

INTRODUCTION
In molecular pathology the emphasis is on assessing gene expression, morphology, and using gene expression analysis to validate large numbers of targets.
Molecular pathology techniques have been used in the clinical laboratory to aid in the diagnosis and monitoring of treatment regimens of many infectious diseases such as HIV, hepatitis B, and tuberculosis (Netterwald 2006). These tests are usually performed on serological or other body fluids such as sputum and seminal fluid. Currently the most well known and most advertised molecular testing is for human papillomavirus (HPV).
Clinical and especially research laboratories may use additional molecular pathology techniques, such as blotting methods that are used to study extracted ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). Blotting methods consist of extracting DNA and/or RNA from homogenized tissues and then analyzing these nucleic acids using dot, Southern and northern blotting filter hybridization methods (Sambrook et al 1989). Blotting techniques such as these are powerful tools for the qualitative analysis of extracted nucleic acid from fresh or frozen cells and frozen tissues.
The polymerase chain reaction (PCR) is included in molecular pathology methods. PCR is a common method of creating copies of specific fragments of DNA. PCR rapidly amplifies a single DNA molecule into many billions of molecules. In one application of the technology, small samples of DNA, such as those found in a strand of hair at a crime scene, can produce sufficient copies to carry out forensic tests. PCR may also be used in addition to in situ hybridization (ISH) to study a specific genome of a tissue (Innis et al 1990).
All of this leads to the role the histology laboratory plays in molecular pathology. In the histology laboratory the main method used in molecular pathology is in situ hybridization. John et al (1969) and Gall and Pardue (1969) described the technique of in situ hybridization almost simultaneously.
In situ hybridization is a method of localizing and detecting specific mRNA sequences in preserved tissue sections or cell preparations by hybridizing the complementary strand of a nucleotide probe to the sequence of interest.
In situ hybridization consists of denaturing (breaking apart) DNA/RNA strands using heat. A probe (a labeled complementary single strand) is incorporated with the DNA/RNA strands of interest. The strands will anneal (nucleotides bonding back together) with their homologous partners when cooled (Fig. 26.1 ). Some will anneal with the original complementary strands, but some will also anneal or hybridize with the probe. As probes increase in length, they become more specific. The chances of a probe finding a homologous sequence other than the target sequence decreases as the number of nucleotides in the probe increases. Optimal probe size for in situ hybridization is small fragments of about 200–300 nucleotides. However, probes may be as small as 20–40 base pairs (bp) or as large as 1000 bp.
Fig. 26.1.
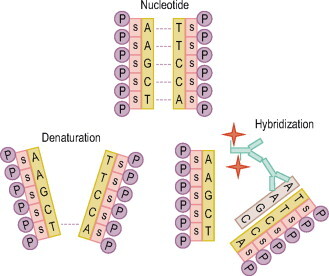
The genetic information for humans is encoded in billions of nucleotides, the building blocks of the DNA code, arranged in a double helix molecule. Nucleotides consist of a base, a sugar (S), and a phosphate (P). The DNA code is written in an alphabet that uses four letters to represent each of the bases. (A) = adenine, (T) = thymine, (C) = cytosine, (G) = guanine. These bases will form pairs. (A) will only pair with (T). (G) will only pair with (C). Therefore, double-stranded DNA consists of two strands of homologous nucleotides. The genetic code in DNA is in triplets such as ATG. The base sequence of that triplet in the partner strand is therefore TAC.
The detection of specific nucleic acid sequences—RNA, viral DNA or chromosomal DNA—in cells, tissues or whole organisms by in situ hybridization has numerous applications in biology, clinical and anatomical pathology, and research.
ISH methods may employ radiolabeled probes that are visualized on a photographic film or photographic emulsion. However, most of these probes do not work well on routinely fixed, processed tissues, require the use of frozen sections, and take around 20–50 days' exposure before seeing results. The development of non-radiolabeled probes that perform well on routine surgical and autopsy specimens has extended the field of anatomic pathology.
Detection of mRNA is particularly useful if the protein product is quickly degraded or rapidly transported out of the target cell.
In ISH detection, immunohistochemistry (IHC)-like methods may be incorporated to detect the labeled (biotin, digoxygenin (DIG)) probe. So the question arises, why not just do IHC? It is well-established, reliable, and less time consuming than ISH. IHC has been employed in the clinical and research arenas for several decades and has become a routine procedure in the histology laboratory. IHC has provided diagnostic procedures and a close look at the proteins within and on the cell membranes. So why do ISH? The advantages of ISH over IHC are:
-
1.
high degree of specificity.
