

Abstract
Discerning the underlying pathological mechanisms and the identification of therapeutic strategies to treat individuals affected with rare neurological diseases has proven challenging due to a host of factors. For instance, rare diseases affecting the nervous system are inherently lacking in appropriate patient sample availability compared to more common diseases, while animal models often do not accurately recapitulate specific disease phenotypes. These challenges impede research that may otherwise illuminate aspects of disease initiation and progression, leading to the ultimate identification of potential therapeutics. The establishment of induced pluripotent stem cells (iPSCs) as a human cellular model with defined genetics has provided the unique opportunity to study rare diseases within a controlled environment. iPSC models enable researchers to define mutational effects on specific cell types and signaling pathways within increasingly complex systems. Among rare diseases, pediatric diseases affecting neurodevelopment and neurological function highlight the critical need for iPSC-based disease modeling due to the inherent difficulty associated with collecting human neural tissue and the complexity of the mammalian nervous system. Rare neurodevelopmental disorders are, therefore, ideal candidates for utilization of iPSC-based in vitro studies. In this review, we address both the state of the iPSC field in the context of both their utility and limitations for neurodevelopmental studies, as well as speculating about the future applications and unmet uses for iPSCs in rare diseases.
Keywords: induced pluripotent stem cell, stem cell, reprogramming, rare disease, disease modeling, neural differentiation, neuronal differentiation, pluripotency, neurodevelopment, embryonic
BACKGROUND
Pluripotent Cellular Models for Rare Diseases - Derivation and Discrepancies
Before the development of iPSCs as a model system, pluripotent cell models were limited to embryonic stem cells (ESCs). ESCs are typically derived during early embryonic development and maintain indefinite pluripotent proliferation in cell culture settings (1). ESCs were first derived from mouse blastocysts in 1981 (2, 3) and then later from human blastocysts in 1998 from the inner cell mass (ICM) of early-stage embryos (1, 4). As ESCs became more mainstream in developmental research, a standardized definition was necessary in characterizing the requirements to classify an ESC. These requirements included the source of harvest being from pre-implanted or peri-implanted embryos, prolonged proliferation without differentiation when grown in vitro, and the ability to generate all three embryonic germ layers (1). Somatic cell nuclear transfer (SCNT) between an egg and somatic cell has also been utilized as an alternative source for ESC generation (5), though genomic instability and poor efficiency have deterred widespread use of this method (6). Human ESCs have great application potential due to their ability to theoretically differentiate into any terminal cell type found in the body, allowing for the study of signaling mechanisms required for maintenance of pluripotency determining how defined lineages, and cell transplantation/replacement therapies for various diseases (7, 8). While blastocyst isolation of ESCs or SCNT are technically difficult, the protocols are defined and provide an unlimited source of cells for use in downstream assays of interest (5, 9). However, a number of technical and ethical hurdles are associated with derivation and usage of human ESCs. These include the limited supply of human embryos available for ESC isolation, the presence of legal restriction on ESC derivation or research within a number of US states and European countries, and the ethical controversy surrounding the destruction of blastocysts or oocytes during ESC derivation (10, 11). Particularly relevant for rare disease research, the availability of disease-carrying embryos for human ESC isolation are extremely limiting and would typically require disease identification in utero (12). While ESCs derived through either traditional blastocyst isolation or SCNT represent outstanding models for the study of human development, an unmet need exists for new disease-specific human cellular models for the study of rare diseases.
The generation of induced pluripotent stem cells (iPSCs) by Kazutoshi Takahashi and Shinya Yamanaka in 2006 created an outstanding cellular model for the study of rare diseases (13). In contrast to being derived from blastocysts, iPSCs were first generated from mouse embryonic and adult fibroblasts before being applied to human cells (13). iPSCs exhibit morphology and growth properties similar to those of ESCs and can be derived from a number of somatic cell types. iPSCs are most commonly derived from fibroblasts, peripheral blood-derived mononuclear cells (PBMCs), or other types of lineage-restricted stem cells (14). iPSCs were first generated through viral transduction of key pluripotency factors including Sox2, Klf4, c-Myc, and Oct3/4, later referred to as the Yamanaka factors (Figure 1). The definition of Klf4 and c-Myc activity in various cancers helped elucidate their role in enhancing cell proliferation and metabolic reprogramming to facilitate iPSC derivation and maintenance, while Sox2 and Oct3/4 activity drive transcriptional reprogramming toward the pluripotent state (13). Additional transcription factors that have roles in early development, such as Nanog, have been supplemented in addition to the original Yamanaka factors to support and fine-tune the maintenance and induction of pluripotency in both ESC cultures and iPSC cultures (15, 16).
Figure 1. Comparison of iPSC and ESC derivation and expansion for downstream differentiation assays.
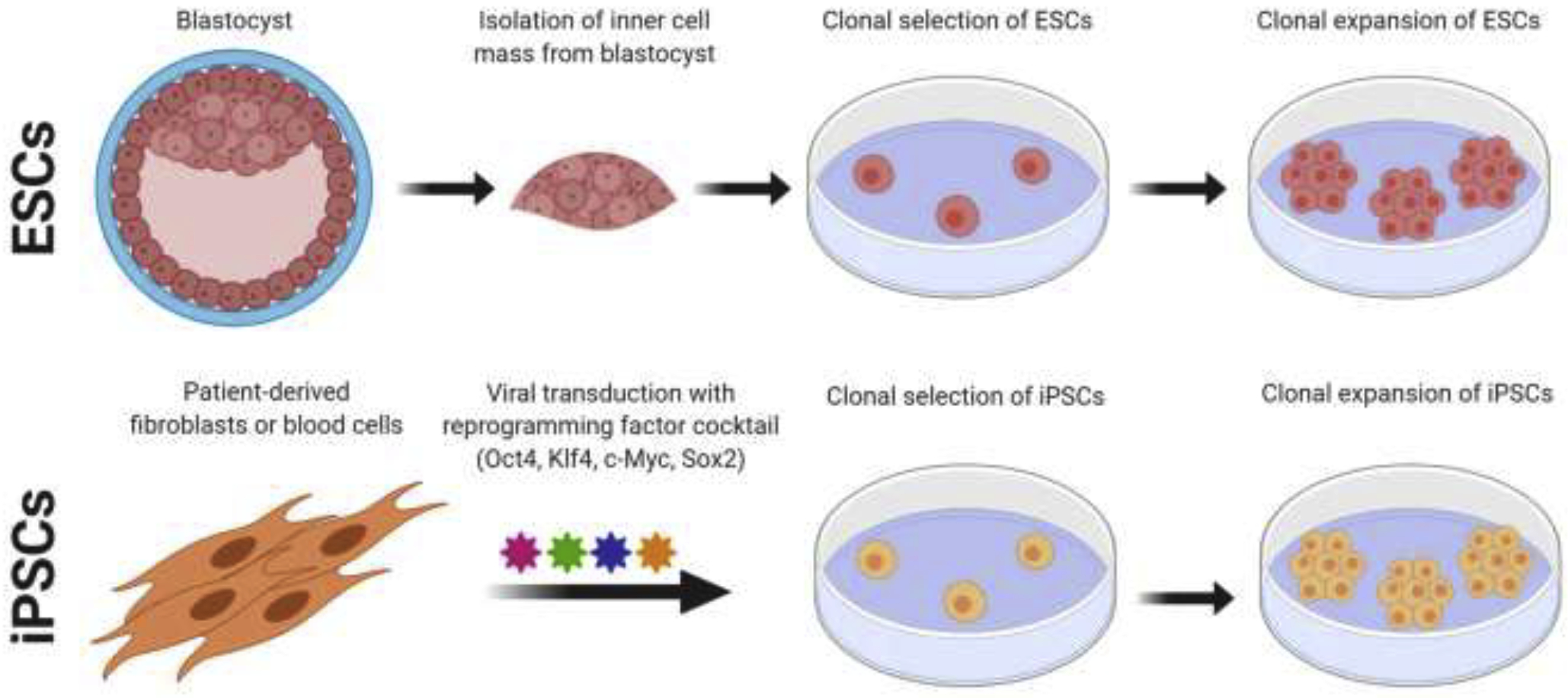
Human ESCs are typically derived from cleavage-stage embryos cultured to the blastocyst stage, followed by inner cell mass mechanoisolation, expansion in vitro, and clonal isolation/expansion of ESC lines. Conversely, iPSC derivation typically begins with the somatic cell of choice being transduced with a reprogramming cocktail to initiate reprogramming. Reprogrammed cells are then allowed to proliferate and expand, prior to clonal expansion and derivation of individual clonal lines. These images were created using BioRender.
Transduction of the aforementioned transcription factor genes for iPSC generation is commonly performed using integration-free delivery systems (non-integrating viruses or episomal vectors) in many different somatic cell types (fibroblasts, adipocyte stem cells, neural stem cells [NSCs], hematopoietic stem cells [HSCs], and PBMCs) with varying degrees of reprogramming efficiencies (17–20). Following transduction, reprogrammed cells undergo positive selection based on morphology, growth properties, and expression of pluripotent surface markers before clonal expansion. Though assays for validation of newly-derived iPSCs are often lab and/or disease specific, widely used standard assays including verification of iPSC differentiation into all three embryonic germ layers through in vitro embryoid body and/or in vivo teratoma formation, expression of pluripotent cellular markers at the transcriptional and protein level, genomic stability over time in culture, normal pluripotent cellular morphology and growth properties, DNA fingerprinting, and ensuring pathogen-free culture confirmation are recommended. Additional considerations regarding both basic research and clinical applications of ESC and iPSC derivatives are available (21).
iPSCs offer a number of outstanding utilities as models for biomedical research. First, inducing pluripotency from somatic cells offers a non-invasive, efficient method to obtain cellular models for genome-specific and disease-relevant in vitro research. Second, iPSCs exhibit comparable pluripotent capabilities to human ESCs while avoiding many of the ethical dilemmas. Furthermore, iPSC models allow the study of human diseases outside of the patient while continuing to offer a patient-specific and disease-centric approach (personalized medicine). From the perspective of neurological disorders, iPSC models have filled an important niche between linking neurobiology, neural function, and disease-associated cellular changes, addressing research questions with translational potential. The opportunity to observe human-derived neural cell types without the need for post-mortem samples or invasive biopsies has opened countless doors for identifying cellular phenotypes that would possibly not otherwise be recognized if utilizing non-neural cell types, animal models, or patient clinical observations, each of which have distinct disadvantages in the case of rare neurological disorders. Additionally, advances in the field of genome editing offer exciting new avenues for disease modeling using iPSCs. In particular, utilizing CRISPR-based genome editing to induce or correct disease-causing mutations allows for the identification of gene-specific functions in the realm of disease pathology. CRISPR and other genome editing technologies also allow for creation of isogenic control lines where the disease-causing mutation has been genetically corrected and can be applied in developmental assays to determine gene-specific deficits. Rescue studies have already been conducted for some of the diseases mentioned in this review, highlighting the causative role of these genetic alterations and opening the door for the discussion of potential therapies (22, 23). For laboratories new to the iPSC field, a number of biobanks and cellular repositories are available for distribution of various control and patient-derived iPSC models (Table 1).
Table 1. Commonly utilized repositories and biobanks for distribution of human iPSC models.
