followed by ZIKV infection (MOI 1) for 48 h. The protein levels of IFNα2, TNFα and IL-6 were quantified in conditioned media using ELISA. ELISA was performed for IFNα2 (BioLegend, San Diego, CA, sensitivity 3 pg/mL) and inflammatory cytokines TNFα and IL-6 (R&D Systems, Minneapolis, MN) as per manufacturer’s instruction and the data were represented as pg/mL of culture media. The experiment was performed in biological triplicates and technical duplicates for statistical analysis.
ATP measurement.
HRvEC cells were pre-treated with AICAR followed by infection at MOI 1 and treated with AICAR for 48 hpi. The cell lysate was collected in luciferase lysis buffer. The standard curve for ATP was prepared according to the manufacturer’s instructions (ThermoFisher Scientific). The ATP levels (nM) in the control and treated and/or infected cell lysates were quantified using the standard curve as per the manufacturer’s protocol.
Glucose uptake assay.
HRvEC cells were pre-treated with AICAR followed by infection with ZIKV at MOI 1 and treated with AICAR for 48 hpi. The cells were washed and kept in 1X PBS. One set of control cells was treated with 2DG for 10 mins, which served as the positive control for the experiment. The level of 2DG-6-P was quantified using the luciferase-based Glucose Uptake-Glo assay kit (Promega) according to the manufacturer’s instructions.
Statistical analysis.
The data used in the study have been expressed as mean ± SD. Statistical differences between the experimental groups were calculated using Graph Pad Prism software. A value of p<0.05 was considered statistically significant. All experiments were performed at least three times unless mentioned otherwise.
RESULTS
ZIKV infection downregulates AMPKα phosphorylation in endothelial cells
Endothelial cells constitute an important barrier for viral entry, including the inner BRB in the eye (1). Retinal endothelial cells have cellular receptors required for ZIKV entry and support efficient replication of the virus (27). We confirmed the ability of HRvEC (Human retinal vascular endothelial cells) and HUVEC (human umbilical cord vein endothelial cells) cells to support replication of the Puerto Rican, American clade ZIKV strain PRVABC59. Similar to previous studies (30, 46), we found that both endothelial cell types were permissive to ZIKV at MOI 1 as evidenced by intracellular staining of viral antigens (Fig. 1A). Moreover, the production of infectious virions was similar in both cell types, with titers produced in HUVEC cells peaking at 48 hpi, followed by a slight decline at 72 hpi (Fig. 1B). Another hallmark of viral infection is the induction of innate antiviral responses in infected cells (Supplementary Fig. S1). Similar to our results in HRvEC cells (27), ZIKV-infected HUVEC exhibited induced expression of multiple pattern recognition receptors, inflammatory mediators, IFNs and ISGs (data not shown). ZIKV thus replicates efficiently in endothelial cells of different anatomic origins and elicits a spectrum of antiviral responses.
Figure 1. ZIKV infection alters AMPK activity in endothelial cells.
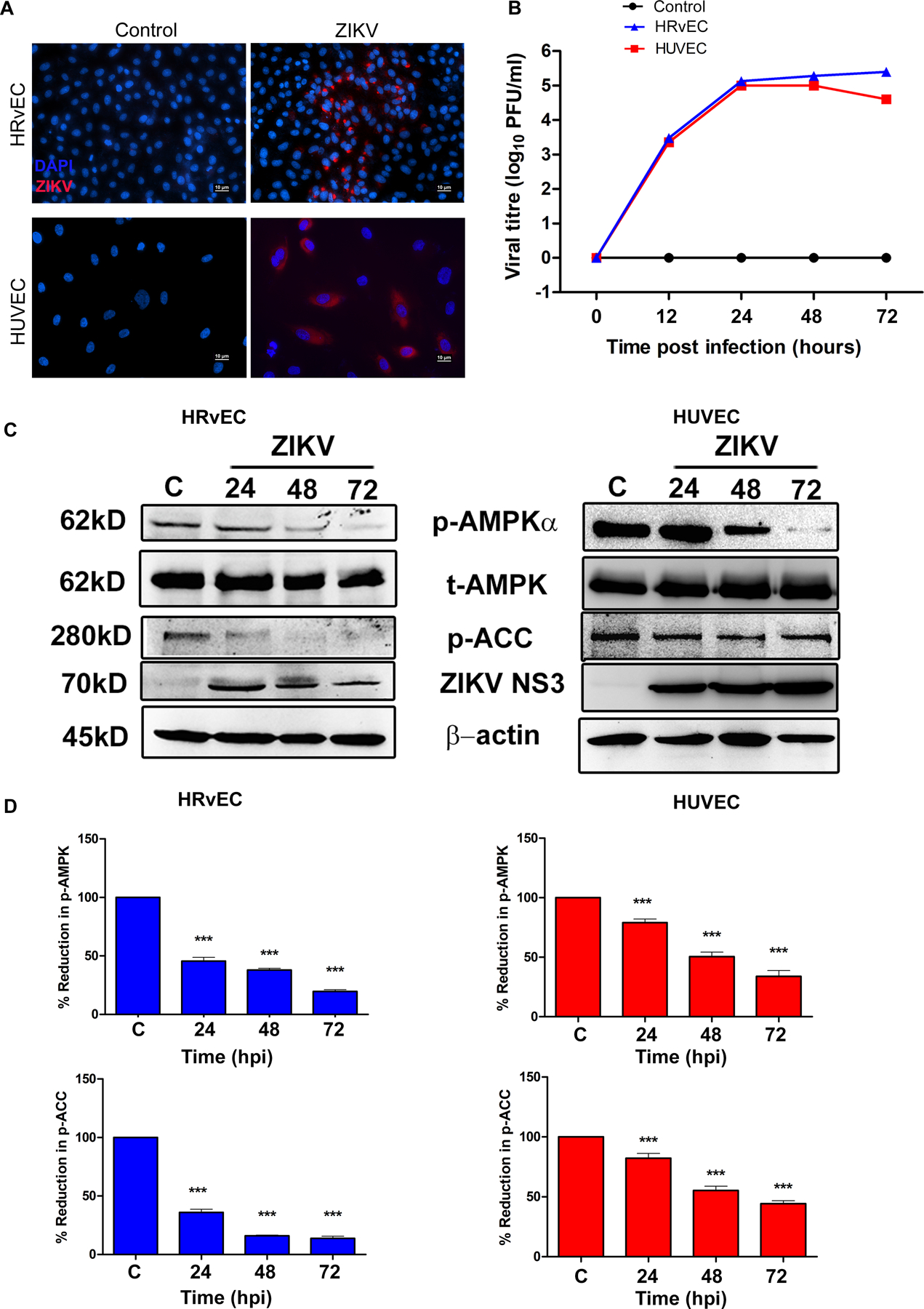
(A). (A) HRvEC (Human retinal vascular endothelial cells) and HUVEC (Human umbilical cord vein endothelial cells) cells were infected with ZIKV strain PRVABC59 at MOI 1 for 48 h and immunostained for ZIKV envelope antigen 4G2 and detected with an Alexa 594-conjugated secondary antibody (red), while the nuclei were stained with DAPI (magnification, 200X). (B) HRvEC and HUVEC cells were infected with ZIKV (MOI 1) for indicated time points and viral titers were determined using plaque assay. (C) Mock-infected control (C) and ZIKV infected whole cell lysates of HRvEC and HUVEC at the indicated time points were immunoblotted for p-AMPKα (Thr172), p-ACC (Ser79), total AMPKα, ZIKV NS3, and β-actin. (D) The expression levels of p-AMPK and p-ACC were quantified by densitometry and plotted as a percentage of expression relative to the mock-infected controls (mean ± SD, n=3). Band densities of the proteins were normalized to the respective loading control, β-actin. The p-value was calculated using one way ANOVA with Dunnett’s test. *** P <0.0001.
To determine whether ZIKV infection alters AMPK activity, a time-course study was performed in both HRvEC and HUVEC cells. Phosphorylation of AMPKα (Thr172) and its main downstream effector acetyl-CoA (coenzyme A) carboxylase (ACC, Ser79), a rate-limiting enzyme in fatty acid synthesis was assessed by immunoblotting. ZIKV NS3 protein level was used as a marker for ZIKV infectivity. Compared to uninfected control cells, ZIKV infection resulted in a decrease in AMPKα phosphorylation at Thr172 in both cell types at all the time points (Fig. 1C, D and Fig. S2). Coincident with the reduction in p-AMPKα, ACC phosphorylation was also reduced at all time-points (Fig. 1C & D). Because ACC is an important downstream target of AMPK (47), this data suggests that AMPK signaling is impaired in ZIKV-infected endothelial cells.
AMPK agonists restrict ZIKV replication and potentiate antiviral responses
Since ZIKV reduced AMPK activity in endothelial cells, we assessed the effect of AMPK activation on ZIKV infectivity. Viral infectivity and progeny virion formation were quantified using immunostaining of viral antigens and plaque assay, respectively. We exposed ZIKV-infected HRvEC and HUVEC cells to known pharmacological activators of AMPK [AICAR (1mM) and metformin (20mM)] in the presence or absence of an AMPK inhibitor, compound C (10μM). Cells exposed to these compounds did not exhibit cytotoxic effects in MTT assay (Supplementary Fig. S3). Similarly, these compounds did not exert direct antiviral activity or potentially impair viral entry/adsorption, as assessed by plaque assays and qRT-PCR for viral RNA (Supplementary Fig. S4A–C). Moreover, AMPK activators exerted a dose-dependent response in inhibiting ZIKV replication in HRvEC (Supplementary Fig. S4D). In both cell types, AICAR or metformin treatment significantly reduced ZIKV infection, as evidenced by reduced ZIKV antigen-positive cells as compared to untreated and ZIKV infected cells (Fig. 2A, B & C). Although the compound C alone did not affect viral replication, it reversed the inhibitory effects of AICAR and metformin. Quantification of viral progeny by plaque assay further confirmed these findings by showing significantly reduced viral titers ranging from 1.5 (metformin) to 2.5 (AICAR) log10 units with AMPK activators, while compound C treatment nullified their effects (Fig. 2D & E).
Figure 2. Pharmacological activation of AMPK inhibits ZIKV replication in endothelial cells.
