

Abstract
Optic fissure closure defects result in uveal coloboma, a potentially blinding condition affecting between 0.5 and 2.6 per 10,000 births that may cause up to 10% of childhood blindness. Uveal coloboma is on a phenotypic continuum with microphthalmia (small eye) and anophthalmia (primordial/no ocular tissue), the so-called MAC spectrum. This review gives a brief overview of the developmental biology behind coloboma and its clinical presentation/spectrum. Special attention will be given to two prominent, syndromic forms of coloboma, namely, CHARGE (Coloboma, Heart defect, Atresia choanae, Retarded growth and development, Genital hypoplasia, and Ear anomalies/deafness) and COACH (Cerebellar vermis hypoplasia, Oligophrenia, Ataxia, Coloboma, and Hepatic fibrosis) syndromes. Approaches employed to identify genes involved in optic fissure closure in animal models and recent advances in live imaging of zebrafish eye development are also discussed.
Introduction
The eye develops starting the third week of human gestation as an evagination of the diencephalic neuroepithelium to form the optic vesicle, which subsequently invaginates to form the optic cup. During evagination, the optic vesicle comes in close proximity to the surface ectoderm, which subsequently thickens, forming the lens placode. As the optic vesicle invaginates to give rise to a bilayered optic cup (innermost neural retina and outermost retinal pigment epithelium, RPE), the lens placode also invaginates in a coordinated morphogenetic process to form the primordial crystalline lens, the lens vesicle (Figure 1). Optic vesicle invagination, however, is asymmetric, such that a ventral opening, the optic fissure, forms around the fifth week of human gestation (O’Rahilly 1983). For the eye to develop normally, the two edges of the fissure must approximate and fuse. If the optic fissure margins fail to fuse, uveal coloboma, a potentially blinding congenital malformation, ensues.
Figure 1.
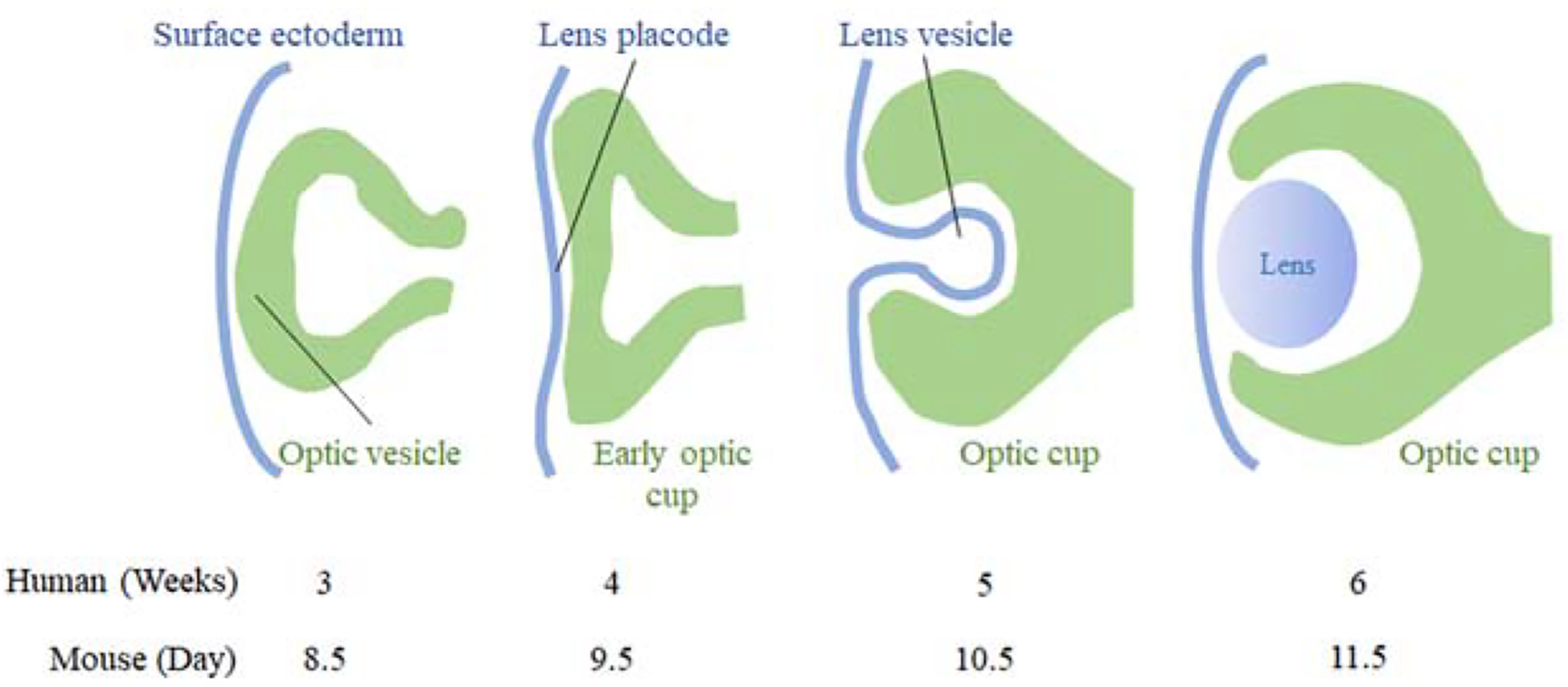
Schematic of embryonic eye development.
Clinically, coloboma presents as lens, iris, neural retina/RPE/choroid and/or optic nerve defects in the inferior (and often slightly nasal) quadrant (Figure 2A, B). The visual impact is largely determined by whether the defect affects macular development. Coloboma may occur in normal-sized eyes but it is also frequently accompanied by small eyes (microphthalmia), or even rudimentary eye tissue or clinically absent (anophthalmia) eyes (i.e., the microphthalmia anophthalmia-coloboma disease or MAC spectrum). In the broadest sense, any eye with an antero-posterior axial length two standard deviations shorter than the age-appropriate mean is microphthalmic, even if this may not be clinically obvious to inspection. Mild forms of optic fissure closure defects include peaking of the pupil towards the inferonasal quadrant and minor RPE defects seen on fundoscopy (Figure 2C, D). Variable expressivity and incomplete penetrance often exist within families where multiple affected individuals are present and the phenotype may be asymmetrically severe within an individual. Both observations suggest that genetic and/or environmental as well as stochastic processes influence the phenotype.
Figure 2.
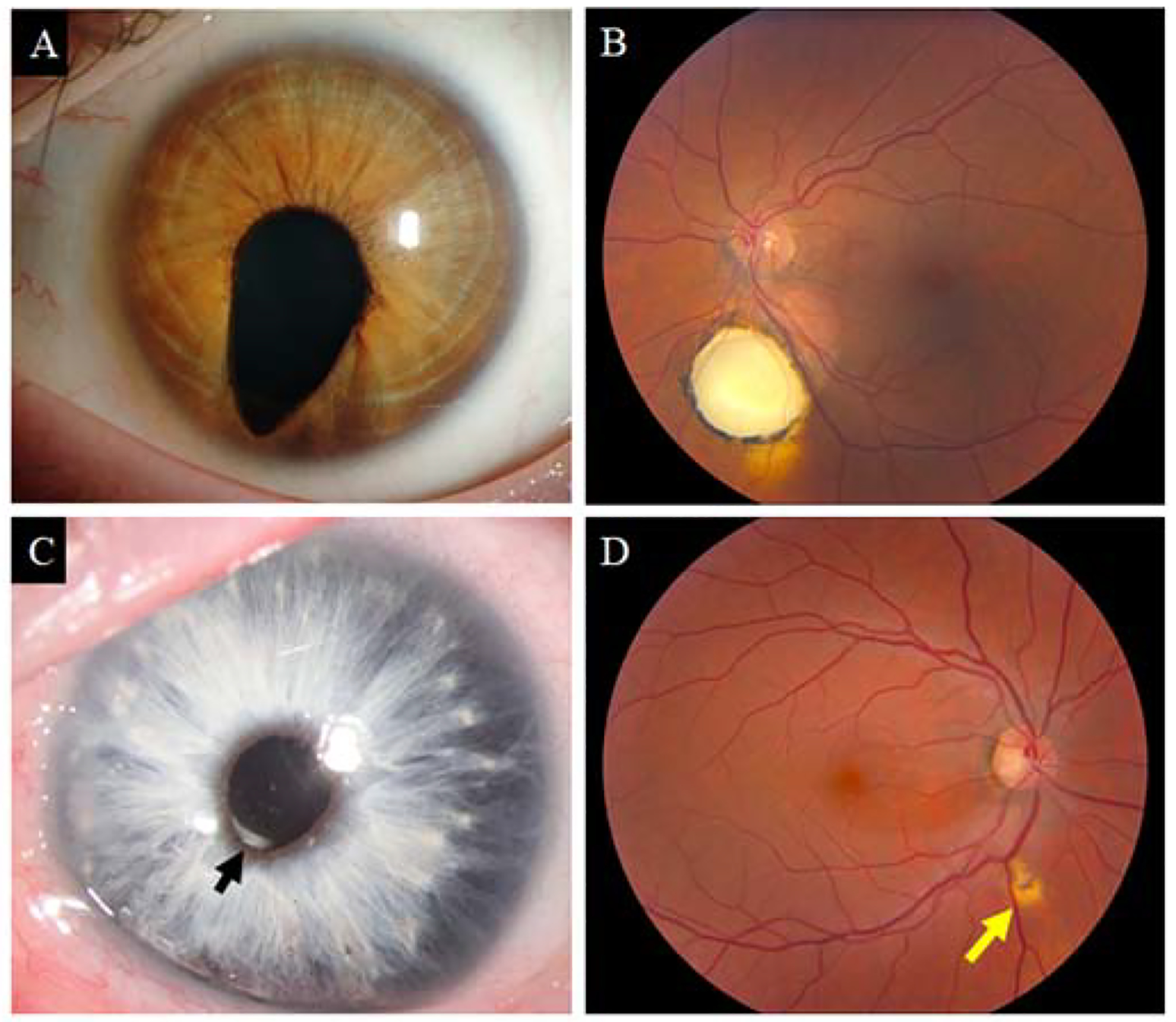
Clinical presentations of uveal coloboma. (A) Typical iris coloboma of a left eye. Note the inferonasal positioning of the coloboma, corresponding to the position of the optic fissure. (B) Typical chorioretinal coloboma inferior to the optic nerve in a patient with excellent visual acuity. (C) Microform of iris coloboma in a patient with Waardenburg syndrome, type 2A. Note slight peaking of the pupil of the inferonasal quadrant (arrow). (D) Microform of a chorioretinal coloboma in the asymptomatic mother of a patient with bilateral nonsyndromic coloboma.
The term “coloboma” is often used in the nomenclature of other eye defects such as the “eyelid coloboma” of Treacher-Collins syndrome and the “macular coloboma” in severe forms of Leber congenital amaurosis; the “morning glory” anomaly is sometimes erroneously referred to as “optic nerve coloboma.” Each of these conditions is characterized by missing and/or dysplastic tissue in an ocular or adnexal structure. However, they all arise from processes quite distinct from optic fissure closure and should not be confused with uveal coloboma. For the purposes of this article, the terms “uveal coloboma” and “coloboma” can be considered synonymous.
Coloboma presents with considerable genetic heterogeneity; it is associated with many chromosomal abnormalities and is likely influenced by environmental factors that are reviewed in detail in the literature (Chang et al., 2006). Mutations in several developmentally-regulated genes encoding transcription factors, cell-cell adhesion proteins, growth factors, cytoskeletal proteins and enzymes have been reported in patients with uveal coloboma. We performed pathway analysis with human MAC associated genes using Enrichr website (https://amp.pharm.mssm.edu/Enrichr/) to identify signaling pathways that significantly contribute towards the human MAC phenotype. To test the accuracy of our analysis we checked the “Disease/Drug” tab and the top entry in the ClinVar option was “anophthalmia microphthalmia syndrome” with an adjusted P-value of 3.856e-24, the top entry in the “Cell Types” tab was retina (P=0.000003883) according to the Human Gene Atlas option (Figure 3). Top ten pathways based on the adjusted P-value in the KEGG 2019 option according to our analysis are show in Figure 3. Some usual suspects like TGF-β signaling, adherens junctions and Hedgehog signaling were observed, providing credence to our analysis. The most significant and rather unexpected was the Hippo signaling pathway, which is involved in the regulation of organ size (Harvey et al., 2003; Wu et al., 2003). Some recent studies have shown the association of Hippo signaling genes like FAT1, RERE and YAP1 with human MAC phenotype (Lahrouchi et al., 2019; Fregeau et al., 2016; Williamson et al., 2014).
Figure 3.