The iPSC collections listed are broadly available for research use following material transfer agreement and minimal investigator cost. Cell lines available include patient-derived iPSC models of various common and rare diseases, as well as age-matched controls, isogenic controls, and gene-edited iPSCs.
| Repository, cell line bank, or vendor | Human iPSC lines available |
|---|---|
| Allen Institute iPSC Cell Collection (via Coriell Insitute) | >30 fluorescently labeled iPSC lines derived from WTC parental iPSC line |
| ATCC | ~20 iPSC lines derived from unaffected individuals of various ethnicities |
| California Institute for Regenerative Medicine (CIRM) iPSC bank | >1500 iPSC lines, including >100 from ASD spectrum, proband, and unaffected siblings |
| European Bank for induced pluripotent Stem Cells (EBiSC) | >800 iPSC lines, including both controls and models of ataxias, Dravet syndrome, ALS |
| New York Stem Cell Foundation | Variety of control and patient-derived iPSC lines, including CLN3 Batten disease |
| NIGMS iPSC Collection (via Coriell Insitute) | >50 iPSC lines, including CLN2 Batten disease, Friedreich Ataxia, NPC1, and SMA |
| NINDS and NIH CRM iPSC Collection | >130 iPSC lines, including controls, reporter lines and models of SMA and Huntington’s disease |
| Riken BRC | >450 rare disease-derived iPSC lines, including Spinocerebellar degeneration, neuropathies |
| Simons Foundation Autism Research Initiative | >100 iPSC lines from ASD subjects, including Phelan-McDermid syndrome |
| WiCell | >1400 iPSC lines, including Rett syndrome, FXS, and SMA models |
As with any model, iPSCs do have recognized limitations that must be taken into account. One issue is a degree of variability that exists between iPSC lines, even those isolated from the same donor. A recent study analyzed >100 human control iPSC lines, including multiple lines from the same donor, and identified a list of genes which correlated with intrinsic or extrinsic variation both between and within cell lines (24). This study applied RNA sequencing and cellular phenotype analysis to detect cell lines where gene expression data did not correlate with their physiological behavior (24). Once differentiation of iPSCs is initiated, variability can also be enhanced due to altered differentiation efficiency. When comparing human iPSCs to ESCs, it has been shown that iPSCs differentiate to neuroepithelium, functional neurons and/or glia at similar rates (25). However, increased variability in efficiency was reported within iPSC models (25). iPSCs displayed a differentiation efficiency rate of 15–79% toward neuroepithelium, in comparison to 90–97% for ESC-derived neuroepithelium (25). It should also be noted that the results from this study were independent of the reprogramming method used. Additional issues related to more general upkeep and maintenance of iPSC integrity and genomic stability have been widely discussed previously. While these issues should be taken into consideration for any iPSC-based project, they do not limit the excitement or applicability of iPSC models for rare neurological disease research.
Current Models and Methods for Generation of Neural Cell Types from iPSCs
When generating disease models of neurodegeneration and neurodevelopment, the process of differentiating iPSCs is a critical step in recapitulating and identifying important aspects of disease onset and progression. Various protocols have been optimized to direct iPSC differentiation toward multipotent NSCs with the capacity to generate terminal neural lineages including defined neuronal populations, astrocytes, and oligodendrocytes. Current iPSC disease models can utilize either two-dimensional (2D) or three-dimensional (3D) systems, each offering advantages and disadvantages depending on the research goals and objectives. For example, more recently developed 3D models are of interest due to their possible recapitulation of naturally occurring spatial organization and complexity reminiscent of human development. On the other hand, 2D differentiation models offer highly efficient and expandable differentiation capacity, as well as a record of achievement in displaying some neurodevelopmental defects and disease processes.
Since the advent of iPSC generation, a host of protocols have been developed to direct pluripotent cells toward neurons and glia that function similar to those found in vivo. However, there are major discrepancies between in vitro protocols and de-novo differentiation due to a limited understanding of environmental cues, as well as poorly defined cell autonomous and non-cell autonomous effects. To help address these discrepancies, 3D models were developed to better mimic an in vivo environment while maintaining use of human models of genetic disease. Advantages and disadvantages of published methods in recapitulating developmental and functional phenotypes associated with rare pediatric diseases of the nervous system are discussed. For any neural differentiation assay to be utilized, a critical issue that must be considered is maintenance of optimal iPSC quality as defined by quality control measures is essential for differentiation assays of choice.
Differentiation toward neural lineages in two-dimensional models
Perhaps the most common method used to generate multipotent NSCs operates via induction of embryoid bodies (EBs) from iPSCs. EBs are 3D, spherical structures generated from aggregated iPSCs that mimic the cell-cell interactions observed during early embryogenesis (26). EBs have the ability to spontaneously differentiate to all primary germ layers (endoderm, ectoderm, and mesoderm) and many different cell lineages depending on 1) the quality of iPSCs, 2) the factors contained within the differentiation medium, and 3) the density and/or size of the EB (27, 28). The establishment of EBs from iPSCs represent a key experimental axis where most iPSC disease models converge before branching into directed differentiation methods for either 2D or 3D models. In traditional 2D neural differentiation assays, EBs are plated onto neural competent substrates, such as laminin or poly-D-lysine, to form neural rosettes. From the neural rosette stage, which has been shown to be representative of primitive human NSCs, standard methodology involves manually or enzymatically isolating NSCs and culturing for downstream differentiation assays (Figure 2). However, the EB formation method does have some drawbacks, including poorly defined culture settings, prolonged differentiation, and poor overall yield due to EB heterogeneity (28). An alternative method of generating neural rosettes utilizes dual-SMAD inhibition, blocking SMAD signaling via Noggin and SB431542 and directing iPSCs and/or ESCs toward a neural fate (29). Dual-SMAD inhibition alleviates the need for embryoid body formation and creates a more direct differentiation protocol. This protocol can produce a significant enrichment of neural precursors when compared to a standard MS5/Noggin protocol (29). Additionally, the MS5 protocol requires low density plating for optimal efficiency, while dual-SMAD inhibition maintains efficiency at higher seeding densities, allowing for more total neural precursor cells in addition to the overall higher efficiency (29). Dual-SMAD differentiation does require additional optimization steps as the starting cell density and timing of differentiation will vary across cell lines.
Figure 2. Differentiation timeline for standard 2D and organoid neuralization assays.
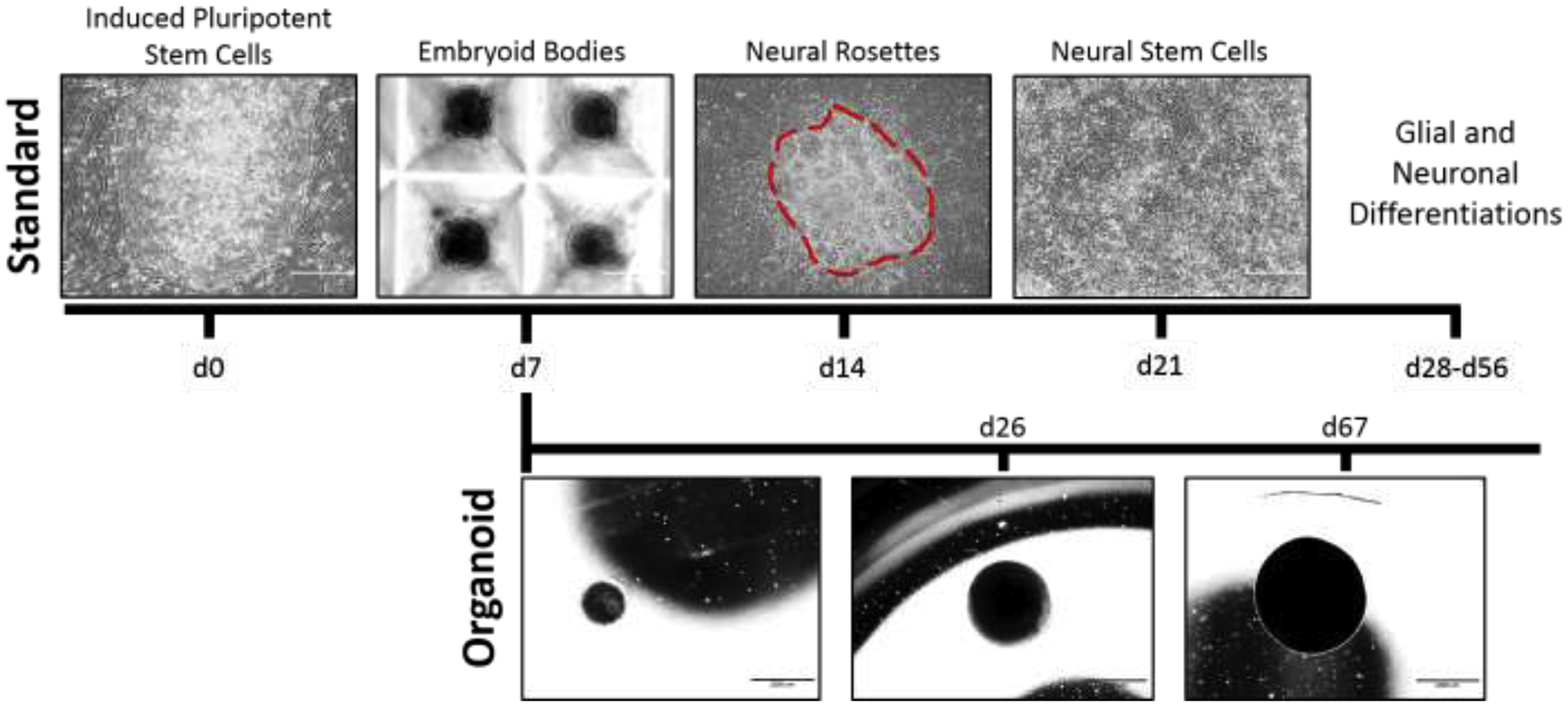
Traditionally, formation of neural stem cells and terminally differentiated neural cell types utilizes an intermediate embryoid body stage induced to neutralize through BMP inhibition. 3D (organoid) differentiation allows for prolonged directed neurodevelopment combined with tissue self-organization. Spatial organization and robust formation of mature neural cell types within cerebral organoids are time dependent, require a minimum of 60 days differentiation, and necessitates immunohistochemical analyses for cellular identification. Images shown were generated by the authors.
Alternatively, direct differentiation protocols have recently been developed to circumvent drawbacks associated with both EB formation and NSC maintenance. Such methods can differentiate iPSC colonies directly to terminal neural subtypes, bypassing EB and NSC formation. Wang et al. utilized an AAVS1-targeted, doxycycline-inducible neurogenin 2 transgene to rapidly convert iPSCs to viable neurons in a two-step protocol (30). This method induces a highly homogenous population of functional glutamatergic neurons amenable to high content analyses and drug screening (30). For diseases where excitatory neuron pathology is of interest, this inducible method creates an system for a relatively quick and efficient differentiation assay with multiple utilities (30). However, many studies are beginning to highlight the importance of other neural subtypes in the pathogenesis of several neurological diseases, creating interest in refining reprogramming methods that generate astrocytes and oligodendrocytes (OLs). A recent publication utilized transcriptional overexpression of gliogenic factors including Sox9 and NFIB to directly induce astrocyte formation from iPSC models in a rapid and efficient manner (~90% astrocytes on d7 post-induction) (31). In the context of rare neurological disease where researchers often focus on specific neuronal or glial subpopulations, direct reprogramming methods such as these would be warranted.