-
2.
DNA and mRNA are not as sensitive to formalin fixatives.
-
3.
probe–target hybrid is stronger than antibody–antigen complex.
-
4.
provides an alternative means of detection when reliable antibodies are not available.
-
5.
provides a diagnosis at the molecular level.
It is important to understand the ‘how and why’ of the different stages in the ISH process in order for the testing to result in a functional outcome. This chapter focuses on the ‘how and why’ of ISH and general procedures on tissue sections.
APPLICATIONS
There are many modifications to ISH methods that relate to the application needs. Although the demonstration of DNA and RNA sequences by in situ hybridization is a valuable research tool, according to Warford and Lauder (1991) and Mitchell et al (1992), it is also used diagnostically in:
-
•
detection of abnormal genes
-
•
identification of viral infection
-
•
tumor phenotyping.
In situ hybridization alone can provide cytological information on the location and alteration of genomic sequences in chromosomes. Traditionally the technique has been applied to metaphase chromosome spreads (Davis et al 1984; Lux et al 1990), but it has been shown to be applicable to interphase nuclei (Hopman et al 1988; Poddighe et al 1992). Routine paraffin wax preparations of tissues can be used and ‘interphase cyto-genetics’, as the method is termed, can provide direct information on chromosomal abnormalities in unselected tumor cell populations. Chromogenic in situ hybridization (CISH) is a method ‘that enables the detection of gene expression in the nucleus using a conventional histochemical reaction’ (White 2005); it is used for the detection of abnormal genes and to identify a gene therapy treatment direction. CISH can be used as an alternative in screening archived breast cancer tissue samples for HER-2/neu (type 1 growth factor receptor gene) (Madrid & Lo 2004).
In situ zymography (ISZ) is a method that uses specific protease substrates to detect and localize protease activities in tissue sections. In the regulation of biolo-gical processes, proteases modulate several cellular functions. Several molecular techniques identify and characterize proteases in cells and tissue, such as a northern blot and reverse transcription–polymerase chain reaction (RT-PCR) but ISZ works as well. One drawback to ISZ is that you must use unfixed fresh frozen tissues. Advantages to using ISZ are that it cost less than conventional ISH methods, it has two approaches (one uses a photographic emulsion, the other uses a fluorescent-labeled substrate), and is applicable to almost any protease (Yan & Blomme 2003).
Viral identification can be undertaken using a variety of methods of which only immunohistochemistry and in situ hybridization provide simultaneous morphological information. The sensitivity of immunohistochemistry for the visualization of viral antigens, and in situ hybridization for the demonstration of cytomegalovirus, correlated well (Van den Berg et al 1989). Most viral in situ hybridization methods use probes against DNA. Others such as in the demonstration of the Epstein–Barr virus, the detection of a virally encoded RNA transcript, provide results that are more sensitive than the use of antibodies and may even approach that of the PCR (Pringle et al 1992).
Immunolabeling electron microscopy (IEM) in combination with ISH has been used in detecting severe acute respiratory syndrome (SARS). Viral immunogold labeling and ultrastructural ISH was used to analyze the morphogenesis of this newly emergent virus. A negative-sense riboprobe was used for the ultrastructural ISH (Goldsmith et al 2004).
Fluorescent in situ hybridization (FISH) has an advantage over chromogenic methods in labeling specific nucleic acid sequences in cells and tissues. It is a ‘direct’ technique so it is faster and in some cases it does not require IHC-like detection. These probes employ fluorescent (fluorescein) tags that glow under ultraviolet light to detect the hybridization. FISH allows the use of multiple probes that may be spatially or spectrally overlapping on the same tissue. The literature suggests that it is possible to distinguish at least four to five different fluorescent signals in a single sample (Haugland & Spence 2005). FISH, also known as molecular cytogenetics, has enabled a huge advance in the diagnostic and prognostic capability of the clinical cytogenetic laboratory (see Chapter 27). FISH can also vividly paint chromosomes or portions of chromosomes with fluorescent molecules, thus, the term ‘chromosome painting’.