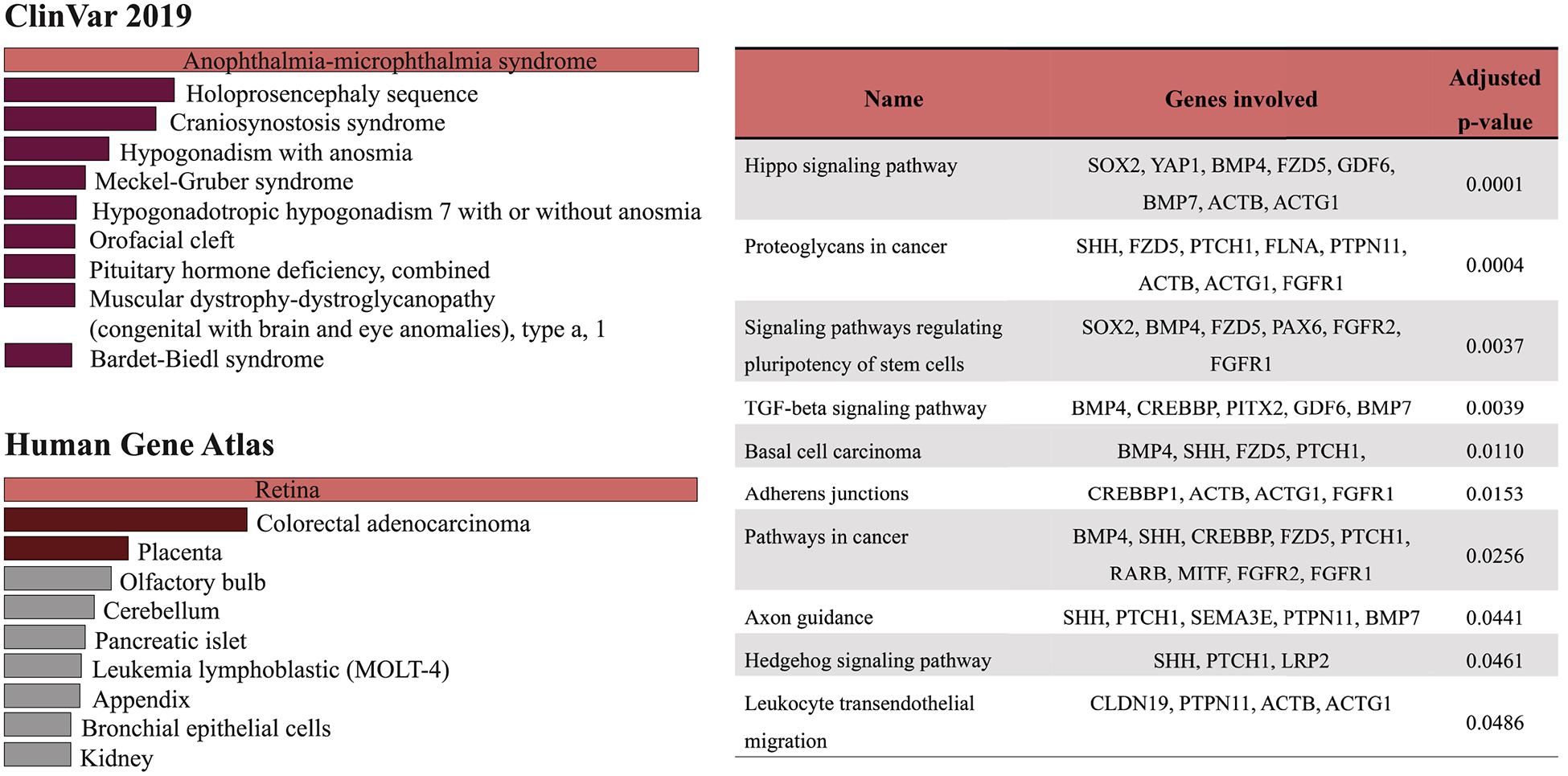
Pathway analysis was performed using online tool Enrichr (https://amp.pharm.mssm.edu/Enrichr/enrich#) where ninety genes described Table 1 and 2 were uploaded to the site. The genes were assigned to the most relevant disease phenotype by the ClinVar and Cell Types by Human Gene Atlas options. KEGG 2019 Human option was used to identify the significant pathways that were arranged in the order of decreasing P value as shown in the table.
Genetic diversity may help explain, in part, the phenotypic complexity of the disease and suggests that multiple developmental mechanisms (e.g., cell autonomous, non-cell autonomous) may be at work to produce a similar human disease presentation. Because developmentally- regulated genes are expressed in discrete anatomical locations at defined times, genetic diversity may also explain specific phenotypic patterns. For example, mutations in the paired box 2 (PAX2) transcription factor gene in Papillorenal Syndrome tend to result in colobomas most often affecting the optic nerve, as this gene is expressed prominently in the optic stalk, the precursor of the optic nerve (Sanyanusin et al., 1995; Pichaud and Desplan, 2002).
MAC may be isolated (non-syndromic) or associated with other systemic abnormalities (syndromic). Nomenclature classifications have been made in the Online Mendelian Inheritance in Man (OMIM) based, in part, on this distinction, as well as on the gene(s) associated with the phenotype (Table 1). The occurrence of coloboma with a myriad of syndromic conditions suggests that the process of optic fissure closure shares the same developmental pathways at play in the development of other organ systems. Using the example of PAX2 in Papillorenal Syndrome again, both the kidney and the eye rely on this transcription factor during development and PAX2 mutations result in congenital ocular and renal defects. The explanations provided by these patterns of expression, however, are not absolute. For example, genes associated with syndromic forms of coloboma may also present with isolated microphthalmia and/or coloboma and some syndromic forms of MAC have circumscribed phenotypes despite a much wider pattern of mutant gene expression.
Table 1:
Classification of coloboma genes based on MAC phenotype. The number preceding the gene name denotes the OMIM nomenclature# (CHX10:MCOP2).
| MAC phenotype | OMIM Nomenclature | OMIM #. Gene | |
|---|---|---|---|
| Isolated microphthalmia | MCOP | 1. ch 14q32, | 5. MFRP |
| 2. CHX10 | 6. PRSS56 | ||
| 3. RAX | 7. GDF3 | ||
| 4. GDF6 | 8. ALDH1A3 | ||
| Syndromic microphthalmia | MCOPS | 1. NAA10 | 8. 6q21 |
| 2. BCOR | 9. STRA6 | ||
| 3. SOX2 | 10. - | ||
| 4. ANOP1 | 11. VAX1 | ||
| 5. OTX2 | 12. RARB | ||
| 6. BMP4 | 13. HMGB3 | ||
| 7. HCCS | 14. MAB21L2 | ||
| Isolated microphthalmia with coloboma | MCOPCB | 1. Chr.X | 5. SHH |
| 2. 15q12-q15 | 6. GDF3 | ||
| 3. CHX10 | 7. ABCB6 | ||
| 4. - | 8. STRA6 | ||
| 9. TENM3 | |||
Excellent, detailed reviews on non-syndromic coloboma have been published (Williamson and FitzPatrick, 2014; Reis and Semina, 2015) and this review only briefly discusses the genetics of non-syndromic coloboma. Most of our attention will be on the etiology and pathogenesis of syndromic coloboma, particularly in CHARGE and COACH syndromes, in which coloboma is highly prevalent.
Genetics of non-syndromic uveal coloboma: isolated microphthalmia (MCOP) and isolated microphthalmia with coloboma (MCOPCB)
Non-syndromic forms of coloboma can present in dominant, recessive, or X-linked patterns, although, most often, coloboma occurs sporadically, and the precise inheritance pattern is difficult to discern. A comprehensive list of genes associated to date with non-syndromic coloboma is presented in Table 2 and we refer the interested readers to the in-depth review of the genetics of non-syndromic coloboma by Williamson and FitzPatrick (2014). Most of the genes associated with non-syndromic coloboma tend to be eye-specific transcription factors that are involved in developmental processes.
Table 2.
Genes associated with non-syndromic coloboma
| Gene | MAC phenotype | OMIM | Inheritance | Syndrome/associated phenotype -* | Reference |
|---|---|---|---|---|---|
| ABCB6 | Microphthalmia, Coloboma of iris, retina and choroid | 605452 | AD | - | Wang et al., 2012 |
| ALDH1A3 | Microphthalmia, retinal coloboma | 600463 | AR | - | Fares-Taie et al., 2013; Yahyavi et al., 2013 |
| ATOH7 | Microphthalmia | 609875 | AR | Persistent hyperplastic primary vitreous | Khan et al., 2012 |
| CRYAA | Coloboma of iris, Cataract | 123580 | AD | - | Beby et al., 2007 |
| FZD5 | Microphthalmia, Coloboma of iris, retina and choroid | 601723 | AD | - | Liu et al., 2016 |
| IPO13 | Microphthalmia, coloboma of iris, cataract | 610411 | AR | - | Huang et al., 2018 |
| LCP1 | Coloboma of iris and choroid | 153430 | AD | - | Rainger et al., 2017 |
| MAF | Coloboma of iris, Cataract | 177075 | AD | Cataract | Jamieson et al., 2002 |
| Mir204 | Coloboma of iris | 610942 | AD | Retinal dystrophy | Conte et al., 2015 |
| PAX6 | Coloboma of iris, retina, choroid and optic nerve Anophthalmia | 607108 | AD AR | Aniridia, Morning glory disc anomaly, Peter’s Anomaly, Anterior segment dysgenesis, Cataract with late-onset corneal dystrophy, Foveal hypoplasia, Keratitis, Optic nerve hypoplasia. | Azuma et al., 1996; Azuma et al., 2003; Glaser et al., 1994 |
| RARB | Microphthalmia | 180220 | AR, AD | diaphragmatic hernia, pulmonary hypoplasia, and cardiac defects | Srour et al., 2013 |
| RAX | Microphthalmia, anophthalmia, coloboma of optic nerve | 601881 | AR | - | Voronina et al., 2004 |
| RBP4 | Microphthalmia, Coloboma of iris, choroid and retina | 180250 | AD AR | Retinal dystrophy, comedogenic acne syndrome | Cukras et al., 2012; Chou et al., 2015. |
| SALL2 | Microphthalmia, Coloboma of iris, retina and choroid | 602219 | AR | - | Kelberman et al., 2014 |
| SHH | Microphthalmia with Coloboma of retina, choroid, iris and retina | 600725 | AD | Holoprosencephaly | Schimmenti et al. 2003; Nanni et al., 1999 |
| SIX6 | Coloboma of iris, choroid and optic nerve | 606326 | AR | Optic disc anomalies, macular atrophy and reduces retinal ganglion cell differentiation | Aldahmesh et al., 2013; Yariz et al., 2015 |
| STRA6 | Microphthalmia, anophthalmia, coloboma | 610745 | AR | - | Casey et al., 2011 |
| TENM3/ODZ3 | Microphthalmia, iris coloboma | 610083 | AR | - | Aldahmesh et al., 2012 |
| VSX2 | Microphthalmia, anophthalmia, iris coloboma | 142993 | AR | cataracts | Kohn et al., 1988; Bar-Yosef et al., 2004 |
No associated phenotype reported with this gene.
AD: Autosomal dominant, AR: Autosomal recessive.
Genetics of syndromic forms of coloboma: syndromic microphthalmia (MCOPS) loci, CHARGE, and COACH
The genes associated with syndromic forms of coloboma tend to be widely expressed and generally have pleiotropic effects. A list of all the syndromes involving coloboma is presented in Table 3. The prevalence varies among syndromes, with CHARGE and COACH being strongly associated with coloboma; coloboma, in fact, is one of their diagnostic criteria. However, genotyped-confirmed cases of CHARGE and COACH in the absence of coloboma are also observed.
Table 3.
Human syndromes with eye coloboma
| Gene | MAC phenotype | OMIM | Inheritance | Syndrome/associated phenotype | Reference |
|---|---|---|---|---|---|
| ACTB | Coloboma of iris and retina | 102630 | AD | Baraitser-Winter syndrome 1, Dystonia, juvenile-onset | Riviere et al., 2012 |
| ACTG1 | Coloboma of iris and choroid | 102560 | AD | Baraitser-Winter syndrome 2, Deafness | Riviere et al., 2012 |
| ALG3 | Coloboma of iris | 608750 | AR | Congenital disorder of glycosylation, type IV (CDGS type IV) | Korner et al., 1999 |
| ALX1 | Microphthalmia, coloboma of iris and eye lid | 601527 | AR | Frontofacionasal dysostosis | Uz et al., 2010 |
| BCOR | Microphthalmia, Coloboma of iris, choroid and optic nerve | 300485 | XL Recessive | Lenz syndrome | Ng et al., 2004 |
| BMP4 | Microphthalmia, anophthalmia, coloboma | 112262 | AD | Orofacial cleft facial dysmorphism | Bakrania et al., 2008; Reis et al., 2011 |
| BMP7 | Microphthalmia, anophthalmia, Coloboma of retina, choroid and optic nerve | 112267 | AD | Developmental delay, deafness, scoliosis, and cleft palate | Wyatt et al., 2010 |
| C12orf57 | Coloboma of iris, retina and choroid | 615140 | AR | Temtamy syndrome | Temtamy et al., 1996 |
| CHD7 | Coloboma of iris, retina, choroid and optic nerve, eye lid (rarely) | 608892 | AD | CHARGE syndrome | Vissers et al., 2004 |
| CLDN19 | Pseudo coloboma? | 610036 | AR | Hypomagnesemia, renal, with ocular involvement | Khan et al., 2018 |
| CREBBP | Microphthalmia, coloboma of iris, choroid and retina | 600140 | AD/del | Rubinstein-T aybi syndrome | Ge et al., 1995 |
| CRIM1 | Coloboma of iris, retina, choroid and optic nerve | 606189 | AD | Toker et al., 2003 | |
| DPYD | Microphthalmia, coloboma of iris and choroid | 612779 | AR | Dihydropyrimidine dehydrogenase deficiency | Van Gennip et al., 1994; Meinsma et al. 1995 |
| FAT1 | Microphthalmia, coloboma of choroid and retina | 600976 | AR | Glomerulonephropathy, cutaneous syndactyly | Lahrouchi et al., 2019 |
| FBN1 | Coloboma of lens; rarely iris, retina and optic disk coloboma | 134797 | AR | Marfan syndrome | Nemet et al., 2006 |
| FBN2 | Coloboma of retina and choroid | 612570 | AD | Congenitalcontractural arachnodactyly | Bard 1979 |
| FGFR1 | Microphthalmia, anophthalmia, coloboma of iris and eye lid | 136350 | Somatic Mosaicism | Oculocerebrocutaneous syndrome Encephalocraniocutaneo us lipomatosis | Prontera et al., 2009 |
| FGFR2 | Coloboma of iris | 176943 | AD | Multiple | Graul-Neumann et al., 2017 |
| FLNA | Coloboma of iris, retina and optic nerve | 300017 | XL Dominant XL Recessive | Multiple | Robertson et al., 2003 |
| FOXA2 | Choroidal coloboma | 600288 | de novo | hypopituitarism, hyperinsulinism and endoderm-derived organ abnormalities | Giri et al., 2017 |
| FOXE3 | Microphthalmia, Coloboma of iris, retina and optic disc | 601094 | AD, AR | Anterior segment dysgenesis, Cataract, aniridia | Khan et al., 2016 |
| FREM1 | Anophthalmia, microphthalmia and coloboma of upper eyelid | 608944 | AR | MOTA syndrome | Marles et al., 1992 |
| GDF3 | - | 606522 | AD | Klippel-Feil Syndrome3, skeletal anomalies | Ye et al., 2010 |
| GDF6 | Microphthalmia, Coloboma of iris, retina, choroid and optic nerve | 601147 | AD AR | Klippel-Feil Syndrome1 Leber congenital amaurosis | Asai-Coakwell et al., 2009 |
| GJA8 | Microphthalmia, | 600897 | AD | Congenital cataracts | Ceroni et al., 2019 |
| HMGB3 | Coloboma of iris, retina and choroid | 300193 | XL | microcephaly, short stature, and intellectual disability | Scott et al., 2014 |
| HMX1 | Coloboma of iris, retina and choroid | 142992 | AR | Oculoauricular syndrome | Schorderet et al., 2008 |
| IGBP1 | Coloboma of iris and optic nerve | 300139 | XL Recessive | Corpus callosum defect, mental retardation, and micrognathia | Graham et al., 2003 |
| KCTD1 | Coloboma of iris and eye lid | 613420 | AD | Scalp-ear-nipple syndrome (Finlay-Marks syndrome) | Sobreira et al., 2006 |
| KMT2D | Coloboma of iris, retina, choroid and optic nerve | 602113 | AD | Kabuki syndrome | Ming et al., 2003 |
| LINC00237 | Coloboma of retina and eye lid | 614992 | AD | MOMO syndrome | Moretti-Ferreira et al., 1993 |
| LRP2 | Coloboma of iris | 600073 | AR | Donnai-Barrow syndrome | Avunduk et al., 2000 |
| MAB21L2 | Anophthalmia, microphthalmia, Coloboma of iris and retina | 604357 | AD AR | skeletal dysplasia | Rainger et al., 2014 |
| MITF | Microphthalmia, coloboma | 156845 | AR | COMMAD syndrome | George et al., 2016 |
| MKS1 | Microphthalmia, coloboma of iris | 609883 | AR | Meckel-Gruber syndrome | Slaats et al., 2016 |
| MSX2 | Coloboma of iris, retina and choroid | 123101 | de novo duplication | Craniosynostosis 2, Parietal foramina 1 | Plaisancie et al., 2015 |
| OTX2 | Microphthalmia | 600037 | AD | Retinal dystrophy, early-onset, with or without pituitary dysfunction | Ragge et al., 2005a |
