

Abstract
Pathogenic mutations in the mitochondrial genome (mtDNA) impair organellar ATP production, requiring mutant cells to activate metabolic adaptations for survival. Understanding how metabolism adapts to clinically relevant mtDNA mutations may provide insight into cellular strategies for metabolic flexibility. In this study, we use 13C isotope tracing and metabolic flux analysis to investigate central carbon and amino acid metabolic reprogramming in isogenic cells containing mtDNA mutations. We identify alterations in glutamine and cystine transport which indirectly regulate mitochondrial metabolism and electron transport chain function. Metabolism of cystine can promote glucose oxidation through the transsulfuration pathway and the production of α-ketobutyrate. Intriguingly, activating or inhibiting α-ketobutyrate production is sufficient to modulate both glucose oxidation and mitochondrial respiration in mtDNA mutant cells. Thus, cystine-stimulated transsulfuration serves as an adaptive mechanism linking glucose oxidation and amino acid metabolism in the setting of mtDNA mutations.
Keywords: mtDNA mutations, Transsulfuration, Metabolic reprogramming
1. Introduction
Mutations in the human mitochondrial genome result in a heterogeneous set of diseases with significant pathology and morbidity (Gorman et al., 2016; McCormack et al., 2017). The fundamental biochemical defects result in reduced electron transport chain (ETC) function and oxidative phosphorylation, and alternative energy-producing pathways are elicited to allow cellular survival even at high mutation loads. The precise strategies of cellular metabolic adaptation in response to mtDNA mutations are not understood. However, recent results suggest that metabolic interventions, either via mTOR inhibition or activation of hypoxia-related pathways, are beneficial in mice suffering from neuronal mitochondrial disease (Ferrari et al., 2017; Jain et al., 2016; Johnson et al., 2013). Similarly, modulating NAD(H) levels in mice and C.elegans with mitochondrial dysfunction can also influence disease progression (Cerutti et al., 2014; Zhang et al., 2013; Khan et al., 2014; McCormack et al., 2015). The success of metabolic treatments which do not rescue the mitochondrial biochemical defect per se, support a model in which the nature of metabolic alterations downstream of the oxidative phosphorylation defect contributes to disease progression.
Several types of metabolic adaptations have been previously observed in cells subject to mitochondrial dysfunction. It is well known that decreased glucose oxidation within the mitochondria, coupled with increased lactic acid production, is classically associated with respiratory inhibition (Dickman and Mandel, 1990). More recently, alterations in fluxes of tricarboxylic acid (TCA) cycle metabolism, one-carbon metabolism, and glutamine utilization have been reported in the context of mitochondrial dysfunction (Bao et al., 2016; Shin et al., 2017; Gaude et al., 2018; Chen et al., 2018; Mullen et al., 2011, 2014; Nikkanen et al., 2016; Lozoya et al., 2018). We recently reported Nrf2- and ATF4-dependent upregulation of the SLC7A11 gene product (xCT) in the context of mtDNA mutations (Shin et al., 2017); xCT is also upregulated in the setting of a large number of physiologic stressors (Shin et al., 2017; Koppula et al., 2018; Albertini et al., 2018; Massie et al., 2015; Parsanathan and Jain, 2018; Chen et al., 2017; Wang et al., 2016; Zhang et al., 2016), and is the functional component of the xc− transport system on the plasma membrane, which imports cystine (the oxidized form of cysteine) while exporting glutamate. Enhanced xc− activity was found to be maladaptive in vitro in the setting of glucose depletion, due to an inability to efficiently utilize glutamate for survival (Shin et al., 2017; Koppula et al., 2018). These results indicate that amino acid transport at the plasma membrane can have important functional consequences in mtDNA mutant cells.
Understanding metabolic flexibility in the setting of mtDNA mutations may ultimately shed light on mechanisms underlying disease progression. In this study, we investigate alterations in central carbon and amino acid metabolism in the context of two common pathogenic mtDNA mutations. Using 13C-isotope tracing techniques, we calculated metabolic flux values for residual glucose oxidation and TCA cycling in mtDNA mutant cells. We find that differential expression of glutamine and glutamate transporters imparts changes in cystine and glucose metabolism, thereby influencing TCA cycle fluxes within mitochondria. Specifically, cystine-stimulated transsulfuration in mtDNA mutant cells maintains residual glucose oxidation via the metabolite α-ketobutyrate. We show that targeting α-ketobutyrate production is sufficient to modulate glucose oxidation and mitochondrial respiration in mtDNA mutant cells, suggesting that linkages between amino acid and glucose metabolism promote flexibility in the setting of genetic perturbations.
2. Methods
2.1. Reagents
Antibodies to the following proteins were used: SLC1A5 (Cell Signaling Technology, #5100), SLC7A11 (Cell Signaling Technology, #12691), SLC7A5 (Santa Cruz Biotechnology, #sc-374232), GLS (Abcam, #ab93434), GLUL (Proteintech, #11037–2-AP), GLUD1 (EMD Millipore, #ABN443), CTH (Santa Cruz Biotechnology, #sc-374249), Histone H2B (Santa Cruz Biotechnology, #sc-515808), Histone H3 (Cell Signaling Technology, #9715). Oligomycin, CCCP, antimycin A, α-ketobutyrate, α-hydroxybutyrate, pyruvate, sodium sulfide and propargylglycine were obtained from Sigma-Aldrich. The following isotope labeled compounds were used in this study:
| Compound | Source | Final concentration |
|---|---|---|
| [U–13C]-glucose | Cambridge Isotope Laboratories #CLM-1396 | 10 mM |
| [U–13C]-glutamine | Cambridge Isotope Laboratories #CLM-1822 | 1 mM |
| [U–13C,15N]-cystine | Cambridge Isotope Laboratories #CNLM-4244 | 0.2 mM |
| [U–13C]-methionine | Cambridge Isotope Laboratories #CLM-893 | 0.2 mM |
| [U–13C,15N]-alanine | Cambridge Isotope Laboratories #CNLM-534 | N/A |
| [2,3,3-D3]-aspartate | Cambridge Isotope Laboratories #DLM-546 | N/A |
2.2. Cell culture and isotopic labeling
143B cybrid cell lines were maintained in DMEM (Sigma D6429) supplemented with 10% FBS, 100U/mL penicillin/streptomycin, 2 mM L-glutamine, and 0.1 mg/mL uridine at 37°C and 5% CO2. Cells with homoplasmic mtDNA mutations are cybrids from the fusion of enucleated patient cells with 143B ρ0 cells (lacking mtDNA), and previously described: ND1 cybrid cells have a G3460A mutation in the mt-ND1 gene of complex I (Mustafa et al., 2009). ATP6 cells have a T8993G mutation in the mt-ATP6 subunit of complex V (Kwong et al., 2007). All experiments were performed in pyruvate-free DMEM supplemented with 10% dialyzed FBS, unless otherwise noted in the text. For isotopic labeling with glucose or glutamine, cells were cultured in 6-well plates in glucose, glutamine, pyruvate free DMEM (Gibco A144301), supplemented with 10% dialyzed FBS, 100U/mL penicillin/streptomycin, 1 mM L-glutamine, and 10 mM glucose. For isotopic labeling with cystine or methionine, cells were cultured in 6-well plates in glutamine, pyruvate, methionine, and cystine free DMEM (Gibco 21013024), supplemented with 10% dialyzed FBS, 100U/mL penicillin/streptomycin, 1 mM L-glutamine, 0.2 mM methionine, and 0.2 mM cystine.
