

Abstract
In November 2018, the American Society for Mass Spectrometry hosted the Annual Fall Workshop on informatic methods in metabolomics. The Workshop included sixteen lectures presented by twelve invited speakers. The focus of the talks was untargeted metabolomics performed with liquid chromatography/mass spectrometry. In this review, we highlight five recurring topics that were covered by multiple presenters: (i) data sharing, (ii) artifacts and contaminants, (iii) feature degeneracy, (iv) database organization, and (v) requirements for metabolite identification. Our objective here is to present viewpoints that were widely shared among participants, as well as those in which varying opinions were articulated. We note that most of the presenting speakers employed different data processing software, which underscores the diversity of informatic programs currently being used in metabolomics. We conclude with our thoughts on the potential role of reference datasets as a step towards standardizing data processing methods in metabolomics.
Keywords: Metabolomics, Informatics, ASMS Fall Workshop, Metabolism
With recent advances in instrumentation, it has become routine to acquire high-quality LC-/MS-based metabolomic data.1,2 Accordingly, the field has grown exponentially and the number of facilities offering metabolomic services continues to rise. At this time, the technology is readily available to most interested researchers at relatively affordable costs around the world. Following the path of its “omic” predecessors, metabolomics is now in high demand among both technological specialists as well as biologists and clinicians who see the power of its application.
Despite the increasing amount of untargeted metabolomic data being acquired, the ability to process and interpret the data is still severely limited. It is typical to detect thousands of metabolomic features in liquid chromatography/mass spectrometry (LC/MS) experiments performed on biological samples.3,4 Yet, at this time, only a small fraction of these features can typically be identified with biochemical names.5 Moreover, in most cases, the process of going from raw metabolomic data to biochemical structures is not automated. The informatic burden can require days, weeks, or even months of time and resources.6 Even then, after extensive data analysis, there may be large numbers of “unknowns” that cannot be characterized. Thus, although many researchers now have access to metabolomic data, the challenge of interpreting the results has created major obstacles that are preventing the full potential of metabolomics from being realized. In this light, the American Society for Mass Spectrometry (ASMS) selected informatic methods in metabolomics to be the focus of the Annual Fall Workshop.
The 2018 Fall Workshop: Metabolomics Informatics
On November 29-30 2018, the ASMS hosted the Annual Fall Workshop on “Metabolomics Informatics” in San Francisco, CA. The Workshop was designed to provide a broad perspective on the state of data processing in metabolomics by leading experts in the field.
The goal of the Fall Workshop was not only to make attendees aware of the latest developments in state-of-the-art informatic resources, but also to educate investigators on proper methods for reliably interpreting datasets. Processing metabolomic data involves multiple steps (e.g., peak detection, database searching, pathway mapping). Each has unique challenges and can produce misleading results when improperly performed. Scientists who have created foundational resources to accomplish these various steps presented their contributions at the Workshop to help attendees maximize the value of their metabolomic data and hopefully prevent misinterpretation.
The Workshop was organized by Erin Baker (North Carolina State University) and Gary Patti (Washington University in St. Louis), and there were approximately 100 attendees (Figure 1). The presenting speakers were Gary Patti, Erin Baker, Jessica Prenni (Colorado State University), Timothy Garrett (University of Florida), Emma Schymanski (University of Luxembourg), David Wishart (by proxy, University of Alberta), Alla Karnovsky (University of Michigan), Oliver Fiehn (University of California-Davis), Shuzhao Li (Emory University), Justin Cross (Memorial Sloan Kettering Cancer Center), Krista Zanetti (National Cancer Institute), and Lloyd Sumner (University of Missouri). Lecture topics included vendor software solutions in metabolomics, principles of peak picking and data alignment, databases and compound identification, annotation of adducts and fragments, characterization of “unknown” metabolites, in silico modeling, pathway analysis, isotope tracer analysis, integrating data from multiple “omic” experiments, using ion mobility in metabolomics, best practices for sharing metabolomic data, automating metabolite identification, and integrating informatic resources in metabolomics. All of the presentations in the Workshop focused on untargeted metabolomics performed with (LC/MS). The utility of (GC/MS) for profiling sugars was briefly noted. Oliver Fiehn raised the question whether we would be using GC/MS for untargeted metabolomics in 10 years. No workflows using direct infusion (i.e., profiling without chromatographic separation) were presented.
Figure 1.