Polymerase chain reaction–ISH (PISH) is another form of ISH. Viral RNA is detected by reverse transcription–polymerase chain reaction (RT–PCR), using formalin-fixed paraffin-embedded tissue (FFPE). PISH results have been compared to IHC on staining for Newcastle disease in veterinary medicine. Newcastle disease is an avian viral infection that has a potential for rapid spread and may cause serious economic impact and inter-national trade restrictions for the poultry industry (Wakamatsu et al 2005). PISH is also used in the detection of human papillomavirus in uterine cervical neoplasias (Xiao et al 2001).
The identification of mRNA sequences by in situ hybridization may be the technique of choice for the rapid and sensitive identification of viral infection. Another advantage of in situ hybridization for viral detection is that some viral coat antigens are not expressed at certain stages of the viral replication cycle, thus negating the use of immunohistochemical methods.
ISH methods have been cultivated throughout the years so that most FFPE tissues, including decalcified tissues, can be used (Janneke et al 1999).
Another area in which in situ hybridization and immunochemistry can be viewed as complementary techniques is in the phenotyping of tumors. Many monoclonal and polyclonal antibodies are available for phenotyping and these may be employed in sensitive and rapid techniques. When problems arise in the interpretation of immunohistochemistry, mRNA phenotyping by in situ hybridization can be helpful (Pringle et al 1990; Kendall et al 1991; Ruprai et al 1991; Pringle et al 1993).
GLOSSARY AND DEFINITIONS OF THE TERMINOLOGY USED IN THIS CHAPTER AND IN ISH TECHNIQUES
Amplification
Production of additional copies of a gene sequences. This multiplication of the sequence makes it easier to identify.
Anneal
To join two strands of complementary nucleic acids.
Antisense RNA
A RNA which is complementary to a mRNA.
Base
A building block of the nucleotide. Four different bases: DNA: (A)—adenine, (T)—thymine, (C)—cytosine, (G)—guanine. In RNA the thymine is replaced by (U)—uracil.
Base pair (bp)
Two bases which are complementary to each other and joined by hydrogen bonds.
cDNA
A complementary sequence of DNA that has been synthesized from an initial template of RNA by the enzyme reverse transcriptase.
Clone
A collection of cells with identical genetic makeup. Population derived from a single cell.
Codon
A set of three consecutive nucleotides in DNA or mRNA that directs the incorporation of an amino acid during protein synthesis or signals the start or stop of translation. Multiple codons will code for the same amino acid.
Complementary sequence
Nucleic acid sequence of bases that can form a double-stranded structure by matching base pairs. For example, the complementary sequence to C-A-T-G (where each letter stands for one of the bases in DNA) is G-T-A-C.
Deletion
The loss of a segment of a gene or a larger portion of a chromosome.
Denature
To dissociate a double-stranded region of nucleic acid into the homologous single strands, by breaking the hydrogen bonds. This is usually achieved by heating.
DNA
Deoxyribonucleic acid is the genetic material in chromosomes.
DNA polymerase
An enzyme that synthesizes new DNA using a parental DNA strand as a template.
Downstream
A region extending from the 3′ end of the gene.
Endonuclease
Any enzyme that starts to degrade DNA or RNA somewhere within the nucleic acid sequence. Used as molecular scalpels to cut DNA or RNA at precise points.
Episome
A plasmid that can exist either independently of the host cell or be incorporated into it.
Exon
A sequence of DNA that codes information for protein synthesis that is transcribed to messenger RNA.
Frameshift mutation
The insertion or deletion of nucleotides in the coding region of a gene causes the reading frame to shift. Shifting of the reading frame results in a change in the translation of the mRNA transcript. Therefore, a one or two base change results in a frameshift, while a three base change does not shift the reading frame, but can change the amino acid added by that codon.
Gene
A DNA segment that codes for a polypeptide.
Gene cloning
The selection, application, and production of an individual nucleotide sequence.
Gene library
A collection of nucleotide sequences that have been artificially inserted into microorganisms or viruses and propagated.
Genome
The total amount of DNA present in a cell.
Hapten
A small non-immunogenic molecule which can elicit an immune response only when attached to a macromolecule carrier such as a protein; the carrier may be one that also does not elicit an immune response by itself. (Generally, only large molecules, infectious agents, or insoluble foreign matter can elicit an immune response in the body.)
Hybridization
The action whereby two complementary single-stranded pieces of nucleic acids are joined to form a double-stranded segment.