To modulate gene expression in a non-clonal manner, we utilized either shRNA-mediated knockdown, or retroviral based overexpression. shRNA-mediated knockdown was achieved via lentiviral infection using the following shRNA plasmids (see Table, purchased from Sigma Aldrich) in the pLKO.1 backbone, followed by selection and maintenance with puromycin (1 μg/mL). To generate shNT (non-targeting shRNA) cells, parent cell lines were infected by pLKO.1 lentiviral vectors expressing shRNA targeting Renilla luciferase. Overexpression was achieved by retroviral infection using pQCXIP expression plasmids containing SLC1A5 or SLC7A11 human cDNA, followed by selection and maintenance with puromycin (1 μg/mL).
| Gene Target | TRC Number |
|---|---|
| SLC7A5 | 0000043011 |
| SLC7A11 | 0000043126 |
| Cystathionine γ-lyase (CTH) | 0000222704 (shCTH1) |
| Cystathionine γ-lyase (CTH) | 000078263 (shCTH2) |
mtDNA genotypes were verified in all cell lines by Sanger sequencing on the relevant regions in mt-ND1 and mt-ATP6, using primers listed below.
| Gene | Primer 1 | Primer 2 |
|---|---|---|
| mt-ND1 | CCCATGGCCAACCTCCTACTCC | CATGATGGCAGGAGTAATCAGAGG |
| mt-ATP6 | CTAACCTCCTCGGACTCCTG | TGTGTTGTCGTGCAGGTAGA |
For proliferation assays, cells were plated at 5000 cells/well in 96-well plates and incubated in pyruvate-free media, at 37 °C with 5% CO2. At timepoints, cells were stained and quantified as previously described (Vichai and Kirtikara, 2006). Briefly, cells were fixed to the plate with 10% (wt/vol) trichloroacetic acid, and stained with sulforhodamine B (0.057% wt/vol) for 30 min. Absorbance was measured at 510 nm using a FLUOstar Omega Microplate Reader (BMG Labtech).
2.3. Metabolite extraction and mass spectrometry analysis
Cells were rapidly washed with cold PBS, scraped into 80% methanol and lysed with three freeze-thaw cycles. For analysis of spent culture media, 50 μL of media was added to cold water:-methanol:chloroform (1:1:1 mixture) and vortexed for 1 min. Precipitated protein was removed by centrifugation, an internal standard (norvaline) was added, and the metabolite supernatant was evaporated (SpeedVac, ThermoFisher). For GC-MS analysis, dried metabolites were derivatized to form methoxime-TBDMS adducts by incubating with 1% methoxyamine hydrochloride (Sigma-Aldrich) in pyridine at 70°C for 15 min followed by addition of N-tert-Butyldimethylsiyl-N-methyltrifluoroacetamide (MTBSTFA, Sigma-Aldrich) for 1 hour. Derivatized samples were analyzed using an Agilent Technologies 7890B gas chromatographer with a HP-5MS 5% phenyl methyl Silox column (Agilent) coupled to an Agilent Technologies 5977A mass spectrometer. For analysis of glutathione (GSH), metabolites were extracted and dried as above, and then analyzed by LC-MS as previously described (Mullen et al., 2014). Retention times of all metabolites analyzed were validated using pure standards. For quantitation of α-hydroxybutyrate (Fig. S5L), standards were spiked into fresh media and then extracted as described above. Isotopologue abundances were determined by integrating peak areas for the indicated metabolite ion fragments and normalizing to total ion count. Mass isotopomer distributions were obtained after correction for natural abundance as previously described (Fernandez et al., 1996). Principal components analysis was performed in MATLAB (Mathworks, Inc.). PLS-DA (Partial Least Squares Discriminant Analysis) was performed using SIMCA software (Sartorius Stedim Biotech).
2.4. Metabolic flux analysis
Steady state metabolic fluxes were calculated by combining extracellular flux measurements (glucose/glutamine consumption, lactate/glutamate/alanine/aspartate secretion) and mass isotopomer distributions using the isotopomer network compartmental analysis (INCA) software (Young, 2014), which simulates mass isotopomer distributions (MIDs) using the elementary metabolite unit (EMU) framework (Young et al., 2008; Antoniewicz et al., 2007). The metabolic flux analysis assumptions are:
Cells are at metabolic and isotopic steady state (Figs. S2A and B).
Produced CO2 is not reincorporated via carboxylation reactions.
Succinate and fumarate are symmetrical, and their metabolism does not favor a particular orientation.
Pyruvate and aspartate are present in both the cytosol and mitochondria; malate, citrate and oxaloacetate freely exchange between the cytosol and mitochondria.
During metabolite extraction, mitochondrial and cytosolic pools are mixed and GC-MS analysis of isotopic enrichments reflects the mixture of different pools.
Biomass requirements (v29) were assumed to be similar to CHO cells (Ahn and Antoniewicz, 2011).