Attendees of the 2018 ASMS Fall Workshop on Informatic methods in metabolomics, held in San Francisco, CA
Rather than comprehensively summarizing individual lectures, here, we provide a list of some recurring themes that were discussed by multiple presenters throughout the Workshop. We use this as an opportunity to highlight ideas that were widely shared by participants, as well as topics in which clear differing opinions were expressed. We (the authors of this Review) note that the text reflects our interpretation of events at the meeting. Additionally, although our objective here is to identify shared and differing perspectives, this was not an explicit goal of the Workshop.
More Data Sharing Is Needed
There was universal enthusiasm among all participants for increased sharing of not only primary data files but also the associated meta-data. Krista Zanetti highlighted the benefits of data sharing by analogy to genomics. In 2007, the NIH introduced the Genome-Wide Association Studies (GWAS) Policy, which applied to grants generating GWAS data after the start of 2008. The NIH also introduced the database of Genotypes and Phenotypes (dbGaP) to facilitate access to GWAS data based on informed consent.7 By 2014, dbGaP had provided 2221 investigators access to 304 studies. Strikingly, this resulted in 924 publications. The example highlighted that data sharing not only allows for replication of results but can also increase the visibility of a study. It was suggested that metabolomics data could similarly be extended beyond single studies with related data-sharing practices. Emma Schymanski described that data exchange can also facilitate identification of unknowns in metabolomics by cross-annotation. In addition to the NIH-supported Metabolomics Workbench for data sharing, MetaboLights was also presented.2,8 Participants voiced enthusiasm for stronger mandates of data sharing from research journals.
High Frequency of Artifacts and Contaminants in Metabolomics Data
A point common to several presentations was the high frequency of features in LC-/MS-based metabolomics data that do not correspond to unique metabolites (Figure 2). The percentage of artifacts from informatic errors and contaminants from chemical background was estimated to be greater than 50% in some experiments.9 Multiple approaches were presented to address the issue ranging from credentialing to blank feature filtering (i.e., using a blank for background subtraction).10,11 It was shown that, because of artifacts and contaminants, total feature number is a poor evaluation metric for metabolome coverage. When metabolites have poor chromatographic peak shapes, for example, they are often reported as multiple artifactual features by software programs. It was also demonstrated that filtering out this noise improves statistics when making global comparisons of the data.
Figure 2.
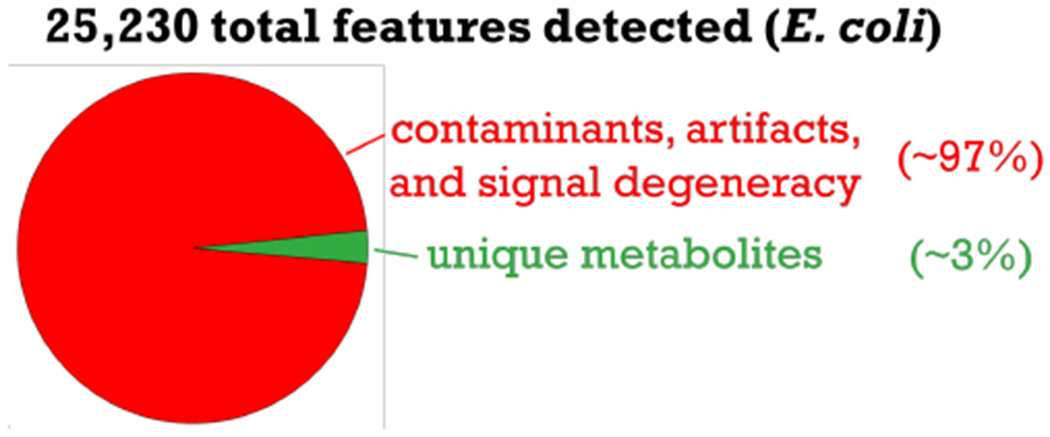
Adapted from slide presented by Gary Patti at the Fall Workshop. Chart illustrates that most features detected in the E. coli dataset shown arise from contaminants, artifacts, and signal degeneracy (i.e., adducts, naturally occurring isotopes, dimers, etc.)