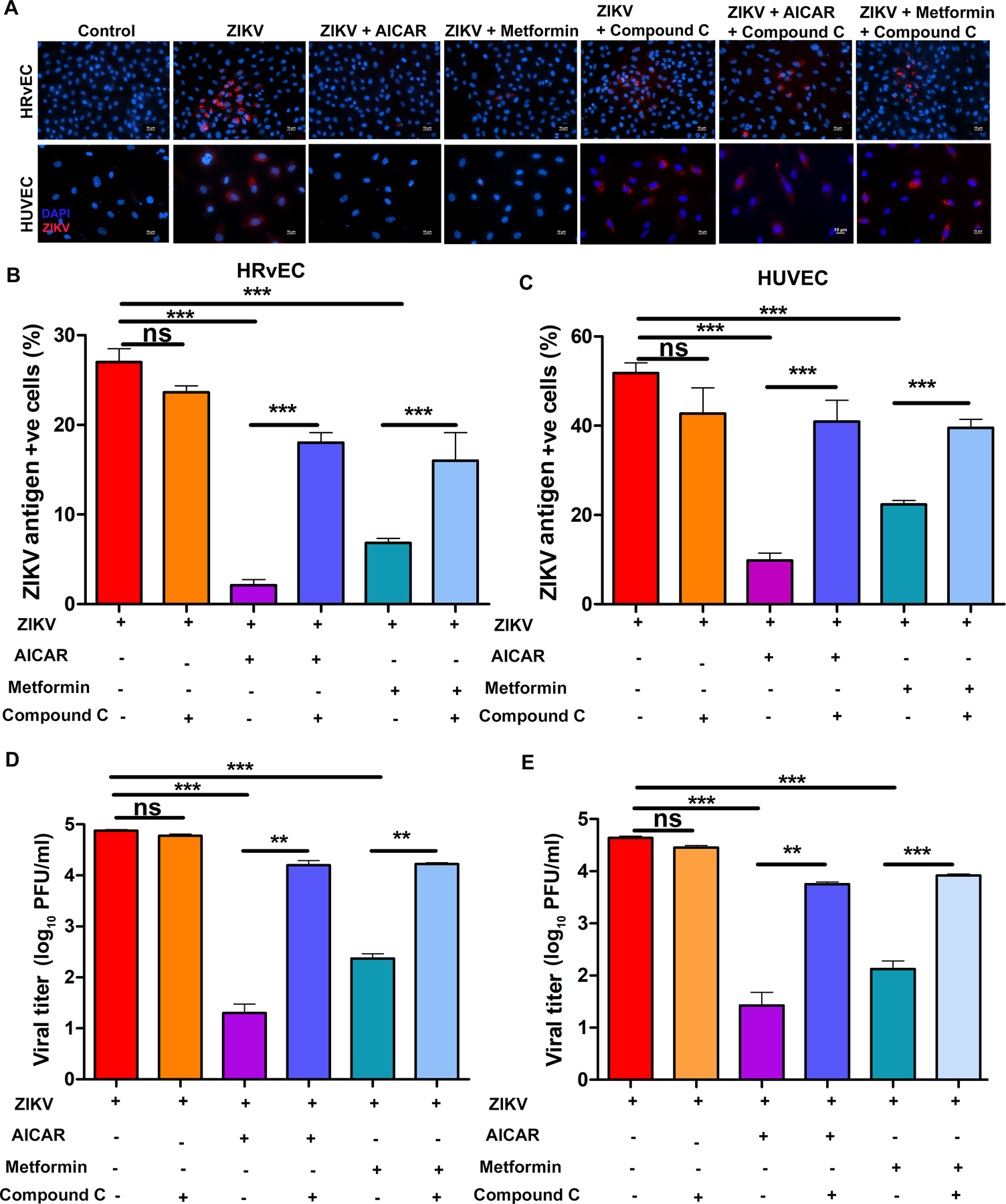
(A) HRvEC and HUVEC cells were infected with ZIKV at MOI 1 for 48 h in the presence or absence of AMPK activators [AICAR (1 mM), Metformin (20 mM)] and AMPK inhibitor (Compound C; 10 μM). The cells were immunostained for ZIKV envelope antigen 4G2 (red) and the nuclei were counterstained with DAPI (magnification, 200X). Culture supernatants were collected, and viral titers were estimated by plaque assay. Values were plotted on a logarithmic scale (mean ± SD, n=3) for (B) HRvEC, and (C) HUVEC cells. At least four independent fields were counted and plotted as a percentage of ZIKV antigen-positive cells (mean ± SD, n=4) relative to the total number of cells in the captured field for (D) HRvEC and (E) HUVEC. The p-value was calculated using one-way ANOVA Dunnett’s multiple comparison tests. ** P <0.001, *** P <0.0001, ns: not significant.
To further assess the effect of AMPK activation, we used AICAR as an AMPK activator in subsequent experiments; because 1) it inhibited ZIKV replication at a significantly higher rate in both endothelial cell types as compared to metformin (Fig. 2), and 2) our prior study demonstrated its therapeutic efficacy in an ocular bacterial infection model (32). We postulated that AMPK activation alters the antiviral innate responses in infected endothelial cells. To test this, cells were pre-treated with AICAR for an hour followed by ZIKV infection in the presence of the drug for 48 hr. qPCR was used to quantitate mRNA expression of classical antiviral and inflammatory genes, respectively. As anticipated, ZIKV challenge induced the expression of interferons (IFNα2, IFNβ1, and IFNγ), IFN inducible genes (OAS2, ISG15, and MX1), and inflammatory mediators (CCL5, TNFα, and CXCL10) in both HRvEC (Fig. 3) and HUVEC cells (Fig. 4). AICAR alone was found to increase the expression of some IFNs and ISGs compared to uninfected control (mock) cells. Interestingly, AICAR treatment potentiated this response in ZIKV-infected cells as compared to ZIKV-infected cells alone. In contrast, levels of inflammatory mediators (TNFα, CCL5, and CXCL10) were drastically reduced in AICAR treated and ZIKV-infected cells (Fig. 3A & 4A). The expression of IFNs, (i.e., IFNα) and inflammatory cytokines (i.e., TNFα, and IL-6) was further confirmed at protein levels by ELISA. These results were corroborated by increased expression of IFNα mRNA upon AICAR treatment in uninfected control cells, and its further enhancement upon ZIKV infection. Similarly, as anticipated, TNFα and IL-6 protein levels were lower in AICAR treated-ZIKV-infected cells. Moreover, the response was similar in both cell types, HRvEC (Fig. 3B) and HUVEC (Fig. 4B). These findings suggest that activation of AMPK enhanced the expression of ZIKV-induced IFNs and their downstream effectors, ISGs.
Figure 3. AICAR treatment potentiates ZIKV-induced interferon response and decreases the inflammatory response in HRvEC cells.
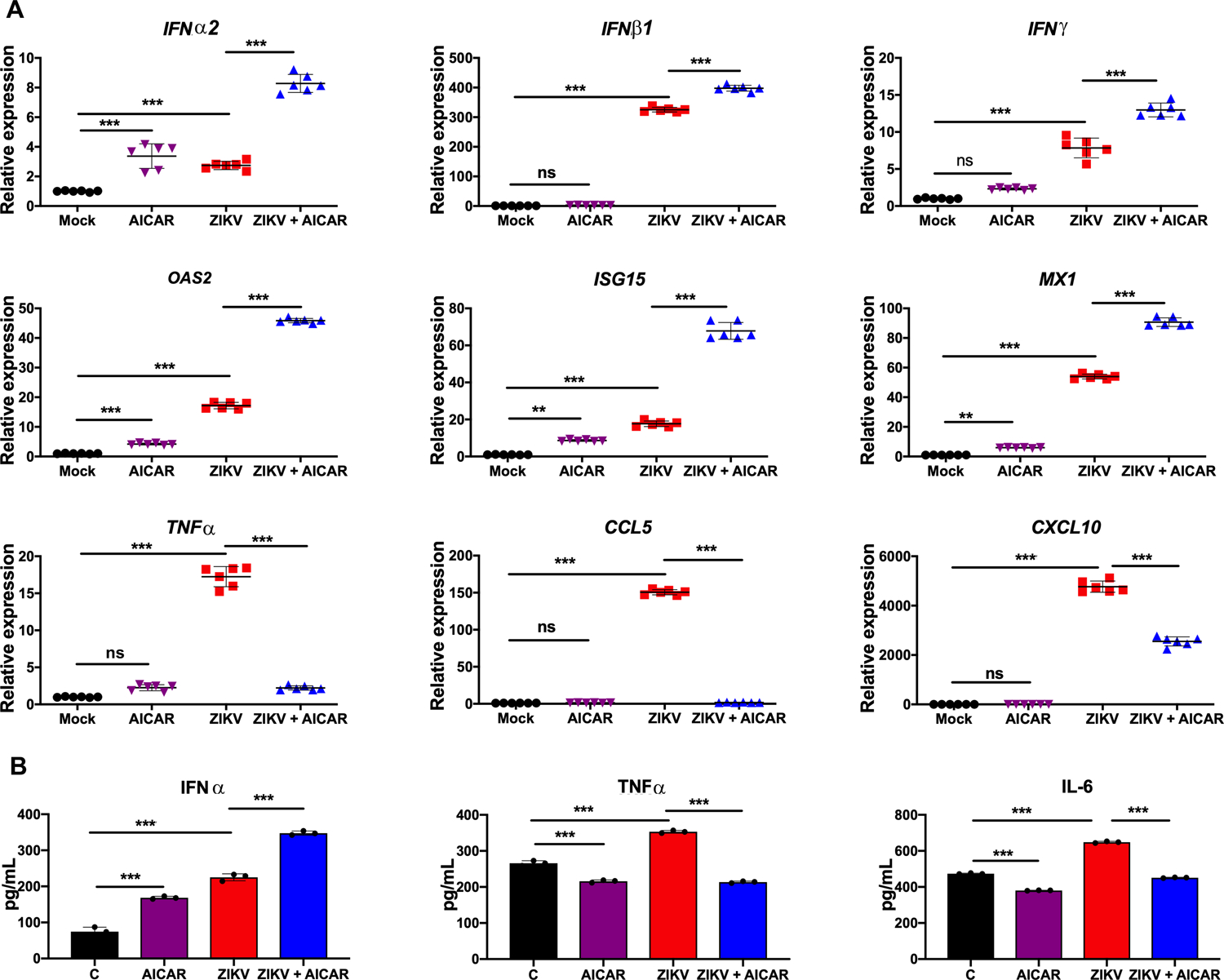
(A) HRvEC cells were pretreated with AMPK activator, AICAR (1 mM) for 1 h followed by ZIKV challenge (MOI 1) for 48 h. Cells were collected in Trizol for RNA isolation. mRNA levels of the indicated genes were quantified using qRT-PCR, normalized relative to β-actin. (B) Indicated cytokine levels were assessed from the culture supernatant using ELISA. The values are plotted as mean ± SD with n=3; p-values were calculated using one-way ANOVA-Bonferroni’s multiple comparison tests. * P <0.05, *** P <0.0001, ns: not significant.