In situ
In the normal location. An ‘in situ’ tumor is one that is confined to its site of origin and has not invaded neighboring tissue or gone elsewhere in the body.
In situ hybridization
A technique that identifies and quantifies nucleic acid sequences within cells.
Integration
The combining of a foreign segment of DNA with cellular DNA sequences, causing the expression and replication of foreign DNA.
Intron
A segment of a gene situated between exons that is removed before translation of messenger RNA and does not function in coding for protein synthesis.
In vitro transcription
Using a DNA template to synthesize a RNA sequence in the presence of RNA polymerase and nucleotide triphosphates.
Kilobase
A measure of length of nucleic acids. One kilobase (kb) equals 1000 nucleotides of single-stranded nucleic acid. (kbp) refers to kilobase pairs of double-stranded DNA.
Melting temperature
The temperature at which the hydrogen bonds between complementary nucleotides will break, causing the dissociation of double-stranded nucleic acid. It is dependent on the G+C content of the DNA.
mRNA
Messenger RNA carries the message of the DNA to the cytoplasm of the cell where protein is made. Single-stranded RNA synthesized from a DNA template during transcription binds to ribosomes and directs protein synthesis.
Missense mutation
A single base substitution in DNA that changes a codon for one amino acid into a codon for a different amino acid.
Mutation
A change in the sequence of nucleotides in DNA.
Nick translation
Incorporating a labeled deoxyribonucleotide triphosphate into DNA.
Nonsense codon
A codon that does not code for an amino acid, but is a signal to terminate protein synthesis.
Northern blot
A cellulose or nylon membrane to which RNA molecules have been attached by capillary action. The transferred RNA is hybridized to single-stranded DNA probes. Northern blot technique is often used to measure expression (transcription) of a gene for which a specific cDNA is available for use as a probe. A gel-based laboratory procedure that locates mRNA sequences on a gel that are complementary to a piece of DNA used as a probe.
Nucleotide
The unit of DNA or RNA that consists of a phosphate group, a sugar, and a base.
Oligonucleotide
A short piece of nucleic acid that can be used as a hybridization probe.
Oncogene
A gene whose activity is associated with the conversion of normal cells to cancer cells.
Operator
The segment of DNA to which the repressor binds; it controls the expression of adjacent genes.
Operon
The sequence of bases in DNA that contain one or more structural genes together with the operator controlling their expression.
Phage
A virus that infects a bacterium. Phages are often used to produce recombinant DNA molecules.
Plasmid
A piece of DNA that can replicate independently of the chromosome or be incorporated into it. A plasmid is inherited, but not required for the host cell's growth or reproduction. Plasmids can be used to produce recombinant DNA.
Probe
A single-stranded piece of labeled DNA or RNA that will bind to a complementary sequence (target).
Promotor
The region of DNA, at the start of a gene, that the RNA polymerase binds to before beginning transcription.
Random priming
A method for labeling double-stranded DNA to produce a probe.
Recombinant DNA
The production of a single piece of DNA from two different sources.
Replication
The process by which an exact copy of parental DNA or RNA is made with the parental molecule serving as a template.
Restriction endonuclease
An endonuclease that is specific for a particular nucleotide sequence.
Reverse transcriptase
An enzyme that will synthesize a complementary sequence of DNA from a RNA template.
RNA
Ribonucleic acid. A nucleic acid that plays an important role in protein synthesis and other cell activities.
RNA polymerase
An enzyme that catalyzes the synthesis of RNA from a DNA template, using nucleotide triphosphates as substrates.
RNases
Ubiquitous RNA degrading enzymes.
rRNA
Ribosomal RNA is a component of ribosomes and functions as a non-specific site for making polypeptides.
Sense strand
The DNA strand that RNA polymerase copies to produce mRNA, rRNA, or tRNA.
Southern blot
The analysis of DNA sequences. It identifies and quantifies the DNA sequences using a specific hybridization protein.
Stringency
Conditions employed in hybridization reactions used to control the specificity of probe binding. The highest stringency conditions ensure the probe will bind only to completely complementary sequences. Lower stringency conditions will allow binding to sequences with some mismatching.
Template
A strand of DNA or RNA that specifies the base sequence of a newly synthesized complementary strand of DNA or RNA.
Transcription
The process by which a DNA sequence is copied into a complementary RNA sequence.
tRNA
Transfer RNA is a short-chain type of RNA present in cells that transfers specific amino acids in the formation of proteins.