In addition to established methods for differentiation of mature neurons, protocols have also been developed for the generation of functional astrocytes and oligodendrocytes. Ehrlich et al. (2017) and Li et al. (2016) separately established 28-day differentiation protocols to differentiate iPSCs to O4+ OLs (32, 33). These protocols utilized viral induction of SOX10, OLIG2, and NKX6.2 to generate OLs that displayed similar morphology, protein marker expression, and myelination properties to those observed in human primary OLs (33). Similarly, OLs have been induced from hiPSC-derived NSCs over a twelve-week timespan. The culture conditions for this differentiation were defined on their ability to promote the expansion of oligodendrocyte progenitor cells (OPCs) through the bulk of this differentiation assay. As with other hiPSC-derived differentiated cells, extensive morphological and functional tests were conducted to validate the generated OLs (34). Wang et al. (2013) presented an OL differentiation protocol spanning 110 and 150 days for oligodendrocyte progenitor cells (OPCs) and OLs respectively. These cells were utilized in functional assays where the hiPSC-derived OLs rescued a congenital hypomyelination phenotype following transplantation (35). Recent findings have highlighted accelerated OL induction when culturing NSCs in media depleted of FGF2, an apparent inhibitor of OPC differentiation (36). Moreover, the presence of immature human astrocytes may enhance the transition of OPCs to OLs, a crucial limiting step in this protocol that can span up to 2/3 the total differentiation timeline (37, 38).
Astrocyte differentiation protocols have focused on expanding neural progenitors before gliogenesis initiates, modeling normal human development (39). Although most glial differentiation protocols fail to induce terminal differentiation at rates and developmental time points similar to humans in vivo, some protocols have attempted to more closely parallel human development by taking advantage of known signaling pathways identified as key initiators of developmental processes. For example, some protocols rely on stimulating the JAK/STAT3 pathway which has been shown to promote astrogenesis (40, 41). Medium containing leukemia inhibitory factor (42), a known activator of the JAK/STAT signaling has shown promise in inducing astrocytes from NCSs (41, 43). Current astrocyte differentiation protocols vary in generation times, ranging from 4–12 weeks depending on culture conditions, cell medium composition, and cell density (39, 43, 44).
Similar to OL differentiation, protocols for astrocyte differentiation from iPSC-derived NSCs are lagging in time and efficiency compared to neuron differentiation. However, new methods relying on transduction of transcription factors such as SOX9 and NFIB can boost the efficiency and speed in generating viable astrocytes (31). Additionally, use of small molecules such as VC6TFZ has been shown to reprogram fibroblasts to functional astrocytes in fewer than four weeks, providing an alternative and efficient method of astrocyte generation for use in downstream applications, including modeling of neurological diseases and therapeutic discovery (45). As more studies continue to improve and expand the current protocols utilized to generate functional astrocytes, it is important to keep in mind the potential transcriptional variability across populations of similarly induced astrocytes. To this end, a recent study has shown that astrocytes behave functionally different depending on the region of neural progenitors from which they were derived (46). Whether this variation occurs during normal development or whether it is an artifact of the model remains unclear. Regardless of the method used, differentiated astrocytes will continue to be an important aspect of studying neurological disorders due to their implicit roles in modulating energy consumption (storage and maintenance of glycogen granules), glutamate (neurotransmitter) uptake, altered calcium signaling in the presence of neurotransmitters, cholesterol cross talk with neurons, and secrete cytokines associated with neuroinflammation (31, 43, 44, 47).
Although microglia arise from the mesoderm as opposed to the ectodermal origin of the three principal neural cell types, protocols for iPSC-derived microglia have been developed to allow further study of this critical CNS resident. Microglia act as the primary immune cell in the brain (48, 49). While they have been widely studied in the realm of neurodegenerative disorders, they potentially play a role in neurodevelopmental diseases as well (49). To generate microglia, iPSCs must first be driven toward a mesodermal, hematopoietic lineage (50). The hematopoietic progenitor cells are non-adherent and can be induced to further differentiate into microglia following supplementation with macrophage colony stimulating factor, IL-34, and TGFβ−1 (50). A study by Douvaras et. al. in 2017 generated iPSC-derived microglial progenitor cells in 30 days, with an additional 15–30 days required for maturation (51). A similar study looking at the role of microglia in neurodegenerative disease generated mature microglia in 38 days (52). Both studies performed cellular and functional analyses to confirm cell maturation, including cell marker analysis and chemokine and cytokine secretion following artificial stimulation (52). While microglia require a lengthy process similar to the aforementioned glial cell types, the published derivation methods appear to be much more well-defined and consistent across labs (50–53).
Three-dimensional neural differentiation using iPSC-derived organoids
Three-dimensional differentiations, such as cerebral organoids, are formed by transferring embryoid bodies generated from iPSC models to non-tissue culture treated low-binding dishes, inhibiting the EBs from adhering to the surface of the dish. These organoids can be cultured for prolonged periods of time, sometimes exceeding 180 days, during which time they can mature and self-organize into structures reminiscent of the developing nervous system. Through assays for both protein and transcript expression, mature cerebral organoids have been shown to exhibit in vivo neurological structures such as cortical layering and sub ventricular zone formation. In the context of neurodevelopmental diseases, organoids offer a way to model the patient’s brain through an in vitro model while providing insight to cell interactions and tissue development (54). A study in 2015 found a dysregulation of excitatory vs. inhibitory neuron differentiation in an autism spectrum disorder organoid model (55). Organoids as a model for disease are still in the developing stages and with time, should become a strong model, especially in the study of neurodevelopmental disorders. Very few studies have successfully captured the ability to direct differentiation of specific neural subtypes in a 3D model, but some labs have shown promise in generating enriched populations of oligodendrocytes (56). Beyond modeling cortical development, protocols for organoids mimicking formation of the hippocampus, midbrain, hypothalamus, and cerebellum have been produced as well (57–62).
While iPSC-derived organoids have attracted significant attention as a complex in vitro model system, organoid-based studies do have caveats to consider prior to initiation. First, organoid-to organoid variability is a common occurrence (63–65). When iPSCs are formed into brain organoids, a multitude of cell types are formed and through maturation of these cells, variability from organoid to organoid can increase. A study utilizing 31 human brain organoids and single-cell RNA sequencing of over 80,000 cells identified several clusters correlating to specific cellular subtypes within the developing brain (66). Through analysis of this data set, it was emphasized that in order to use brain organoids as in vitro models of the human brain, a more thorough understanding of their composition and diversity is required (66). As with any iPSC-based differentiation model, lower iPSC quality at the initiation of organoid assays will dramatically impact the differentiation potential and ultimate quality of organoid maturation. Another technical hurdle related to organoid development and health is poor nutrient availability impacting organoid growth and viability. Use of tissue culture systems like bioreactors to boost nutrient flow and cause surface tension pressure upon differentiating cells, as well as endothelial co-culture to improve nutrient transport, have been successfully utilized (67–69). The development of organoid production methods which minimize variability is key to take full advantage of this powerful tool (63–65).
Direct reprogramming of somatic cells toward neural subtypes
In addition to the growing number of methods being established to differentiate iPSCs toward multipotent or terminal cell types, several groups have repurposed reprogramming methodology to transdifferentiate terminal cells directly to another terminal cell type of interest without the need for iPSC generation. This method is known as direct reprogramming or transdifferentiation and is still being optimized for defined cell types. Transdifferentiation to cells of interest dramatically decreases the time required for generation, eliminates the costs associated with iPSC maintenance and differentiation, and decreases the potential tumorigenic risks of stem cell intermediates. Transdifferentiation of MEFs to neurons has been accomplished via transduction of transcription factors Brn2, Ascl1, and BAM (70). Since then, several labs have attempted transdifferentiation of fibroblasts to dopaminergic and motor neural subtypes with varying success (71–73). To avoid transgene uptake, chemical compounds alone have begun to be used in specific concentrations to achieve similar results to transdifferentiating MEFs. In 2015, Li et al. induced transdifferentiation of MEFs via supplementing growth media with ISX9, Forskolin, CHIR99021, and I-BET151 (74). The resulting cell conversion resulted in TUJ-1-positive neurons capable of producing action potentials and functional synapses (74). Hu et al. reported a similar direct reprogramming protocol in which fibroblasts from Alzheimer’s patients were converted into neuronal cells via small molecules VCRFSGY, including VPA, CHIR99021, RepSox, Forskolin, SP600125, GO6983, and Y-27632 (75). Heinrich et al. (2010) first reported direct conversion of neurons from astrocytes in vitro via overexpression of exogenous transcriptions factors in vitro (76), and soon thereafter, Guo et al (2014) reprogrammed astrocytes to neurons in vivo as a potential treatment for regenerating functional neurons in Alzheimer’s disease (77). In 2015, Caiazzo et al. published a method of inducing directed astrocyte transdifferentiation from MEFs via induction of transcription factors NFIA, NFIB, and SOX9 (78).
Pluripotent Stem Cell Models of Rare Neurodevelopmental Disorders – Successes and Challenges
Within the United States, any condition affecting less than 200,000 individuals is classified as a rare disease according to the Orphan Drug Act. Estimates from various sources suggest of the 7,000 rare diseases believed to be present globally, 80% are believed to be of genetic origin. It is further estimated that approximately 10% of the world’s population is afflicted with some form of a rare disease, many of whom are pediatric cases due to the developmental origins of most rare diseases (79). Further, a high percentage (~20%) of infant deaths are associated with malformations associated with rare diseases (80). Rare diseases thus represent a global health burden further compounded by poor accessibility to affected patient tissue, animal models that are not characteristic of clinical phenotypes, and poorly understood disease pathology. Due to the sheer number of rare diseases impacting the nervous system, we have selected a small number of conditions to discuss in some detail below. For these selected disorders, iPSC models have provided significant insight into disease pathogenesis and the cellular mechanisms likely impacting patients.
Rett syndrome -
Rett syndrome (RTT) is a rare, X-linked dominant neurodevelopmental disease that is caused by mutations in MECP2 (81). MECP2 is a transcriptional repressor that binds to methylated CpG dinucleotides and recruits co-repressors and chromatin remodeling proteins. MECP2 is located on the X chromosome and when mutated, widely disrupts chromatin organization (82). Previous studies have implemented iPSC models of Rett syndrome to identify key developmental defects, therapeutic windows, and genetic alterations (83). For example, experiments conducted utilizing reprogrammed patient RTT fibroblasts revealed X chromosome re-activation occurs upon full reprogramming to a pluripotent state, while neuronal differentiation leads to X-inactivation (83–85). Additional studies utilizing RTT patient-derived iPSCs have generated differentiated RTT cortical neurons, revealing synaptic deficits defined by fewer glutamatergic synapses, smaller somas, reduced dendritic arborization, and fewer dendritic spines (22, 82, 83, 86–88). Interesting work by Cheung et. al. utilized the non-random X-inactivation of iPSC clones to model MECP2 effects on neuronal phenotypes from an RTT patient (86). Due to the mosaic expression of mutant MECP2 in a cell-specific manner based upon X-chromosome silencing of the mutant versus wild-type MECP2 allele, this study was able to demonstrate reduced neuronal soma size in a RTT iPSC clone expressing mutant MECP2 compared to a wild-type MECP2 expressing iPSC clone derived from the same patient (86). These data defined novel and critical cellular deficits in RTT that can now be further explored for mechanisms of progression and drug targets.