Figure 4. AICAR treatment potentiates ZIKV-induced interferon response and decreases the inflammatory response in HUVEC cells.
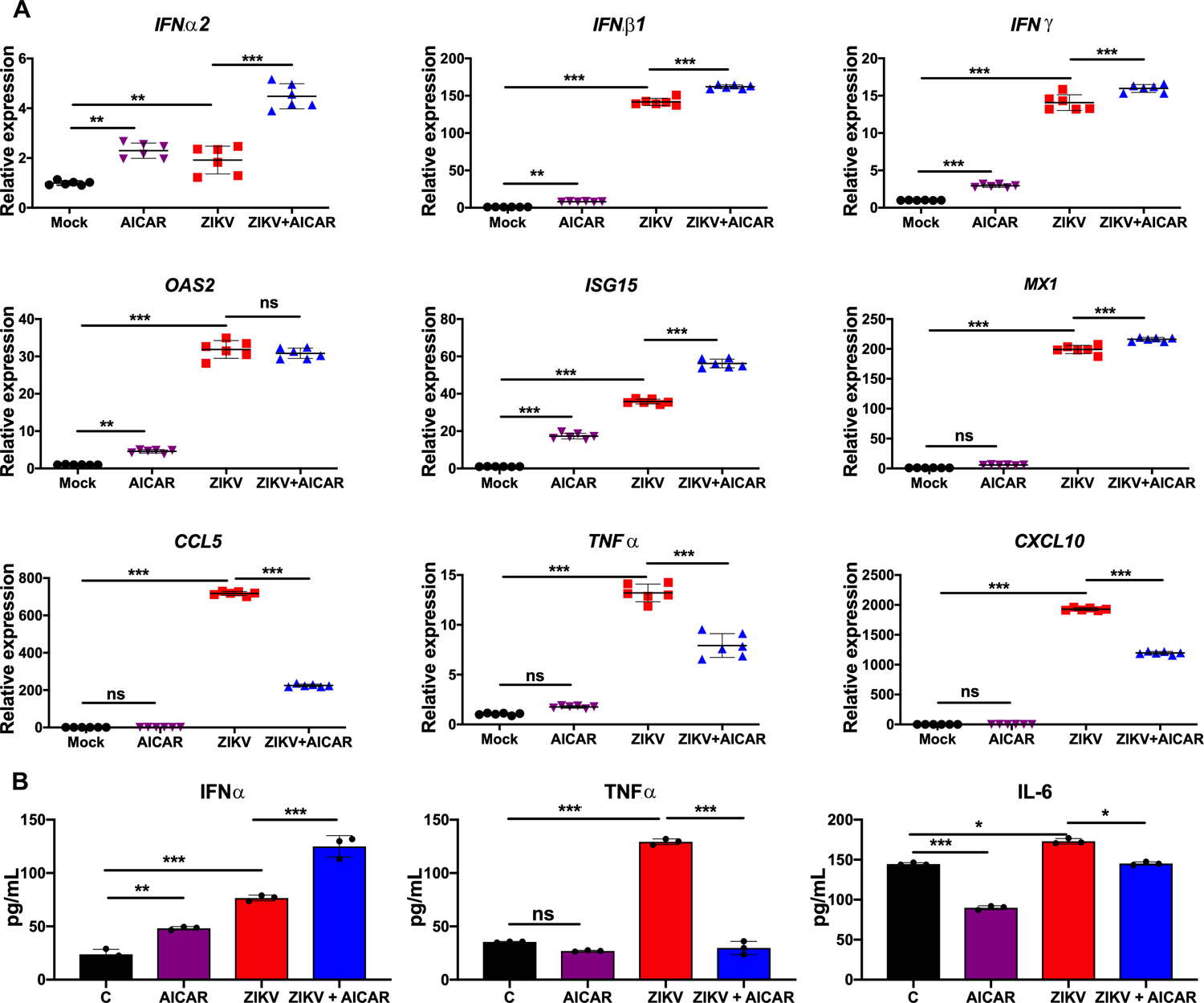
HUVEC cells were pretreated with AMPK activator, AICAR (1 mM) for 1 h followed by ZIKV challenge (MOI 1) for 48 h. Cells were collected in Trizol for RNA isolation. mRNA levels of the indicated genes were quantified using qRT-PCR, normalized relative to β-actin. (B) Indicated cytokine levels were assessed from the culture supernatant using ELISA. The values are plotted as mean ± SD with n=3; p-values were calculated using one-way ANOVA-Bonferroni’s multiple comparison tests. * P <0.05 **, P <0.001, *** P <0.0001, ns: not significant.
AMPK activation attenuated the ZIKV-evoked glycolytic response
Because AMPK is the master regulator of energy metabolism, we postulated that the antiviral effect of AMPK activation could be due to modulation of ZIKV-induced metabolic changes in infected cells. We performed bioenergetics analyses of ZIKV-infected HRvEC and HUVEC cells in the presence or absence of the AMPK activator, AICAR. An extracellular flux analyzer (Seahorse Bioscience) was used to determine cellular glycolytic activity by measuring the extracellular acidification rate (ECAR) (48). ZIKV-infected HRvEC (Fig. 5A & C) and HUVEC (Fig. 5B & D) exhibited increased ECAR, while AICAR treatment reduced ECAR levels.
Figure 5: AICAR treatment inhibits ZIKV-induced glycolysis in endothelial cells.
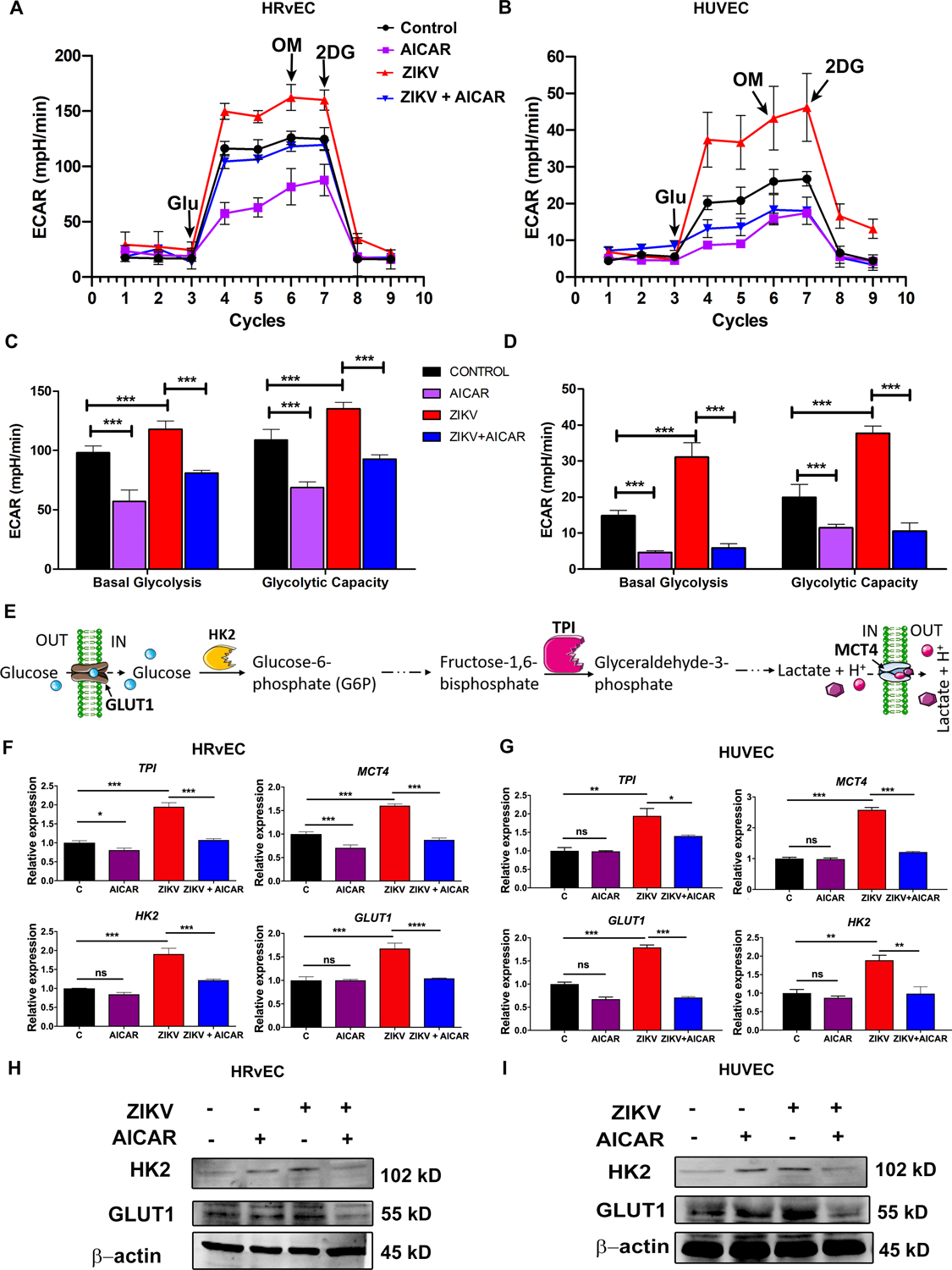
HRvEC (A, C, F & H) and HUVEC (B, D, G & I) cells were pretreated with AICAR (1 mM) for one hour followed by ZIKV infection for 12 h. (A-D) The extracellular acidification rate (ECAR), an indicator of glycolytic activity, was examined using a Seahorse XFe96 analyzer. The data are presented as mean ± SD. (E) A schematic representation of the glycolysis pathway showing metabolic intermediates and their respective enzymes. (F) HRvEC and (G) HUVEC cells were challenged with ZIKV for 12 h in the presence or absence of AICAR, as described above, and total RNA was extracted and subjected to qRT-PCR analysis of key metabolic genes (HK2, GLUT1, TPI, and MCT4) regulating glycolysis. The gene expression was normalized with ribosomal L-27 as a housekeeping gene. The whole cell lysate of mock-infected, AICAR-treated, ZIKV-infected, and AICAR treated / ZIKV infected HRvEC (H) and HUVEC (I) cells were immunoblotted for the glycolytic enzyme proteins Hexokinase 2 (HK2), GLUT1, and β-actin. The results represent the mean ± SD from three independent experiments, and the statistical analysis was performed using two-way (C & D) and one-way (F & G) ANOVA with Bonferroni’s multiple comparison tests. * P <0.05 **, P <0.001, *** P <0.0001, ns: not significant.