Translation
The process by which the genetic message carried by the mRNA directs the synthesis of polypeptides; protein synthesis.
REAGENT PREPARATION
Listed here are formulas and methods on how to prepare the reagents for most ISH methods. The majority of these reagents can be purchased pre-mixed or in a kit for easy mixing. Keep in mind that different reagents may be suggested with some ISH methods, as you will see further in this chapter. The reagents listed here are for use in a universal no-frills or special equipment ISH procedure. Purchasing the reagents pre-mixed or in kits is convenient and safer to use, and provides some comfort that they are mixed according to manufacturer's specification and guaranteed by the vendor. This may cut down the possibility of human error. Some laboratories have limited budgets and find that pre-mixed reagents and kits are not an option. They can be expensive and expire before all of the reagents are used. To aid those laboratories in getting started, a reagent preparation list follows.
Diethylpyrocarbonate (DEPC) treated water
| Diethylpyrocarbonate | 1 ml |
| Distilled water | 1000 ml |
Stir while bringing to a boil for 10 min (in fume hood). Autoclave to expel DEPC.
2% aminoalkylsilane (positively charged slides)
| Aminoalkylsilane (AAS)* stored at 4°C | 5 ml |
| Dry acetone | 250 ml |
Dip clean slides in 2% AAS for 1 min. Rinse in three changes of deionized water.
Note
These slides may be purchased pre-coated. Make sure they are RNA/DNA free.
Proteinase K
| Proteinase K | 100mg |
| Buffer #1 | 5 ml |
Aliquot and freeze below −20°C.
Hyaluronidase
| Hyaluronidase | 20 mg |
| Buffer #1 | 20 ml |
0.1 M triethanolamine (make fresh)
| Triethanolamine | 0.1 ml |
| DEPC water | 100 ml |
| Acetic anhydride | 0.25 ml* |
Add just prior to use. Stir for 5 minutes, then add an additional 0.25 ml, and stir for another 5 min.
1 M Tris (stock)
| Trizma base | 60.55g |
| DEPC water | 500 ml |
| Adjust pH to 8.0 with conc. HCl | 20 ml* |
Autoclave.
1 M magnesium chloride (stock)
| Magnesium chloride | 20.34g |
| DEPC water | 100 ml* |
Autoclave.
5 M sodium chloride (stock)
| Sodium chloride | 29.22g |
| DEPC water | 100 ml* |
Autoclave.
Maleic acid buffer
| Maleic acid | 100mM |
| Sodium chloride | 150mM |
Mix 1:10 with water and adjust pH to 7.5, or add Tween 20 (0.3% v/v) for a washing buffer.
Buffer #1: Tris buffered saline, pH 7.5
| 1 M Tris (stock) | 10 ml |
| 5 M sodium chloride (stock) | 3.3 ml |
| 1 M magnesium chloride (stock) | 0.2 ml |
| Deionized water | 86.7 ml |
Adjust pH to 7.5 with HCl.
Buffer #2: Tris buffered saline, pH 9.5
| 1 M Tris (stock) | 10 ml |
| 5 M sodium chloride (stock) | 2 ml |
| 1 M magnesium chloride (stock) | 5 ml |
| Deionized water | 83 ml |
Adjust pH to 9.5 with sodium hydroxide (NaOH).
20× Saline sodium citrate (SSC) buffer
| Sodium chloride | 348 g |
| Sodium citrate | 167.4g |
| DEPC water | 1600 ml |
Adjust to pH 7.4 with dilute acetic acid, stirring vigorously. Autoclave.
2× SSC
| 20× SSC | 10 ml |
| DEPC water | 100 ml |
| 1× SSC | |
| 20× SSC | 5 ml |
| DEPC water | 95 ml |
Denhart's solution
May cause increase in background.