| PACS1 | Coloboma of iris and optic nerve | 615009 | de novo (c.607C>T) | Schuurs-Hoeijmakers syndrome | Pefkianaki et al., 2018 |
| PAX2 | Coloboma of optic nerve | 167409 | AD | Papillorenal Syndrome | Sanyanusin et al., 1995 |
| PDE6D | Coloboma of optic nerve | 602676 | AR | Joubert syndrome 22 | Thomas et al., 2014 |
| PIGL | Coloboma of retina and choroid | 605947 | AR | CHIME syndrome | Ng et al., 2012 |
| PITX2 | Microphthalmia, coloboma of iris | 601542 | AD | Rieger syndrome, type1 | Ozeki et al., 1999 |
| POMT1 | Microphthalmia, coloboma | 607423 | AR | W alker-W arburg syndrome | Beltran-Valero de Bernabe et al., 2002 |
| PQBP1 | Coloboma of retina, choroid and optic disc | 300463 | XL Recessive | Renpenning syndrome | Martinez-Garay et al., 2007 |
| PRR12 | Coloboma of iris. | 616633 | de novo (LOF) | Leduc et al., 2018 | |
| PTCH1 | Coloboma of iris | 601309 | AD/Sporadic | Holoprosencephaly, Basal cell nevus syndrome | Chassaing et al., 2016 |
| PTPN11 | Coloboma of iris, retina and optic nerve | 176876 | AD | Noonan syndrome | Lee et al., 1992 |
| PUF60 | Coloboma of iris, retina and choroid | 604819 | de novo | Verheij syndrome | Graziano et al., 2017 |
| RAB3GAP1 | Microphthalmia, anophthalmia, coloboma of iris, choroid, retina, optic nerve | 602536 | AR | Warburg micro syndrome 1 | Aligianis et al., 2005 |
| RERE | Coloboma | 605226 | de novo | Neurodevelopmental disorder with or without anomalies of the brain, eye, or heart | Fregeau et al., 2016 |
| SALL1 | Coloboma of retina and choroid | 602218 | AD | Townes-Brocks syndrome | Botzenhart et al., 2005 |
| SALL4 | Microphthalmia, coloboma of iris, choroid and optic nerve | 607343 | AD | Duane-radial ray syndrome (Okihiro syndrome; acroreno-ocular syndrome) | Borozdin et al., 2004 |
| SCLT1/TBC1D32 | Retinochoroideal lacunae of colobomatous | 611399 | XL | Orofaciodigital syndrome IX | Gurrieri et al., 1992 |
| SEMA3E | Coloboma of iris, retina and optic nerve | 608166 | AD | CHARGE syndrome | Lalani et al., 2004 |
| SIX3 | Microphthalmia, coloboma of iris, retina, choroid and macula | 603714 | AD | Holoprosencephaly2, Schizencephaly | Wallis et al., 1999 |
| SMOC1 | Microphthalmia, Coloboma of retina and limb anomalies | 608488 | AR | limb anomalies | Abouzeid et al., 2011; Okada et al., 2011 |
| SMO | Microphthalmia, coloboma of iris | 601500 | AD | Curry-Jones syndrome | Twigg et al., 2016 |
| SOX2 | Anophthalmia, microphthalmia, coloboma of iris, retina and choroid | 184429 | AD | Optic nerve hypoplasia and abnormalities of the central nervous system | Fantes et al., 2003; Ragge et al., 2005b |
| SOX3 | Microphthalmia, Coloboma | 313430 | XL (germline mosaicism) | Panhypopituitarism Mental retardation | Jelsig et al., 2018 |
| SPINT2 | Coloboma of optic nerve | 605124 | AR | Congenital sodium diarrhea | Hirabayashi et al., 2018 |
| SRD5A3 | Coloboma of iris | 611715 | AR | Congenital disorder of glycosylation, Kahrizi Syndrome | Cantagrel et al., 2010 |
| TCOF1 | Coloboma of iris, choroid, optic nerve and eye lid | 606847 | AD | Treacher Collins Syndrome Collaborative Group (1996) | |
| TFAP2A | Microphthalmia, coloboma of iris, choroid and optic nerve | 107580 | AD | Branchio oculofacial syndrome | Milunsky et al., 2008 |
| TGDS | Coloboma of iris | 616146 | XL recessive | Catel-Manzke syndrome | Ehmke et al., 2014 |
| TMEM67 | Coloboma of iris, retina and choroid | 609884 | AR | COACH syndrome | Verloes et al., 1989 |
| VAX1 | Microphthalmia | 604294 | AR | small optic nerves, orofacial clefting, agenesis of corpus callosum | Slavotinek et al., 2012 |
| WDR11 | Coloboma of iris. | 606417 | AD | Hypogonadotropic hypogonadism with or without anosmia | Kim et al., 2010 |
| WASHC5 | Coloboma of iris and retina | 610657 | AR | Ritscher-Schinzel syndrome (3C syndrome) | Leonardi et al., 2001 |
| YAP1 | Coloboma of iris, retina and choroid | 606608 | AD | hearing impairment, cleft lip/palate, and/or mental retardation | Williamson et al., 2014 |
| ZEB2 | Coloboma of iris and retina | 605802 | AD | Mowat-Wilson syndrome | Wakamatsu et al., 2001 |
AD: Autosomal dominant, AR: Autosomal recessive, XL: X-linked, LOF: Loss of function.
Syndromic coloboma associated with transcription factor mutations
Transcription factors known to play a key role in early eye development (e.g. SOX2, OTX2, and MITF) are strongly associated with syndromic MAC phenotypes. These are among the earliest eye field transcription factors and play multiple roles in several aspects of eye development. SOX2 (OMIM *184429) is one of the most frequently identified genes associated with bilateral anophthalmia and severe microphthalmia. The majority of known cases of SOX2 are de novo mutations (Fantes et al., 2003), but familial, autosomal dominant transmission has also been observed (Chassaing et al., 2007; Williamson and FitzPatrick, 2014). Germline mosaicism is reported in five cases where the SOX2 mutations were maternally transmitted (Chassaing et al., 2007, Faivre et al., 2006, Schneider et al., 2008, Schneider et al., 2009, Stark et al., 2011) and germline transmission of a SOX2 loss-of-function allele has been reported only for one family (Gerth-Kahlert et al., 2013). Mosaicism is important to look for clinically, as it potentially affects genetic counseling, changing a rare, de novo mutation event unlikely to recur into a recurrence risk of up to 50% with each pregnancy. Numerous systemic abnormalities including pituitary/hypothalamus dysfunction (Kelberman et al., 2006), intellectual disability (Zenteno et al., 2005), esophageal atresia (Williamson et al., 2006; Zenteno et al., 2006), microcephaly (Faivre et al., 2006) and (possibly) dental abnormalities (Numakura et al., 2010) have been reported.
Mutations in OTX2 (OMIM *600037) were first reported by Ragge et al. (2005a) in a cohort of patients with microphthalmia/clinical anophthalmia. Additional ocular (retinal degeneration, microcornea, cataract, optic nerve hypoplasia, coloboma) and non-ocular (developmental delay, structural brain abnormalities, hypotonia, seizures, pituitary dysfunction) abnormalities were also noted in this report and largely confirmed in subsequent studies (Dateki et al, 2008; Tajima et al., 2009; Chassaing et al., 2012). Mutations can appear de novo or are transmitted in an autosomal dominant fashion. Unlike SOX2, there is no evident transmission bias between paternal vs. maternal alleles. Non-penetrance and variable expressivity have been reported for non-synonymous or clear loss-of-function alleles. Two confirmed and one suspected case of gonadal mosaicism have been reported concomitant with OTX2 mutation (Ragge et al., 2005a, Wyatt et al., 2008).
Autosomal dominant mutations in PAX2 (OMIM *167409) cause Papillorenal Syndrome and are strongly associated with optic nerve coloboma/congenital excavation of the optic nerve and renal dysfunction (Sanyanusin et al., 1995). Microphthalmia is not a common presentation with the Papillorenal Syndrome. De novo mutations and familial cases are approximately equally reported. Non-penetrance and highly variable expressivity can be present within the same family as well as maternal and paternal germline mosaicism (Amiel et al., 2000; Cheong et al., 2007).
We recently reported two cases of MITF (OMIM *156845) compound heterozygosity (George et al., 2016) resulting in colobomatous microphthalmia, macrocephaly, severe albinism, sensorineural deafness and osteopetrosis (COMMAD syndrome). Both probands with a strong phenotypic overlap were born of non-consanguineous unions and parents were diagnosed with classic Waardenburg syndrome, type 2a (WS2A) due to heterozygous MITF mutations. Each parent in both families had a unique variant described in the literature to be present in all the known MITF isoforms. In both probands, there was complete lack of melanin pigment in the hair, skin, and eyes, severe colobomatous microphthalmia, profound congenital sensorineural hearing loss, and osteopetrosis.
Syndromic coloboma associated with secreted growth factors
Genes of the transforming growth factor beta (TGFβ) superfamily signaling pathway play important roles in many aspects of eye development. For example, bone morphogenetic protein (BMP) ligands are involved in the formation of the retina, lens, iris, and ciliary body (for review see Wang et al., 2014). Furthermore, growth differentiation factors (GDFs) are involved in determining the dorso-ventral symmetry of the optic cup (Yang, 2004). Dominant mutations in genes encoding growth factors from these two families are known to cause coloboma. Heterozygous mutations in BMP4 (OMIM*112262, Reis et al., 2011) and BMP7 (OMIM*112267, Wyatt et al., 2010) are associated with bilateral anophthalmia, microphthalmia and/or coloboma. MAC phenotypes ranging from anophthalmia (bilateral or unilateral) to uveal coloboma are associated with incompletely penetrant autosomal dominant GDF3 and GDF6 mutations (Asai-Coakwell et al, 2007; Asai-Coakwell et al., 2009; Ye et al., 2010).
Colobomatous microphthalmia in CHARGE syndrome due to CHD7 mutation
CHARGE syndrome (OMIM 214800) is a rare genetic condition arising during early fetal development that affects multiple organ systems. Diagnosis is classically clinically based, relying on the presence of major and minor criteria (Table 4) (Blake et al., 1998; Verloes, 2005; Hale et al., 2016). CHARGE syndrome has an estimated prevalence of approximately 1:10,000 births worldwide and presents with considerable clinical variability (Issekutz et al., 2005; Sanlaville and Verloes, 2007). The term CHARGE itself is an acronym first coined in 1982 for the association of Coloboma, Heart defect, Atresia choanae, Retarded growth and development, Genital hypoplasia, and Ear anomalies/deafness (Pagon et al., 1981). Coloboma is present in 75–81% of patients; conversely, approximately 15–30% of patients diagnosed with microphthalmia/coloboma present with CHARGE syndrome (Traboulsi, 1999). Additional consistent clinical observations in CHARGE syndrome patients include semicircular canal hypoplasia, external ear abnormalities, and cranial nerve dysfunction (over 90% of the patients); choanal atresis (i.e., blockage of the posterior nasal apertures found between the nasal cavity and the throat), resulting from failure of recanalization of the nasal fossae during fetal development (38–55%); congenital heart defects (76–77%); genital hypoplasia (62–81%); cleft lip and/or palate (33–48%) and tracheoesophageal anomalies (19–29%) (Bergman et al., 2011; Zentner et al., 2010b).
Table 4:
Diagnosis criteria for CHARGE syndrome
| Major criteria | Minor criteria |
|---|---|
| Coloboma | Cranial nerves dysfunction including hearing loss |
| Choanal atresia and/or cleft lip or palate | Dysphagia (feeding difficulties) |
| Abnormal external, middle or inner ears, including hypoplastic semicircular canals | Structural brain anomalies |
| Pathogenic CHD7 variant | Developmental delay/intellectual disabilities/autism |
| Hypothalamo-hypophyseal dysfunction (gonadotropin or growth hormone deficiency) and genital anomalies | |
| Heart or esophagus malformation | |
| Renal anomalies | |
| Skeletal/limb anomalies |
In 2004, Vissers and colleagues identified an overlapping 2.3Mb de novo microdeletion on chromosome 8q12 by using comparative genomic hybridization (CGH) in patients with CHARGE syndrome (Vissers et al., 2004). This region includes the Chromodomain Helicase DNA-binding 7 (CHD7) gene, which encodes a 2997 amino acid helicase-domain-containing protein, localizing to both the nucleoplasm and nucleolus (Schnetz et al., 2009). Point mutations were further identified in CHD7 in unrelated CHARGE patients, establishing this gene as causative for the syndrome (Jongmans et al., 2006; Lalani et al., 2006). Although most mutations are de novo, autosomal dominant loss-of-function mutations or deletions have been reported in families with more than one affected member (Lalani et al., 2006). Known CHD7 mutations are scattered throughout the gene with no clear genotype-phenotype correlation (Jongmans et al., 2006); this becomes further apparent by the differences in clinical phenotype of sib pairs with identical mutations and mode of inheritance.