High Frequency of Redundant Signals in Metabolomics Data
It was agreed that an additional source of considerable complexity when analyzing metabolomics data is redundant signals arising from the same metabolite. Redundant signals result from naturally occurring isotopes, in-source fragmentation, adducts from salts such as sodium, analyte-analyte dimers, etc. (Figure 3). Similar to artifacts and contaminants, signal redundancies complicate compound identification, statistical analyses, and pathway mapping. Multiple software solutions were presented for annotating redundant signals including CAMERA, RamClust, MS-FLO, Mz.Unity, Binner, and various vendor options.12,15 It was noted that CAMERA (and its predecessor16) has been one of the mostly widely used software programs for the analysis of feature degeneracies. It was also brought up that rigorous comparisons of the software platforms for annotating feature degeneracies, some of which are conceptually similar, have not been performed. The question was raised whether different laboratories need to reimplement analogous algorithms. Notwithstanding, most workflows presented highlighted that it is critical to filter redundant features when processing datasets. The percentage of metabolites represented in a dataset after filtering varied by presenter and sample type.
Figure 3.
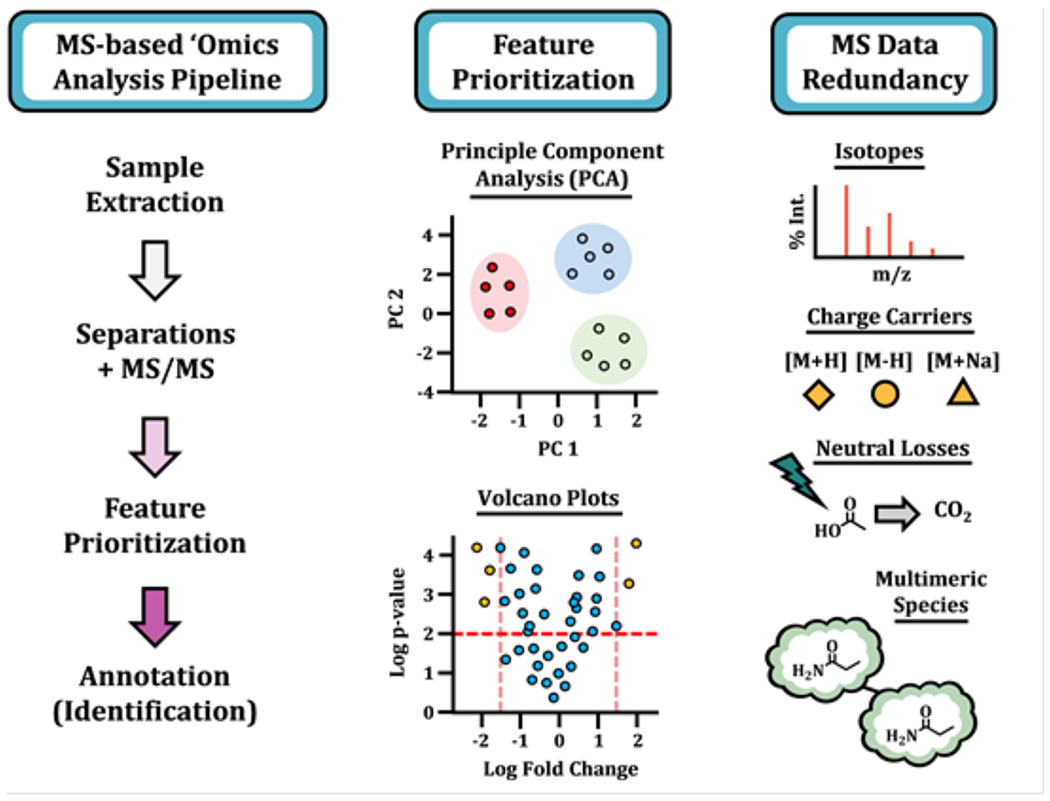
A schematic of an untargeted “omics” workflow, illustrating the analysis pipeline, two methods for feature prioritization, and several associated sources of signal degeneracy.
Organization of Reference Data: Global or Specialized Databases?
Although it is common to perform untargeted metabolomics by searching m/z values in comprehensive databases, some argued that this was a mistake. It was stated that many compounds in databases such as ChemSpider may not occur in the biological system being investigated. Accordingly, searching these databases may result in a high number of false positives. Similarly, it was highlighted that databases like METLIN, MoNA, and GNPS mix compounds from many different organisms like plants, fungi, and bacteria as well as compounds that do not occur naturally in cells such as drugs and pesticides.17,18 As David Wishart argued, “yeast don’t wear cosmetics.” These databases may therefore similarly yield a high rate of false hits. As an alternative, it was suggested that databases be developed for specific applications. KNApSAck, KEGG, MetaboLights, MetaCyc, for example, were presented as databases with species or source information.19–21 There was disagreement, however, about whether m/z values from untargeted metabolomics should be searched in general databases or specialized databases. The argument against the latter was that it might limit discovery of unexpected compounds, which is a major goal of untargeted metabolomics.