Prehybridization solution
| Deionized formamide | 5 ml*1 |
| 20× SSC | 2 ml |
| Denhart's solution | 0.10 ml*2 |
| 50% dextran sulfate | 2 ml |
| Salmon sperm DNA (10 mg/ml) | 0.30 ml*3 |
| Yeast tRNA (10 mg/ml) | 25 ml*4 |
purified = less non-specific staining
reduces non-specific probe binding
denature by boiling for 10 min
blocks non-specific staining
Hybridization solution
| Prehybridization solution | 1 ml |
| Labeled probe (500 ng/25 μl) | 10 ml |
Detection method reagents: pick one
|
0.01 ml |
| Buffer #1 | 5 ml |
|
0.01 ml |
| Buffer #1 | 2.50 ml |
|
0.01 ml |
| Buffer #1 | 5 ml |
Colorimetric detection reagents: pick one
| |
| 5-bromo-4-chloro-3-indolyl | |
| phosphate (BCIP) | 0.5 mg/ml |
| nitro-blue tetrazolium salt (NBT) | 0.3 mg/ml |
|
|
| 3-amino-9-ethylcarbazole (AEC) | 0.08g |
| acetone | 10 ml |
| 0.05 M acetate buffer | 200 ml |
| hydrogen peroxide (30%) | 0.10 ml |
|
|
| Diaminobenzidine (DAB) | 22mg |
| Tris buffer | 50 ml |
| Hydrogen peroxide (30%) | 0.2 ml |
PROBES AND THEIR CHOICE
Probe choice is based on the type of sequence you are trying to detect. The technologist needs to optimize the conditions he or she uses as much as possible. The strength of the bonds between the probe and the target plays an important role. The strength decreases in the order RNA–RNA to DNA–RNA. Various hybridization conditions such as concentration of formamide, salt concentration, hybridization temperature, and pH influence this stability.
A probe is a labeled bit of DNA or RNA used to find its complementary sequence or locate a particular clone. The choice of probes will depend on availability, sensitivity, and resolution required. The sensitivity of the probe will depend on the degree of substitution and the size of the labeled fragments. Degree of substitution refers to the original nucleotide substituted by the labeled analogues. The sensitivity of detection correlates with the amount of label substituted. In general, probes with 25–32% substitution yield the highest sensitivity. There are several different types of probe. Each has unique characteristics that must be considered for each application.
Probe type and means of synthesis
There are essentially four types of probe that can be used in performing in situ hybridization.
Oligonucleotide probes are usually 20–50 bases in length. They are produced synthetically by an automated chemical synthesis employing a specific DNA nucleotide sequence (of your choice). These probes are resistant to RNases and are small which allows for easy penetration into the cells or tissue of interest. However, the small size has it disadvantage in that it covers fewer targets. The label should be positioned at the 3′ or the 5′ end. To increase sensitivity one can use a mixture of oligonucleotides that are complementary to different regions of the target molecule. Oligonucleotide protocols can be standardized for many different probes regardless of the target genes being measured. Another advantage of oligonucleotide probes is that they are single stranded, therefore excluding the possibility of renaturation.
Single-stranded DNA probes are a much larger size range (=200–500 bp) than oligonucleotide probes. Single-stranded DNA probes can be prepared by a primer extension on single-stranded templates by RT-PCR of RNA, or by an amplified primer extension of a PCRgenerated fragment in the presence of a single antisense primer, or by the chemical synthesis of oligonucleotides. PCR-based methods are much easier and probes can be synthesized from small amounts of starting material. Moreover, PCR allows great flexibility in the choice of probe sequences by the use of appropriate primers.
Double-stranded DNA probes can be prepared by nick-translation, random primer, or PCR in the presence of a labeled nucleotide, and denatured prior to hybridization in order for one strand to hybridize with the mRNA of interest. They can also be produced by the inclusion of the sequence of interest in bacteria, which is replicated, lysed, and then the DNA is extracted and purified. The sequence of interest is removed with restriction enzymes. Random priming and PCR give the highest specific activities. These probes are less sensitive than single-stranded probes, since the two strands have a tendency to rehybridize to each other, thus reducing the concentration of probe available for hybridization to the target. Nevertheless, the sensitivity obtained with double-stranded probes is sufficient for many purposes, although they are not widely used today.
RNA probes (cRNA probes or riboprobes) are thermostable and are resistant to digestion by RNases. These probes are single stranded and are the most widely used probes with ISH. RNA probes are generated by in vitro transcription from a linearized template using a promoter for RNA polymerase that must be available on the vector DNA containing the template (SP6, T7, or T3). RNA polymerase is used to synthesize RNA complementary to the DNA substrate. Most commonly, the probe sequence is cloned into a plasmid vector so that it is flanked by two different RNA polymerase initiation sites enabling either sense-strand (control) or antisense (probe) RNA to be synthesized. The plasmid is linearized with a restriction enzyme so that plasmid sequences are not transcribed, since these may cause high backgrounds.