In CHARGE syndrome, coloboma is commonly bilateral, and can involve choroid, retina and optic nerve (Tellier et al., 1998; Aramaki et al., 2006; Jongmans et al., 2006; Alazami et al., 2008; McMain et al., 2008); iris colobomas are described less frequently (Aramaki et al., 2006; Jongmans et al., 2006; McMain et al., 2008); and eyelid colobomas are rare. Microphthalmos, optic nerve hypoplasia, myopia, and strabismus are also reported (Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Delahaye et al., 2007; Alazami et al., 2008; Wincent et al., 2008; Jyonouchi et al., 2009) in CHARGE syndrome patients. Here, we focus on colobomatous-microphthalmic findings in genotype-confirmed CHARGE syndrome patients.
In a large cohort of individuals with the CHD7 mutations reported by Lalani et al., 2006, coloboma of the iris, retina, or optic disc/nerve was observed in 55 out of 62 affected individuals (89%). The study also included a pair of female monozygotic twins carrying the mutation p.(E1271X), both of whom displayed bilateral coloboma in the presence of other phenotypic variation; only one of them, however, survived. Delahaye et al., (2007) reported two familial cases of CHARGE syndrome exhibiting extreme intrafamilial variability and CHD7 mutations. Each family was comprised of a single mildly affected parent and severely affected children (two boys of each parent). In the first family, the mother carrying the p.(S834F) mutation as well as one of the sons had unilateral (left) optic nerve coloboma whereas, hypoplastic optic nerves were observed in the other son. The father in the second family, who carried a truncating mutation p.(R157X), had no coloboma, whereas one son carrying the same mutation had unilateral retinal coloboma and the coloboma present in the deceased son was not specified. Both families included unaffected siblings without CHD7 mutations.
Nishina et al., 2012, provided an in-depth ophthalmic evaluation from a multicenter study of 19 Japanese affected individuals who were confirmed for CHD7 mutations and were retrospectively studied. Coloboma affected the posterior segment of 92.1% of the eyes examined. Fifteen patients presented with retinochoroidal and optic nerve coloboma bilaterally (78.9%) and three unilaterally (7.9%). Coloboma affecting the macula was observed in eight patients bilaterally (42.1%) and five patients unilaterally (13.2%). Only one patient displayed iris coloboma bilaterally and another patient presented with lens coloboma unilaterally. Microphthalmos was present bilaterally in three patients and unilaterally in two patients. The article also reports the patient’s refractive errors and BCVA. The authors suggest a correlation between the location of the protein truncation and the severity of the anatomical abnormality in the eye. However, an earlier study had not found any significant genotype-phenotype correlation in a cohort of 107 European patients (Jongmans et al., 2006).
Husu et al., 2013, reported that out of 18 Danish patients with CHD7 mutations, coloboma and microphthalmia were observed in 17 of the cases. Of 17 affected, one individual presented with unilateral and all others had bilateral coloboma. Coloboma was observed in all structures of the eye i.e., retina (73%), iris and choroid (7%), optic disc (13%). No information was available for one of the affected individuals. Four cases of unilateral and one of bilateral microphthalmia accounted for 28% of the subjects.
In a large French cohort of 92 patients carrying mutations in the CHD7 gene (Legendre et al., 2017), 67 (73%) were observed to have coloboma. As reported for earlier studies, coloboma of the retina (80%) was more common than iris coloboma (15%).
Recently, prenatal diagnosis of CHARGE syndrome has been conducted in three studies (Busa et al., 2016; Hoch et al., 2017 and Millischer et al., 2019) in which ocular defects have been investigated prenatally. This is particularly important as it would enable better care and management of affected individuals after birth. Busa et al., (2016) screened 12 pregnancies with CHARGE syndrome diagnosis and CHD7 mutation. At birth, coloboma was observed in 11/12 patients and two had severe microphthalmia along with coloboma. Although the authors were not able to detect ocular abnormalities during the prenatal screening by ultrasound and MRI with the help of a dysmorphologist, they argue that the microphthalmia and coloboma could have been identified by careful examination of the ocular region. Recently, Millischer et al., 2019 performed a retrospective study of 26 suspected cases of CHARGE syndrome, with 20 out of 26 patients positive for the CHD7 mutation. The MRI features that were most consistently detected were arhinencephaly, dysplasia of the semicircular canals, agenesis and posterior fossa anomalies, whereas, ocular anomalies were observed in only four individuals out of 11 and were overlooked in five cases. Coloboma was identified as a posterior focal bulging of the eyeball, in the MRI axial plane. Ocular asymmetry suggested microphthalmia. Note that the current resolution of MRI does not allow for the imaging of the optic fissure closure defect per se. The literature suggests that CHD7 functions by controlling gene expression programs through ATP-dependent chromatin remodeling (Bajpai et al., 2010; Schnetz et al., 2009) and rearrangement of nucleosomes on the DNA (Jiang and Pugh 2009). Zentner et al. have suggested that CHD7 may also play a role in the nucleolus, where it promotes ribosomal RNA biogenesis (Zentner et al, 2010a). Experiments with human pluripotent stem cells have suggested a role for CHD7 in specification of neural crest fate (Bajpai et al. 2010). Chai et al., (2018) reported that CHD7 is essential in maintaining neuro-epithelium identity and CNS lineage development by indirectly suppressing the induction of neural crest fates. Furthermore, important roles for CHD7 in neurogenesis and oligodendrocyte maturation and myelination (He et al. 2016) have been identified using the mouse as a model. In the developing mouse eye, CHD7 is expressed in the neural ectoderm and surface ectoderm but not in the peri-ocular mesenchymal tissue, and is required for eye morphogenesis, lens development and optic fissure closure (Gage et al., 2015). Finally, Bajpai demonstrated that, in Xenopus, CHD7 is necessary for normal neural crest development and is essential for activating several components of the neural crest transcriptional circuitry (e.g., Twist, Slug and Sox9) (Bajpai et al., 2010). Taken together, these studies suggest that CHD7 may play multiple roles in optic fissure closure and these may vary in different species, as CHD7 expression, for example, was not observed in the mouse peri-ocular mesenchyme, which is comprised of neural crest and mesenchymal tissue.
Coloboma in COACH syndrome due to mutations in TMEM67
Joubert syndrome (JS, OMIM#213300) is a developmental disorder characterized by brainstem malformation, cerebellar vermis hypoplasia/dysplasia, ataxia, hypotonia, mental retardation, neonatal breathing abnormalities, and oculomotor apraxia. It is caused by mutations in more than 35 genes that encode proteins involved in primary cilia and cilium basal body establishment. Additional clinical features in JS subtypes include nephronophthisis, renal cystic dysplasia, hepatic fibrosis, ocular coloboma, retinal dystrophy, and polydactyly. One of the subsets of JS, sometimes referred to as Joubert syndrome related disorders - JSRD, is COACH syndrome, caused by mutations in the transmembrane protein 67 (TMEM67)/MKS3 gene (Verloes and Lambotte, 1989 and Smith et al., 2006). An autosomal recessive condition, COACH syndrome is characterized by Cerebellar vermis hypoplasia, Oligophrenia (developmental delay/mental retardation), Ataxia, Coloboma, and Hepatic fibrosis. Multiple transcript variants encoding different protein isoforms have been found for this gene. Chorioretinal coloboma, a partially penetrant phenotype of COACH syndrome, is also strongly associated with TMEM67 mutations. Mutations causing COACH syndrome are spread throughout the TMEM67 gene. Missense mutations in TMEM67 largely cause COACH syndrome, whereas truncating mutations are more likely to cause a related condition, Meckel syndrome, type 3 (OMIM#607361) (Otto et al, 2009). No correlation exists, however, between mutation position/type and the coloboma phenotype.
Otto et al., 2009, performed mutation screening of 120 unrelated individuals with JS and identified TMEM67 recessive mutations in five patients belonging to four independent families. Retinal coloboma was observed in three of these patients and a blind sibling pair. Brancati et al., (2009) reported eight patients carrying recessive TMEM67/MKS3 mutations out of 12 affected individuals diagnosed with COACH syndrome. Of these eight, chorioretinal or optic nerve colobomas were detected in five patients (42%) and two other cases revealed abnormalities (enlarged optic cup or pale optic disc) upon fundoscopy. Subsequently, Doherty et al., (2010), showed that TMEM67 mutations account for 9% of families in a large JSRD cohort, and that TMEM67 mutations are observed almost exclusively in the COACH syndrome subtype of JSRD.
Iannicelli et al., (2010), performed mutation analysis of TMEM67 in 341 probands (265 were JSRD and 76 Meckel syndrome fetuses). Among the 265 JSRD patients, TMEM67 compound heterozygous mutations were identified in eight out of ten probands with a phenotype of JS plus liver disease and six of these eight probands had coloboma (unilateral or bilateral). No coloboma was reported in 12 of the fetuses from terminated pregnancies carrying TMEM67 mutation out of 76 Meckel syndrome cases.
In a Northern European patient cohort (51 cases) diagnosed with JS, Kroes et al., (2016) identified five (~10%) patients with colobomatous microphthalmia carrying recessive TMEM67 mutations. Bachmann-Gagescu et al., (2015), also reported a significant genotype-phenotype correlation between TMEM67 mutations causing JS and coloboma. Suzuki et al., 2016 analyzed a cohort of 27 families of Japanese descent diagnosed with JS and found TMEM67 gene mutation in seven of them, however none displayed coloboma. Kang et al. (2016) identified four Korean patients with recessive mutations in TMEM67 out of seven nephronophthisis patients with hepatic fibrosis. All four of these patients had JS and congenital hepatic fibrosis compatible with COACH diagnosis (Gentile et al., 1996).
Recently, Brooks et al., (2018) reported a comprehensive ophthalmic evaluation of JS patients and observed high prevalence of coloboma in those patients carrying mutations in TMEM67 gene. Out of 22 patients genotypically confirmed for TMEM67 mutation, retinal coloboma was observed in 13, retina and optic nerve coloboma in four, and optic nerve coloboma alone in one, for a total of 18 coloboma affected individuals. Conversely, coloboma was observed only rarely in JSRD due to mutations in other genes.
The TMEM67 gene product MECKELIN, is predicted to be a 7-transmembrane receptor- like protein and contains an extracellular cysteine-rich region (Smith et al., 2006). MECKELIN (also referred to as MKS or MKS3) is localized to the transition zone of the cilium in protein complexes with other JS and NePHronoPhthisis (NPHP) proteins. The protein complexes localized at ciliary transition zone can be categorized into three main protein families- MKS, NPHP and CEP290 group. These protein complexes extensively collaborate for the proper assembly and functioning of the transition zone and are further composed of multiple interacting proteins including – MKS, B9D1, B9D2, TCTN1, TCTN2, TCTN3, CC2D2A, TMEM17, TMEM67, TMEM107, TMEM216, TMEM231, TMEM237, NPHP1, NPHP4, CEP290, NPHP5, RPGRIP1L, RPGRIP1, RPGR, LCA5, AHI1, CEP162, TMEM138, JBTS17 and TMEM80 (Gonçalves and Pelletier 2017). MECKELIN has also been detected at the plasma membrane in cell lines and primary cells. Silencing of TMEM67/MKS3 and of the related MKS1 gene was shown to regulate centrosome number and cilia length in IMCD-3 cell cultures (Dawe et al., 2007). The role of TMEM67 in eye development has not yet been studied and more importantly, the functional relationship between primary cilium defects and optic fissure fusion defects remains unknown.
Molecular and cellular studies of optic fissure closure
Identification of genes involved in optic fissure closure using animal models
One of the first efforts to identify genes involved in optic fissure closure in an unbiased fashion was reported by Brown et al., 2009. The authors performed gene expression microarray analysis of mouse tissue samples micro-dissected from the fusing margins of the optic fissure corresponding to before (embryonic day (E)10.5), during (E11.5) and after (E12.5) optic fissure fusion. Data analysis identified 250 probe sets that represent 168 annotated genes and 54 predicted genes. Of the 168 annotated candidate genes, 83 have been experimentally mutated in mouse or zebrafish models, out of which, five exhibit coloboma, 22 have other eye phenotypes and 21 have no reported eye phenotype. Association with eye or eye development for 35 of the mutated genes could not be determined from published reports. A high percentage of annotated genes from the screen were confirmed to be expressed during eye development. Of the 78/168 genes for which in situ hybridization (ISH) studies exist at relevant time points, 70 are expressed in the eye, four are not expressed in the eye, and expression in the eye is undetermined for the remaining four.
Similar efforts have been reported in mouse (Cao et al., 2018), chick (Hardy et al., 2019) and zebrafish (Richardson et al., 2019) to identify genes involved in optic fissure closure. Cao et al., (2018), used laser-assisted microdissection to collect optic cup tissue samples from E11.5 mouse embryos from central-nasal, central-temporal retina and ventral optic fissure, to provide a comparative gene expression profile of the three different regions. Microarray data analysis highlighted 36 probe sets corresponding to 32 genes that showed significantly different expression levels between nasal and temporal retina and include many known genes (e.g., Foxg1, Foxd1, Hmx1, Efna5, Epha5, and Tenm3), validating the robustness of the technique. These data also revealed several new nasal-temporal differentially expressed genes, including those encoding cell adhesion molecules (Cdh9, Cdh11 and Sema5a), transcription factors (Sall1, Zfhx4, Onecut2, Tfec, and Glis3), and a predicted noncoding RNA (3110039M20Rik). The authors further performed differential gene expression analysis comparing the optic fissure transcription profile with nasal and temporal retinal gene expression to identify optic fissure- specific genes. Using this strategy, novel and already known optic fissure signature genes were identified, including Vax1, Vax2, Vax2os, Ntn1, Smoc1, Aldh1a3, Cyp1b1, Ptchd1, Zfp503, Laminins, Bmpr1b, Bmp7 and Tenm3. A number of genes that had not been reported previously to be expressed in the optic fissure were confirmed by ISH and 11 were conclusively established as new optic fissure-specific genes: Afap1l2, Adamts16, Bmf, Slitrk1, Cp, Ror2, Tfec, Cplx3, Neto1, Shtn1, and Flrt2.