Data Required to Structurally Identify Metabolites
There was a strong consensus among participants that accurate mass data alone are insufficient for structural identification of a metabolite. It was agreed that additional data (such as MS/MS fragmentation spectra, chromatographic retention times, collisional cross sections from ion mobility, and NMR results) increase identification confidence. Several participants discussed quantifying confidence in metabolite identification by using levels, such as those suggested by the Compound Identification work group of the Metabolomics Society.5,22–24 Notably, however, there was not a clear agreement on what data are needed for each level or whether levels suggested by the Compound Identification work group should be refined. Some members of the audience expressed concern that matching accurate mass, MS/MS data, retention time, collisional cross section, and NMR spectra to an authentic reference standard was often impractical.
Attendee Feedback
Attendees had an opportunity to provide anonymous feedback after the Workshop via an online survey. Multiple respondents noted the wide range of expertise among the audience, which complicated question and answer sessions. It was also suggested that the addition of hands-on exercises for data analysis might be useful for future ASMS Workshops. Overall, most attendees expressed a high level of enthusiasm about their experiences at the meeting.
Perspective on the Future
The success of any big-data science relies upon establishing standardized practices for sample handling, data acquisition, data processing, and data sharing. Such standardized practices are important for several reasons. First, they facilitate interpretation of the data by other laboratories. Second, they enable metacomparisons of existing datasets. Third, they promote efficiency by preventing multiple laboratories from having to repeat the same experiment. Finally, standardized practices make data more accessible to researchers from other fields with less expertise.
A major challenge in standardizing data processing in metabolomics is the large amount of available software with overlapping functionality. Counting only the commonly used programs that are freely available, there are over 175 pieces of software designed to accomplish various functions within the metabolomics data processing workflow (e.g., peak picking, de-isotoping, adduct determinations, etc.).25 Considering the number of different ways in which many of these modular tools can be combined, it amounts to an even larger number of unique processing pipelines. The diversity in data processing algorithms was evident at the Fall Workshop, with each of the twelve speakers presenting unique software solutions. With such a heterogeneous landscape, a critical question for standardization is how software tools designed to accomplish the same function compare. Ideally, despite the application of different software platforms, users would obtain similar results from the processing of the same dataset. Yet, recent side-by-side comparisons of some of the most widely used metabolomic software indicate that concordance is less than 50%.26,28 At the current time, however, comparisons of existing software tools have been limited. Although a rigorous evaluation of informatic options would certainly benefit the community, such efforts are challenging because most research groups do not have expertise across multiple software platforms. Most importantly, without an accurate understanding of input parameters, a software output may not reflect optimal performance and conclusions based on the program’s results will therefore be of minimal or no value.
A potential path to standardization may be to establish reference datasets for rigorous benchmarking of metabolomic software performance. The reference dataset could be processed by different researchers having extensive expertise with specific software programs, thereby enabling accurate head-to-head comparison of output files. A critical question will be what sample(s) should be used to create the reference dataset(s). An advantage of using chemical standards is that the composition of the dataset will be well defined. On the other hand, chemical standards alone may not reflect the complexity of a biological sample. A challenge of using a biological sample to generate a reference dataset will be deciding which specimen to use (e.g., bacteria, plants, humans, biofluid, tissue, etc.). It is likely that different software tools are better suited for specific sample types, extraction methods, separation strategies, and instrument platforms, all of which cannot be captured in a single reference dataset. Notwithstanding, even a non-optimal reference dataset that was broadly adopted by the community would represent a step forward in the standardization of data processing methods in metabolomics.
We note that the perspective expressed in this section reflects only that of the authors and not necessarily the attendees or other speakers at the Fall Workshop.