When comparing double-stranded to single-stranded probes, single-stranded probes provide a few advantages over double-stranded probes such as:
-
•
The probe does not self-anneal in solution so the probe is not exhausted.
-
•
Large probe chains are not formed in solution, thus probe penetration is not affected.
If high sensitivity is required, single-stranded probes should be used (Table 26.1 ).
Table 26.1.
Probe types
| Probe | Labeling | Advantages | Disadvantages |
|---|---|---|---|
| dsDNA | Random primers |
|
|
| ssDNA | Primer extension |
|
|
| ssRNA | DNA polymerase transcription |
|
|
| Oligo |
|
|
|
PROBE PREPARATION AND LABELING
To visualize where the probe has bound within your tissue section or within your cells, you must attach a detectable label to your probe before hybridization. Two major choices must be made for the preparation of a probe.
-
•
What type of nucleic acid is to be used (DNA or RNA, single or double stranded)?
-
•
What type of label is to be incorporated into the probe?
A vital consideration is the length of the probe, and the means by which this is controlled depends on the type and the method of synthesis. There are two methods of probe labeling. They are:
-
•
Direct: The reporter molecules (enzyme, radioisotope or fluorescent marker) are directly attached to the DNA or RNA.
-
•
Indirect: A hapten (biotin, digoxygenin, or fluorescein) is attached to the probe and detected by a labeled binding protein (typically an antibody).
Methods for incorporation of labels into DNA are nick translation and random primer methods.
Oligonucleotide probe labeling
-
•
5′-end labeling:
The 5′ end of DNA or RNA undergoes direct phosphorylation of the free 5′-terminal OH groups. The free 5′-OH substrates can be labeled using T4 polynucleotide kinase. This method is usually used for radiolabeling. Non-radiolabels use a covalent linker.
-
•
3′-end labeling:
Terminal dexoxynucleotidyl transferase (TdT) is used to add a labeled residue to the 3′ end of a synthetic oligonucleotide that is approximately 14–100 nucleotides in length. These probes provide excellent specificity but only moderate sensitivity. See oligonucleotide 3′-end labeling procedure in this chapter (see Procedure 26.1).
-
•
3′ tailing:
A tail containing labeled nucleotides is added to the free 3′ end of double- or single-stranded DNA using TdT. These probes are more sensitive than the 3′-end labeled but can produce more non-specific background. Oligonucleotide tailing kits are commercially available.
It should be noted that the use of commercially available labeling kits can greatly assist in making methods simpler to undertake while providing results of an assured standard.
Purification of labeled probes
Sephadex G-50 column
Sephadex matrix traps unincorporated labeled probes. The labeled probes are excluded from the pores of the matrix and spun through the column. The labeled probes are collected in the eluent. Other brand name spin columns are commercially available.
Sephadex G-50 chromatography
Purifies probes, but fractions of the eluent have to be collected and tested for the presence of label. There are smaller sized columns available than the ones described above that fit into ordinary microcentrifuges.
Selective precipitation
This can be used to remove interferences from a mixture. A chemical reagent is added to the solution, and it selectively reacts with the interference to form a precipitate. The precipitate can then be physically separated from the mixture by filtration or centrifugation. Under this premise, unincorporated labeled nucleotides can be eliminated by ethanol precipitation in the presence of a DNA carrier. Mononucleotides remain in the supernatant. Labeled and/or purified probes can be concentrated by ethanol precipitation and can be stored at −20°C. Do not purify biotinylated probes by phenol/chloroform extraction, as this will destroy the biotin.
Spin columns provide a simple way of purifying nucleic acid sequences from labeling reagents. This is achieved by centrifuging the labeling solution through a column of packed G-50 Sephadex, Sephacryl S-400, or Probe Quant G-50, in which the labeling reagents are retained but the larger nucleic acid sequences are excluded and, therefore, are recovered in the eluent.
METHOD
Preparation of columns
-
1.
Plug the barrel of a 1.0 ml disposable syringe with sterile siliconized glass wool (GE) or polyallomer wool. Fill the syringe with a slurry of 5 g Sephadex G-50 (Roche 1273973), Sephacryl S-400 (GE 17-0609-10), or Probe Quant G-50 (GE 27-5335-01), pre-swollen (this may take a couple of hours) in 100 ml of 1× Tris buffer, pH 8.0.