To further confirm increased glycolytic activity in ZIKV-infected endothelial cells, we determined the expression of key enzyme genes involved in the glycolysis pathway (Fig. 5E). We found increased mRNA expression of glucose transporter 1 (GLUT1), hexokinase 2 (HK2), triosephosphate isomerase (TPI), and monocarboxylate transporter 4 (MCT4) in both ZIKV-infected HRvEC (Fig. 5F) and HUVEC (Fig. 5G). In contrast, the expression of these genes was markedly reduced in cells treated with AICAR. Similarly, the protein levels of HK2 and GLUT1 in HRvEC (Fig. 5H) and HUVEC (Fig. 5I) were increased upon ZIKV infection and decreased upon AICAR treatment. The data demonstrate that the AMPK agonist, AICAR, inhibits the expression of key glycolytic enzymes and thereby reduces glycolysis in ZIKV-infected endothelial cells.
Glycolysis is needed for ZIKV replication in endothelial cells
Because of the observed increased glycolytic activity in ZIKV-infected endothelial cells, and the fundamental role of glycolysis is to provide energy and biochemical building blocks needed for efficient viral replication, including flaviviruses (49), we directly examined the role of glycolysis in ZIKV replication. To do this, we used 2-DG (2-deoxyglucose), a pharmacological analog of D-glucose that is a well-established inhibitor of glycolysis. In HRvEC treated with 2-DG (1mM), ZIKV replication was markedly inhibited, as evidenced by significantly reduced number of ZIKV antigen-positive cells (Fig. 6A & B) as well as viral titer (Fig. 6C) relative to untreated controls. Under similar conditions, 2-DG treatment increased the phosphorylation of AMPK (Fig. 6D, lane 2) in mock-infected cells; most importantly, 2-DG restored (lane 4) the reduced AMPK activation (lane 3) upon ZIKV challenge. Moreover, in agreement with immunostaining and virus titration, the expression of the ZIKV NS3 protein was undetected with 2-DG treatment. Because glucose uptake is required for activation of glycolysis, we then assessed the effect of ZIKV infection on glucose uptake and its modulation by AICAR treatment (Fig. 6E). As expected, in comparison to uninfected cells, ZIKV infection increased glucose uptake, while treatment with either 2-DG or AICAR reduced the ZIKV-induced glucose uptake. In parallel with increased glucose uptake, ATP production was higher in ZIKV-infected cells; this response was attenuated by AICAR treatment (Fig. 6F). Taken together, these results indicate that ZIKV-induced glycolysis is necessary for its replication and inhibition of glycolysis attenuates ZIKV replication.
Figure 6. Inhibition of glycolytic response attenuates ZIKV replication by restoring AMPK activity.
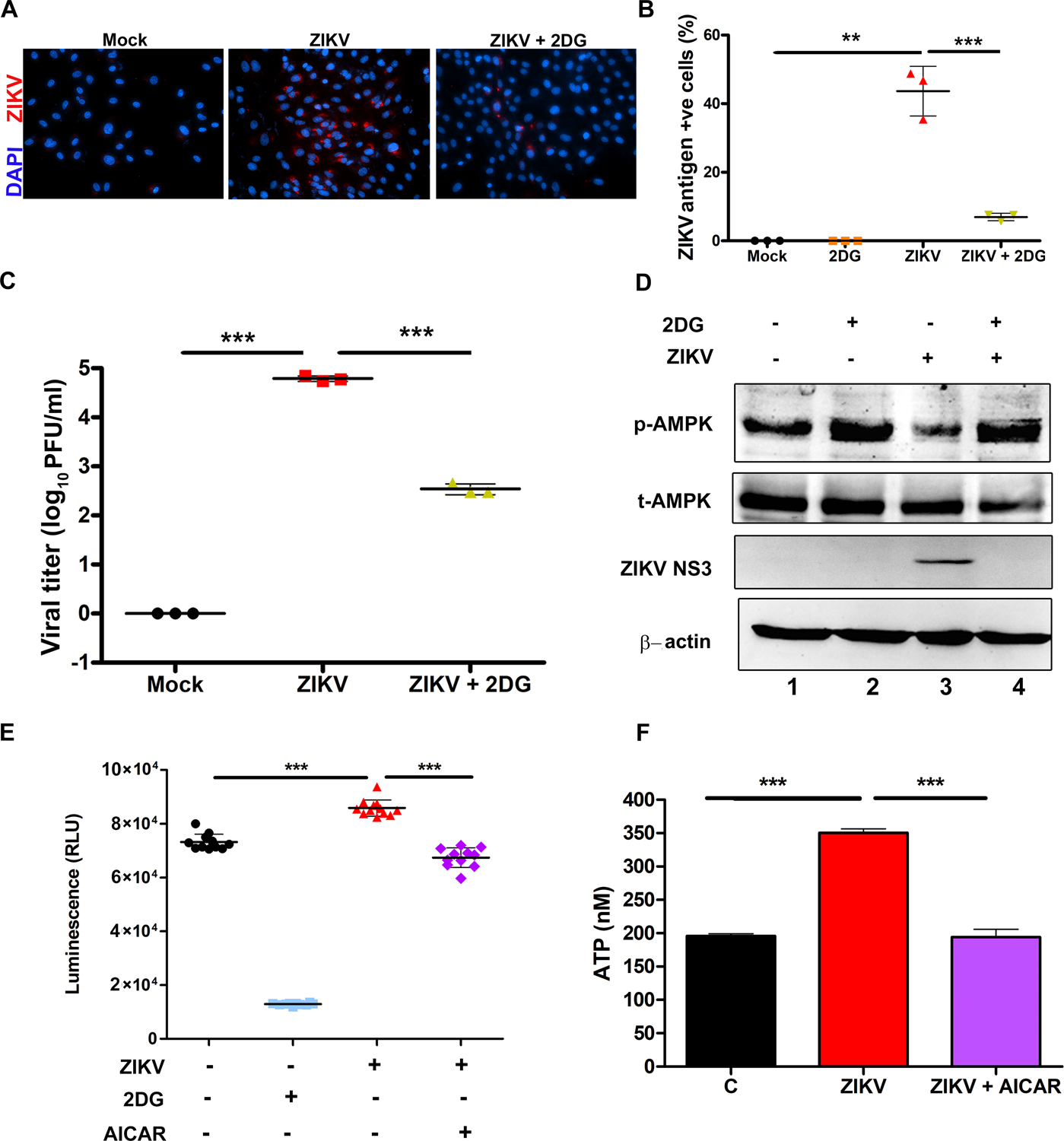
(A) HRvEC cells were treated with a glycolytic inhibitor, 2-deoxyglucose (2-DG), for 1 h followed by infection with ZIKV at MOI 1 or mock infection for 48 h. The cells were fixed and immunostained for ZIKV envelope antigen (red) and nuclei were counterstained using DAPI (magnification 200X). (B) The percentage of ZIKV antigen-positive cells were determined from at least four fields (mean ± SD). (C) HRvEC cells were treated with 2-DG and infected with ZIKV at MOI 1 and the culture supernatant was collected at 48 hpi for virus titration. (D) HRvEC cell lysates from mock-infected, 2-DG treated, ZIKV infected and ZIKV + 2-DG treated cells were immunoblotted for p-AMPK and total AMPK, ZIKV NS3, and β-actin. (E) Glucose uptake was measured in HRvEC cells infected with ZIKV for 48 h in the presence or absence of 1 mM AICAR. 2DG was used as a positive control for inhibition of glucose uptake. The values were plotted as Relative Luminescence units (RLU). (F) ATP levels were measured in HRvEC cells infected with ZIKV in the presence or absence of AICAR (1 mM). The values represent mean ±SD from three independent experiments. The statistical analysis was performed using one-way ANOVA- Bonferroni’s multiple comparison tests. ** P <0.001, *** P <0.0001.
Specific activation and inhibition of AMPK established its antiviral role in ZIKV infection
While aforementioned results using a well-studied AMPK agonist, AICAR, clearly demonstrated that AMPK activation evoked antiviral response against ZIKV, in part, by inhibiting virus-induced glycolysis, some studies have shown AMPK-independent effects of AICAR (50, 51). To confirm the antiviral role of AMPK in our experiments, we used a specific and potent AMPKα activator, GSK621 (52). We found that similar to AICAR (Fig. 2), GSK621 (10μM) treatment completely abolished ZIKV infection in HRvEC cells as assessed by immunostaining for ZIKV antigens (Fig. 7A) and titration of infectious progeny by plaque assay (Fig. 7B). GSK621-mediated AMPK activation was confirmed by western blotting of phospho-AMPKα and ACC (Fig. 7C). The reduction in ZIKV NS3 protein levels in cell lysates provides further evidence that antiviral activity can be induced by AMPK activation (GSK621 treatment) in endothelial cells. Moreover, the antiviral activity of GSK621 against ZIKV showed a dose-dependent response (Fig. S4D).
Figure 7. Specific modulation of AMPK activity alters ZIKV replication and antiviral responses in endothelial cells.