Additional functional deficits, including altered calcium signaling and electrophysiological defects were also detected in RTT patient-derived neurons. Intracellular calcium activity, when increased, is implicated in the activation of various signaling pathways, including neuronal excitation. iPSC-based studies have demonstrated that calcium oscillations are reduced in RTT neurons comparison to unaffected controls (83). Calcium signaling deficits are suggestive of a decrease in neuronal network connectivity and network dynamics, providing additional functional support for significant functional attenuation in RTT patient-derived neurons (82, 83). Electrophysiological studies also noted that RTT neurons feature diminished action potential firing amplitude and frequency (83). Differentiation assays have also highlighted an increase in in vitro astrocyte formation, which was supported by findings in post-mortem patient samples (82, 89). Astrocytes are known for their role in neuronal support in regard to synapse formation and maintenance, and neurotransmitter reuptake from synapses. Increases in astrocyte formation in RTT cultures suggest a possible role for glial MECP2 in regulating neuronal maturation and dendritic arborization, further supporting the previously mentioned findings highlighting reduced neuronal network complexity in mature RTT cultures (82, 90).
Fragile X syndrome -
Fragile X syndrome (FXS) results from trinucleotide CGG repeat expansion in the fragile X mental retardation 1 (FMR1) gene, producing a loss-of-function (42). The CGG repeats must exceed 200 copies for disease pathology to present, though the number of repeats does correlate to a range of disease severity in patients (91, 92). The FMR1 gene codes for the FMRP protein which functions as an RNA-binding protein that controls the location and translation of mRNA into proteins involved in synaptic plasticity and connectivity in the central nervous system (CNS) (93). FXS is inherited as an X-linked trait and clinically is known as the most prevalent inherited form of cognitive impairment as well as the leading genetic cause of autism (92). Modeling FXS using iPSCs is essential due to poor correlation between human clinical findings and animal models of the disease. Unfortunately, cloning and generating animal models for long, repetitive segments has proven difficult and none of the current animal models available precisely or fully imitate the molecular and cellular phenotypes observed in FXS patients (92). These iPSC models have allowed for characterization and highlighting of neurodevelopmental phenotypes through in vitro models. Previous studies from FMR1 knockout mice have reported shortened neurites and fewer neurite branch points in comparison to controls (94). However, some in vitro studies have shown contrasting results. This is possibly due to variance in origin of cells, neuronal subtype being assayed, culture conditions, and analysis methods. When studying forebrain neurons specifically, fewer and shorter processes were observed, supporting the previous in vivo findings (93). It should also be noted that FMR1 is known to be localized in the growth cone of developing axons. Assays examining motility and extension of neurites found that FXS neurons showed constrained motility and a decreased rate of neurite extension (93).
Autism spectrum disorders -
Autism spectrum disorders (ASDs) are characterized by impaired social interactions, communication deficits, and repetitive behaviors (95–98). A recent study published by the Centers for Disease Control and Prevention (CDC) stated that ASD now affects 1 in 59 children. This recorded prevalence has climbed quickly, jumping from 1 in 150 children from 2000–2002 to its most recent count (99). While ASDs would not be considered a rare disease when clustered together based upon clinical presentation (patients commonly present with accompanying conditions including epilepsy, depression, anxiety, intellectual deficits, and gastrointestinal complications (98, 100)), ASDs are defined genetically as syndromic or idiopathic cases. Syndromic cases are caused by a known genetic disorder, such as Rett or Fragile X syndrome (95). In contrast, idiopathic cases have an unknown genetic cause. While animal models are challenging for use in autism modeling due to the inherently complex behavioral and social phenotypes present in ASD patients (101), poorly defined genetic causes further complicates the study of ASDs using animal models. While past studies were able to obtain post-mortem samples from patients, this limited the study of ASD at the cellular and molecular level and did not allow for experimental manipulation to test hypotheses. Given the ever-growing prevalence, broad phenotypic range, and variance in disease progression from patient to patient, iPSCs offer a patient-specific approach that allows for the study of ASDs that can better encompass the variability between patients. Through this model, researchers have discovered novel developmental and functional deficits in a variety of neural cell types. Using time-dependent neural differentiation of ASD iPSCs, researchers can utilize developmental snapshots at different stages to correlate patient-specific genetic variation to any changes observed (102). With neurons as the focus of many initial studies, it was identified that decreased synaptic activity and fewer synapses were present in patients with syndromic ASD (103). iPSCs have also provided insight into the dynamic changes that occur during development. Through these studies, it was observed that ASD patient iPSC-derived neurons exhibit both reduced connectivity and reduced function of glutamatergic synapses (95). Alternatively, a recent study by Zaslavsky et. al in 2019 demonstrated that iPSC-derived cortical neurons from ASD individuals with mutations in the gene SHANK2 exhibit increased dendrite length, increased dendrite complexity, and increased synapse numbers (104). These findings support a hyperconnected phenotype present in a unique subset of ASD patients and demonstrate ASD neuronal deficits are not limited to reduced function (104). More recent studies have generated iPSC-derived neurons and astrocytes and conducted functional and molecular assays both individually and in co-culture settings. It was noted that the interplay between these two cell types contributes to the pathophysiology observed in ASD patients (96).
Other Neurodevelopmental Disorders of Particular Interest
Timothy syndrome -
Timothy syndrome (TS) is a rare disorder caused by a missense mutation in CACNA1C, which encodes for the L-type calcium channel Cav1.2 (105). Regarding the observed neurological phenotypes, TS patients present with autism, developmental delay, intellectual deficits, and seizures (106). Given the rare nature of Timothy syndrome, iPSCs offer a promising alternative model in comparison to previous studies. The use of iPSCs to study TS allow for the identification of defects present at various developmental stages. Splice variants associated with Cav1.2 mutations in TS induce a loss of inactivation which is believed to have prominent effects on neuronal signaling (105). iPSCs revealed mutations in Cav1.2 increase the amplitude of Ca2+ elevations in NPCs and neurons derived from TS patients (105).
Smith-Lemli-Opitz syndrome -
Smith-Lemli-Opitz syndrome (SLOS), is an autosomal recessive inborn error of metabolism caused by mutations in DHCR7, which encodes for the enzyme responsible for reducing 7-dehydrocholesterol to cholesterol in the final step of the cholesterol biosynthesis pathway. Patients with SLOS present with a wide range of neurological phenotypes, including intellectual disability, ASD, self-injurious behaviors, and structural brain abnormalities (107). In the most severe cases, patients die within the first week of life due to a failure to thrive, while many patients survive into adulthood. Animal models of SLOS are limited due to lethality and cholesterol metabolism differences between species, while limited patient tissue is available for study (108, 109). In the modeling of neurodevelopmental deficits of SLOS, challenge with cholesterol deficient conditions induces a loss of iPSC pluripotency, NSC multipotency and an accelerated drive toward neural differentiation (110).
Zika virus -
Following in utero infection of developing human fetuses with Zika virus, neurodevelopmental defects were reported in a number of two-year old children (111). In a comprehensive study performed after the 2015–16 Zika epidemic, 125 pregnant ZIKV+ women were monitored for gestational and child effects (111). Common findings in affected individuals included low birth weight, premature birth, and dramatic microcephaly (111). As ZIKV infected infants aged, speech impairments and abnormal eye and hearing assessments were identified (111). Neurodevelopmental deficits were also noted to occur with increasing severity based upon the gestational stage the mother when infection occurred (111). iPSCs can therefore serve as a model to identify the cellular and signaling mechanisms associated with this emerging disease. Using iPSCs as a model, human neural progenitor cells (hNPCs) were used to highlight the robust impact of the Brazilian ZIKV strain on hNPCs and neurodevelopment, demonstrating hNPC targeted cell death is likely a contributing factor to the microcephaly observed in ZIKV infected patients (112). In addition to two-dimensional iPSC-derived neural differentiation models, three-dimensional models have provided novel insight into the impact of Zika virus infection in utero on neurodevelopment (113). To take this model a step further, brain region-specific organoids were produced and infected with ZIKV, again demonstrating hNPC-targeted cell death upon ZIKV infection and recapitulation of microcephalic phenotypes, supporting previous ZIKV studies (61).
Defining Disease-Associated Neurological Phenotypes Using iPSC Models – Commonly Utilized Functional and Phenotypic Assays
Differentiating neural cell types from iPSC models is an ever increasingly complex procedure with many emerging protocols yielding varied populations of neurons, astrocytes, and oligodendrocytes. While these specified cell types may display pathological markers and morphology observed in naturally occurring cell types in vivo, it is critically necessary to test the functional capacity of differentiated cells as a measure of appropriate cell-specific behaviors. Differentiated cell types that exhibit basic neural function in comparison to fetal-derived or rodent primary culture cells are a critical requirement for validation of the differentiation process. An additional issue for consideration is potential batch-to-batch variation for iPSC-derived neurons and neuronal subtypes. When characterizing and quantifying connectivity and signaling or analyzing cell expression profiles, conditions such as cell density and cell subtype should be accounted for and kept consistent between assays (114, 115). Below, we highlight assays that are broadly applicable to rare neurodevelopmental disorders. However, the analysis of specific functional assays of high relevance for of a particular disease should be determined by individual laboratories based upon available clinical symptoms and previously published findings from other models.
Neurite Outgrowth -
Neurite outgrowth assays can be used to measure the number, width, arborization, and length of neurites that branch off the soma of neurons, offering valuable insight into neuron development, health, and maturity. Numerous publications have observed the difference between human pluripotent stem cell-derived neurons and primary neurons from rodent models (116, 117). Neurite outgrowth assays are an established method to examine the effectiveness and/or neurotoxicity of new treatment options for various rare diseases (116, 118). Further, neurite retraction can be utilized as a readout for establishing the neurotoxicity of novel drugs and small molecules. In regard to neurodevelopmental studies, neurite length, neurite arborization, and neurite width are each measures of neuronal connectivity and maturation. High-content image analysis and neurite tracing is a consistent and unbiased method of imaging as well as image analysis, with most analysis software being highly automated and refined by adjusting many variables as needed to improve the sensitivity and coverage of multiple analyses (116, 118, 119). A recent study performed a high-throughput drug screen in control iPSC-derived neurons to identify novel molecules which inhibit or promote neurite extension (120). The application of similar high-throughput neurite-based drug screens or the evaluation of neurite impacting compounds represents a promising avenue for normalization of neurite parameters in rare disease iPSC models where neurite defects are likely contributors to disease pathology (121, 122).