In 2019, Hardy et al., reported on a similar developmental profiling strategy applied to the chick eye, albeit using a distinct and complementary approach to laser capture microdissection (Hardy et al, 2019). The authors manually microdissected tissue in/around the optic fissure margins, the ventral eye, the dorsal eye and the eye as a whole from Hamburger-Hamilton stage (HH.St) 25–26 (pre-fusion), HH.St27–28 (initiation of fusion) and HH.St28–30 (during active optic fissure fusion) embryos. After RNA sequencing (RNA-Seq) profiling and analysis, they compiled a list of genes enriched in the optic fissure; these include both known coloboma genes (e.g., PAX2, VAX1) and genes not well established to be important in optic fissure closure. Among the latter, the gene with the highest expression at all stages is Netrin-1 (NTN1), which is also differentially regulated during mouse optic fissure closure (Brown et al, 2009). NTN1 belongs to a family of laminin-related secreted proteins important for axonal guidance. Knockout of Ntn1/ntn1a function in mouse and zebrafish produces optic fissure closure defects— an additional proof that optic fissure gene profiling strategies can identify genes of potential clinical relevance for coloboma.
Lastly, Richardson et al., (2019) performed RNA-Seq of dorsal retina versus tissue around the optic fissure and identified candidate genes that are differentially expressed at developmental time points corresponding to before (32 hpf), during (48 hpf), and after (56 hpf) optic fissure closure in zebrafish. This screen also suggests an important role for ntn1a in optic fissure closure, that was further confirmed via morpholino-mediated depletion and ocular coloboma appearance in zebrafish morphants. Notably, ntn1a expression is completely downregulated at the optic fissure margins prior to fusion.
A major difference (other than the obvious species difference) between the Richardson et al., (2019) and the Hardy et al., (2019) data sets compared Brown et al., (2009) and Cao et al., (2018) is the use of RNA-Seq technology instead of microarrays. Unlike hybridization-based approaches, RNA-Seq is not limited to detecting transcripts that correspond to known genomic sequences and is applicable to non-model organisms with genomic regions that are yet to be sequenced.
Studies of optic cup morphogenesis and optic fissure margins fusion using live zebrafish
Investigation of the morphogenetic dynamics of optic cup formation and optic fissure margins fusion was initiated as early as the 1980s (Hero 1989, 1990 and Hero et al., 1991). In seminal studies performed on the cinnamon mouse, Hero used transmission electron microscopy (TEM) to show how early appositional junctions are established, followed by subsequent creation of cell-cell junctions in apposing RPE cells during optic fissure margins fusion. Given the limitations of working with developing mouse embryos, the focus has shifted towards the use of developing zebrafish. Imaging optic cup morphogenesis in live zebrafish presents with many advantages over fixed tissues. In one of the earliest efforts, Kwan et al., (2012) showed how live imaging eliminates fixation artifacts, yields relatively accurate size and shape measurements, and allows concomitant tracking of many cell types. These “4-dimensional” data sets, coupled with advanced cell tracking and the use of segmentation software, have revealed several unexpected findings: 1) although cell division contributes to the growth of the optic cup, it is largely dispensable for eye formation; 2) optic vesicle evagination persists longer than it had been previously appreciated and cells move in a “pinwheel” fashion, with retinal precursors involuting around the rim of the optic cup; and 3) cells that are adjacent early in the process and, presumably, subject to similar extracellular cues, can settle in disparate locations in the final optic cup structure.
Recent studies have shed light for the first time on morphogenetic events shaping the optic cup (Heermann et al., 2015 and Bryan et al., 2016), optic stalk (Gordon et al., 2018) and fusion of optic fissure margins (Bernstein et al., 2018), in real time. These studies have been instrumental in understanding optic cup morphogenesis up to the point of the optic fissure margins organizing around the fissure. Such understanding would not have been possible studying fixed tissues at different “static” time points.
Heermann et al., (2015) describe the lens-averted epithelium as a source of presumptive stem cells that flow around the distal rims of the optic cup to their destination in the ciliary marginal zone and contribute to the growing neuroretina. This epithelial flow contributes to optic cup morphogenesis and, when inhibited by BMP, ectopic neuroretina forms in the RPE domain, leading to failed fissure closure and coloboma. Using live imaging, Bryan et al., (2016) determined the important role of laminin extracellular matrix in optic cup morphogenesis. They showed that in lama1 mutant zebrafish, optic cup morphogenesis, optic stalk constriction, invagination, and formation of a spherical lens are affected. Also, lama1 mutants exhibit loss of epithelial polarity and altered adhesion leading to defective tissue architecture and disorganized retina. Similarly, Nicolás-Pérez et al., (2016) provided evidence for the role of contractile actomyosin network dynamics mediated by lamc1 and extracellular matrix in shaping the optic cup. They elegantly demonstrated that optic cup morphogenesis requires clustered myosin accumulation inside basal neuroepithelial cells and attachment of these cells to an underlying extracellular matrix. In addition, Sidhaye and Norden (2017) have shown that active migration of connected epithelial cells into the retinal neuroepithelium, driven by cell-matrix contacts, together with basal shrinkage of retinal neuroepithelium are crucial for optic cup formation. Finally, Gordon et al., (2018) for the first time employed a combination of four-dimensional imaging, cell tracking, and molecular genetics in zebrafish to understand the morphogenesis of the optic stalk in relation to the optic fissure. Using the ptch2 mutant in which the Hedgehog (Hh) signaling is overactive, they show how cell motility required for optic fissure and stalk formation is impaired via non-cell-autonomous and cell-autonomous mechanisms.
More recent studies are focused on understanding the actual fusion process of the optic fissure margins. Although Drosophila dorsal lip/pore closure has been the system of choice for studying epithelial sheet fusion, investigations of optic fissure margin fusion have been gaining traction in vertebrate models. Bernstein et al., (2018) suggest that optic fissure closure is accomplished by breaking down of the basement membrane along the fissure margins, and by subsequently establishing basement membrane continuity along the dorsal and ventral surfaces of the fissure. Fissure closure is finally accomplished by cell protrusions and movements of partially polarized retinal cells into the fissure space to initiate the fusion and intercalation of various tissues into the fissure space.
Conclusion and future directions
One of the most important aspects of understanding the genetics of human optic fissure closure defects is to disentangle the primary drivers of fusion per se and other, more “secondary” causes such as morphogenetic defects and non-cell autonomous effects from surrounding tissues. Tools such as in vivo imaging (especially if this were to extend into a mammalian system), tissue-specific genetic manipulations, and complementary in vitro cell culture models will likely be crucial in disentangling these potential mechanisms. We anticipate that the knowledge gained by studying optic cup formation and optic fissure closure will have important implications for human disease and will likely be generalizable to other developmental processes involving epithelial sheet fusion.
Highlights.
Uveal coloboma, a rare potentially blinding condition affecting between 0.5 and 2.6 per 10,000 births.
Isolated and syndromic cases of uveal coloboma.
Current knowledge on the genetics of human uveal coloboma with a specific focus on CHARGE and COACH syndromes.
Approaches to identify genes involved in optic fissure closure using animal models.
Live imaging of zebrafish eye development to understand optic fissure closure.
Funding
Supported by the intramural program of the National Eye Institute, EY000469.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abouzeid H, Boisset G, Favez T, Youssef M, Marzouk I, Shakankiry N, Bayoumi N, Descombes P, Agosti C, Munier FL, Schorderet DF, 2011. Mutations in the SPARC-related modular calcium-binding protein 1 gene, SMOC1, cause waardenburg anophthalmia syndrome. Am J Hum Genet 88, 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami MA, Alzahrani F, Alkuraya FS, 2008. Expanding the “E” in CHARGE. Am J Med Genet A 146A, 1890–1892. [DOI] [PubMed] [Google Scholar]
- Aldahmesh MA, Khan AO, Hijazi H, Alkuraya FS, 2013. Homozygous truncation of SIX6 causes complex microphthalmia in humans. Clinical Genetics 84, 198–199. [DOI] [PubMed] [Google Scholar]
- Aldahmesh MA, Mohammed JY, Al-Hazzaa S, Alkuraya FS, 2012. Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genetics In Medicine 14, 900. [DOI] [PubMed] [Google Scholar]
- Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, Maina EN, Morgan NV, Tee L, Morton J, Ainsworth JR, Horn D, Rosser E, Cole TRP, Stolte-Dijkstra I, Fieggen K, Clayton-Smith J, Mégarbané A, Shield JP, Newbury-Ecob R, Dobyns WB, Graham JM, Kjaer KW, Warburg M, Bond J, Trembath RC, Harris LW, Takai Y, Mundlos S, Tannahill D, Woods CG, Maher ER, 2005. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nature Genetics 37, 221–224. [DOI] [PubMed] [Google Scholar]
- Amiel J, Audollent S, Joly D, Dureau P, Salomon R, Tellier A-L, Augé J, Bouissou F, Antignac C, Gubler M-C, Eccles MR, Munnich A, Vekemans M, Lyonnet S, Attié-Bitach T, 2000. PAX2 mutations in renal–coloboma syndrome: mutational hotspot and germline mosaicism. European Journal of Human Genetics 8, 820–826. [DOI] [PubMed] [Google Scholar]
- Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, Oki H, Nanao K, Moriyama N, Oku S, Hasegawa T, Takahashi T, Fukushima Y, Kawame H, Kosaki K, 2006. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr 148, 410–414. [DOI] [PubMed] [Google Scholar]
- Asai-Coakwell M, French CR, Berry KM, Ye M, Koss R, Somerville M, Mueller R, van Heyningen V, Waskiewicz AJ, Lehmann OJ, 2007. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet 80, 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai-Coakwell M, French CR, Ye M, Garcha K, Bigot K, Perera AG, Staehling-Hampton K, Mema SC, Chanda B, Mushegian A, Bamforth S, Doschak MR, Li G, Dobbs MB, Giampietro PF, Brooks BP, Vijayalakshmi P, Sauvé Y, Abitbol M, Sundaresan P, van Heyningen V, Pourquié O, Underhill TM, Waskiewicz AJ, Lehmann OJ, 2009. Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Human Molecular Genetics 18, 1110–1121. [DOI] [PubMed] [Google Scholar]
- Avunduk AM, Aslan Y, Kapicioglu Z, Elmas R, 2000. High myopia, hypertelorism, iris coloboma, exomphalos, absent corpus callosum, and sensorineural deafness: report of a case and further evidence for autosomal recessive inheritance. Acta Ophthal Scand 78, 221–222. [DOI] [PubMed] [Google Scholar]
- Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M, 1996. PAX6 missense mutation in isolated foveal hypoplasia. Nature Genetics 13, 141–142. [DOI] [PubMed] [Google Scholar]
- Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M, 2003. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet 72, 1565–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi AR, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla À, Ozyürek H, Haliloğlu G, Tuysuz B, Topçu M, University of Washington Center for Mendelian G, Chance P, Parisi MA, Glass IA, Shendure J, Doherty D, 2015. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 52, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, Wysocka J, 2010. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Martin A, Williams S, Lindley V, Gilmore J, Restori M, Robson AG, Neveu MM, Holder GE, Collin JRO, Robinson DO, Farndon P, Johansen-Berg H, Gerrelli D, Ragge NK, 2008. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 82, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard LA, 1979. Congenital contractural arachnodactyly and intraocular colobomas. Birth Defects Orig Artic Ser 15, 189–205. [PubMed] [Google Scholar]
- Bar-Yosef U, Abuelaish I, Harel T, Hendler N, Ofir R, Birk OS, 2004. CHX10 mutations cause non-syndromic microphthalmia/anophthalmia in Arab and Jewish kindreds. Human Genetics 115, 302–309. [DOI] [PubMed] [Google Scholar]
- Beby F, Commeaux C, Bozon M, Denis P, Edery P, Morlé L, 2007. New Phenotype Associated With an Arg116Cys Mutation in the CRYAA Gene: Nuclear Cataract, Iris Coloboma, and Microphthalmia. JAMA Ophthalmology 125, 213–216. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki A-E, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG, 2002. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 71, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA, 2011. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 48, 334–342. [DOI] [PubMed] [Google Scholar]
- Bernstein CS, Anderson MT, Gohel C, Slater K, Gross JM, Agarwala S, 2018. The cellular bases of choroid fissure formation and closure. Developmental Biology 440, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KD, Davenport SL, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JM Jr., 1998. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr 37, 159–173 [DOI] [PubMed] [Google Scholar]
- Borozdin W, Wright MJ, Hennekam RC, Hannibal MC, Crow YJ, Neumann TE, Kohlhase J, 2004. Novel mutations in the gene SALL4 provide further evidence for acro-renal-ocular and Okihiro syndromes being allelic entities, and extend the phenotypic spectrum. J Med Genet. 41, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botzenhart EM, Green A, Ilyina H, König R, Lowry RB, Lo IFM, Shohat M, Burke L, McGaughran J, Chafai R, Pierquin G, Michaelis RC, Whiteford ML, Simola KOJ, Rösler B, Kohlhase J, 2005. SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum Mutat 26, 282–282. [DOI] [PubMed] [Google Scholar]