References
- (1).Cho K; Mahieu NG; Johnson SL; Patti GJ Curr Opin Biotechnol 2014, 28, 143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS; Sumner S; Subramaniam S Nucleic Acids Res 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Johnson CH; Ivanisevic J; Benton ΗP; Siuzdak G Anal Chem 2015, 87, 147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mahieu NG; Genenbacher JL; Patti GJ CurrOpin Chem Biol 2016, 30, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blazenovic I; Kind T; Ji J; Fiehn O Metabolites 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tautenhahn R; Cho K; Uritboonthai W; Zhu Z; Patti GJ; Siuzdak G Nat Biotechnol 2012, 30, 826–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mailman MD; Feolo M; Jin Y; Kimura M; Tryka K; Bagoutdinov R; Hao L; Kiang A; Paschall J; Phan L; Popova N; Pretel S; Ziyabari L; Lee M; Shao Y; Wang ZY; Sirotkin K; Ward M; Kholodov M; Zbicz K; Beck J; Kimelman M; Shevelev S; Preuss D; Yaschenko E; Graeff A; Ostell J; Sherry ST Nat Genet 2007, 39, 1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kale NS; Flaug K; Conesa P; Jayseelan K; Moreno P; Rocca-Serra P; Nainala VC; Spicer RA; Williams M; Li X; Salek RM; Griffin JL; Steinbeck C Curr Protoc Bioinformatics 2016, 53, 14 13, 1–18. [DOI] [PubMed] [Google Scholar]
- (9).Mahieu NG; Patti GJ Anal Chem 2017, 89, 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mahieu NG; Huang X; Chen YJ; Patti GJ Anal Chem 2014, 86, 9583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Patterson RE; Kirpich AK; P. KJ; S. K; M. MA; K. C; N. S; M. ML; T. G; A. YR Metabolomics 2017, 13.29249917 [Google Scholar]
- (12).Kuhl C; Tautenhahn R; Bottcher C; Larson TR; Neumann S .Anal Chem 2012, 84, 283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Broeckling CD; Afsar FA; Neumann S; Ben-Hur A; Prenni JE Anal Chem 2014, 86, 6812–7. [DOI] [PubMed] [Google Scholar]
- (14).DeFelice BC; Mehta SS; Samra S; Cajka T; Wancewicz B; Fahrmann JF; Fiehn O Anal Chem 2017, 89, 3250–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mahieu NG; Spalding JL; Gelman SJ; Patti GJ Anal Chem 2016, 88, 9037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tautenhahn R; Bottcher C; Neumann S; Springer Berlin Fleidelberg: Berlin, Heidelberg, 2007, pp 371–380. [Google Scholar]
- (17).Smith CA; O’Maille G; Want EJ; Qin C; Trauger SA; Brandon TR; Custodio DE; Abagyan R; Siuzdak G Ther Drug Monit 2005, 27, 747–51. [DOI] [PubMed] [Google Scholar]
- (18).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu WT; Crusemann M; Boudreau PD; Esquenazi E; Sandoval-Calderon M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu CC; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw CC; Yang YL; Humpf HU; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; P CAB; Torres-Mendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining ΟB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodriguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard PM; Phapale P; Nothias LF; Alexandrov T; Litaudon M; Wolfender JL; Kyle JE; Metz TO; Peryea T; Nguyen DT; VanLeer D; Shinn P; Jadhav A; Muller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BO; Pogliano K; Linington RG; Gutierrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N Nat Biotechnol 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nakamura K; Shimura N; Otabe Y; Hirai-Morita A; Nakamura Y; Ono N; Ul-Amin MA; Kanaya S Plant Cell Physiol 2013, 54, e4. [DOI] [PubMed] [Google Scholar]
- (20).Kanehisa M; Furumichi M; Tanabe M; Sato Y; Morishima K Nucleic Acids Res 2017, 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Caspi R; Billington R; Fulcher CA; Keseler IM; Kothari A; Krummenacker M; Latendresse M; Midford PE; Ong Q; Ong WK; Paley S; Subhraveti P; Karp PD Nucleic Acids Res 2018, 46, D633–D639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schymanski EL; Jeon J; Guide R; Fenner K; Ruff M; Singer ΗP; Hollender J Environ Sci Technol 2014, 48, 2097–8. [DOI] [PubMed] [Google Scholar]
- (23).Creek DJ; Dunn WB; Fiehn O; Griffin JL; Hall RD; Lei Z; Mistrik R; Neumann S; Schymanski EL; Sumner LW; Trengove R; Wolfender JL Metabolomics 2014, 10, 350–353. [Google Scholar]
- (24).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Metabolomics 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Spicer R; Salek RM; Moreno P; Canueto D; Steinbeck C Metabolomics 2017, 13, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Myers OD; Sumner SJ; Li S; Barnes S; Du X Anal Chem 2017, 89, 8689–8695. [DOI] [PubMed] [Google Scholar]
- (27).Li Z; Lu Y; Guo Y; Cao H; Wang Q; Shui W Anal Chim Acta 2018, 1029, 50–57. [DOI] [PubMed] [Google Scholar]
- (28).Rafiei A; Sleno L Rapid Commun Mass Spectrom 2015, 29, 119–27. [DOI] [PubMed] [Google Scholar]