Patch-Clamp and Current Clamp Electrophysiology –
Patch-clamp electrophysiology is a commonly used method to quantify the functional activity of neurons in relation to either neurodevelopmental stages or disease states. While patch-clamp electrophysiology is a technically demanding technique, this method has been the gold standard for establishment of basic neuronal function in neurons differentiated from control or patient-derived iPSC models (123, 124). However, only a handful of outstanding publications have utilized patch-clamp to demonstrate functional neuronal deficits within rare disease iPSC models. In FXS murine models, neuronal hyperexcitability has been identified (125). Unfortunately, there have been few iPSC-derived neuron electrophysiology assays conducted for FXS. Within FXS hESC-derived neurons, a lack of action potential maturation, characterized by small amplitude over an extended period of time, were observed (126). Patch-clamp electrophysiology has also demonstrated that RTT neurons fire fewer action potentials of decreased amplitude along with reduced inward current peaks, suggesting defective Na+ channel function (127). This functional deficit in RTT neurons persisted in both excitatory and inhibitory synaptic transmission, where impaired long-term potentiation (LTP) has also been observed in RTT (128). Interestingly, electrophysiological functional changes resulting from rare diseases are not limited to neuronal deficits. Zaslavsky et. al. recently identified an increase in the frequency of spontaneous excitatory postsynaptic currents within SHANK2 neurons, a novel finding of interest (104). Electrophysiology is also a critical assay for defining functional deficits in rare diseases caused by mutations within genes responsible for neuronal function. Within Timothy syndrome, which results due to mutations within a voltage-gated L-type calcium channel, iPSC models demonstrated a distinctive increase in action potential width characteristic of a neuron’s inability to inactivate (105). In addition, studies within Dravet syndrome iPSCs demonstrate impaired action potential frequency in GABAergic neurons due to mutation within the voltage-gated sodium channel gene SCN1A (129). Measurement of neuronal function will continue to be a critical assay for assessment of both robust and functional differentiation of iPSCs, as well as a tool for measurement of disease-specific functional defects.
Multi-Electrode Arrays -
Multi-electrode arrays (MEA) act as a non-invasive interface between neurons and electric circuitry to detect network-level activity and connectivity in neuronal circuits. This method provides a large-scale readout of neuronal function. These arrays transduce a change in neuronal membrane voltage to electronic current. Multi-electrode arrays measure electrical firing activity by converting electrical field potential recordings to raster plots. These plots highlight each spike above the set threshold in an easy-to-read format. Multi-electrode arrays can vary in the number of electrodes present, ranging anywhere from tens to thousands of electrodes per array. The more electrodes per assay, the greater the sensitivity due to decreased distance between the cells and the electrodes. In Rett syndrome, it has been shown that neuronal network synchronization is altered in iPSC-derived neurons and that RTT iPSC-derived neurons have an increased number of glutamatergic synapses in comparison to controls (130). Multi-electrode arrays have shown a higher frequency of synchronized burst events in RTT neurons, although this has been disputed in previous studies (130). In Fragile X Syndrome, multi-electrode arrays have highlighted abnormal network development that contributes to clustered burst firing. This assay observed that differences in firing is dynamic throughout development (130). After 7 DIV, there was no significant differences in firing rate, however that changed by DIV 21 when the cultures were more mature. After 21 DIV, the FXS neurons began to present with a unique firing pattern which consists of strong clustered bursts (131). A recent study has exquisitely demonstrated the MEA can be used to measure organoid maturation in a repeated measures experiment over months of (132). MEA is quickly developing into a functional method complementary to traditional patch clamp electrophysiology to ascertain network-level neural activity in iPSC-derived models.
Proteomic and Transcriptomic Analyses -
Transcriptome assays have become a vital tool in defining the global developmental stage and suggestive of functional activity in iPSC models. For rare diseases specifically, RNA sequencing has become an indispensable assay to highlight heterogeneity between patients and controls, or in the case of single-cell RNA sequencing, cellular heterogeneity. These assays are highly sensitive and are becoming more cost effective as sequencers and protocols become more efficient. For iPSC-derived neurons and glia, RNA sequencing provides insight to differences occurring at the transcriptomic level in comparison to better defined and homogenous populations of fetal, post-mortem, or rodent cell types. A detailed transcriptional profile is able to determine differentially expressed or enriches pathways expressed in discrete populations of cells, providing a detailed transcriptional highlighting developmental, functional, or disease-causing mechanisms associated with pathogenesis. In Rett syndrome, transcriptome analysis of a pluripotent stem cell-derived neuronal model showed genome-wide transcriptional down regulation, supported by the function of MECP2 as a transcriptional regulator (133). A separate study, looking at RTT iPSC undifferentiated samples analyzed global gene expression patterns and observed that some sets of genes were humbly differentially expressed. However, MECP2 is expressed at lower levels in iPSCs than differentiated neurons, demonstrating cell type-specific transcriptional effects and the necessity for studying relevant cell types. This study also highlighted distinct transcriptional profiles between samples corresponding to patient-specific mutations (134). Transcriptome analysis of Fragile X syndrome iPSC-derived neurons showed upregulation of genes relating to neuron differentiation, cell fate specification, and regulation of transcription. The same studies observed decreased expression of genes related to synaptic transmission, memory, cell-to-cell signaling, telencephalon development, and transmission of nerve impulses (135, 136). Recently, RNA sequencing at the single cell level has become a commonly utilized method to detail cellular heterogeneity within differentiated neural cultures, identifying distinct developmental programs and cell specific pathways of interest. For example, application of single cell analyses to rare disease iPSC models has helped define radial glial deficits in a NRXN1 autism model (137). As the availability of single cell technology becomes more accessible, this technology offers a profound capacity for discovery-based analyses for the study of rare disease populations.
In additional to transcriptional analyses, establishing defined cellular proteomes can help delineate the impact of a rare disease on cellular function. Proteomics analyses can detail differences in protein levels across various cell types, as well as to generate quantitative comparisons between healthy and diseased samples. Different mutations in the same gene can produce differential effects downstream and present with protein differences. In a Rett syndrome model, one study utilized quantitative multidimensional protein identification technology (MudPIT) mass spectrometry (MS)-based shotgun proteomics to identify proteome differences suspected to be caused by MECP2 transcriptional or post-transcriptional regulation. This study observed at least a two-fold decrease in ALDOC, S100B, and GFAP. These are three key astrocytic markers, which supports an observed defect in astrogenesis (138). Fragile X syndrome proteomic studies have observed differences in proteins related to presynaptic specialization, vesicle recycling, neurotransmitter release, and neuron excitability (91, 94, 139). Though currently less utilized in rare disease iPSC work in comparison to RNA sequencing, future proteomic analyses may be utilized as a complementary technique to fully characterize signaling mechanisms within iPSC models.
Fused and Multi-System Organoids -
Fused organoid models build upon the traditional 3D organoid model by culturing two organoids in a confined space to induce organoid fusion. Individual organoids can be cultured separately prior to fusion to drive each toward a specific regional fate. Fused organoids offer unique insight to the development of connections between complex brain regions during neurodevelopment. While connectivity and neuronal maturation can be analyzed in traditional two-dimensional models, brain organoids offer a more physiological accurate representation of brain development. Recently developed techniques to fuse organoids can be used to assay neuronal migration from one specified region to another, as well as measure neuronal projections and connectivity between regional models. A 2017 article investigated complex interactions between dorsal-mimicking cortical organoids and ventral-mimicking medial ganglionic eminence organoids (140). Following parallel differentiation protocols, including utilizing ventralizing factors SHH and purmorphamine to generate an MGE-like organoid, it was shown that migration of MGE-derived interneuron progenitors remarkably migrated into the cortical organoid, replicating the ventral-to-dorsal migration of interneuron progenitors observed in vivo (141). Another study utilized a similar approach to demonstrate reduced interneuron motility in a Timothy syndrome model utilizing fusion of dorsalized and ventralized organoids (142). In addition, organoids feature mature cell types due to their prolonged differentiation timeline. In addition to neurons, formation and maturation of defined astrocyte populations with the capacity to accentuate synaptic formation and neuronal calcium flux in organoid models (143). These fused organoid models allow for the study of extraordinarily intricate neurodevelopmental mechanisms previously only visible within animal models. Exciting recent work combining neuralized organoids with endothelium, either of primary isolation or iPSC-differentiated sources, has the potential to produce organoids of enhanced function and relevance for defining cell-cell interactions and blood barrier transport (144). This work has laid the foundation for future detailed studies of additional brain region models and disease mechanisms of interest for rare disease researchers in physiologically relevant human tissues.
Conclusions
iPSCs offer a genetically unique, developmentally plastic, and niche model system for studying rare neurological diseases where patient neural tissue has limited availability or rodent models offer less than ideal utility. This model has been used for disease-causing mechanism discovery, differentiation assays, and drug developmental studies. The theoretically infinite proliferative capabilities of iPSCs, as well as their ability to generate cells from all three germ layers, makes them ideal candidates for rare disease studies of any kind. In the field of rare neurological diseases specifically, iPSCs are attractive due to their non-invasive isolation method, the robust assays available for differentiation to all three neural cell types, the ability to define functional deficits in patient-derived neurons, and the availability of complex three-dimensional models of neurodevelopment. As iPSCs continue to become a reliable and mainstream model for the study of human diseases, the integration of newer methodologies in future studies will likely produce exciting new discoveries and hopefully novel therapies for various rare neurological diseases.
Highlights.