Glucose, lactate, glutamine and glutamate concentrations were measured in fresh and spent culture medium using a BioProfile Basic-4 analyzer (NOVA). Alanine and aspartate concentrations in media were quantitated using GC-MS by spiking a known concentration of labeled standard into samples prior to extraction. Secretion and consumption rates for metabolites were calculated by normalizing to total protein amount and time. Reaction networks describing the stoichiometry and atom transitions of central carbon metabolism are listed in Table S1. Extracellular flux reactions (v1, v8, v10, v23, v24, v26) were constrained based on measured values and uncertainties. Cytosolic and mitochondrial pools are assumed to mix during metabolite extraction, and mixing reactions are incorporated into the reaction network (v31–v36). Steady state (12hr labeling) mass isotopomer distributions from [U–13C]-glucose and [U–13C]-glutamine experiments were used to fit the model and estimate flux values. Flux estimations were initiated from random values at least 50 times to ensure a global minimum was reached. Goodness-of-fit was assessed by a chi-squared test. 95% confidence intervals (Table S1) were calculated by assessing the sensitivity of the sum of squared residuals to individual variations of each flux parameter. Fluxes within the malate-aspartate shuttle were found to be unconstrained, and the network model was therefore simplified to ignore compartmentalization of malate, aspartate, and oxaloacetate. (Table S1).
2.5. RT-qPCR screen
Primer sequences targeting exon boundaries of SLC family members were obtained from qPrimerDepot (Cui et al., 2007), and synthesized by Sigma-Aldrich. We targeted known amino-acid transporters on the plasma membrane (see Table S2 for full list). RNA from wild-type and mtDNA mutant cell lines was prepared using TRIzol Reagent (Thermo Fisher) according to manufacturer instructions, and used to measure transcript levels by RT-qPCR using iScript Reverse Transcription Supermix (Bio-Rad). Only PCR products which match the expected size and contained single peak melting curves were analyzed; other products were considered undetectable. Transcript levels were normalized to β-actin levels in triplicate measurements, and are reported in Table S2. Genes were sorted based on those having the largest absolute change (Δ) in transcript levels.
2.6. Oxygen consumption rate (OCR)
Cells were plated at 15,000 cells per well in an XFe96 well plate (Seahorse Bioscience) and incubated overnight. The next day cells were washed three times into assay media (pyruvate-free DMEM (Sigma D5030) supplemented with 2 mM L-glutamine, 10 mM glucose, 100 U/mL penicillin/streptomycin, pH7.4), and incubated at 37°C in a non-CO2 incubator for 45 min before starting the assay. Oxygen consumption rate (OCR) was measured in a Seahorse XFe96 instrument using consecutive 3 min measurements, separated by 3 min mixing periods. Oligomycin (final concentration 2 μM), CCCP (final concentration 1 μM), and antimycin A (final concentration 2 μM) were injected as indicated. Mitochondrial OCR (mito-OCR) was calculated by subtracting antimycin-inhibited OCR from basal OCR (prior to drug injections). Cell plates were fixed with trichloroacetic acid after assay completion and stained with sulforhodamine B as described (Vichai and Kirtikara, 2006) for normalization.
2.7. NAD+/NADH measurements
NAD+/NADH measurements were performed as previously described (Sullivan et al., 2015) in pyruvate-free media. Briefly, cells were washed 3 times in cold PBS, extracted in 100 μL cold lysis buffer (1% Dodecyltrimethylammonium bromide (DTAB) in 0.2N NaOH diluted 1:1 with PBS) and immediately placed on ice. 20 μL of the sample was incubated at 75°C for 30 min to selectively degrade NAD+, while 20 μL of sample was moved to tubes containing 20 μL of lysis buffer plus 20 μL 0.4N HCl and incubated at 60°C for 15 min to selectively degrade NADH. Following incubations, samples were equilibrated to room temperature (10 min), then quenched with 0.2M Tris in 0.2N HCl (to measure NADH) or 0.5M Tris base (to measure NAD+). The NAD/NADH-Glo assay kit (Promega G9071) was then used to quantitate NAD+ and NADH levels per manufacturer’s instructions.
3. Results
3.1. Quantitative flux analysis identifies altered central carbon metabolism in mtDNA mutant cells
We focused on isogenic 143B cybrid cell lines harboring human pathogenic mtDNA mutations which retain residual respiratory activity; this model allows us to evaluate the metabolic compensation to mtDNA mutations in a common nuclear background. Mutant cells contained homoplasmic mutations in either mt-ND1 (G3460A) or mt-ATP6 (T8993G) (Figs. S1A and B). Oxygen consumption measurements (in pyruvate-free media) indicate that these mutant cell lines retain ~10% respiratory activity (Fig. 1A). To assess central carbon metabolism, cells were pulsed for 1 hour with media containing uniformly 13C-labeled ([U–13C]) glucose or glutamine, and labeled isotopologue abundance of mitochondrial TCA cycle metabolites was measured (Fig. 1B). mtDNA-mutant cell lines displayed significantly decreased entry of labeled glucose into TCA cycle metabolites (Fig. 1C). We also observed significant decreases in the abundance of glutamine-derived isotopologues of the TCA cycle metabolite citrate (Fig. 1C), despite adequate labeling of the upstream metabolite glutamate, consistent with previous reports assessing glutamine metabolism in cells with oxidative phosphorylation defects (Mullen et al., 2011).
Fig. 1. Metabolic flux analysis of ND1 and ATP6 mutant cell lines.