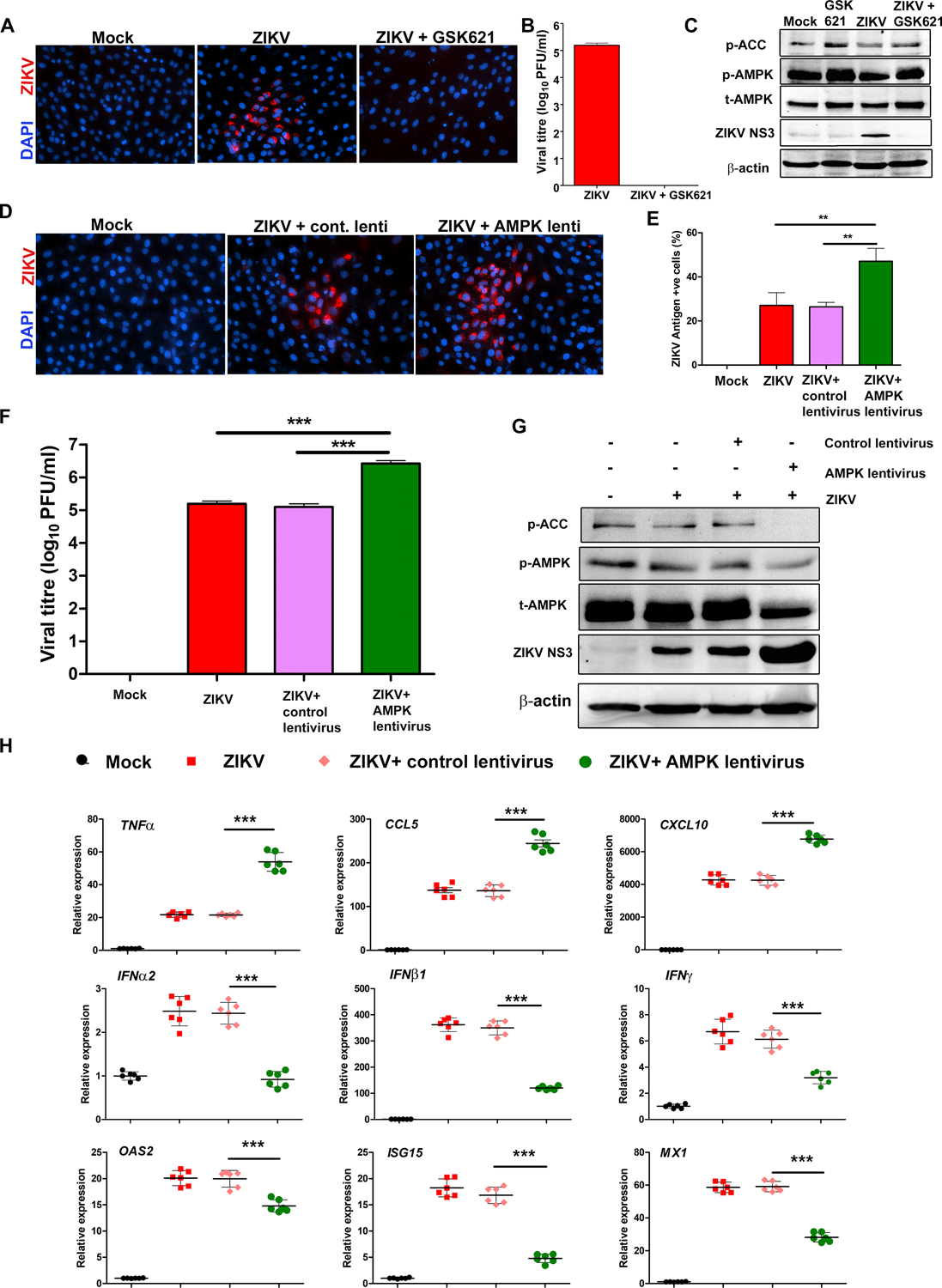
(A) HRvEC cells were infected with ZIKV at MOI 1 for 48 h in the presence or absence of 10 μM GSK621. Infected and treated cells were immunostained for ZIKV envelope antigen 4G2 and images were captured at 200X magnification. (B) The culture supernatant from 48 h infected HRvEC cells were used for viral titer determination by plaque assay, and (C) cell lysate were subjected to western blot for p-AMPK, p-ACC, total AMPK, ZIKV NS3, and β-actin. (D & E) Cells transfected with control or AMPK shRNA lentivirus were challenged with ZIKV at MOI 1 for 48 h. ZIKV replication was assessed by immunostaining of ZIKV 4G2 antigen, and (F) plaque assay. (G) Under similar experimental conditions, the whole cell lysate was used for western blot to assess the expression of p-AMPK, p-ACC, total AMPK, ZIKV NS3, and β-actin. (H) The quantitative PCR analysis was performed to quantify the relative expression of inflammatory mediators (TNFα, CCL5, and CXCL10), IFNs (IFNα2, IFNβ1, IFNγ), and IFN-inducible genes (OAS2, ISG15, and MX1) normalized to β-actin. The values represent mean ± SD from three independent experiments, and the statistical analysis was performed using one way ANOVA with Bonferroni’s multiple comparison tests. ** P <0.001, *** P <0.0001.
To further investigate the role of AMPK in ZIKV infection, we used a lentivirus-mediated knockdown approach (Santa Cruz Biotechnologies). Knockdown of AMPK resulted in enhanced ZIKV replication, as evidenced by an increase in the percentage of ZIKV antigen-positive cells (Fig. 7D & E) and higher infectious yields (Fig. 7F) in AMPKα versus control shRNA lentivirus-transfected and ZIKV-challenged HRvEC cells. Similarly, cells transfected with AMPKα lentivirus produced higher levels of ZIKV NS3 protein (Fig. 7G). The knockdown of AMPK and its downstream signaling ACC with lentivirus was confirmed by Western blotting (Fig. 7G).
Because AMPK activation by AICAR treatment (Fig. 3) resulted in increased antiviral and decreased pro-inflammatory mediators in response to ZIKV challenge, we postulated that shRNA lentivirus-mediated knockdown of AMPK would reverse this phenotype. Consistent with this, AMPK knockdown cells expressed higher mRNA expression of inflammatory mediators (TNFα, CCL5, CXCL10) and there was a significant reduction in expression of mRNAs of antiviral genes (OAS2, ISG15, MX1, IFNα2, IFNβ1, IFNγ) (Fig. 7H). Collectively, these findings indicate an antiviral role of AMPK in ZIKV infection.
AMPK−/− mouse embryonic fibroblast (MEF) are permissive to ZIKV infection
In addition to pharmacological activation or inhibition, we used a genetic approach [AMPK α1/α2 knock-out (KO) MEFs] to confirm the antiviral role of AMPK during ZIKV infection. Our results show that AMPK KO MEFs were susceptible towards ZIKV infection as evidenced by positive immunostaining for ZIKV antigens (Fig. 8A & B) as well as increased viral titer by plaque assay (Fig. 8C), in contrast, Wild-type (WT) MEFs were highly resistant to ZIKV infection. Moreover, ZIKV infection of AMPK α1/α2 KO MEFs induced antiviral responses, as evidenced by increased expression of viral recognition receptors (Rig-I), interferons (Ifnβ1, Ifnγ) and interferon-stimulated genes (Oas2, Isg15, Mx1) (Fig. 8D).
Figure 8. AMPK deficient mouse embryonic fibroblasts (MEFs) are susceptible to ZIKV infection.
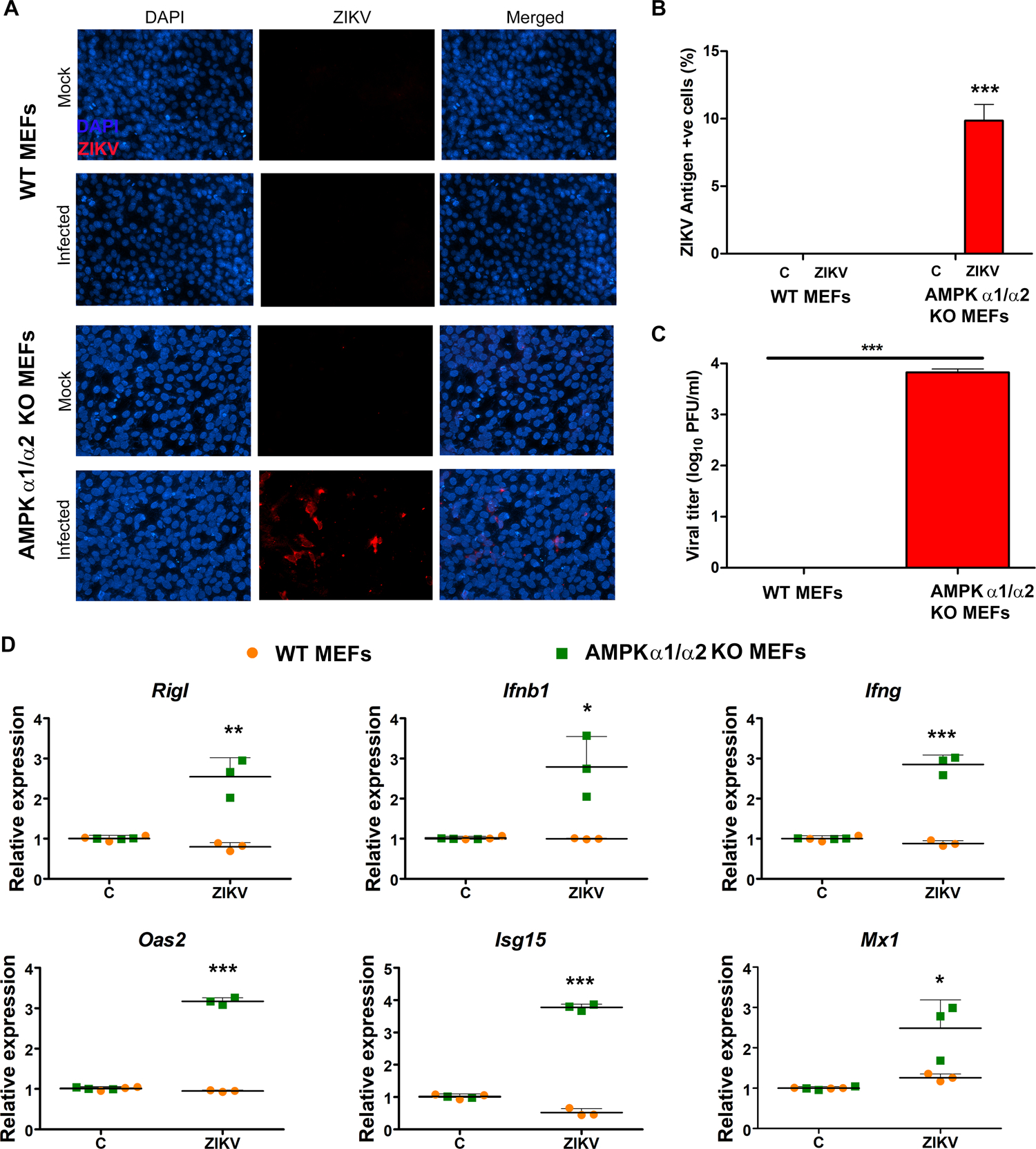
Wild-type (WT) and AMPK α1/α2 knockout (KO) MEFs were infected or mock-infected with ZIKV at MOI 1 for 48 h. (A) Viral infectivity was assessed by immunostaining for ZIKV envelop antigen 4G2 (red) and the nuclei were counterstained using DAPI. Images were captured at 200X magnification. (B) ZIKV antigen-positive cells were counted relative to the total number of cells and plotted as a percentage of ZIKV antigen-positive cells. (C) Culture supernatants from ZIKV infected WT and AMPK KO MEFs were collected at 48 hpi and used for viral titer determination by plaque assay. (D) qRT-PCR was performed on ZIKV-infected (48 h) WT and AMPK KO MEFs to quantify the relative expression of indicated genes. A house-keeping gene, β-actin, was used for normalization. Values represent mean ± SD from three independent experiments. The statistical analysis was performed using one-way ANOVA with Bonferroni’s correction for multiple comparison tests. * P <0.05 **, P <0.001, *** P <0.0001.