Human induced pluripotent stem cells (iPSCs) are an established model system
Rare neurological diseases exert a profound burden on the pediatric population
Neurodevelopmental effects due to rare genetic changes can utilize iPSC models
iPSCs allow functional analyses and spatial disease modeling in human neurons
Acknowledgements
This research was supported by the National Institutes of Health (grant numbers P20GM103620 and P20GM103548).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7. Epub 1998/11/06. [DOI] [PubMed] [Google Scholar]
- 2.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–6. Epub 1981/07/09. [DOI] [PubMed] [Google Scholar]
- 3.Jones JM, Thomson JA. Human embryonic stem cell technology. Semin Reprod Med. 2000;18(2):219–23. Epub 2001/03/21. doi: 10.1055/s-2000-12560. [DOI] [PubMed] [Google Scholar]
- 4.Odorico JS, Kaufman DS, Thomson JA. Multilineage differentiation from human embryonic stem cell lines. Stem Cells. 2001;19(3):193–204. Epub 2001/05/22. doi: 10.1634/stemcells.19-3-193. [DOI] [PubMed] [Google Scholar]
- 5.Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, Kang E, Fulati A, Lee HS, Sritanaudomchai H, Masterson K, Larson J, Eaton D, Sadler-Fredd K, Battaglia D, Lee D, Wu D, Jensen J, Patton P, Gokhale S, Stouffer RL, Wolf D, Mitalipov S. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153(6):1228–38. Epub 2013/05/21. doi: 10.1016/j.cell.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chia G, Agudo J, Treff N, Sauer MV, Billing D, Brown BD, Baer R, Egli D. Genomic instability during reprogramming by nuclear transfer is DNA replication dependent. Nat Cell Biol. 2017;19(4):282–91. Epub 2017/03/07. doi: 10.1038/ncb3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gearhart J New potential for human embryonic stem cells. Science. 1998;282(5391):1061–2. Epub 1998/12/05. doi: 10.1126/science.282.5391.1061. [DOI] [PubMed] [Google Scholar]
- 8.Amit M, Carpenter MK, Inokuma MS, Chiu CP, Harris CP, Waknitz MA, Itskovitz-Eldor J, Thomson JA. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227(2):271–8. Epub 2000/11/10. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- 9.Bongso A, Fong CY, Ng SC, Ratnam S. Isolation and culture of inner cell mass cells from human blastocysts. Hum Reprod. 1994;9(11):2110–7. Epub 1994/11/01. doi: 10.1093/oxfordjournals.humrep.a138401. [DOI] [PubMed] [Google Scholar]
- 10.Robertson JA. Human embryonic stem cell research: ethical and legal issues. Nat Rev Genet. 2001;2(1):74–8. Epub 2001/03/17. doi: 10.1038/35047594. [DOI] [PubMed] [Google Scholar]
- 11.de Wert G, Mummery C. Human embryonic stem cells: research, ethics and policy. Hum Reprod. 2003;18(4):672–82. Epub 2003/03/28. doi: 10.1093/humrep/deg143. [DOI] [PubMed] [Google Scholar]
- 12.Colman A, Dreesen O. Pluripotent stem cells and disease modeling. Cell Stem Cell. 2009;5(3):244–7. Epub2009/09/08. doi: 10.1016/j.stem.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. Epub 2006/08/15. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 14.Sterneckert JL, Reinhardt P, Schöler HR. Investigating human disease using stem cell models. Nature Reviews Genetics. 2014;15:625. doi: 10.1038/nrg3764. [DOI] [PubMed] [Google Scholar]
- 15.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–7. Epub 2007/06/08. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 16.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17(1):42–9. Epub 2007/01/11. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 17.Hester ME, Song S, Miranda CJ, Eagle A, Schwartz PH, Kaspar BK. Two factor reprogramming of human neural stem cells into pluripotency. PLoS One. 2009;4(9):e7044. doi: 10.1371/journal.pone.0007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T, Zou G, Gao Y, Zhao X, Wang H, Huang Q, Jiang L, Guo L, Cheng W. High efficiency of reprogramming CD34(+) cells derived from human amniotic fluid into induced pluripotent stem cells with Oct4. Stem Cells Dev. 2012;21(12):2322–32. doi: 10.1089/scd.2011.0715. [DOI] [PubMed] [Google Scholar]
- 19.Loh YH, Hartung O, Li H, Guo C, Sahalie JM, Manos PD, Urbach A, Heffner GC, Grskovic M, Vigneault F, Lensch MW, Park IH, Agarwal S, Church GM, Collins JJ, Irion S, Daley GQ. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7(1):15–9. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umegaki-Arao N, Pasmooij AM, Itoh M, Cerise JE, Guo Z, Levy B, Gostynski A, Rothman LR, Jonkman MF, Christiano AM. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci Transl Med. 2014;6(264):264ra164. doi: 10.1126/scitranslmed.3009342. [DOI] [PubMed] [Google Scholar]
- 21.Daley GQ, Hyun I, Apperley JF, Barker RA, Benvenisty N, Bredenoord AL, Breuer CK, Caulfield T, Cedars MI, Frey-Vasconcells J, Heslop HE, Jin Y, Lee RT, McCabe C, Munsie M, Murry CE, Piantadosi S, Rao M, Rooke HM, Sipp D, Studer L, Sugarman J, Takahashi M, Zimmerman M, Kimmelman J. Setting Global Standards for Stem Cell Research and Clinical Translation: The 2016 ISSCR Guidelines. Stem Cell Reports. 2016;6(6):787–97. Epub 2016/05/18. doi: 10.1016/j.stemcr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6(9):e25255 Epub 2011/10/04. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vershkov D, Fainstein N, Suissa S, Golan-Lev T, Ben-Hur T, Benvenisty N. FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo. Cell Rep. 2019;26(10):2531–9 e4. Epub 2019/03/07. doi: 10.1016/j.celrep.2019.02.026. [DOI] [PubMed] [Google Scholar]
- 24.Vigilante A, Laddach A, Moens N, Meleckyte R, Leha A, Ghahramani A, Culley OJ, Kathuria A, Hurling C, Vickers A, Wiseman E, Tewary M, Zandstra PW, HipSci C, Durbin R, Fraternali F, Stegle O, Birney E, Luscombe NM, Danovi D, Watt FM. Identifying Extrinsic versus Intrinsic Drivers of Variation in Cell Behavior in Human iPSC Lines from Healthy Donors. Cell Rep. 2019;26(8):2078–87 e3. Epub 2019/02/21. doi: 10.1016/j.celrep.2019.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107(9):4335–40. Epub 2010/02/18. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurosawa H Methods for inducing embryoid body formation: in vitro differentiation system of embryonic stem cells. J Biosci Bioeng. 2007;103(5):389–98. Epub 2007/07/05. doi: 10.1263/jbb.103.389. [DOI] [PubMed] [Google Scholar]
- 27.Khoo ML, McQuade LR, Smith MS, Lees JG, Sidhu KS, Tuch BE. Growth and differentiation of embryoid bodies derived from human embryonic stem cells: effect of glucose and basic fibroblast growth factor. Biol Reprod. 2005;73(6):1147–56. doi: 10.1095/biolreprod.104.036673. [DOI] [PubMed] [Google Scholar]
- 28.Pettinato G, Wen X, Zhang N. Formation of well-defined embryoid bodies from dissociated human induced pluripotent stem cells using microfabricated cell-repellent microwell arrays. Sci Rep. 2014;4:7402 Epub 2014/12/11. doi: 10.1038/srep07402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–80. Epub 2009/03/03. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, Karch CM, Ding S, Gan L. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports. 2017;9(4):1221–33. Epub 2017/10/03. doi: 10.1016/j.stemcr.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canals I, Ginisty A, Quist E, Timmerman R, Fritze J, Miskinyte G, Monni E, Hansen MG, Hidalgo I, Bryder D, Bengzon J, Ahlenius H. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat Methods. 2018;15(9):693–6. Epub 2018/08/22. doi: 10.1038/s41592-018-0103-2. [DOI] [PubMed] [Google Scholar]
- 32.Li P, Li M, Tang X, Wang S, Zhang YA, Chen Z. Accelerated generation of oligodendrocyte progenitor cells from human induced pluripotent stem cells by forced expression of Sox10 and Olig2. Sci China Life Sci. 2016;59(11):1131–8. Epub 2016/10/28. doi: 10.1007/s11427-016-0165-3. [DOI] [PubMed] [Google Scholar]
- 33.Ehrlich M, Mozafari S, Glatza M, Starost L, Velychko S, Hallmann AL, Cui QL, Schambach A, Kim KP, Bachelin C, Marteyn A, Hargus G, Johnson RM, Antel J, Sterneckert J, Zaehres H, Scholer HR, Baron-Van Evercooren A, Kuhlmann T. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc Natl Acad Sci U S A. 2017;114(11):E2243–E52. Epub 2017/03/02. doi: 10.1073/pnas.1614412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu BY, Du ZW, Zhang SC. Differentiation of human oligodendrocytes from pluripotent stem cells. Nat Protoc. 2009;4(11):1614–22. Epub 2009/10/17. doi: 10.1038/nprot.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, Goldman SA. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12(2):252–64. Epub 2013/02/12. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, Zhang SC. Specification of neuronal and glial subtypes from human pluripotent stem cells. Cell Mol Life Sci. 2011;68(24):3995–4008. Epub 2011/07/26. doi: 10.1007/s00018-011-0770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao Y, Zhang SC. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell. 2016;19(5):573–86. Epub 2016/11/05. doi: 10.1016/j.stem.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang P, Deng W. Regenerating white matter using human iPSC-derived immature astroglia. Neurogenesis (Austin). 2016;3(1):e1224453 Epub 2016/09/22. doi: 10.1080/23262133.2016.1224453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Tao Y, Bradley R, Du Z, Tao Y, Kong L, Dong Y, Jones J, Yan Y, Harder CRK, Friedman LM, Bilal M, Hoffmann B, Zhang SC. Fast Generation of Functional Subtype Astrocytes from Human Pluripotent Stem Cells. Stem Cell Reports. 2018;11(4):998–1008. Epub 2018/10/03. doi: 10.1016/j.stemcr.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6(6):547–54. Epub 2004/05/25. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- 41.He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8(5):616–25. Epub 2005/04/27. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14. Epub 1991/05/31. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 43.Santos R, Vadodaria KC, Jaeger BN, Mei A, Lefcochilos-Fogelquist S, Mendes APD, Erikson G, Shokhirev M, Randolph-Moore L, Fredlender C, Dave S, Oefner R, Fitzpatrick C, Pena M, Barron JJ, Ku M, Denli AM, Kerman BE, Charnay P, Kelsoe JR, Marchetto MC, Gage FH. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Reports. 2017;8(6):1757–69. Epub 2017/06/08. doi: 10.1016/j.stemcr.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lundin A, Delsing L, Clausen M, Ricchiuto P, Sanchez J, Sabirsh A, Ding M, Synnergren J, Zetterberg H, Brolen G, Hicks R, Herland A, Falk A. Human iPS-Derived Astroglia from a Stable Neural Precursor State Show Improved Functionality Compared with Conventional Astrocytic Models. Stem Cell Reports. 2018;10(3):1030–45. Epub 2018/02/20. doi: 10.1016/j.stemcr.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian E, Sun G, Sun G, Chao J, Ye P, Warden C, Riggs AD, Shi Y. Small-Molecule-Based Lineage Reprogramming Creates Functional Astrocytes. Cell Rep. 2016;16(3):781–92. Epub 2016/07/12. doi: 10.1016/j.celrep.2016.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradley RA, Shireman J, McFalls C, Choi J, Canfield SG, Dong Y, Liu K, Lisota B, Jones JR, Petersen A, Bhattacharyya A, Palecek SP, Shusta EV, Kendziorski C, Zhang SC. Regionally specified human pluripotent stem cell-derived astrocytes exhibit different molecular signatures and functional properties. Development. 2019;146(13). Epub 2019/06/14. doi: 10.1242/dev.170910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valenza M, Marullo M, Di Paolo E, Cesana E, Zuccato C, Biella G, Cattaneo E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015;22(4):690–702. Epub 2014/10/11. doi: 10.1038/cdd.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andjelkovic AV, Nikolic B, Pachter JS, Zecevic N. Macrophages/microglial cells in human central nervous system during development: an immunohistochemical study. Brain Res. 1998;814(1–2):13–25. Epub 1998/12/05. doi: 10.1016/s0006-8993(98)00830-0. [DOI] [PubMed] [Google Scholar]