(A) Oxygen consumption rates in wild-type (wt; n = 8), ND1 (n = 6) and ATP6 (n = 8) mutant cell lines. (B) Schematic of carbon incorporation from glucose (red) or glutamine (blue) into TCA cycle metabolites. (C) Relative isotopologue abundances of the indicated metabolites following 1 h pulses with labeled glucose or glutamine. Abundances (n = 3 for each group) were normalized to total ion count. Same color scheme as (A). (D) Fractional contribution (1-M0) of glucose or glutamine to the indicated metabolites after steady-state labeling with glucose or glutamine. n = 3 for each group. Same color scheme as (A). (E) Flux ratios for individual enzymatic steps between ND1 and wt cells following metabolic flux analysis. Thickness of the line indicates the magnitude of the ratio. Blue represents fluxes upregulated in ND1 cells; red represents fluxes downregulated in ND1 cells (relative to wt). ala, alanine; glc, glucose; lac, lactate; pyr, pyruvate; acCoA, acetyl-CoA; cit, citrate; αKG, α-ketoglutarate; glu, glutamate; gln, glutamine; suc, succinate; fum, fumarate; mal, malate; oaa, oxaloacetate; asp, aspartate; ser, serine; GAP; glyceraldehyde 3-phosphate; DHAP, dihydroxyacetone phosphate; 3 PG, 3-phosphoglycerate; pro; proline. (F) Same as E, but comparing ATP6 and wt cells. Representative data from a single experiment is shown, and all experiments were repeated at least in triplicate. Error bars represent standard deviations from mean of independent samples. Adjusted p-values calculated by 1-way ANOVA, followed by Tukey’s test for multiple comparisons. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Steady-state labeling experiments indicated that the relative carbon contribution to TCA cycle metabolites shifts from glucose towards glutamine in ND1 and ATP6-mutant cell lines (Fig. 1D), consistent with previous reports in mtDNA mutant cell lines (Gaude et al., 2018; Chen et al., 2018). Concomitant with this shift, proliferation of mutant cell lines (in pyruvate-free media) displayed increased sensitivity to glutamine-withdrawal (Fig. S1C). We utilized steady-state 13 C-glucose and 13C-glutamine-derived mass isotopomer distributions (MIDs) from parallel labeling experiments at isotopic steady state (Figs. S2A–D) to perform metabolic flux analysis (MFA) surrounding TCA cycle metabolism (Table S1) and determine absolute flux values for individual biochemical reactions (Table 1). Net transport rates were constrained based on measured values (Table S1), and flux values (Fig. S3, Table 1) were inferred by minimizing sum of squared residuals to experimentally measured MIDs, as previously described (Young, 2014). Parameter sensitivity and 95% confidence intervals were assessed for each flux value (see Methods and Table S1 and Fig. S3). Flux ratios between mutant and wild-type cells are presented in Fig. 1E and F. Both ND1 and ATP6 mutant cells display enhanced import of glucose and glutamine, as well as enhanced secretion of lactate and glutamate at the expense of further substrate oxidation. Thus, in vitro metabolic reprogramming in response to these particular pathogenic mtDNA mutations involves enhanced secretion of carbons from glucose and glutamine. Intriguingly, the net metabolic reprogramming observed in these mtDNA mutant cells limits efficient contribution of both glucose and glutamine-derived carbons to the TCA cycle.
Table 1.
Calculated metabolic fluxes using the networks illustrated in Fig. 1E and F and Table S1. Fluxes were determined using measured mass isotopomer distributions (Figs. S2A and B), and measured net cellular transport of glucose, lactate, glutamine and glutamate, alanine and aspartate. All values are expressed in nmol/hr/μg protein. Blue and red values were upregulated and downregulated, respectively, in mutant cells (relative to wild-type cells), based on non-overlapping 95% confidence intervals.
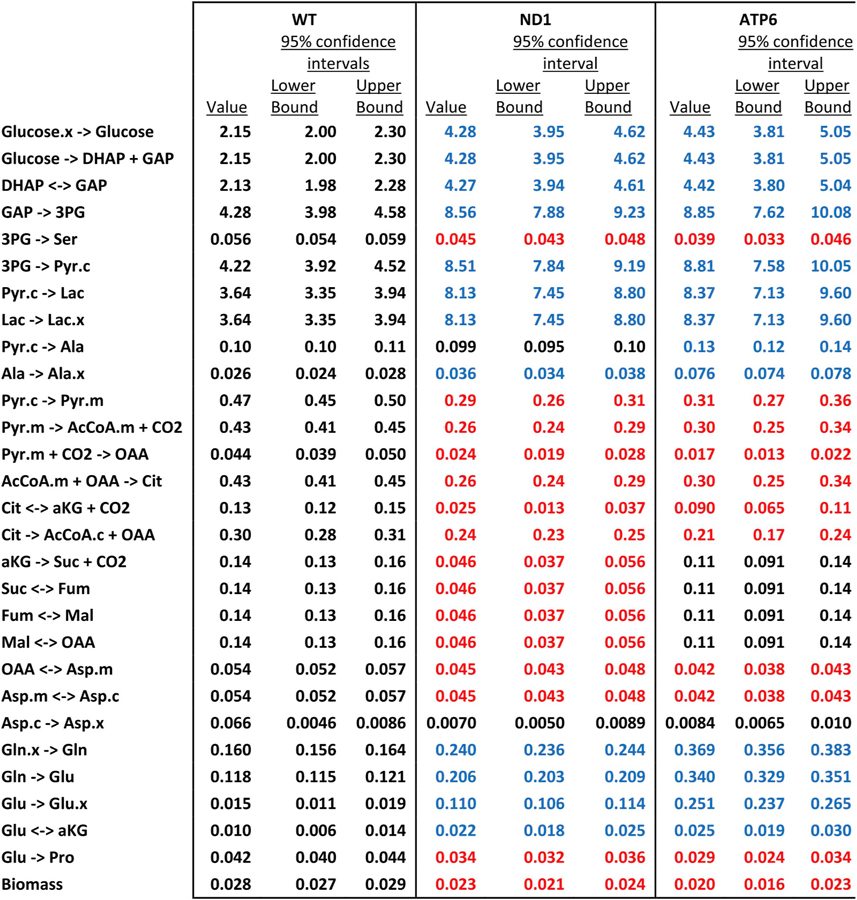
|
Metabolites: DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde 3-phosphate; 3 PG, 3-phosphoglycerate; Pyr, pyruvate; Lac, lactate; Ala, alanine; AcCoA, acetyl-CoA; OAA, oxaloacetate; Cit, citrate; aKG, α-ketoglutarate; Suc, succinate; Fum, fumarate; Mal, malate; Asp, aspartate; Gln, glutamine; Glu, glutamate; Ser, serine; Pro, proline;.x, extracellular;.c, cytosolic;.m, mitochondrial.
3.2. Altered amino acid transport in mtDNA mutant cells
The metabolic flux analysis above indicates increased extracellular shunting of glucose and glutamine-derived carbons; therefore, we decided to assess alterations in the metabolism of other amino acids. We first profiled relative amino acid levels in spent media from wild-type and mutant cells using targeted metabolomics. Principal component analysis (PCA) reliably distinguished spent media from mutant versus wild-type cells (Fig. 2A). Variable importance in projection (VIP) scores > 1.0 indicated that levels of glutamine (Q), glutamate (E), aspartate (D), serine (S) and cystine (CC) were the largest contributors to discrimination between genotypes (Fig. 2B). Alterations in aspartate and serine metabolism have been previously reported in the setting of mitochondrial dysfunction (Bao et al., 2016; Sullivan et al., 2015; Birsoy et al., 2015), while changes in glutamine and glutamate utilization are consistent with our metabolic flux measurements (Fig. 1E and F).