Since ZIKV did not infect WT MEFs, therefore, we concluded that the observed effect of increased antiviral response in AMPK α1/α2 KO MEFs could be due to differences in susceptibility of these MEFs against ZIKV. To address this question, we challenged MEFs with poly I:C (a synthetic analog of dsRNA) to simulate viral infection, and measured the antiviral response using qRT-PCR. We found that poly I:C challenge of WT MEFs but not the AMPK KO MEFs elicited a strong time-dependent antiviral immune response (Fig. 9) characterized by increased mRNA expression of Rig-I, Ifnβ1, Ifnγ, Oas2, Isg15, and Mx1. These findings provide further evidence that AMPK regulates innate antiviral responses.
Figure 9. AMPK deficient MEFs elicit diminished antiviral response upon poly I:C challenge.
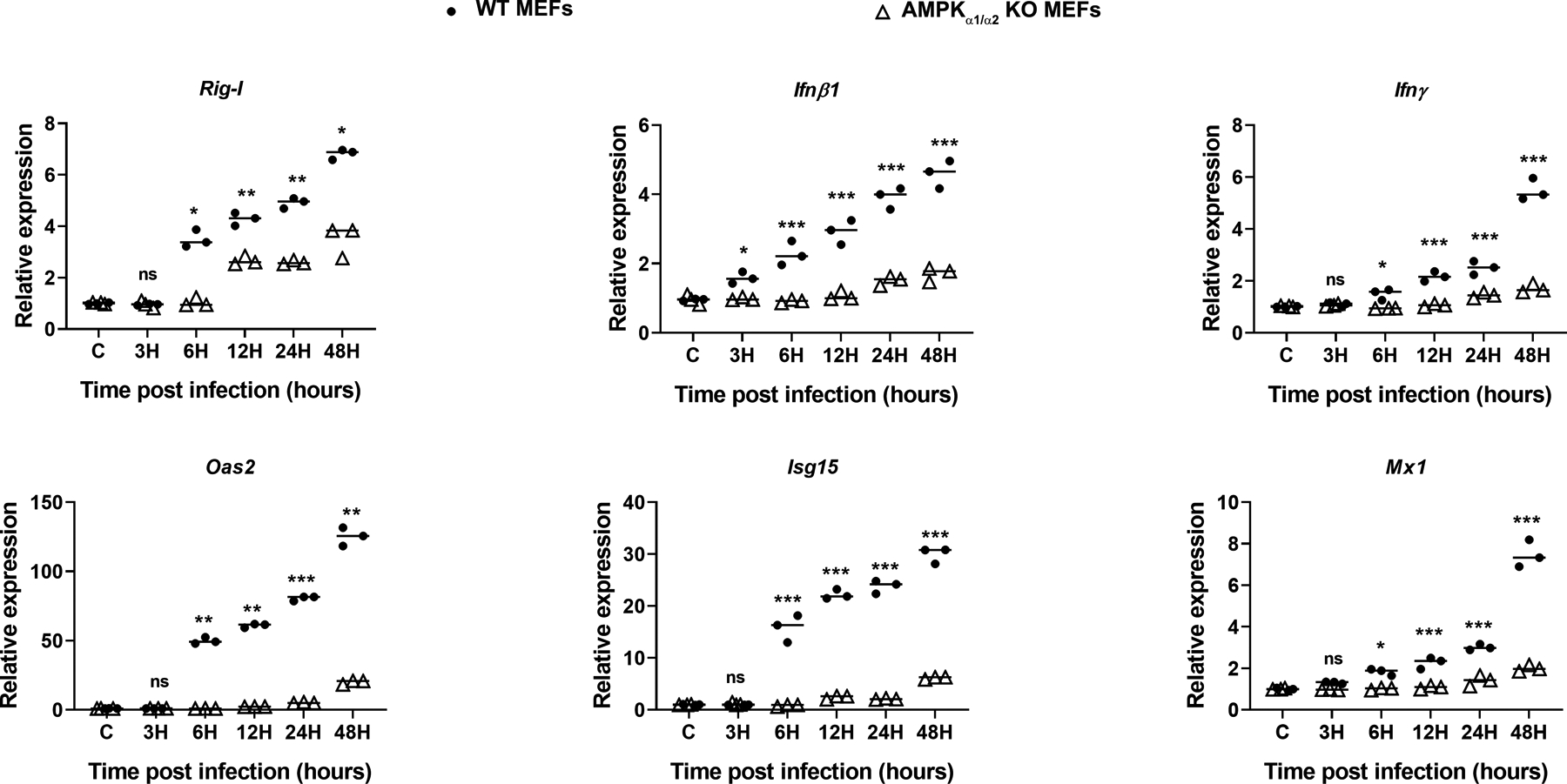
Wild-type (WT) and AMPKα1/α2 knockout (KO) MEFs were challenged with poly I:C (100 ng/ml) for indicated time points (3 to 48h). Cells were collected in Trizol for RNA isolation and cDNA preparation. qRT-PCR was performed to quantify the relative expression of PRRs, antiviral genes (Rig-I, Ifnβ1, and Ifnγ), and IFN-inducible genes (Oas2, Isg15, and Mx1), with normalization to GAPDH mRNA. The values represent mean ± SD from three independent experiments, and the statistical analysis has been performed using one-way ANOVA with Bonferroni’s correction for multiple comparison tests. *** P < 0.0001.
DISCUSSION
This study identifies the role of host cell AMPK signaling in altering antiviral innate immune response and glucose metabolism as relevant to ZIKV pathogenesis. Using multiple approaches (pharmacological, shRNA, & genetic ablation), we found that while suppression of AMPK favored ZIKV infection, its activation exerted a potent antiviral response that restricted viral replication in infected endothelial cells. The novelty of our study lies in defining the specific role of AMPK in regulating antiviral activities via its ability to 1) inhibit ZIKV-induced glycolysis, an essential source of energy and building blocks for viral replication and 2) to enhance antiviral response (IFNs and ISGs) in ZIKV-infected cells. Moreover the use of primary human retinal vascular endothelial cells (HRvEC), which form the inner blood-retinal barrier in the eye enables their use for evaluation of antiviral therapeutic modalities for combating ocular complications due to ZIKV and other flaviviruses (1).
Endothelial cells are an important barrier for the entry of blood-borne pathogens such as ZIKV. In addition to their barrier properties, endothelial cells actively participate in evoking and enhancing innate inflammatory response upon infectious stimuli, including viruses (53). We previously reported that retinal endothelial cells express AXL, TAM receptors, implicated in the entry of ZIKV (27). We hypothesized that ZIKV could directly infect retinal endothelial cells as a potential mechanism to breach the BRB. In support of that, our data here demonstrate that ZIKV-infected HRvEC can produce infectious virions, indicating the potential of this cell type to promote the spread of the virus into the retina/eye. Interestingly, ZIKV infected HRvEC and HUVEC at almost a similar rate and produced viral progeny consistent with prior studies (27, 39, 46, 54). Given our interest in studying the potential transmission of ZIKV from blood to the eye, most of our experiments were performed in HRvEC. However, in some experiments the widely used HUVEC cell line, HUV-EC-C (ATCC CRL-1730) was employed to test the generality of our results. Although the in vivo relevance of ZIKV infectivity of endothelial cells is still not clear, autopsy specimens from dengue virus-infected patients show the presence of virus in endothelial cells (55, 56). Hence, given the ability of ZIKV to infect multiple organs and tissues, endothelial cell biology appears to be an important part of ZIKV biology.
Our recent transcriptomic study of retinal pigment epithelial (RPE) cells indicated the dysregulation of lipid metabolic pathways upon ZIKV infection (26). Moreover, supplementation of cholesterol enhances ZIKV infectivity in RPE cells and pharmacological inhibition of a cholesterol transporter, ABCG1, attenuates this response. These findings indicate the role of lipids (e.g., cholesterol) or fatty acids in the pathogenesis of ZIKV in retinal cells. Since, AMPK maintains cellular lipid homeostasis through activation or inhibition of its downstream effectors, including ACC and has been shown to affect viral infection (35, 40), we investigated its specific mechanism in modulating ZIKV replication in HRvEC. Our data showing reduced activity (phosphorylation) of AMPK and ACC upon ZIKV infection implies an increase in the lipid and fatty acid synthesis, which could provide energy and lipids needed to facilitate efficient viral replication. Several enveloped viruses, including flavivirus family members, decrease AMPK activation to enhance their replication (33, 57). ACC catalyzes the irreversible conversion of malonyl-CoA, a key metabolite that plays multiple roles in fatty acid metabolism and is the main substrate for fatty acid biogenesis (58). ZIKV infection, therefore, might be decreasing the activation of AMPK to decrease the phosphorylation of ACC, leading to enhanced fatty acid synthesis (59). During viral replication, enveloped viruses most likely require increased biosynthesis of diverse biomolecules because virus replication is dependent on the synthesis of nucleic acids, viral proteins, and lipids. Dengue virus (60) and Hepatitis C virus (HCV) (33) inhibit AMPK activation, elevating lipid synthesis machinery in infected cells. Additionally, suppression of AMPK activity through its inhibition of autophagy (61) may create a permissive environment for ZIKV (62). ZIKV encodes three structural and seven non-structural proteins (1); studies are underway to determine which of these ZIKV-encoded proteins is responsible for modulation of AMPK activity in infected cells. Several studies have shown that AMPK levels are inversely related to inflammation and that it is downregulated under inflammatory conditions (32, 35, 63, 64). It remains to be determined whether the observed reduction in AMPK activity is due to ZIKV-induced inflammation (65, 66) and/or inactivation of upstream kinases such as LKB1.