- 49.Lenz KM, Nelson LH. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front Immunol. 2018;9:698 Epub 2018/05/01. doi: 10.3389/fimmu.2018.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton-Jones M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol Neurodegener. 2018;13(1):67 Epub 2018/12/24. doi: 10.1186/s13024-018-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, Terrenoire C, Zhang B, Gandy S, Schadt E, Freytes DO, Noggle S, Fossati V. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Reports. 2017;8(6):1516–24. Epub 2017/05/23. doi: 10.1016/j.stemcr.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, Caraway CA, Fote GM, Madany AM, Agrawal A, Kayed R, Gylys KH, Cahalan MD, Cummings BJ, Antel JP, Mortazavi A, Carson MJ, Poon WW, Blurton-Jones M. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 2017;94(2):278–93 e9. Epub 2017/04/21. doi: 10.1016/j.neuron.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammond TR, Stevens B. Increasing the neurological-disease toolbox using iPSC-derived microglia. Nat Med. 2016;22(11):1206–7. Epub 2016/11/09. doi: 10.1038/nm.4226. [DOI] [PubMed] [Google Scholar]
- 54.Lee CT, Bendriem RM, Wu WW, Shen RF. 3D brain Organoids derived from pluripotent stem cells: promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci. 2017;24(1):59 Epub 2017/08/22. doi: 10.1186/s12929-017-0362-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M, Gerstein M, Grigorenko EL, Chawarska K, Pelphrey KA, Howe JR, Vaccarino FM. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162(2):375–90. Epub 2015/07/18. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodrigues GMC, Gaj T, Adil MM, Wahba J, Rao AT, Lorbeer FK, Kulkarni RU, Diogo MM, Cabral JMS, Miller EW, Hockemeyer D, Schaffer DV. Defined and Scalable Differentiation of Human Oligodendrocyte Precursors from Pluripotent Stem Cells in a 3D Culture System. Stem Cell Reports. 2017;8(6):1770–83. doi: 10.1016/j.stemcr.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di Lullo E, Kriegstein AR. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 2017;18(10):573–84. Epub 2017/09/08. doi: 10.1038/nrn.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10(4):537–50. Epub 2015/02/03. doi: 10.1016/j.celrep.2014.12.051. [DOI] [PubMed] [Google Scholar]
- 59.Monzel AS, Smits LM, Hemmer K, Hachi S, Moreno EL, van Wuellen T, Jarazo J, Walter J, Bruggemann I, Boussaad I, Berger E, Fleming RMT, Bolognin S, Schwamborn JC. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Reports. 2017;8(5):1144–54. Epub 2017/04/19. doi: 10.1016/j.stemcr.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, Takahashi J, Eiraku M, Sasai Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat Commun. 2015;6:8896 Epub 2015/11/18. doi: 10.1038/ncomms9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, Ho CY, Wen Z, Christian KM, Shi PY, Maher BJ, Wu H, Jin P, Tang H, Song H, Ming GL. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165(5):1238–54. Epub 2016/04/28. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang ZN, Freitas BC, Qian H, Lux J, Acab A, Trujillo CA, Herai RH, Nguyen Huu VA, Wen JH, Joshi-Barr S, Karpiak JV, Engler AJ, Fu XD, Muotri AR, Almutairi A. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc Natl Acad Sci U S A. 2016;113(12):3185–90. Epub 2016/03/06. doi: 10.1073/pnas.1521255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qian X, Song H, Ming GL. Brain organoids: advances, applications and challenges. Development. 2019;146(8). Epub 2019/04/18. doi: 10.1242/dev.166074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quadrato G, Arlotta P. Present and future of modeling human brain development in 3D organoids. Curr Opin Cell Biol. 2017;49:47–52. Epub 2017/12/12. doi: 10.1016/j.ceb.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 65.Quadrato G, Brown J, Arlotta P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat Med. 2016;22(11):1220–8. Epub 2016/11/01. doi: 10.1038/nm.4214. [DOI] [PubMed] [Google Scholar]
- 66.Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP, Boyden ES, Lichtman JW, Williams ZM, McCarroll SA, Arlotta P. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545(7652):48–53. Epub 2017/04/27. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, Chapeton K, Patterson B, Yuan Y, He CS, Raredon MSB, Dengelegi J, Kim KY, Sun P, Zhong M, Lee S, Patra P, Hyder F, Niklason LE, Lee SH, Yoon YS, Park IH. Engineering of human brain organoids with a functional vascular-like system. Nat Methods. 2019. Epub 2019/10/09. doi: 10.1038/s41592-019-0586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329–40. Epub 2014/09/05. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9. Epub 2013/09/03. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463(7284):1035–41. Epub 2010/01/29. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, Sotnikova TD, Menegon A, Roncaglia P, Colciago G, Russo G, Carninci P, Pezzoli G, Gainetdinov RR, Gustincich S, Dityatev A, Broccoli V. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature. 2011;476(7359):224–7. Epub 2011/07/05. doi: 10.1038/nature10284. [DOI] [PubMed] [Google Scholar]
- 72.Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476(7359):220–3. Epub 2011/05/28. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, Woolf CJ, Eggan K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell. 2011;9(3):205–18. Epub 2011/08/20. doi: 10.1016/j.stem.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, Zuo X, Jing J, Ma Y, Wang J, Liu D, Zhu J, Du X, Xiong L, Du Y, Xu J, Xiao X, Wang J, Chai Z, Zhao Y, Deng H. Small-Molecule-Driven Direct Reprogramming of Mouse Fibroblasts into Functional Neurons. Cell Stem Cell. 2015;17(2):195–203. Epub 2015/08/09. doi: 10.1016/j.stem.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 75.Hu W, Qiu B, Guan W, Wang Q, Wang M, Li W, Gao L, Shen L, Huang Y, Xie G, Zhao H, Jin Y, Tang B, Yu Y, Zhao J, Pei G. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell. 2015;17(2):204–12. Epub 2015/08/09. doi: 10.1016/j.stem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 76.Heinrich C, Blum R, Gascon S, Masserdotti G, Tripathi P, Sanchez R, Tiedt S, Schroeder T, Gotz M, Berninger B. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8(5):e1000373 Epub 2010/05/27. doi: 10.1371/journal.pbio.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell. 2014;14(2):188–202. Epub 2013/12/24. doi: 10.1016/j.stem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caiazzo M, Giannelli S, Valente P, Lignani G, Carissimo A, Sessa A, Colasante G, Bartolomeo R, Massimino L, Ferroni S, Settembre C, Benfenati F, Broccoli V. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Reports. 2015;4(1):25–36. Epub 2015/01/06. doi: 10.1016/j.stemcr.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melnikova I Rare diseases and orphan drugs. Nat Rev Drug Discov. 2012;11(4):267–8. Epub 2012/03/31. doi: 10.1038/nrd3654. [DOI] [PubMed] [Google Scholar]
- 80.Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: Final Data for 2014. Natl Vital Stat Rep. 2016;65(4):1–122. Epub 2016/07/06. [PubMed] [Google Scholar]
- 81.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8. Epub 1999/10/03. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 82.Andoh-Noda T, Akamatsu W, Miyake K, Matsumoto T, Yamaguchi R, Sanosaka T, Okada Y, Kobayashi T, Ohyama M, Nakashima K, Kurosawa H, Kubota T, Okano H. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol Brain. 2015;8:31 Epub 2015/05/28. doi: 10.1186/s13041-015-0121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–39. Epub 2010/11/16. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dhara SK, Benvenisty N. Gene trap as a tool for genome annotation and analysis of X chromosome inactivation in human embryonic stem cells. Nucleic Acids Res. 2004;32(13):3995–4002. Epub 2004/07/31. doi: 10.1093/nar/gkh746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, Plath K, Hochedlinger K. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1(1):55–70. Epub 2008/03/29. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 86.Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20(11):2103–15. Epub 2011/03/05. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dajani R, Koo SE, Sullivan GJ, Park IH. Investigation of Rett syndrome using pluripotent stem cells. J Cell Biochem. 2013;114(11):2446–53. Epub 2013/06/08. doi: 10.1002/jcb.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim KY, Hysolli E, Park IH. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011;108(34):14169–74. Epub 2011/08/03. doi: 10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagai K, Miyake K, Kubota T. A transcriptional repressor MeCP2 causing Rett syndrome is expressed in embryonic non-neuronal cells and controls their growth. Brain Res Dev Brain Res. 2005;157(1):103–6. Epub 2005/06/09. doi: 10.1016/j.devbrainres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 90.Kishi N, Macklis JD. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp Neurol. 2010;222(1):51–8. Epub 2009/12/23. doi: 10.1016/j.expneurol.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev. 2012;22(3):256–63. Epub 2012/03/03. doi: 10.1016/j.gde.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mor-Shaked H, Eiges R. Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. Genes (Basel). 2016;7(10). Epub 2016/10/01. doi: 10.3390/genes7100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23(15):1777–87. Epub 2014/03/25. doi: 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Klemmer P, Meredith RM, Holmgren CD, Klychnikov OI, Stahl-Zeng J, Loos M, van der Schors RC, Wortel J, de Wit H, Spijker S, Rotaru DC, Mansvelder HD, Smit AB, Li KW. Proteomics, ultrastructure, and physiology of hippocampal synapses in a fragile X syndrome mouse model reveal presynaptic phenotype. J Biol Chem. 2011;286(29):25495–504. Epub 2011/05/21. doi: 10.1074/jbc.M110.210260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Habela CW, Song H, Ming GL. Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol Cell Neurosci. 2016;73:52–62. Epub 2015/12/15. doi: 10.1016/j.mcn.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, Beltrao-Braga PCB. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol Psychiatry. 2018;83(7):569–78. Epub 2017/11/14. doi: 10.1016/j.biopsych.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 97.Acab A, Muotri AR. The Use of Induced Pluripotent Stem Cell Technology to Advance Autism Research and Treatment. Neurotherapeutics. 2015;12(3):534–45. Epub 2015/04/09. doi: 10.1007/s13311-015-0354-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ardhanareeswaran K, Coppola G, Vaccarino F. The use of stem cells to study autism spectrum disorder. Yale J Biol Med. 2015;88(1):5–16. Epub 2015/03/10. [PMC free article] [PubMed] [Google Scholar]
- 99.Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, Kurzius-Spencer M, Zahorodny W, Robinson Rosenberg C, White T, Durkin MS, Imm P, Nikolaou L, Yeargin-Allsopp M, Lee LC, Harrington R, Lopez M, Fitzgerald RT, Hewitt A, Pettygrove S, Constantino JN, Vehorn A, Shenouda J, Hall-Lande J, Van Naarden Braun K, Dowling NF. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018;67(6):1–23. Epub 2018/04/28. doi: 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Volkmar FR, McPartland JC. From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu Rev Clin Psychol. 2014;10:193–212. Epub 2013/12/18. doi: 10.1146/annurev-clinpsy-032813-153710. [DOI] [PubMed] [Google Scholar]