Fig. 2. Alterations amino acid transport in mtDNA mutant cells.

(A) Principal Component Analysis of amino acid levels in fresh and spent media from the indicated cell types. Samples are plotted based on the first two principal components (PC1, PC2) with the indicated contributions to total variance. n = 3 independent samples per group. (B) Variable importance in projection (VIP) scores identified by PLS-DA for amino acids levels in wt, ND1 and ATP6 spent media (n = 3 independent samples per group). VIP scores > 1.0 are indicated by an asterisk (*). (C) qRT-PCR screen of amino acid transporters: Absolute changes from wild-type cells in transcript levels of the indicated transporter are indicated in a heatmap where colors indicate the magnitude and direction of the change in transcript amount (normalized to actin levels). Genes are ordered based on the average absolute magnitude of change in mutant cells. (D) Western blots of indicated transporters and metabolic enzymes in the relevant cell lines. (E) Measurements of glutamine uptake and glutamate secretion in the indicated cell type. Error bars indicate standard deviations from mean of n = 3 independent samples per group. Adjusted p-values calculated by 1-way ANOVA, followed by Tukey’s test for multiple comparisons. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Alterations in amino acid transport represent a potential mechanism underlying our observations from spent media. We therefore screened transcript levels of known plasma membrane amino acid transporters in wild-type and mutant cybrid cell lines (Table S2). An analysis of genes with the largest changes in transcript levels revealed a set of six transporters exhibiting significant deviations in both mutant cell lines (Fig. 2C, red dashed line). First, glutamine importers (encoded by the SLC1A5, SLC38A1 and SLC38A2 genes) were significantly upregulated in mutant cell lines. Second, the glutamine-neutral amino acid antiporter (SLC7A5, also known as LAT1) was downregulated in mutant cell lines. Third, the glutamate-cystine antiporter (SLC7A11, also known as xCT) was upregulated in mutant cell lines, consistent with a previous report (Shin et al., 2017). In addition, SLC3A2, the heavy chain dimerization partner of SLC7A5 and SLC7A11 was upregulated in mutant cell lines. Western blot analysis indicated similar trends at the protein level for SLC1A5, SLC7A11 and SLC7A5 (Fig. 2D).
The functions of these identified transporters largely center around glutamine or glutamate transport. Levels of downstream glutamate processing enzymes (e.g., glutaminase (GLS), glutamine synthetase (GLUL), and glutamate dehydrogenase (GLUD)) were unperturbed in mtDNA mutant cells (Fig. 2D), suggesting that changes in glutamine metabolism may be mediated by altered levels of plasma membrane transporters. Direct measurement of transport flux in wild-type and mutant cells were consistent with changes in transporter levels: ND1 and ATP6 mutant cells display increased glutamine uptake, as well as increased glutamate secretion (Fig. 2E).
3.3. SLC7A11 upregulation promotes glucose oxidation in wild-type cells
We hypothesized that the altered amino acid transport and expression of SLC transporters surrounding glutamine/glutamate transport (Fig. 2) might contribute to the reprogramming of central carbon metabolism observed in mtDNA mutant cells (Fig. 1). We therefore manipulated SLC1A5, SLC7A5 and SLC7A11 levels in wild-type cells (Fig. S4A) via stable shRNA-mediated knockdown (for SLC7A5), or stable overexpression (for SLC1A5 and SLC7A11). Overexpression of SLC1A5 had no effects on net glutamine/glutamate transport; however, modulating SLC7A11 or SLC7A5 levels had significant effects on glutamine/glutamate transport (Fig. 3A), consistent with their transport functions. We assessed central carbon metabolism by measuring steady-state [U–13C]glucose and [U–13C]glutamine-derived MIDs in these cell lines, and determined metabolic flux values as before (Table S1, Figs. 3B and S4B). As expected, we detected significant changes in glutamine transport and processing, indicating that altered transporter expression can regulate glutamine utilization. However, we also observed significantly enhanced flux of pyruvate into the TCA cycle, specifically in the presence of SLC7A11 overexpression (Fig. 3B, Fig. S4C). Direct measurement of [U–13C] glucose labeling reveals enhanced citrate M+2 levels in a 1 h pulse experiment (Fig. 3C). Consistent with these results, the reduced glucose contribution to TCA cycle intermediates in mtDNA mutant cells (Fig. 1C) declined further in the setting of SLC7A11 knockdown in mtDNA mutant cells (Figs. 3D and S4D). Thus, elevations in xc− activity are important to maintain residual pyruvate oxidation in the setting of mtDNA mutations. More generally, despite its direct activity on amino acid substrates, enhanced xc− transporter activity indirectly stimulates glucose oxidation within mitochondria, indicating a link between amino acid transport and central carbon metabolism.
Fig. 3. Overexpression of SLC7A11 promotes pyruvate oxidation.