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D’Arrigo S, Emma F, Fazzi E, Gallizzi R, Gentile M, Loncarevic D, Mejaski-Bosnjak V, Pantaleoni C, Rigoli L, Salpietro CD, Signorini S, Stringini GR, Verloes A, Zabloka D, Dallapiccola B, Gleeson JG, Valente EM, International JSG, 2009. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat 30, E432–E442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BP, Zein WM, Thompson AH, Mokhtarzadeh M, Doherty DA, Parisi M, Glass IA, Malicdan MC, Vilboux T, Vemulapalli M, Mullikin JC, Gahl WA, Gunay-Aygun M, 2018. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology 125, 1937–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Dutta S, Bharti K, Bonner RF, Munson PJ, Dawid IB, Akhtar AL, Onojafe IF, Alur RP, Gross JM, Hejtmancik JF, Jiao X, Chan W-Y, Brooks BP, 2009. Expression profiling during ocular development identifies 2 Nlz genes with a critical role in optic fissure closure. Proc Natl Acad Sci U S A 106, 1462–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan CD, Chien C-B, Kwan KM, 2016. Loss of laminin alpha 1 results in multiple structural defects and divergent effects on adhesion during vertebrate optic cup morphogenesis. Developmental biology 416, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busa T, Legendre M, Bauge M, Quarello E, Bretelle F, Bilan F, Sigaudy S, Gilbert-Dussardier B, Philip N, 2016. Prenatal findings in children with early postnatal diagnosis of CHARGE syndrome. Prenatal Diagnosis 36, 561–567. [DOI] [PubMed] [Google Scholar]
- Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, Lehle L, Hombauer H, Adamowicz M, Swiezewska E, De Brouwer AP, Blümel P, Sykut-Cegielska J, Houliston S, Swistun D, Ali BR, Dobyns WB, Babovic-Vuksanovic D, van Bokhoven H, Wevers RA, Raetz CRH, Freeze HH, Morava E, Al-Gazali L, Gleeson JG, 2010. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Ouyang J, Liang H, Guo J, Lin S, Yang S, Xie T, Chen S, 2018. Regional Gene Expression Profile Comparison Reveals the Unique Transcriptome of the Optic FissureOptic Fissure Specific Transcriptome Analysis. Investigative Ophthalmology & Visual Science 59, 5773–5784. [DOI] [PubMed] [Google Scholar]
- Casey J, Kawaguchi R, Morrissey M, Sun H, McGettigan P, Nielsen JE, Conroy J, Regan R, Kenny E, Cormican P, Morris DW, Tormey P, Chróinín MN, Kennedy BN, Lynch S, Green A, Ennis S, 2011. First implication of STRA6 mutations in isolated anophthalmia, microphthalmia, and coloboma: a new dimension to the STRA6 phenotype. Hum Mutat 32, 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni F, Aguilera-Garcia D, Chassaing N, Bax DA, Blanco-Kelly F, Ramos P, Tarilonte M, Villaverde C, da Silva LRJ, Ballesta-Martínez MJ, Sanchez-Soler MJ, Holt RJ, Cooper-Charles L, Bruty J, Wallis Y, McMullan D, Hoffman J, Bunyan D, Stewart A, Stewart H, Lachlan K, Fryer A, McKay V, Roume J, Dureau P, Saggar A, Griffiths M, Calvas P, Ayuso C, Corton M, Ragge NK, Study D, 2019. New GJA8 variants and phenotypes highlight its critical role in a broad spectrum of eye anomalies. Human Genetics 138, 1027–1042. [DOI] [PubMed] [Google Scholar]
- Chai M, Sanosaka T, Okuno H, Zhou Z, Koya I, Banno S, Andoh-Noda T, Tabata Y, Shimamura R, Hayashi T, Ebisawa M, Sasagawa Y, Nikaido I, Okano H, Kohyama J, 2018. Chromatin remodeler CHD7 regulates the stem cell identity of human neural progenitors. Genes Dev 32, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Blain D, Bertuzzi S, Brooks BP, 2006. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol 17, 447–470. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Davis EE, McKnight KL, Niederriter AR, Causse A, David V, Desmaison A, Lamarre S, Vincent-Delorme C, Pasquier L, Coubes C, Lacombe D, Rossi M, Dufier J-L, Dollfus H, Kaplan J, Katsanis N, Etchevers HC, Faguer S, Calvas P, 2016. Targeted resequencing identifies PTCH1 as a major contributor to ocular developmental anomalies and extends the SOX2 regulatory network. Genome Res 26, 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing N, Gilbert-Dussardier B, Nicot F, Fermeaux V, Encha-Razavi F, Fiorenza M, Toutain A, Calvas P, 2007. Germinal mosaicism and familial recurrence of a SOX2 mutation with highly variable phenotypic expression extending from AEG syndrome to absence of ocular involvement. Am J Med Genet A 143, 289–291. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Sorrentino S, Davis EE, Martin-Coignard D, Iacovelli A, Paznekas W, Webb BD, Faye-Petersen O, Encha-Razavi F, Lequeux L, Vigouroux A, Yesilyurt A, Boyadjiev SA, Kayserili H, Loget P, Carles D, Sergi C, Puvabanditsin S, Chen CP, Etchevers HC, Katsanis N, Mercer CL, Calvas P, Jabs EW, 2012. OTX2 mutations contribute to the otocephaly-dysgnathia complex. J Med Genet 49, 373–379. [DOI] [PubMed] [Google Scholar]
- Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS, Choi Y, 2007. A clinico-genetic study of renal coloboma syndrome in children. Pediatric Nephrology 22, 1283–1289. [DOI] [PubMed] [Google Scholar]
- Chou CM, Nelson C, Tarlé SA, Pribila JT, Bardakjian T, Woods S, Schneider A, Glaser T, 2015. Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell 161, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte I, Hadfield KD, Barbato S, Carrella S, Pizzo M, Bhat RS, Carissimo A, Karali M, Porter LF, Urquhart J, Hateley S, O’Sullivan J, Manson FDC, Neuhauss SCF, Banfi S, Black GCM, 2015. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc Natl Acad Sci U S A 112, E3236–E3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras C, Gaasterland T, Lee P, Gudiseva HV, Chavali VRM, Pullakhandam R, Maranhao B, Edsall L, Soares S, Reddy GB, Sieving PA, Ayyagari R, 2012. Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PLoS One 7, e50205–e50205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dateki S, Fukami M, Sato N, Muroya K, Adachi M, Ogata T, 2008. OTX2 mutation in a patient with anophthalmia, short stature, and partial growth hormone deficiency: functional studies using the IRBP, HESX1, and POU1F1 promoters. J Clin Endocrinol Metab 93, 3697–3702. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA, 2007. The Meckel–Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Human Molecular Genetics 16, 173–186. [DOI] [PubMed] [Google Scholar]
- Delahaye A, Sznajer Y, Lyonnet S, Elmaleh-Bergès M, Delpierre I, Audollent S, Wiener-Vacher S, Mansbach A-L, Amiel J, Baumann C, Bremond-Gignac D, Attié-Bitach T, Verloes A, Sanlaville D, 2007. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clinical Genetics 72, 112–121. [DOI] [PubMed] [Google Scholar]
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA, 2010. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet 47, 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmke N, Caliebe A, Koenig R, Kant SG, Stark Z, Cormier-Daire V, Wieczorek D, Gillessen-Kaesbach G, Hoff K, Kawalia A, Thiele H, Altmuller J, et al. , 2014. Homozygous and compound-heterozygous mutations in TGDS cause Catel-Manzke syndrome. Am J Hum Genet 95, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Williamson KA, Faber V, Laurent N, Grimaldi M, Thauvin-Robinet C, Durand C, Mugneret F, Gouyon JB, Bron A, Huet F, Hayward C, Heyningen VV, Fitzpatrick DR, 2006. Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. Am J Med Genet 140A, 636–639. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch S-A, McGill NI, Collin JRO, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR, 2003. Mutations in SOX2 cause anophthalmia. Nature Genetics 33, 462–463. [DOI] [PubMed] [Google Scholar]
- Fares-Taie L, Gerber S, Chassaing N, Clayton-Smith J, Hanein S, Silva E, Serey M, Serre V, Gérard X, Baumann C, Plessis G, Demeer B, Brétillon L, Bole C, Nitschke P, Munnich A, Lyonnet S, Calvas P, Kaplan J, Ragge N, Rozet J-M, 2013. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am J Hum Genet 92, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregeau B, Kim BJ, Hernández-García A, Jordan VK, Cho MT, Schnur RE, Monaghan KG, Juusola J, Rosenfeld JA, Bhoj E, Zackai EH, Sacharow S, Barañano K, Bosch DGM, de Vries BBA, Lindstrom K, Schroeder A, James P, Kulch P, Lalani SR, van Haelst MM, van Gassen KLI, van Binsbergen E, Barkovich AJ, Scott DA, Sherr EH, 2016. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am J Hum Genet 98, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PJ, Hurd EA, Martin DM, 2015. Mouse Models for the Dissection of CHD7 Functions in Eye Development and the Molecular Basis for Ocular Defects in CHARGE Syndrome. Invest Ophthalmol Vis Sci 56, 7923–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge N, Crandall BF, Shuler JD, Bateman JB, 1995. Coloboma associated with Rubinstein-Taybi syndrome. J Pediatr Ophthalmol Strabismus 32, 266–268. [DOI] [PubMed] [Google Scholar]
- Gentile M, Di Carlo A, Susca F, Gambotto A, Caruso ML, Panella C, Vajro P, Guanti G, 1996. COACH syndrome: Report of two brothers with congenital hepatic fibrosis, cerebellar vermis hypoplasia, oligophrenia, ataxia, and mental retardation. American Journal of Medical Genetics 64, 514–520. [DOI] [PubMed] [Google Scholar]
- George A, Zand DJ, Hufnagel RB, Sharma R, Sergeev YV, Legare JM, Rice GM, Scott Schwoerer JA, Rius M, Tetri L, Gamm DM, Bharti K, Brooks BP, 2016. Biallelic Mutations in MITF Cause Coloboma, Osteopetrosis, Microphthalmia, Macrocephaly, Albinism, and Deafness. Am J Hum Genet 99, 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth-Kahlert C, Williamson K, Ansari M, Rainger JK, Hingst V, Zimmermann T, Tech S, Guthoff RF, van Heyningen V, Fitzpatrick DR, 2013. Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Mol Genet Genomic Med 1, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, Didi M, Gaston-Massuet C, Senniappan S, 2017. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Human Molecular Genetics 26, 4315–4326. [DOI] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL, 1994. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nature Genetics 7, 463–471. [DOI] [PubMed] [Google Scholar]
- Gonçalves J, Pelletier L, 2017. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol Cells 40, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon HB, Lusk S, Carney KR, Wirick EO, Murray BF, Kwan KM, 2018. Hedgehog signaling regulates cell motility and optic fissure and stalk formation during vertebrate eye morphogenesis. Development 145, dev165068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JM Jr., Wheeler P, Tackels-Horne D, Lin AE, Hall BD, May M, Short KM, Schwartz CE, Cox TC, 2003. A new X-linked syndrome with agenesis of the corpus callosum, mental retardation, coloboma, micrognathia, and a mutation in the alpha 4 gene at Xq13. Am J Med Genet 123A, 37–44. [DOI] [PubMed] [Google Scholar]
- Graul-Neumann LM, Klopocki E, Adolphs N, Mensah MA, Kress W, 2017. Mutation c.943G>T (p.Ala315Ser) in FGFR2 Causing a Mild Phenotype of Crouzon Craniofacial Dysostosis in a Three-Generation Family. Mol Syndromol 8, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano C, Gusson E, Severi G, Isidori F, Wischmeijer A, Brugnara M, Seri M, Rossi C, 2017. A de novo PUF60 mutation in a child with a syndromic form of coloboma and persistent fetal vasculature. Ophthalmic Genetics 38, 590–592. [DOI] [PubMed] [Google Scholar]
- Gurrieri F, Sammito V, Ricci B, Iossa M, Bellussi A, Neri G, 1992. Possible new type of oral–facial–digital syndrome with retinal abnormalities: OFDS type VIII. Am J Med Genet 42, 789–792. [DOI] [PubMed] [Google Scholar]
- Hale CL, Niederriter AN, Green GE, Martin DM, 2016. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A 170A, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy H, Prendergast JG, Patel A, Dutta S, Trejo-Reveles V, Kroeger H, Yung AR, Goodrich LV, Brooks B, Sowden JC, Rainger J, 2019. Detailed analysis of chick optic fissure closure reveals Netrin-1 as an essential mediator of epithelial fusion. Elife 8, e43877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK, 2003. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell, 114, 457–467. [DOI] [PubMed] [Google Scholar]
- He D, Marie C, Zhao C, Kim B, Wang J, Deng Y, Clavairoly A, Frah M, Wang H, He X, Hmidan H, Jones BV, Witte D, Zalc B, Zhou X, Choo DI, Martin DM, Parras C, Lu QR, 2016. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat Neurosci 19, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heermann S, Schütz L, Lemke S, Krieglstein K, Wittbrodt J, 2015. Eye morphogenesis driven by epithelial flow into the optic cup facilitated by modulation of bone morphogenetic protein. Elife 4, e05216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hero I, 1989. The optic fissure in the normal and microphthalmic mouse. Exp Eye Res 49, 229–239. [DOI] [PubMed] [Google Scholar]
- Hero I, 1990. Optic fissure closure in the normal cinnamon mouse. An ultrastructural study. Investigative Ophthalmology & Visual Science 31, 197–216. [PubMed] [Google Scholar]
- Hero I, Farjah M, Scholtz CL, 1991. The prenatal development of the optic fissure in colobomatous microphthalmia. Investigative Ophthalmology & Visual Science 32, 2622–2635. [PubMed] [Google Scholar]
- Hirabayashi KE, Moore AT, Mendelsohn BA, Taft RJ, Chawla A, Perry D, Henry D, Slavotinek A, 2018. Congenital sodium diarrhea and chorioretinal coloboma with optic disc coloboma in a patient with biallelic SPINT2 mutations, including p.(Tyr163Cys). Am J Med Genet A 176, 997–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch MJ, Patel SH, Jethanamest D, Win W, Fatterpekar GM, Roland JT, Hagiwara M, 2017. Head and neck MRI findings in CHARGE syndrome. AJNR Am J Neuroradiol. 38, 2357–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X-F, Xiang L, Cheng W, Cheng F-F, He K-W, Zhang B-W, Zheng S-S, Han R-Y, Zheng Y-H, Xu X-T, Yu H-Y, Zhuang W, Leung YF, Jin Z-B, 2018. Mutation of IPO13 causes recessive ocular coloboma, microphthalmia, and cataract. Experimental & Molecular Medicine 50, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husu E, Hove H, Farholt S, Bille M, Tranebjærg L, Vogel I, Kreiborg S, 2013. Phenotype in 18 Danish subjects with genetically verified CHARGE syndrome. Clinical Genetics 83, 125–134. [DOI] [PubMed] [Google Scholar]
- Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, Fischetto R, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, Gentilin B, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P, International JSG, Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM, 2010. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat 31, E1319–E1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issekutz KA, Graham JM Jr, Prasad C, Smith IM, Blake KD, 2005. An epidemiological analysis of CHARGE syndrome: Preliminary results from a Canadian study. American Journal of Medical Genetics Part A 133A, 309–317. [DOI] [PubMed] [Google Scholar]
- Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E, Wirth MG, van Heyningen V, Donnai D, Munier F, Black GCM, 2002. Domain disruption and mutation of the bZIP transcription factor, MAF,associated with cataract, ocular anterior segment dysgenesis and coloboma. Human Molecular Genetics 11, 33–42. [DOI] [PubMed] [Google Scholar]
- Jelsig AM, Diness BR, Kreiborg S, Main KM, Larsen VA, Hove H, 2018. A complex phenotype in a family with a pathogenic SOX3 missense variant. European Journal of Medical Genetics 61, 168–172. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF, 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE, 2009. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics 123, 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MCJ, Admiraal RJ, van der Donk KP, Vissers LELM, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BBA, Brunner HG, Hoefsloot LH, van Ravenswaaij CMA, 2006. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 43, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HG, Lee HK, Ahn YH, Joung J-G, Nam J, Kim NKD, Ko JM, Cho MH, Shin JI, Kim J, Park HW, Park YS, Ha I-S, Chung WY, Lee D-Y, Kim SY, Park WY, Cheong HI, 2016. Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Experimental & molecular medicine 48, e251–e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, Islam L, Lakowski J, Bacchelli C, Chanudet E, Lescai F, Patel A, Stupka E, Buck A, Wolf S, Beales PL, Jacques TS, Bitner-Glindzicz M, Liasis A, Lehmann OJ, Kohlhase J, Nischal KK, Sowden JC, 2014. Mutation of SALL2 causes recessive ocular coloboma in humans and mice. Human molecular genetics 23, 2511–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT, 2006. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest 116, 2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AO, Patel N, Ghazi NG, Alzahrani SS, Arold ST, Alkuraya FS, 2018. Familial non-syndromic macular pseudocoloboma secondary to homozygous CLDN19 mutation. Ophthalmic Genetics 39, 577–583. [DOI] [PubMed] [Google Scholar]
- Khan K, Logan CV, McKibbin M, Sheridan E, Elçioglu NH, Yenice O, Parry DA, Fernandez-Fuentes N, Abdelhamed ZIA, Al-Maskari A, Poulter JA, Mohamed MD, Carr IM, Morgan JE, Jafri H, Raashid Y, Taylor GR, Johnson CA, Inglehearn CF, Toomes C, Ali M, 2012. Next generation sequencing identifies mutations in Atonal homolog 7 (ATOH7) in families with global eye developmental defects. Human molecular genetics 21, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SY, Vasanth S, Kabir F, Gottsch JD, Khan AO, Chaerkady R, Lee M-CW, Leitch CC, Ma Z, Laux J, Villasmil R, Khan SN, Riazuddin S, Akram J, Cole RN, Talbot CC, Pourmand N, Zaghloul NA, Hejtmancik JF, Riazuddin SA, 2016. FOXE3 contributes to Peters anomaly through transcriptional regulation of an autophagy-associated protein termed DNAJB1. Nat Commun 7, 10953–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-G, Ahn J-W, Kurth I, Ullmann R, Kim H-T, Kulharya A, Ha K-S, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, Ozata M, Bick DP, Sherins RJ, Nagase T, Tekin M, Kim S-H, Kim C-H, Ropers H-H, Gusella JF, Kalscheuer V, Choi CY, Layman LC, 2010. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 87, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn G, Shawwa RE, Rayyes EE, 1988. Isolated “clinical anophthalmia” in an extensively affected Arab kindred. Clinical Genetics 33, 321–324. [PubMed] [Google Scholar]
- Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijman IJ, Kloosterman WP, Giles RH, Knoers NVAM, van Haaften G, 2016. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet 24, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner C, Knauer R, Stephani U, Marquardt T, Lehle L, von Figura K, 1999. Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-PMan:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. EMBO J. 18, 6816–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Otsuna H, Kidokoro H, Carney KR, Saijoh Y, Chien CB, 2012. A complex choreography of cell movements shapes the vertebrate eye. Development 139, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahrouchi N, George A, Ratbi I, Schneider R, Elalaoui SC, Moosa S, Bharti S, Sharma R, Abu-Asab M, Onojafe F, Adadi N, Lodder EM, Laarabi F-Z, Lamsyah Y, Elorch H, Chebbar I, Postma AV, Lougaris V, Plebani A, Altmueller J, Kyrieleis H, Meiner V, McNeill H, Bharti K, Lyonnet S, Wollnik B, Henrion-Caude A, Berraho A, Hildebrandt F, Bezzina CR, Brooks BP, Sefiani A, 2019. Homozygous frameshift mutations in FAT1 cause a syndrome characterized by colobomatous-microphthalmia, ptosis, nephropathy and syndactyly. Nat Commun 10, 1180–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SLH, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW, 2006. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet 78, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW, 2004. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 41, e94–e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc MS, Mcguire M, Madan-Khetarpal S, Ortiz D, Hayflick S, Keller K, Eng CM, Yang Y, Bi W, 2018. De novo apparent loss-of-function mutations in PRR12 in three patients with intellectual disability and iris abnormalities. Human Genetics 137, 257–264. [DOI] [PubMed] [Google Scholar]
- Lee NB, Kelly L, Sharland M, 1992. Ocular manifestations of Noonan syndrome. Eye 6, 328–334. [DOI] [PubMed] [Google Scholar]
- Legendre M, Abadie V, Attié-Bitach T, Philip N, Busa T, Bonneau D, Colin E, Dollfus H, Lacombe D, Toutain A, Blesson S, Julia S, Martin-Coignard D, Geneviève D, Leheup B, Odent S, Jouk P-S, Mercier S, Faivre L, Vincent-Delorme C, Francannet C, Naudion S, Mathieu-Dramard M, Delrue M-A, Goldenberg A, Héron D, Parent P, Touraine R, Layet V, Sanlaville D, Quélin C, Moutton S, Fradin M, Jacquette A, Sigaudy S, Pinson L, Sarda P, Guerrot A-M, Rossi M, Masurel-Paulet A, El Chehadeh S, Piguel X, Rodriguez-Ballesteros M, Ragot S, Lyonnet S, Bilan F, Gilbert-Dussardier B, 2017. Phenotype and genotype analysis of a French cohort of 119 patients with CHARGE syndrome. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175, 417–430. [DOI] [PubMed] [Google Scholar]
- Leonardi ML, Pai GS, Wilkes B, Lebel RR, 2001. Ritscher-Schinzel cranio-cerebello-cardiac (3C) syndrome: report of four new cases and review. Am J Med Genet 102, 237–42. [PubMed] [Google Scholar]
- Liu C, Widen SA, Williamson KA, Ratnapriya R, Gerth-Kahlert C, Rainger J, Alur RP, Strachan E, Manjunath SH, Balakrishnan A, Floyd JA, Consortium UK, Li T, Waskiewicz A, Brooks BP, Lehmann OJ, FitzPatrick DR, Swaroop A, 2016. A secreted WNT-ligand-binding domain of FZD5 generated by a frameshift mutation causes autosomal dominant coloboma. Human molecular genetics 25, 1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marles SL, Greenberg CR, Persaud TVN, Shuckett EP, Chudley AE, 1992. New familial syndrome of unilateral upper eyelid coloboma, aberrant anterior hairline pattern, and anal anomalies in Manitoba Indians. American Journal of Medical Genetics 42, 793–799. [DOI] [PubMed] [Google Scholar]
- Martinez-Garay I, Tomas M, Oltra S, Ramser J, Molto MD, Prieto F, Meindl A, Kutsche K, Martinez F, 2007. A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly, and mental retardation. Europ J Hum Genet 15, 29–34. [DOI] [PubMed] [Google Scholar]
- McMain K, Robitaille J, Smith I, Johnson J, Wood E, Tremblay F, Blake K, 2008. Ocular features of CHARGE syndrome. Journal of American Association for Pediatric Ophthalmology and Strabismus 12, 460–465. [DOI] [PubMed] [Google Scholar]
- Meinsma R, Fernandez-Salguero P, Kuilenburg ABPV, Gennip AHV, Gonzalez FJ, 1995. Human Polymorphism in Drug Metabolism: Mutation in the Dihydropyrimidine Dehydrogenase Gene Results in Exon Skipping and Thymine Uracilurea. DNA and Cell Biology 14, 1–6. [DOI] [PubMed] [Google Scholar]
- Millischer A-E, Sonigo P, Attie T, Spaggiari E, O’Gorman N, Bessieres B, Kermorvant E, Boddaert N, Salomon L-J, Grevent D, 2019. Fetal MRI findings in a retrospective cohort of 26 cases of prenatally diagnosed CHARGE syndrome individuals. Prenat Diagn. 39, 781–791. [DOI] [PubMed] [Google Scholar]
- Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, Burch MN, Clemens M, Mulliken JB, Smith R, Lin AE, 2008. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet 82, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Russell KL, Bason L, McDonald-McGinn DM, Zackai EH, 2003. Coloboma and other ophthalmologic anomalies in Kabuki syndrome: distinction from charge association. Am J Med Genet 123A, 249–252. [DOI] [PubMed] [Google Scholar]
- Moretti-Ferreira D, Koiffmann CP, Listik M, Setian N, Wajntal A, 1993. Macrosomia, obesity, macrocephaly and ocular abnormalities (MOMO syndrome) in two unrelated patients: delineation of a newly recognized overgrowth syndrome. Am J Med Genet 46, 555–558. [DOI] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, de Die-Smulders C, Giannotti A, Imaizumi K, Jones KL, Del Campo M, Martin RA, Meinecke P, Pierpont MEM, Robin NH, Young ID, Roessler E, Muenke M, 1999. The Mutational Spectrum of the Sonic Hedgehog Gene in Holoprosencephaly: SHH Mutations Cause a Significant Proportion of Autosomal Dominant Holoprosencephaly. Human Molecular Genetics 8, 2479–2488. [DOI] [PubMed] [Google Scholar]
- Nemet AY, Assia EI, Apple DJ, Barequet IS, 2006. Current concepts of ocular manifestations in Marfan syndrome. Surv Ophthalmol. 51, 561–575. [DOI] [PubMed] [Google Scholar]
- Ng BG, Hackmann K, Jones MA, Eroshkin AM, He P, Wiliams R, Bhide S, Cantagrel V, Gleeson JG, Paller AS, Schnur RE, Tinschert S, Zunich J, Hegde MR, Freeze HH, 2012. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 90, 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AOM, van der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GCM, Biesecker LG, 2004. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nature Genet. 36, 411–416. [DOI] [PubMed] [Google Scholar]
- Nicolás-Pérez M, Kuchling F, Letelier J, Polvillo R, Wittbrodt J, Martínez-Morales JR, 2016. Analysis of cellular behavior and cytoskeletal dynamics reveal a constriction mechanism driving optic cup morphogenesis. Elife 5, e15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina S, Kosaki R, Yagihashi T, Azuma N, Okamoto N, Hatsukawa Y, Kurosawa K, Yamane T, Mizuno S, Tsuzuki K, Kosaki K, 2012. Ophthalmic features of CHARGE syndrome with CHD7 mutations. Am J Med Genet 158A, 514–518. [DOI] [PubMed] [Google Scholar]
- Numakura C, Kitanaka S, Kato M, Ishikawa S, Hamamoto Y, Katsushima Y, Kimura T, Hayasaka K, 2010. Supernumerary impacted teeth in a patient with SOX2 anophthalmia syndrome. Am J Med Genet 152A, 2355–2359. [DOI] [PubMed] [Google Scholar]
- Okada I, Hamanoue H, Terada K, Tohma T, Megarbane A, Chouery E, Abou-Ghoch J, Jalkh N, Cogulu O, Ozkinay F, Horie K, Takeda J, Furuichi T, Ikegawa S, Nishiyama K, Miyatake S, Nishimura A, Mizuguchi T, Niikawa N, Hirahara F, Kaname T, Yoshiura K-I, Tsurusaki Y, Doi H, Miyake N, Furukawa T, Matsumoto N, Saitsu H, 2011. SMOC1 is essential for ocular and limb development in humans and mice. Am J Hum Genet 88, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rahilly R The timing and sequence of events in the development of the human eye and ear during the embryonic period proper. Anat Embryol (Berl). 1983;168(1):87–99. [DOI] [PubMed] [Google Scholar]
- Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MTF, Utsch B, Becker C, Nürnberg G, Nürnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F, 2009. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 46, 663–670. [DOI] [PubMed] [Google Scholar]
- Ozeki H, Shirai S, Ikeda K, Ogura Y, 1999. Anomalies associated with Axenfeld-Rieger syndrome. Graefes Arch Clin Exp Ophthalmol. 237, 730–734. [DOI] [PubMed] [Google Scholar]
- Pagon RA, Graham JM Jr., Zonana J, Yong SL, 1981. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE Association. J Pediatr 99, 223–227. [DOI] [PubMed] [Google Scholar]
- Pefkianaki M, Schneider A, Capasso JE, Wasserman BN, Bardakjian T, Levin AV, 2018. Ocular manifestations of PACS1 mutation. Journal of American Association for Pediatric Ophthalmology and Strabismus 22, 323–325. [DOI] [PubMed] [Google Scholar]
- Pichaud F, Desplan C, 2002. Pax genes and eye organogenesis. Current Opinion in Genetics & Development 12, 430–434. [DOI] [PubMed] [Google Scholar]
- Plaisancié J, Collet C, Pelletier V, Perdomo Y, Studer F, Fradin M, Schaefer E, Speeg-Schatz C, Bloch-Zupan A, Flori E, Dollfus H, 2015. MSX2 Gene Duplication in a Patient with Eye Development Defects. Ophthalmic Genet 36, 353–358. [DOI] [PubMed] [Google Scholar]
- Prontera P, Stangoni G, Manes I, Mencarelli A, Donti E, 2009. Encephalocraniocutaneous lipomatosis (ECCL) in a patient with history of familial multiple lipomatosis (FML). American Journal of Medical Genetics Part A 149A, 543–545. [DOI] [PubMed] [Google Scholar]
- Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, Ruddle P, Hurst J, Collin JRO, Salt A, Cooper ST, Thompson PJ, Sisodiya SM, Williamson KA, Fitzpatrick DR, van Heyningen V, Hanson IM, 2005a. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 76, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JRO, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, FitzPatrick DR, 2005b. SOX2 anophthalmia syndrome. American Journal of Medical Genetics Part A 135A, 1–7. [DOI] [PubMed] [Google Scholar]