To investigate the physiological role of reduced AMPK activity in ZIKV-infected endothelial cells, we used two commonly used pharmacological activators of AMPK, metformin, and AICAR (32). AICAR is converted into an AMP analog, 5-amino-4-imidazole carboxamide ribonucleotide, a naturally occurring intermediate in purine synthesis that directly binds to AMPK (35). Both AMPK activators reduced ZIKV infectivity and the production of infectious virions. Consistent with observations for other enveloped viruses, including flaviviruses, such as West Nile virus and Hepatitis C virus, our finding demonstrated the antiviral role of AMPK activation on ZIKV replication (39–41). The observed antiviral effects of AMPK activators can be attributed to inhibition of several stages of ZIKV replication (1). While we cannot completely exclude the possibility of a direct effect of AICAR on ZIKV gene expression or replication, our data clearly establish that the used drugs/compounds do not interfere viral entry, adsorption and viral infectivity (Fig. S4). Moreover, our dose-response study indicates that the anti-ZIKV activities of these drugs are likely due to AMPK activation. However, some studies have shown their AMPK-independent effects, depending on the cell type and specific experimental conditions (39, 50, 51). For example, Cheng et al. showed that compound C, AICAR, and metformin treatments reduced ZIKV replication in endothelial cells, leading to the conclusion that ZIKV replication is AMPK-independent and study the importance of protein kinase A inhibitor on virus replication. (39). Similarly, AMPK has been shown to possess pro as well as antiviral role during dengue virus (DENV) infection (60, 67). Thus, to exclude the pleiotropic off-target effects of the drugs, we used several complementary approaches, 1) a specific AMPKα activator, GSK621, 2) lentivirus-mediated knockdown of AMPK, and 3) AMPKα deficient MEFs. Collectively, these experiments confirmed our observations (using AICAR) and demonstrate the specific role of AMPK in regulating antiviral innate response to ZIKV infection.
AMPK activation results in phosphorylation of a number of downstream targets, thereby affecting glucose metabolism and fatty acid oxidation to restore energy (ATP) balance in mammalian cells (35). During infection, the increased energy demand due to the activation of innate immune signaling stimulates the production of inflammatory cytokines, antimicrobial, and chemotactic molecules (32, 68). To meet the associated energy demand, cells rely on glycolysis, which not only produces ATP but also provides metabolic intermediates for various biosynthetic pathways, including amino acid, lipid, and nucleic acid production. Since viruses also need these metabolites for their replication (69), we hypothesized that ZIKV could trigger a glycolytic response. Indeed, our bioenergetics analysis revealed increased ECAR levels upon ZIKV infection, suggesting glycolysis may be a source of energy in infected endothelial cells. Concomitant with higher ECAR levels, ZIKV-infected endothelial cells exhibited induced expression of key glycolytic enzymes, such as GLUT1, and HK2 and increased glucose uptake (Fig. 5). These findings are consistent with previous studies implicating an essential role of glycolysis in viral replication (49). We observed that a therapeutic blockade of glucose utilization with 2-deoxy-D-glucose (2-DG) inhibited ZIKV replication in infected endothelial cells. Moreover, 2-DG treatment resulted in augmented AMPK activity. To our knowledge, our study is the first to demonstrate that ZIKV induces glycolysis, and the inhibition of glycolysis reduces ZIKV replication. Because AICAR treatment reduced ECAR levels in ZIKV-infected cells, attenuated the expression of glycolytic enzymes, and reduced glucose uptake, our study suggests that antiviral activity of AMPK, in part, could be due to its ability to inhibit the glycolytic response.
In response to viral infections, host cells trigger an innate immune response, predominantly the production of type I and type II interferons and antiviral ISGs (Interferon-stimulated genes). These innate antiviral responses promote inflammation, immune cell activation, and viral clearance. Importantly, we showed that ZIKV-infected endothelial cells expressed IFNs and ISGs that could aid in restricting viral spread. Indeed, our prior studies demonstrated an essential role of ISG15 in exerting antiviral response against ZIKV infection in retinal (27) as well as corneal cells (28). While AMPK is classically viewed as a cellular energy sensor, studies from our and other laboratories have demonstrated the role of AMPK in immunomodulation, specifically as a negative regulator of inflammation (32, 70, 71). To our knowledge, our laboratory was the first to demonstrate the anti-inflammatory role of AMPK in ocular infection (32). Therefore, we postulated that AMPK might modulate ZIKV-evoked innate responses in infected endothelial cells. Consistent with our previous study (32), we observed that while AICAR treatment reduced the inflammatory response (e.g., TNFα, CCL5), it potentiated the expression of IFNs and ISGs, including ISG15, in ZIKV-infected endothelial cells (Fig. 3 & 4). Interestingly, AICAR treatment alone induced the expression of IFNs (38), including IFNα which has not been reported previously. Although, we assessed the modulation of various ISGs upon AMPK activation in vitro, the in vivo relevance of these observations need to be evaluated using AMPK KO mice (32, 72). Similarly, the silencing of RIG-I, MDA5, and TLR3 might provide the potential contributions of these signaling pathways in inducing IFNs, including IFN-γ.
In agreement with this observation, lentivirus-mediated knockdown of AMPK in endothelial cells resulted in increased inflammatory and reduced antiviral molecules (Fig. 7H). Further confirmation of an antiviral role of AMPK came from the use of AMPK α1/α2 KO MEFs, wherein the AMPK deficiency facilitated ZIKV replication while the wild type MEFs were resistant to ZIKV infection (Fig. 8). In support, ZIKV infectivity correlated with an increase in IFNs and ISGs in AMPK α1/α2 KO but not in the WT MEFs. We also assessed the expression of potential receptors for ZIKV entry (Axl, Tyro3 and Mertk) and observed no significant difference in their basal mRNA levels in WT vs. AMPK KO MEFs. Therefore, the reduced innate antiviral responses in WT MEFs are unlikely due to differences in the expression of viral entry receptors (data not shown). Moreover, it is well known that wild type C57BL/6 (B6) mice are resistant to ZIKV infection. Therefore, most studies have utilized both, immunocompromised mice, with type I and II IFN receptor knockout or neutralizing interferon receptors (21–23, 73). Consistent with these observations, we found that wild type MEFs are not susceptible to ZIKV infection, hence relatively reduced induction of innate immune response (Figure. 8D).
Since there was almost no ZIKV infectivity in WT MEFs, modulation of the antiviral response cannot be compared in WT versus AMPK α1/α2 KO MEFs. Hence, we challenged the MEFs with poly I:C, a synthetic dsRNA, and a classic TLR3 ligand, to evoke antiviral responses (74). Similar to AMPK knockdown in endothelial cells, our data showed that reduced IFNs and ISGs in AMPK α1/α2 KO MEFs treated with poly I:C (Fig. 9). Together, these findings suggest that AMPK promotes an antiviral response and that the downregulation of AMPK in infected endothelial cells could be a strategy used by ZIKV to subvert the host cell antiviral innate defense.
In summary, our study demonstrates the role of AMPK in regulating the innate antiviral response and cellular bioenergetics (glucose metabolism) of endothelial cells during ZIKV infection. Although the interplay between viruses and host cell metabolism is very complex, our findings elucidated mechanisms underlying antiviral effects of AMPK signaling against ZIKV infections.
Supplementary Material
Key points:
ZIKV inhibits AMPK signaling and increases glycolysis to promote viral replication.
AMPK potentiates antiviral immune response and inhibits ZIKV-induced glycolysis.
AMPK ablation increases ZIKV replication and reduces innate antiviral responses.
Acknowledgment
The authors would like to thank Dr. Benoit Viollet (INSERM, France) for providing the AMPKα1/α2 knockout MEFs. We thank Katherine R. Spindler (University of Michigan) for helpful discussion and reading a draft of the manuscript. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Funding source: NIH (R21AI135583, R01EY026964, & R01 EY027381)
Footnotes
Conflict of interest
The authors declare no financial conflicts of interest.
References
- 1.Singh S, and Kumar A. 2018. Ocular Manifestations of Emerging Flaviviruses and the Blood-Retinal Barrier. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliver GF, Carr JM, and Smith JR. 2019. Emerging infectious uveitis: Chikungunya, dengue, Zika and Ebola: A review. Clin Exp Ophthalmol 47: 372–380. [DOI] [PubMed] [Google Scholar]
- 3.Vogel G 2016. INFECTIOUS DISEASE. Experts fear Zika’s effects may be even worse than thought. Science 352: 1375–1376. [DOI] [PubMed] [Google Scholar]
- 4.Moshfeghi DM, de Miranda HA, and Costa MC. 2016. Zika Virus, Microcephaly, and Ocular Findings. JAMA Ophthalmol 134: 945. [DOI] [PubMed] [Google Scholar]
- 5.Jamil Z, Waheed Y, and Durrani TZ. 2016. Zika virus, a pathway to new challenges. Asian Pac J Trop Med 9: 626–629. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro LS, Marques RE, Jesus AM, Almeida RP, and Teixeira MM. 2016. Zika crisis in Brazil: challenges in research and development. Curr Opin Virol 18: 76–81. [DOI] [PubMed] [Google Scholar]
- 7.Watts AG, Huber C, Bogoch II, Brady OJ, Kraemer MUG, and Khan K. 2018. Potential Zika virus spread within and beyond India. J Travel Med 25. [DOI] [PubMed] [Google Scholar]
- 8.Lee BY, Alfaro-Murillo JA, Parpia AS, Asti L, Wedlock PT, Hotez PJ, and Galvani AP. 2017. The potential economic burden of Zika in the continental United States. PLoS Negl Trop Dis 11: e0005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansson MA, Mier-y-Teran-Romero L, Reefhuis J, Gilboa SM, and Hills SL. 2016. Zika and the Risk of Microcephaly. N Engl J Med 375: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mysorekar IU, and Diamond MS. 2016. Modeling Zika Virus Infection in Pregnancy. N Engl J Med 375: 481–484. [DOI] [PubMed] [Google Scholar]
- 11.Panchaud A, Stojanov M, Ammerdorffer A, Vouga M, and Baud D. 2016. Emerging Role of Zika Virus in Adverse Fetal and Neonatal Outcomes. Clin Microbiol Rev 29: 659–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Araujo AQ, Silva MT, and Araujo AP. 2016. Zika virus-associated neurological disorders: a review. Brain 139: 2122–2130. [DOI] [PubMed] [Google Scholar]
- 13.Abbasi AU 2016. Zika Virus Infection; Vertical Transmission and Foetal Congenital Anomalies. J Ayub Med Coll Abbottabad 28: 1–2. [PubMed] [Google Scholar]
- 14.Ventura CV, Maia M, Bravo-Filho V, Gois AL, and Belfort R Jr. 2016. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 387: 228. [DOI] [PubMed] [Google Scholar]
- 15.Ventura CV, Maia M, Dias N, Ventura LO, and Belfort R Jr. 2016. Zika: neurological and ocular findings in infant without microcephaly. Lancet 387: 2502. [DOI] [PubMed] [Google Scholar]
- 16.Ventura CV, Maia M, Travassos SB, Martins TT, Patriota F, Nunes ME, Agra C, Torres VL, van der Linden V, Ramos RC, Rocha MA, Silva PS, Ventura LO, and Belfort R Jr. 2016. Risk Factors Associated With the Ophthalmoscopic Findings Identified in Infants With Presumed Zika Virus Congenital Infection. JAMA Ophthalmol 134: 912–918. [DOI] [PubMed] [Google Scholar]