(A) Measurements of glutamine uptake and glutamate secretion in wild-type cells either overexpressing SLC family members (empty, SLC1A5, SLC7A11) or with shRNA-mediated knockdown of target genes (shNT (non-targeting), shSLC7A5). n = 3 independent samples per group. (B) Flux ratios for individual enzymatic steps between SLC7A11-overexpressing and control (empty vector) cells following metabolic flux analysis. Thickness and color of the lines indicates the magnitude of the ratio, similar to Fig. 1E. (C) Relative isotopologue abundance of M+2 labeled citrate following a 1hr [U–13C] glucose pulse in control (empty) or SLC7A11-overexpressing wild-type cells. Abundances were normalized by total ion count. n = 3 per group. (D) Same as (C), but in control (shNT) and shRNA-mediated knockdown of SLC7A11 (shSLC7A11) mutant cells. n = 3 per group. Representative data from a single experiment is shown, and all experiments were repeated at least in triplicate. Error bars indicate standard deviations from mean for independent samples. Adjusted p-values calculated from 1-way ANOVA, followed by Tukey’s test for multiple comparisons. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Stimulated transsulfuration promotes glucose oxidation via production of α-ketobutyrate
Stimulated glucose oxidation was not observed in the setting of SLC7A5 knockdown, despite similar effects on glutamine utilization (Fig. S4B). We therefore evaluated the role of cystine, which is imported by SLC7A11 but not SLC7A5. Cystine potentially contributes to multiple pathways, including production of glutathione, pyruvate and taurine (Stipanuk, 1986); thus, we investigated the fate of [U–13C,15N] cystine in wild-type and mtDNA mutant cells. As expected, a large fraction of glutathione is labeled by cystine at steady-state in both wild-type and mtDNA mutant cells (Fig. 4A). In contrast, we did not observe significant contribution of cystine to pyruvate and taurine (Fig. S5A). Interestingly, we detected significant fractions of cystine contribution to cystathionine, with substantially increased labeling in mtDNA mutant cells (Fig. 4B). Cystathionine is a metabolite within the transsulfuration pathway (Fig. 4C); typically, homocysteine (derived from methionine) and serine are condensed to cystathionine, followed by cleavage to produce de novo α-ketobutyrate and cysteine (Fig. 4C). Cysteine (derived from cystine) can biochemically substitute for serine (Chen et al., 2004), thereby stimulating conversion of methionine to α-ketobutyrate, while recycling cysteine.
Fig. 4. α-ketobutyrate production from transsulfuration promotes glucose oxidation.

(A) Fractional contribution (1-M0) of cystine to glutathione (GSH) at steady state in the indicated cell lines. (B) Steady-state mass isotopomer distributions of cystathionine after labeling with [U–13C,15N] cystine in the indicated cell lines. (C) Schematic of transsulfuration pathway and potential fates of α-ketobutyrate. CTH, cystathionine γ-lyase. (D) Isotopologue abundance of M+4 α-hydroxybutyrate in spent media from the indicated cell type after labeling with [U–13C] methionine. (E) NAD+/NADH ratios in the indicated cell lines. ρ0, mtDNA-depleted 143B cells. (F) Isotopologue abundance of the indicated metabolites in ND1 and ATP6 cells incubated with vehicle or 1 mM α-ketobutyrate. Cells were subjected to a 1 h pulse with [U–13C] glucose; abundance was normalized to total ion count. (G) Same as F, but with cells expressing non-targeting (shNT) or CTH-targeted (shCTH1, shCTH2) shRNAs. Representative data from a single experiment is shown, and all experiments were repeated at least in triplicate. All error bars indicate standard deviations from mean for n = 3 independent samples. Adjusted p-values calculated from 1-way ANOVA, followed by Tukey’s or Dunnett’s test for multiple comparisons.
We therefore assessed transsulfuration activity in cells by measuring incorporation of [U–13C] methionine. As expected, cystathionine was predominantly labeled by methionine at steady state in wild-type and mtDNA mutant cells (Fig. S5B). α-ketobutyrate, the methionine-derived product of transsulfuration, was not detectable in our assays, but is proposed to have two major intracellular fates (Fig. 4C): It can be metabolized within the TCA cycle, or it can be reduced (via cellular dehydrogenases) to α-hydroxybutyrate (Sullivan et al., 2015; Ciman and Siliprandi, 1968; Rosalki and Wilkinson, 1960). [U–13C] methionine tracing in our cell lines indicated no measurable labeling in TCA cycle intermediates (Fig. S5C). However, we observed significantly increased amounts of M+4 labeled α-hydroxybutyrate in the spent media of mutant cells (Fig. 4D). Consistent with enhanced transsulfuration activity, CTH (cystathionine γ-lyase) protein levels were elevated in mtDNA mutant cells (Fig. S5D), and α-hydroxybutyrate labeling was eliminated following knockdown of CTH (Figs. S5F and G).
Cystine-stimulated transsulfuration is expected to stimulate production of α-ketobutyrate, as well as produce hydrogen sulfide (H2S) as a by-product (Fig. 4C). We therefore tested whether either of these metabolites can stimulate glucose oxidation in cells. Incubating cells with H2S does increase contribution of [U–13C] glucose to early glycolytic intermediates (Fig. S5E), consistent with the reported activation of GAPDH by H2S (Mustafa et al., 2009). However, inhibition of pyruvate oxidation within the TCA cycle was noted with H2S treatment (Fig. S5E).
The production of α-hydroxybutyrate (from α-ketobutyrate) is linked to regeneration of NAD+ from NADH (Fig. 4C). It has been reported that exogenously supplied α-ketobutyrate can serve as an electron acceptor to oxidize NADH in the setting of severe mitochondrial dysfunction (Sullivan et al., 2015), which is classically associated with a reduced [NAD+]/[NADH] ratio due to loss of complex I activity. However, despite significant impairment of respiratory function (Fig. 1A), ND1 and ATP6 mutant cells do not display alterations in steady-state [NAD+]/[NADH] ratios (Fig. 4E) or pyruvate-auxotrophic growth (Fig. S5H), as compared with respiration-deficient ρ0 (mtDNA-depleted) cell lines. Similar elevations in transsfuluration enzymes and activity were observed in ρ0 cells, which are known to have altered redox balance (Figs. S5I and J).
As efficient glucose oxidation is well-known to require continual NAD+ regeneration, we hypothesized that exogenous α-ketobutyrate production should stimulate glucose oxidation. Indeed, mtDNA mutant cells supplemented with exogenous α-ketobutyrate displayed significant production rates of α-hydroxybutyrate (Fig. S5L), and corresponding increases in glucose contribution to TCA cycle metabolites (Fig. 4F). Inversely, pharmacologic (propargylglycine (PAG) treatment) or genetic (CTH knockdown from two independent shRNAs) reduction of transsulfuration activity resulted in decreased glucose oxidation within the mitochondria of mtDNA mutant cells (Figs. 4G and S5K), although the two mutant cell lines displayed some diversity in their response. This latter result is surprising since the measured endogenous α-hydroxybutyrate production rate is significantly lower than lactate production, and is not expected to significantly contribute to the total cellular [NAD+]/[NADH] ratio (Fig. S5L). Consistent with this, CTH knockdown does not inhibit proliferation of mutant cells in pyruvate-free media (Fig. S5M). The results therefore suggest a model in which exogenous α-ketobutyrate directly impacts mitochondrial NAD+ availability and promotes pyruvate oxidation, while endogenously produced α-ketobutyrate is linked to pyruvate oxidation through an unknown mechanism.