- 101.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11(7):490–502. Epub 2010/06/19. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vaccarino FM, Urban AE, Stevens HE, Szekely A, Abyzov A, Grigorenko EL, Gerstein M, Weissman S. Annual Research Review: The promise of stem cell research for neuropsychiatric disorders. J Child Psychol Psychiatry. 2011;52(4):504–16. Epub 2011/01/06. doi: 10.1111/j.1469-7610.2010.02348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ilieva M, Fex Svenningsen A, Thorsen M, Michel TM. Psychiatry in a Dish: Stem Cells and Brain Organoids Modeling Autism Spectrum Disorders. Biol Psychiatry. 2018;83(7):558–68. Epub 2018/01/04. doi: 10.1016/j.biopsych.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 104.Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, Zhao M, Ross PJ, El Hajjar J, Romm A, Thompson T, Piekna A, Wei W, Wang Z, Khattak S, Mufteev M, Pasceri P, Scherer SW, Salter MW, Ellis J. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22(4):556–64. Epub 2019/03/27. doi: 10.1038/s41593-019-0365-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–62. Epub 2011/11/29. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dufendach KA, Timothy K, Ackerman MJ, Blevins B, Pflaumer A, Etheridge S, Perry J, Blom NA, Temple J, Chowdhury D, Skinner JR, Johnsrude C, Bratincsak A, Bos JM, Shah M. Clinical Outcomes and Modes of Death in Timothy Syndrome: A Multicenter International Study of a Rare Disorder. JACC Clin Electrophysiol. 2018;4(4):459–66. Epub 2018/08/02. doi: 10.1016/j.jacep.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 107.Bianconi SE, Cross JL, Wassif CA, Porter FD. Pathogenesis, Epidemiology, Diagnosis and Clinical Aspects of Smith-Lemli-Opitz Syndrome. Expert Opin Orphan Drugs. 2015;3(3):267–80. Epub 2015/03/04. doi: 10.1517/21678707.2015.1014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tortelli B, Fujiwara H, Bagel JH, Zhang J, Sidhu R, Jiang X, Yanjanin NM, Shankar RK, Carillo-Carasco N, Heiss J, Ottinger E, Porter FD, Schaffer JE, Vite CH, Ory DS. Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum Mol Genet. 2014;23(22):6022–33. Epub 2014/06/27. doi: 10.1093/hmg/ddu331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pipalia NH, Cosner CC, Huang A, Chatterjee A, Bourbon P, Farley N, Helquist P, Wiest O, Maxfield FR. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc Natl Acad Sci U S A. 2011;108(14):5620–5. Epub 2011/03/26. doi: 10.1073/pnas.1014890108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francis KR, Ton AN, Xin Y, O’Halloran PE, Wassif CA, Malik N, Williams IM, Cluzeau CV, Trivedi NS, Pavan WJ, Cho W, Westphal H, Porter FD. Modeling Smith-Lemli-Opitz syndrome with induced pluripotent stem cells reveals a causal role for Wnt/beta-catenin defects in neuronal cholesterol synthesis phenotypes. Nat Med. 2016;22(4):388–96. Epub 2016/03/22. doi: 10.1038/nm.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nielsen-Saines K, Brasil P, Kerin T, Vasconcelos Z, Gabaglia CR, Damasceno L, Pone M, Abreu de Carvalho LM, Pone SM, Zin AA, Tsui I, Salles TRS, da Cunha DC, Costa RP, Malacarne J, Reis AB, Hasue RH, Aizawa CYP, Genovesi FF, Einspieler C, Marschik PB, Pereira JP, Gaw SL, Adachi K, Cherry JD, Xu Z, Cheng G, Moreira ME. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med. 2019. Epub 2019/07/10. doi: 10.1038/s41591-019-0496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, Figueira CP, Paredes BD, Nonaka CK, Azevedo CM, Rocha VP, Bandeira AC, Mendez-Otero R, Dos Santos RR, Soares MB. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep. 2016;6:39775 Epub 2016/12/23. doi: 10.1038/srep39775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Majolo F, Marinowic DR, Moura AA, Machado DC, da Costa JC. Use of induced pluripotent stem cells (iPSCs) and cerebral organoids in modeling the congenital infection and neuropathogenesis induced by Zika virus. J Med Virol. 2019;91(4):525–32. Epub 2018/10/26. doi: 10.1002/jmv.25345. [DOI] [PubMed] [Google Scholar]
- 114.Baker M 1,500 scientists lift the lid on reproducibility. Nature. 2016;533(7604):452–4. Epub 2016/05/27. doi: 10.1038/533452a. [DOI] [PubMed] [Google Scholar]
- 115.Volpato V, Smith J, Sandor C, Ried JS, Baud A, Handel A, Newey SE, Wessely F, Attar M, Whiteley E, Chintawar S, Verheyen A, Barta T, Lako M, Armstrong L, Muschet C, Artati A, Cusulin C, Christensen K, Patsch C, Sharma E, Nicod J, Brownjohn P, Stubbs V, Heywood WE, Gissen P, De Filippis R, Janssen K, Reinhardt P, Adamski J, Royaux I, Peeters PJ, Terstappen GC, Graf M, Livesey FJ, Akerman CJ, Mills K, Bowden R, Nicholson G, Webber C, Cader MZ, Lakics V. Reproducibility of Molecular Phenotypes after Long-Term Differentiation to Human iPSC-Derived Neurons: A Multi-Site Omics Study. Stem Cell Reports. 2018;11(4):897–911. Epub 2018/09/25. doi: 10.1016/j.stemcr.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ryan KR, Sirenko O, Parham F, Hsieh JH, Cromwell EF, Tice RR, Behl M. Neurite outgrowth in human induced pluripotent stem cell-derived neurons as a high-throughput screen for developmental neurotoxicity or neurotoxicity. Neurotoxicology. 2016;53:271–81. Epub 2016/02/09. doi: 10.1016/j.neuro.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 117.Sirenko O, Hesley J, Rusyn I, Cromwell EF. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev Technol. 2014;12(1):43–54. Epub 2013/11/16. doi: 10.1089/adt.2013.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sirenko O, Hesley J, Rusyn I, Cromwell EF. High-content high-throughput assays for characterizing the viability and morphology of human iPSC-derived neuronal cultures. Assay Drug Dev Technol. 2014;12(9–10):536–47. Epub 2014/12/17. doi: 10.1089/adt.2014.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Druwe I, Freudenrich TM, Wallace K, Shafer TJ, Mundy WR. Sensitivity of neuroprogenitor cells to chemical-induced apoptosis using a multiplexed assay suitable for high-throughput screening. Toxicology. 2015;333:14–24. Epub 2015/04/07. doi: 10.1016/j.tox.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 120.Sherman SP, Bang AG. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis Model Mech. 2018;11(2). Epub 2018/01/24. doi: 10.1242/dmm.031906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chailangkarn T, Muotri AR. Modeling Williams syndrome with induced pluripotent stem cells. Neurogenesis (Austin). 2017;4(1):e1283187 Epub 2017/02/24. doi: 10.1080/23262133.2017.1283187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marchetto MC, Winner B, Gage FH. Pluripotent stem cells in neurodegenerative and neurodevelopmental diseases. Hum Mol Genet. 2010;19(R1):R71–6. Epub 2010/04/27. doi: 10.1093/hmg/ddq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7(10):1836–46. Epub 2012/09/15. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 124.Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, Garnes J, Dang V, Lievers J, Shoukat-Mumtaz U, Martinez R, Gai H, Blake R, Vaisberg E, Grskovic M, Johnson C, Irion S, Bright J, Cooper B, Nguyen L, Griswold-Prenner I, Javaherian A. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–64. Epub 2013/07/31. doi: 10.1016/j.mcn.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Contractor A, Klyachko VA, Portera-Cailliau C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron. 2015;87(4):699–715. Epub 2015/08/21. doi: 10.1016/j.neuron.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Telias M, Segal M, Ben-Yosef D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol. 2013;374(1):32–45. Epub 2012/12/12. doi: 10.1016/j.ydbio.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 127.Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012;17(12):1261–71. Epub 2012/01/11. doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fink JJ, Levine ES. Uncovering True Cellular Phenotypes: Using Induced Pluripotent Stem Cell-Derived Neurons to Study Early Insults in Neurodevelopmental Disorders. Front Neurol. 2018;9:237 Epub 2018/05/02. doi: 10.3389/fneur.2018.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim HW, Quan Z, Kim YB, Cheong E, Kim HD, Cho M, Jang J, Yoo YR, Lee JS, Kim JH, Kim YI, Kim DS, Kang HC. Differential effects on sodium current impairments by distinct SCN1A mutations in GABAergic neurons derived from Dravet syndrome patients. Brain Dev. 2018;40(4):287–98. Epub 2018/01/04. doi: 10.1016/j.braindev.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 130.Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E, Vanderhaeghen P, Verfaillie CM, Raitano S, Kumar A, Carvalho CM, Bagni C, Ramocki MB, Araujo BH, Torres LB, Lupski JR, Van Esch H, Muotri AR. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry. 2016;21(2):178–88. Epub 2015/09/09. doi: 10.1038/mp.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cao Z, Hulsizer S, Tassone F, Tang HT, Hagerman RJ, Rogawski MA, Hagerman PJ, Pessah IN. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet. 2012;21(13):2923–35. Epub 2012/04/03. doi: 10.1093/hmg/dds118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, Wang A, Wu W, Haddad GG, Chaim IA, Domissy A, Vandenberghe M, Devor A, Yeo GW, Voytek B, Muotri AR. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell. 2019. Epub 2019/09/03. doi: 10.1016/j.stem.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Loven J, Kwok SM, Feldman DA, Bateup HS, Gao Q, Hockemeyer D, Mitalipova M, Lewis CA, Vander Heiden MG, Sur M, Young RA, Jaenisch R. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13(4):446–58. Epub 2013/10/08. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tanaka Y, Kim KY, Zhong M, Pan X, Weissman SM, Park IH. Transcriptional regulation in pluripotent stem cells by methyl CpG-binding protein 2 (MeCP2). Hum Mol Genet. 2014;23(4):1045–55. Epub 2013/10/17. doi: 10.1093/hmg/ddt500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu P, Chen X, Feng Y,Zeng Q, Jiang C, Zhu X, Fan G, and Xue Z. Integrated transcriptome analysis of human iPS cells derived from a fragile X syndrome patient during neuronal differentiation. Sci China Life Sci. 2016;59:1093–105. doi: 10.1007/s11427-016-0194-6. [DOI] [PubMed] [Google Scholar]
- 136.Halevy T, Czech C, Benvenisty N. Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports. 2015;4(1):37–46. Epub 2014/12/09. doi: 10.1016/j.stemcr.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lam M, Moslem M, Bryois J, Pronk RJ, Uhlin E, Ellstrom ID, Laan L, Olive J, Morse R, Ronnholm H, Louhivuori L, Korol SV, Dahl N, Uhlen P, Anderlid BM, Kele M, Sullivan PF, Falk A. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal functionality. Exp Cell Res. 2019;383(1):111469 Epub 2019/07/16. doi: 10.1016/j.yexcr.2019.06.014. [DOI] [PubMed] [Google Scholar]
- 138.Kim JJ, Savas JN, Miller MT, Hu X, Carromeu C, Lavallee-Adam M, Freitas BCG, Muotri AR, Yates JR 3rd, Ghosh A Proteomic analyses reveal misregulation of LIN28 expression and delayed timing of glial differentiation in human iPS cells with MECP2 loss-of-function. PLoS One. 2019;14(2):e0212553 Epub 2019/02/23. doi: 10.1371/journal.pone.0212553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deng PY, Sojka D, Klyachko VA. Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci. 2011;31(30):10971–82. Epub 2011/07/29. doi: 10.1523/JNEUROSCI.2021-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14(7):743–51. Epub 2017/05/16. doi: 10.1038/nmeth.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, Cakir B, Kim KY, Lombroso AP, Hwang SM, Zhong M, Stanley EG, Elefanty AG, Naegele JR, Lee SH, Weissman SM, Park IH. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell. 2017;21(3):383–98 e7. Epub 2017/08/02. doi: 10.1016/j.stem.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N, O’Rourke NA, Steinmetz LM, Bernstein JA, Hallmayer J, Huguenard JR, Pasca SP. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–9. Epub 2017/04/27. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sloan SA, Darmanis S, Huber N, Khan TA, Birey F, Caneda C, Reimer R, Quake SR, Barres BA, Pasca SP. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron. 2017;95(4):779–90 e6. Epub 2017/08/18. doi: 10.1016/j.neuron.2017.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pham MT, Pollock KM, Rose MD, Cary WA, Stewart HR, Zhou P, Nolta JA, Waldau B. Generation of human vascularized brain organoids. Neuroreport. 2018;29(7):588–93. Epub 2018/03/24. doi: 10.1097/WNR.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]