- Rainger J, Pehlivan D, Johansson S, Bengani H, Sanchez-Pulido L, Williamson KA, Ture M, Barker H, Rosendahl K, Spranger J, Horn D, Meynert A, Floyd JAB, Prescott T, Anderson CA, Rainger JK, Karaca E, Gonzaga-Jauregui C, Jhangiani S, Muzny DM, Seawright A, Soares DC, Kharbanda M, Murday V, Finch A, Uk10K, Baylor-Hopkins Center for Mendelian G, Gibbs RA, van Heyningen V, Taylor MS, Yakut T, Knappskog PM, Hurles ME, Ponting CP, Lupski JR, Houge G, FitzPatrick DR, 2014. Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am J Hum Genet 94, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainger J, Williamson KA, Soares DC, Truch J, Kurian D, Gillessen-Kaesbach G, Seawright A, Prendergast J, Halachev M, Wheeler A, McTeir L, Gill AC, van Heyningen V, Davey MG, Uk10K, FitzPatrick DR, 2017. A recurrent de novo mutation in ACTG1 causes isolated ocular coloboma. Hum Mutat 38, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, Semina EV, 2015. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today 105, 96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Schilter KF, Abdul-Rahman O, Innis JW, Kozel BA, Schneider AS, Bardakjian TM, Lose EJ, Martin DM, Broeckel U, Semina EV, 2011. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Human genetics 130, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R, Owen N, Toms M, Young RM, Tracey-White D, Moosajee M, 2019. Transcriptome profiling of zebrafish optic fissure fusion. Sci Rep 9, 1541–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière J-B, van Bon BWM, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, Gijsen S, Sullivan CT, Christian SL, Abdul-Rahman OA, Atkin JF, Chassaing N, Drouin-Garraud V, Fry AE, Fryns J-P, Gripp KW, Kempers M, Kleefstra T, Mancini GMS, Nowaczyk MJM, van Ravenswaaij-Arts CMA, Roscioli T, Marble M, Rosenfeld JA, Siu VM, de Vries BBA, Shendure J, Verloes A, Veltman JA, Brunner HG, Ross ME, Pilz DT, Dobyns WB, 2012. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nature genetics 44, 440–S442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SP, Twigg SRF, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, Ørstavik KH, Quarrell OWJ, Schwartz CE, Shears DJ, Suri M, Kendrick-Jones J, Bacino C, Becker K, Clayton-Smith J, Giovannucci-Uzielli M, Goh D, Grange D, Krajewska-Welasek M, Lacombe D, Morris C, Odent S, Savarirayan R, Stratton R, Superti-Furga A, Verloes A, Vigneron J, Wilcox W, Winter R, Young K, Wilkie AO, OPD-spectrum Disorders Clinical Collaborative Group., 2003. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nature Genetics 33, 487–491. [DOI] [PubMed] [Google Scholar]
- Sanlaville D, Verloes A, 2007. CHARGE syndrome: an update. European Journal of Human Genetics 15, 389–399. [DOI] [PubMed] [Google Scholar]
- Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont MEM, Sullivan MJ, Dobyns WB, Eccles MR, 1995. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nature Genetics 9, 358–364. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, Muenke M, 2003. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. American Journal of Medical Genetics 116A, 215–221. [DOI] [PubMed] [Google Scholar]
- Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, Kherani F, Katowitz J, Schimmenti LA, Hummel M, FitzPatrick DR, Young TL, 2008. Familial recurrence of SOX2 anophthalmia syndrome: Phenotypically normal mother with two affected daughters. Am J Med Genet 146A, 2794–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Bardakjian T, Reis LM, Tyler RC, Semina EV, 2009. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet 149A, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T, Crawford GE, Scacheri PC, 2009. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res 19, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorderet DF, Nichini O, Boisset G, Polok B, Tiab L, Mayeur H, Raji B, de la Houssaye G, Abitbol MM, Munier FL, 2008. Mutation in the human homeobox gene NKX5–3 causes an oculo-auricular syndrome. Am J Hum Genet 82, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AF, Mohr DW, Kasch LM, Barton JA, Pittiglio R, Ingersoll R, Craig B, Marosy BA, Doheny KF, Bromley WC, Roderick TH, Chassaing N, Calvas P, Prabhu SS, Jabs EW, 2014. Identification of an HMGB3 Frameshift Mutation in a Family With an X-linked Colobomatous Microphthalmia Syndrome Using Whole-Genome and XExome Sequencing. JAMA Ophthalmology 132, 1215–1220. [DOI] [PubMed] [Google Scholar]
- Sidhaye J, Norden C, 2017. Concerted action of neuroepithelial basal shrinkage and active epithelial migration ensures efficient optic cup morphogenesis. Elife 6, e22689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaats GG, Isabella CR, Kroes HY, Dempsey JC, Gremmels H, Monroe GR, Phelps IG, Duran KJ, Adkins J, Kumar SA, Knutzen DM, Knoers NV, Mendelsohn NJ, Neubauer D, Mastroyianni SD, Vogt J, Worgan L, Karp N, Bowdin S, Glass IA, Parisi MA, Otto EA, Johnson CA, Hildebrandt F, van Haaften G, Giles RH, Doherty D, 2016. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J Med Genet 53, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM, Chao R, Vacik T, Yahyavi M, Abouzeid H, Bardakjian T, Schneider A, Shaw G, Sherr EH, Lemke G, Youssef M, Schorderet DF, 2012. VAX1 mutation associated with microphthalmia, corpus callosum agenesis, and orofacial clefting: the first description of a VAX1 phenotype in humans. Hum Mutat 33, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, AlGazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH, Harris PC, Johnson CA, 2006. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nature Genetics 38, 191–196. [DOI] [PubMed] [Google Scholar]
- Sobreira NL de M, Brunoni D, Cernach MCSP, Perez ABA, 2006. Finlay-Marks (SEN) syndrome: a sporadic case and the delineation of the syndrome. Am J Med Genet 140A, 300–302. [DOI] [PubMed] [Google Scholar]
- Srour M, Chitayat D, Caron V, Chassaing N, Bitoun P, Patry L, Cordier M-P, Capo-Chichi J-M, Francannet C, Calvas P, Ragge N, Dobrzeniecka S, Hamdan FF, Rouleau GA, Tremblay A, Michaud JL, 2013. Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am J Hum Genet 93, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, Storen R, Bennetts B, Savarirayan R, Jamieson RV, 2011. Isolated hypogonadotropic hypogonadism with SOX2 mutation and anophthalmia/microphthalmia in offspring. Eur J Hum Genet 19, 753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, Ieda D, Saitoh S, Hiyane M, Suzumura H, Yagyu K, Shiraishi H, Nakajima M, Fueki N, Habata Y, Ueda Y, Komatsu Y, Yan K, Shimoda K, Shitara Y, Mizuno S, Ichinomiya K, Sameshima K, Tsuyusaki Y, Kurosawa K, Sakai Y, Haginoya K, Kobayashi Y, Yoshizawa C, Hisano M, Nakashima M, Saitsu H, Takeda S, Matsumoto N, 2016. Molecular genetic analysis of 30 families with Joubert syndrome. Clin Genet 90, 526–535. [DOI] [PubMed] [Google Scholar]
- Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, Fujieda K, 2009. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab 94, 314–319. [DOI] [PubMed] [Google Scholar]
- Tellier AL, Cormier-Daire V, Abadie V, Amiel J, Sigaudy S, Bonnet D, de Lonlay-Debeney P, Morrisseau-Durand MP, Hubert P, Michel JL, Jan D, Dollfus H, Baumann C, Labrune P, Lacombe D, Philip N, LeMerrer M, Briard ML, Munnich A, Lyonnet S, 1998. CHARGE syndrome: Report of 47 cases and review. Am J Med Genet 76, 402–409. [DOI] [PubMed] [Google Scholar]
- Temtamy SA, Salam MA, Aboul-Ezz EHA, Hussein HA, Helmy SAH, Shalash BA, 1996. New autosomal recessive multiple congenital abnormalities/mental retardation syndrome with craniofacial dysmorphism absent corpus callosum, iris colobomas and connective tissue dysplasia. Clin. Dysmorph 5, 231–240. [PubMed] [Google Scholar]
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, Saada J, Perrault I, Amiel J, Litzler J, Filhol E, Elkhartoufi N, Kwong M, Casanova J-L, Boddaert N, Baehr W, Lyonnet S, Munnich A, Burglen L, Chassaing N, Encha-Ravazi F, Vekemans M, Gleeson JG, Valente EM, Jackson PK, Drummond IA, Saunier S, Attié-Bitach T, 2014. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat 35, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker E, Elcioglu N, Ozcan E, Yenice O, Ogut M, 2003. Colobomatous macrophthalmia with microcornea syndrome: report of a new pedigree. Am J Med Genet 121A, 25–30. [DOI] [PubMed] [Google Scholar]
- Traboulsi EI, 1999. Colobomatous microphthalmia, anophthalmia, and associated malformation syndromes In: Traboulsi EI, editor. Genetic diseases of the eye. New York: Oxford University Press; pp. 51–80. [Google Scholar]
- Twigg SRF, Hufnagel RB, Miller KA, Zhou Y, McGowan SJ, Taylor J, Craft J, Taylor JC, Santoro SL, Huang T, Hopkin RJ, Brady AF, et al. , 2016. A recurrent mosaic mutation in SMO, encoding the Hedgehog signal transducer smoothened, is the major cause of Curry-Jones syndrome. Am J Hum Genet 98, 1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, Ozdag H, Liehr T, Balci S, Alikasifoglu M, Wollnik B, Akarsu NA, 2010. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet 86, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gennip AH, Abeling NGGM, Stroomer AEM, van Lenthe H, Bakker HD, 1994. Clinical and biochemical findings in six patients with pyrimidine degradation defects. J Inherit Metab Dis 17, 130–132. [DOI] [PubMed] [Google Scholar]
- Verloes A, 2005. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 15, 306–308. [DOI] [PubMed] [Google Scholar]
- Verloes A, Lambotte C, Neri G, Reynolds JF, 1989. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. American Journal of Medical Genetics 32, 227–232. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG, 2004. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature Genetics 36, 955–957. [DOI] [PubMed] [Google Scholar]
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, Linberg JV, Schneider AS, Mathers PH, 2004. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Human Molecular Genetics 13, 315–322. [DOI] [PubMed] [Google Scholar]
- Wakamatsu N, Yamada Y, Yamada K, Ono T, Nomura N, Taniguchi H, Kitoh H, Mutoh N, Yamanaka T, Mushiake K, Kato K, Sonta S. i., Nagaya M, 2001. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nature Genetics 27, 369–370. [DOI] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M, 1999. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nature Genetics 22, 196–198. [DOI] [PubMed] [Google Scholar]
- Wang L, He F, Bu J, Zhen Y, Liu X, Du W, Dong J, Cooney JD, Dubey SK, Shi Y, Gong B, Li J, McBride PF, Jia Y, Lu F, Soltis KA, Lin Y, Namburi P, Liang C, Sundaresan P, Paw BH, Li W, Li DY, Phillips JD, Yang Z, 2012. ABCB6 mutations cause ocular coloboma. Am J Hum Genet 90, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, Zhang Q, Ye J, Yan Z, Denduluri S, Idowu O, Li M, Shen C, Hu A, Haydon RC, Kang R, Mok J, Lee MJ, Luu HL, Shi LL, 2014. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis 1, 87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson KA, FitzPatrick DR, 2014. The genetic architecture of microphthalmia, anophthalmia and coloboma. European Journal of Medical Genetics 57, 369–380. [DOI] [PubMed] [Google Scholar]
- Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, FitzPatrick DR, 2006. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet 15, 1413–1422. [DOI] [PubMed] [Google Scholar]
- Williamson KA, Rainger J, Floyd JA, Ansari M, Meynert A, Aldridge KV, Rainger JK, Anderson CA, Moore AT, Hurles ME, Clarke A, van Heyningen V, Verloes A, Taylor MS, Wilkie AO, UK10K Consortium, Fitzpatrick DR, 2014. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am J Hum Genet 94, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincent J, Holmberg E, Stromland K, Soller M, Mirzaei L, Djureinovic T, Robinson K, Anderlid B, Schoumans J, 2008. CHD7 mutation spectrum in 28 Swedish patients diagnosed with CHARGE syndrome. Clin Genet 74, 31–38. [DOI] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D, 2003. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456. [DOI] [PubMed] [Google Scholar]
- Wyatt A, Bakrania P, Bunyan DJ, Osborne RJ, Crolla JA, Salt A, Ayuso C, Newbury-Ecob R, Abou-Rayyah Y, Collin JR, Robinson D, Ragge N, 2008. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia, microphthalmia and coloboma. Hum Mutat 29, E278–83. [DOI] [PubMed] [Google Scholar]
- Wyatt AW, Osborne RJ, Stewart H, Ragge NK, 2010. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum Mutat 31, 781–787. [DOI] [PubMed] [Google Scholar]
- Yahyavi M, Abouzeid H, Gawdat G, de Preux A-S, Xiao T, Bardakjian T, Schneider A, Choi A, Jorgenson E, Baier H, El Sada M, Schorderet DF, Slavotinek AM, 2013. ALDH1A3 loss of function causes bilateral anophthalmia/microphthalmia and hypoplasia of the optic nerve and optic chiasm. Human molecular genetics 22, 3250–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, 2004. Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Seminars in Cell & Developmental Biology 15, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariz KO, Sakalar YB, Jin X, Hertz J, Sener EF, Akay H, Özbek MN, Farooq A, Goldberg J, Tekin M, 2015. A homozygous SIX6 mutation is associated with optic disc anomalies and macular atrophy and reduces retinal ganglion cell differentiation. Clinical Genetics 87, 192–195. [DOI] [PubMed] [Google Scholar]
- Ye M, Berry-Wynne KM, Asai-Coakwell M, Sundaresan P, Footz T, French CR, Abitbol M, Fleisch VC, Corbett N, Allison WT, Drummond G, Walter MA, Underhill TM, Waskiewicz AJ, Lehmann OJ, 2010. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Human Molecular Genetics 19, 287–298. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Gascon-Guzman G, Tovilla-Canales JL, 2005. Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clin Genet 68, 564–566. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Perez-Cano HJ, Aguinaga M, 2006. Anophthalmia-esophageal atresia syndrome caused by an SOX2 gene deletion in monozygotic twin brothers with markedly discordant phenotypes. Am J Med Genet 140A, 1899–1903. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, Wei C, Tesar PJ, Hatzoglou M, Martin DM, Scacheri PC, 2010a. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Human molecular genetics 19, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Layman WS, Martin DM, Scacheri PC, 2010b. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 152A, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]