3.5. Transsulfuration activity regulates residual mitochondrial respiration in mtDNA mutant cell lines
Based on our above results, we initially hypothesized that enhanced pyruvate oxidation within mitochondria should promote residual electron transport chain activity, and we therefore examined the influence of α-ketobutyrate and transsulfuration on residual respiratory activity in ND1 and ATP6 mutant cells. Surprisingly, we observed the opposite effects: Addition of exogenous α-ketobutyrate immediately suppressed residual respiratory activity in both ND1 and ATP6 mutant cell lines (Fig. 5A,B,E). Injection of pyruvate had similar inhibitory effects on oxygen consumption (Fig. S6A), suggesting that the inhibition of respiratory activity is due to the presence of an exogenous electron acceptor, and not a direct inhibitory effect of α-ketobutyrate. Inversely, we found that inhibiting transsulfuration (by knockdown of CTH with two independent shRNAs) significantly stimulated residual respiration in mtDNA mutant cell lines (Fig. 5C,D,F). This stimulation occurs despite the continued presence of homoplasmic pathogenic mutations in ND1 or ATP6 genes (Figs. S1A and B). Addition of α-ketobutyrate was sufficient to suppress respiratory activity in CTH-knockdown cell lines, consistent with α-ketobutyrate’s proposed function downstream of CTH activity (Fig. S5B). Thus, we conclude that although enhanced transsulfuration promotes glucose oxidation in mitochondria, this particular metabolic adaptation inhibits residual respiratory activity in these mtDNA mutant cells.
Fig. 5. α-ketobutyrate production from transsulfuration regulates respiratory activity in mtDNA mutant cells.

(A) Oxygen consumption rates (OCR) in ND1 cells injected with vehicle (n = 5) or 1 mM (final concentration) α-ketobutyrate (αKB; n = 6) at the indicated time (arrow). (B) Same as (A), but in ATP6 cells injected with vehicle (n = 5) or 1 mM αKB (n = 6). (C) Oxygen consumption rates in ND1 mutant cells expressing non-targeting (shNT; n = 10) or CTH-targeting (shCTH1; n = 8) shRNAs. Oligomycin, CCCP, antimycin A were injected at the indicated times (arrows). (D) Same as (C), but in ATP6 mutant cells expressing shNT (n = 12) or shCTH1 (n = 14). (E) Mitochondrial oxygen consumption rates in ND1 and ATP6 cells incubated for 1 h with vehicle (n = 16,16) or 1 mM α-ketobutyrate (n = 16,14). (F) Mitochondrial oxygen consumption rates in ND1 and ATP6 cells expressing control (shNT; n = 14,16) or CTH-targeted (shCTH1 (n = 23,24), shCTH2 (n = 22,21) shRNAs. (G) Model describing how enhanced transsulfuration might regulate mitochondrial respiration in mtDNA mutant cell lines. Representative data from a single experiment is shown, and all experiments were repeated at least in triplicate. All error bars indicate standard deviations from mean for independent samples. Adjusted p-values calculated from 1-way ANOVA, followed by Tukey’s test for multiple comparisons.
4. Discussion
mtDNA defects and electron transport chain (ETC) dysfunction are largely characterized by decreased pyruvate oxidation within the mitochondrion, coupled with decreased TCA cycling. As the TCA cycle provides intermediates for various biosynthetic pathways in vitro, it is not surprising that metabolic adaptations in response to genetic defects may serve to maintain residual TCA cycling. We show that xc− and transsulfuration activity are induced in cells with particular mtDNA mutations, and promote glucose oxidation within the mitochondrion. The two mutations examined here (mt-ND1 G3460A and mt-ATP6 T8993G) are causative for distinct mitochondrial disorders with partially overlapping phenotypes; yet some similarities in the metabolic response between these mutations are observed here. In addition, enhanced transsulfuration has been previously reported in cells with inhibition of mtDNA replication (Bao et al., 2016; Nikkanen et al., 2016), and α-hydroxybutyrate has been proposed as a metabolic biomarker in patients with Leigh syndrome secondary to LRPPRC (Leucine Rich Pentatricopeptide Repeat Containing) gene mutations (Thompson Legault et al., 2015). Here, we provide evidence for increased transsulfuration in the setting of pathogenic human mtDNA mutations, and outline its effects in promoting glucose oxidation within the mitochondria via the production of α-ketobutyrate. Thus, enhanced cystine metabolism and decreased glutamine utilization are an adaptive mechanism to preserve glucose utilization in ETC-dysfunctional mitochondria. It will be important to assess the generality of this adaptation in the setting of other pathogenic mtDNA mutations and cell types, as well as in vivo, where glucose and amino acid utilization varies significantly between tissue types.
Surprisingly, the glucose oxidation from stimulated transsulfuration appears to be maladaptive for residual mitochondrial respiration in mtDNA mutant cells, and we are able to increase oxygen consumption by inhibiting α-ketobutyrate production. Since NADH is the immediate substrate for complex I of the electron transport chain, we propose that exogenously provided electron acceptors (such as α-ketobutyrate or pyruvate) may functionally compete with complex I for available NADH (Fig. 5G). However, the low endogenous rate of α-hydroxybutyrate production (Fig. S5L) is not consistent with the observed pyruvate oxidation (Fig. 4G) and respiratory (Fig. 5F) differences on a stoichiometric basis. Alternative explanations include a further metabolic adaptation in response to the loss of transsulfuration, perhaps mediated by changes in H2S production rates. Intriguingly, H2S is produced by cystine-stimulated transsulfuration (Fig. 4C), and is a non-competitive inhibitor of complex IV (Petersen, 1977), suggesting a direct link between xC−, transsulfuration activity and mitochondrial respiration that will be tested in future work.