- 17.Jampol LM, and Goldstein DA. 2016. Zika Virus Infection and the Eye. JAMA Ophthalmol 134: 535–536. [DOI] [PubMed] [Google Scholar]
- 18.Miranda HA, Costa MC, Frazão MAM, Simão N, Franchischini S, and Moshfeghi DM. 2016. Expanded Spectrum of Congenital Ocular Findings in Microcephaly with Presumed Zika Infection. Ophthalmology 123: 1788–1794. [DOI] [PubMed] [Google Scholar]
- 19.de Paula Freitas B, de Oliveira Dias JR, Prazeres J, Sacramento GA, Ko AI, Maia M, and Belfort R Jr. 2016. Ocular Findings in Infants With Microcephaly Associated With Presumed Zika Virus Congenital Infection in Salvador, Brazil. JAMA Ophthalmol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, Van den Pol A, Lindenbach BD, Horvath TL, and Iwasaki A. 2016. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 166: 1247–1256.e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah A, and Kumar A. 2016. Zika Virus Infection and Development of a Murine Model. Neurotox Res 30: 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossi SL, and Vasilakis N. 2016. Modeling Zika Virus Infection in Mice. Cell Stem Cell 19: 4–6. [DOI] [PubMed] [Google Scholar]
- 23.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, and Diamond MS. 2016. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 165: 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, Weger-Lucarelli J, Manzella F, Rückert C, Govero J, Noguchi KK, Ebel GD, Diamond MS, and Apte RS. 2016. Zika Virus Infection in Mice Causes Panuveitis with Shedding of Virus in Tears. Cell Rep 16: 3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Z, Yang M, Azar SR, Soong L, Weaver SC, Sun J, Chen Y, Rossi SL, and Cai J. 2017. Viral Retinopathy in Experimental Models of Zika Infection. Invest Ophthalmol Vis Sci 58: 4355–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh PK, Khatri I, Jha A, Pretto CD, Spindler KR, Arumugaswami V, Giri S, Kumar A, and Bhasin MK. 2018. Determination of system level alterations in host transcriptome due to Zika virus (ZIKV) Infection in retinal pigment epithelium. Sci Rep 8: 11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh PK, Guest JM, Kanwar M, Boss J, Gao N, Juzych MS, Abrams GW, Yu FS, and Kumar A. 2017. Zika virus infects cells lining the blood-retinal barrier and causes chorioretinal atrophy in mouse eyes. JCI Insight 2: e92340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh PK, Singh S, Farr D, and Kumar A. 2019. Interferon-stimulated gene 15 (ISG15) restricts Zika virus replication in primary human corneal epithelial cells. Ocul Surf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salinas S, Erkilic N, Damodar K, Moles JP, Fournier-Wirth C, Van de Perre P, Kalatzis V, and Simonin Y. 2017. Zika Virus Efficiently Replicates in Human Retinal Epithelium and Disturbs Its Permeability. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roach T, and Alcendor DJ. 2017. Zika virus infection of cellular components of the blood-retinal barriers: implications for viral associated congenital ocular disease. J Neuroinflammation 14: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cortese M, Goellner S, Acosta EG, Neufeldt CJ, Oleksiuk O, Lampe M, Haselmann U, Funaya C, Schieber N, Ronchi P, Schorb M, Pruunsild P, Schwab Y, Chatel-Chaix L, Ruggieri A, and Bartenschlager R. 2017. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep 18: 2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar A, Giri S, and Kumar A. 2016. 5-Aminoimidazole-4-carboxamide ribonucleoside-mediated adenosine monophosphate-activated protein kinase activation induces protective innate responses in bacterial endophthalmitis. Cell Microbiol 18: 1815–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mankouri J, and Harris M. 2011. Viruses and the fuel sensor: the emerging link between AMPK and virus replication. Rev Med Virol 21: 205–212. [DOI] [PubMed] [Google Scholar]
- 34.Hardie DG 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774–785. [DOI] [PubMed] [Google Scholar]
- 35.Hardie DG, Schaffer BE, and Brunet A. 2016. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol 26: 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang BB, Zhou G, and Li C. 2009. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9: 407–416. [DOI] [PubMed] [Google Scholar]
- 37.Fay JR, Steele V, and Crowell JA. 2009. Energy homeostasis and cancer prevention: the AMP-activated protein kinase. Cancer Prev Res (Phila) 2: 301–309. [DOI] [PubMed] [Google Scholar]
- 38.Prantner D, Perkins DJ, and Vogel SN. 2017. AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling. J Biol Chem 292: 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng F, Ramos da Silva S, Huang IC, Jung JU, and Gao SJ. 2018. Suppression of Zika Virus Infection and Replication in Endothelial Cells and Astrocytes by PKA Inhibitor PKI 14–22. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jimenez de Oya N, Blazquez AB, Casas J, Saiz JC, and Martin-Acebes MA. 2018. Direct Activation of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by PF-06409577 Inhibits Flavivirus Infection through Modification of Host Cell Lipid Metabolism. Antimicrob Agents Chemother 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silwal P, Kim JK, Yuk JM, and Jo EK. 2018. AMP-Activated Protein Kinase and Host Defense against Infection. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han J, Li Y, Liu X, Zhou T, Sun H, Edwards P, Gao H, Yu FS, and Qiao X. 2018. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS One 13: e0193031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Liu X, Zhou T, Kelley MR, Edwards PA, Gao H, and Qiao X. 2014. Suppression of choroidal neovascularization through inhibition of APE1/Ref-1 redox activity. Invest Ophthalmol Vis Sci 55: 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He M, Zhang H, Li Y, Wang G, Tang B, Zhao J, Huang Y, and Zheng J. 2018. Cathelicidin-Derived Antimicrobial Peptides Inhibit Zika Virus Through Direct Inactivation and Interferon Pathway. Front Immunol 9: 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tebbe C, Chhina J, Dar SA, Sarigiannis K, Giri S, Munkarah AR, and Rattan R. 2014. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget 5: 4746–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, DeLalio LJ, Isakson BE, and Wang TT. 2016. AXL-Mediated Productive Infection of Human Endothelial Cells by Zika Virus. Circ Res 119: 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardie DG, Ross FA, and Hawley SA. 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dar S, Chhina J, Mert I, Chitale D, Buekers T, Kaur H, Giri S, Munkarah A, and Rattan R. 2017. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci Rep 7: 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontaine KA, Sanchez EL, Camarda R, and Lagunoff M. 2015. Dengue virus induces and requires glycolysis for optimal replication. J Virol 89: 2358–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirchner J, Brune B, and Namgaladze D. 2018. AICAR inhibits NFkappaB DNA binding independently of AMPK to attenuate LPS-triggered inflammatory responses in human macrophages. Sci Rep 8: 7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao E, Zhang Y, Li Q, Hao J, Egilmez NK, Suttles J, and Li B. 2016. AMPK-dependent and independent effects of AICAR and compound C on T-cell responses. Oncotarget 7: 33783–33795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM, Nicodeme E, Decrooqc J, Nepstad I, Green AS, Mondesir J, Hospital MA, Jacque N, Christodoulou A, Desouza TA, Hermine O, Foretz M, Viollet B, Lacombe C, Mayeux P, Weinstock DM, Moura IC, Bouscary D, and Tamburini J. 2015. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep 11: 1446–1457. [DOI] [PubMed] [Google Scholar]
- 53.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, and Rosen H. 2011. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146: 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, and Choe H. 2017. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci U S A 114: 2024–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dalrymple N, and Mackow ER. 2011. Productive dengue virus infection of human endothelial cells is directed by heparan sulfate-containing proteoglycan receptors. J Virol 85: 9478–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jessie K, Fong MY, Devi S, Lam SK, and Wong KT. 2004. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis 189: 1411–1418. [DOI] [PubMed] [Google Scholar]
- 57.Moser TS, Schieffer D, and Cherry S. 2012. AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog 8: e1002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saggerson D 2008. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr 28: 253–272. [DOI] [PubMed] [Google Scholar]
- 59.Hardie DG, and Pan DA. 2002. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30: 1064–1070. [DOI] [PubMed] [Google Scholar]
- 60.Soto-Acosta R, Bautista-Carbajal P, Cervantes-Salazar M, Angel-Ambrocio AH, and Del Angel RM. 2017. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: A potential antiviral target. PLoS Pathog 13: e1006257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J, Kundu M, Viollet B, and Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao B, Parnell LA, Diamond MS, and Mysorekar IU. 2017. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med 214: 2303–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan K, Lin L, Ai Q, Wan J, Dai J, Liu G, Tang L, Yang Y, Ge P, Jiang R, and Zhang L. 2018. Lipopolysaccharide-Induced Dephosphorylation of AMPK-Activated Protein Kinase Potentiates Inflammatory Injury via Repression of ULK1-Dependent Autophagy. Front Immunol 9: 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreira D, Silvestre R, Cordeiro-da-Silva A, Estaquier J, Foretz M, and Viollet B. 2016. AMP-activated Protein Kinase As a Target For Pathogens: Friends Or Foes? Curr Drug Targets 17: 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W, Li G, De W, Luo Z, Pan P, Tian M, Wang Y, Xiao F, Li A, Wu K, Liu X, Rao L, Liu F, Liu Y, and Wu J. 2018. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat Commun 9: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng Y, Liu Q, Wu Y, Ma L, Zhang Z, Liu T, Jin S, She Y, Li YP, and Cui J. 2018. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jordan TX, and Randall G. 2017. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fox CJ, Hammerman PS, and Thompson CB. 2005. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 5: 844–852. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez EL, and Lagunoff M. 2015. Viral activation of cellular metabolism. Virology 479–480: 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sag D, Carling D, Stout RD, and Suttles J. 2008. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 181: 8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giri S, Nath N, Smith B, Viollet B, Singh AK, and Singh I. 2004. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 24: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mangalam AK, Rattan R, Suhail H, Singh J, Hoda MN, Deshpande M, Fulzele S, Denic A, Shridhar V, Kumar A, Viollet B, Rodriguez M, and Giri S. 2016. AMP-Activated Protein Kinase Suppresses Autoimmune Central Nervous System Disease by Regulating M1-Type Macrophage–Th17 Axis. The Journal of Immunology. [DOI] [PubMed] [Google Scholar]
- 73.Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, Van den Pol A, Lindenbach BD, Horvath TL, and Iwasaki A. 2016. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 166: 1247–1256 e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar A, Zhang J, and Yu FS. 2006. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 117: 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.