We conclude that, in the cell lines studied here, linkages between cystine metabolism and glucose oxidation constitute a portion of the metabolic adaptation to mitochondrial ETC inhibition. These observations may be specific to the cultured carcinogenic cells studied herein, which likely have oncogenic liabilities to maintain proliferation, and xCT has been previously reported to influence metabolic reprogramming in the setting of NSCLC (Non-small-cell lung carcinoma) (Ji et al., 2018). However, increased serum α-hydroxybutyrate levels have been reported in patients with Leigh’s syndrome (Thompson Legault et al., 2015), as well as insulin resistance (Gall et al., 2010), suggesting that the metabolic adaptations discussed herein may be more commonly relevant to human diseases. More generally, our findings emphasize the flexibility of cellular metabolism in responding to genetic perturbations, and establish a mechanism whereby reprogramming of amino acid metabolism can be used to influence utilization of other substrates.
Supplementary Material
{kind=link}
Acknowledgments
We thank Drs. Valerio Carelli, Giovanni Manfredi and David Chan for providing 143B cybrid and 293T cell lines; members of the Mishra laboratory for comments on the manuscript; and members of the DeBerardinis laboratory and the CRI Metabolomics Facility for helpful discussions and technical help with mass spectrometry. This work was supported by the United Mitochondrial Disease Foundation (research grant to P.M.) and the National Institutes of Health (NIAMS R01AR073217 grant to P.M.). N.P.L. acknowledges financial support from a National Institutes of Health T32 grant (T32GM008203–30). The funding sources had no role in study design or manuscript preparation. The graphical abstract was created with Biorender.com.
Footnotes
Data availability
All data supporting the findings of this study are available within the article, extended data files, or available from the corresponding author upon reasonable request.
Declaration of competing interest
R.J.D. is an advisor to Agios Pharmaceuticals.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymben.2020.03.010.
References
- Ahn WS, Antoniewicz MR, 2011. Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab. Eng 13 (5), 598–609. [DOI] [PubMed] [Google Scholar]
- Albertini G, et al. , 2018. Genetic deletion of xCT attenuates peripheral and central inflammation and mitigates LPS-induced sickness and depressive-like behavior in mice. Glia 66 (9), 1845–1861. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, Kelleher JK, Stephanopoulos G, 2007. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng 9 (1), 68–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XR, et al. , 2016. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, et al. , 2015. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162 (3), 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti R, et al. , 2014. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metabol 19 (6), 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Jhee KH, Kruger WD, 2004. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem 279 (50), 52082–52086. [DOI] [PubMed] [Google Scholar]
- Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N, 2017. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 8 (31), 51164–51176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, et al. , 2018. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metabol 27 (5), 1007–1025 e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciman M, Siliprandi N, 1968. On the oxidation of alpha-oxobutyrate by isolated mammalian mitochondria. Biochim. Biophys. Acta 162 (2), 164–169. [DOI] [PubMed] [Google Scholar]
- Cui W, Taub DD, Gardner K, 2007. qPrimerDepot: a primer database for quantitative real time PCR. Nucleic Acids Res 35 (Database issue), D805–D809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman KG, Mandel LJ, 1990. Differential effects of respiratory inhibitors on glycolysis in proximal tubules. Am. J. Physiol 258 (6 Pt 2), F1608–F1615. [DOI] [PubMed] [Google Scholar]
- Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H, 1996. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. : JMS 31 (3), 255–262. [DOI] [PubMed] [Google Scholar]
- Ferrari M, et al. , 2017. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. U.S.A 114 (21), E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall WE, et al. , 2010. alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PloS One 5 (5), e10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, et al. , 2018. NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol. Cell 69 (4), 581–593 e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, et al. , 2016. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2, 16080. [DOI] [PubMed] [Google Scholar]
- Jain IH, et al. , 2016. Hypoxia as a therapy for mitochondrial disease. Science 352 (6281), 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, et al. , 2018. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 37 (36), 5007–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, et al. , 2013. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342 (6165), 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, et al. , 2014. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med 6 (6), 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P, Zhang Y, Zhuang L, Gan B, 2018. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Canc. Commun 38 (1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Henning MS, Starkov AA, Manfredi G, 2007. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol 179 (6), 1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozoya OA, et al. , 2018. Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol 16 (4), e2005707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie A, Boillee S, Hewett S, Knackstedt L, Lewerenz J, 2015. Main path and byways: non-vesicular glutamate release by system xc(−) as an important modifier of glutamatergic neurotransmission. J. Neurochem 135 (6), 1062–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack S, et al. , 2015. Pharmacologic targeting of sirtuin and PPAR signaling improves longevity and mitochondrial physiology in respiratory chain complex I mutant Caenorhabditis elegans. Mitochondrion 22, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack SE, et al. , 2017. Hospitalizations for mitochondrial disease across the lifespan in the. U.S. Mol. Gene. Metabol 121 (2), 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, et al. , 2011. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481 (7381), 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, et al. , 2014. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7 (5), 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, et al. , 2009. H2S signals through protein S-sulfhydration. Sci. Signal 2 (96), ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkanen J, et al. , 2016. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metabol 23 (4), 635–648. [DOI] [PubMed] [Google Scholar]
- Parsanathan R, Jain SK, 2018. Hydrogen sulfide increases glutathione biosynthesis, and glucose uptake and utilisation in C2C12 mouse myotubes. Free Radic. Res 52 (2), 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen LC, 1977. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim. Biophys. Acta 460 (2), 299–307. [DOI] [PubMed] [Google Scholar]
- Rosalki SB, Wilkinson JH, 1960. Reduction of alpha-ketobutyrate by human serum. Nature 188, 1110–1111. [DOI] [PubMed] [Google Scholar]
- Shin CS, et al. , 2017. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun 8, 15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH, 1986. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr 6, 179–209. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, et al. , 2015. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162 (3), 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Legault J, et al. , 2015. A metabolic signature of mitochondrial dysfunction revealed through a monogenic form of Leigh syndrome. Cell Rep 13 (5), 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V, Kirtikara K, 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc 1 (3), 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wang SF, et al. , 2016. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2alpha-ATF4-xCT pathway. Oncotarget 7 (45), 74132–74151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, 2014. INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30 (9), 1333–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Walther JL, Antoniewicz MR, Yoo H, Stephanopoulos G, 2008. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng 99 (3), 686–699. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. , 2013. Primary respiratory chain disease causes tissue-specific dysregulation of the global transcriptome and nutrient-sensing signaling network. PloS One 8 (7), e69282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, et al. , 2016. xCT expression modulates cisplatin resistance in Tca8113 tongue carcinoma cells. Oncol. Lett 12 (1), 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.