-
2.
Let the Tris buffer drain out, fill the syringe again with Sephadex slurry. Continue addition of Sephadex until the syringe is filled with gel when the Tris buffer has drained out.
-
3.
Insert the syringe into a polypropylene 10 ml centrifuge tube. Centrifuge at 3000× g for 5 min in a bench centrifuge. Do not be alarmed at the appearance of the column. Continue to add Sephadex until the packed column volume after centrifugation is 0.9 ml.
-
4.
Add 100–200 μl of 1× Tris buffer, pH 8.0, to the top of the column and re-centrifuge. Do this three times. Collect the last eluent in an uncapped 1.5 μl Eppendorf tube and measure its volume, which should be 100 μl. If significantly different, add another 100 μl of Tris buffer and centrifuge again.
Use the column immediately or cap with laboratory film (parafilm) and leave at 4°C until required.
Note
If the columns are not used immediately, it will be necessary to repeat step 4 before adding the labeling solution for purification.
Spin columns can also be prepared following the same method, but using a 2 ml disposable syringe, when the purification of a scaled-up labeling reaction is required.
Use of columns
-
1.
Place a 1.5 ml sterile Eppendorf tube uncapped under the column. Add the probe solution to the top of the column and centrifuge at 3000× g for 5 min.
-
2.
Measure volume, cap Eppendorf, and transfer to −20°C for storage.
Estimating the labeling efficiency and testing the probe
It is always good practice to estimate the yield of labeled nucleic acids. This confirms a successful labeling reaction before performing the staining.
Before using a labeled probe, it is useful to prepare and demonstrate test strips to gauge the degree of label incorporation. This may be done by using the normal detection procedure for the ISH method on dots of labeled nucleic acid sequence and a labeled control applied at matching descending concentrations to a positively charged nylon membrane. Some technicians will prepare several test strips from one labeled sequence to compare the sensitivity of different detection systems. The nylon membrane is subjected to an immunological detection which can be either a colorimetric or chemiluminescent method depending on which protocol is used. Direct comparison of the signal intensities of sample and control allows estimation of labeling yield. Kits for this technique are commercially available where the labeled control is already on a test strip. A much quicker method to estimate the yield of your labeled nucleic acids is to use a bioanalyzer that can give you quantitative results in as little as 30 minutes.
Described below is a method for estimating labeling efficiency of the nucleic acid using a dilution series followed by a spot test.
PREPARATION OF THE DILUTION SERIES
-
1.
Dilute the labeling probe using dilution buffer to a starting concentration of 2.5 pmol/μl.
-
2.
Make a dilution series in Eppendorf tubes of purified probe to give nucleic acid concentrations of 300 pg/μl, 100 pg/μl, 30 pg/μl, 10 pg/μl, 3 pg/μl, and one tube containing diluent only. Ensure that all tube volumes are equal. Repeat the same dilution series with your control or used pre-labeled (with control) test strips.
-
3.
Apply 1 μl drops from each tube onto the nylon membrane (Roche). The control dilutions should be lined up with the test sample dilution concentration. For an example of placing spots, see Fig. 26.2 .
-
4.
Label the position of each application with a pencil on the side of the strip (not on the strip).
-
5.
Fix the nucleic acid to the membrane by either baking the membrane for 30 min at 120°C or using a UV light.
-
6.
Wash the membrane briefly in washing buffer.
-
7.
Immerse in blocking solution for 10 min.
-
8.
Incubate with reagents used in ISH detection technique.
Fig. 26.2.
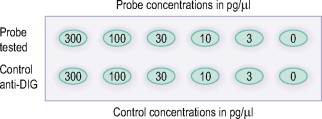
Note
Dilute reagents in blocking solution and use this solution for washing.
-
9.
Detect enzyme using the same solutions and procedure as for ISH method.
-
10.
Rinse in double distilled water and blot dry.
COMMERCIALLY MADE PROBES
Custom designed, pre-made cloned DNA and oligonucleotide sequences and labeling kits are commercially available and their use can greatly assist in making methods simpler to undertake while providing results of an assured standard. Depending on the type of laboratory you have and how many ISH requests you receive, premixed reagents and pre-labeled probes can be cost effective. The kits, reagents, or ordering the labeled probes can cut down on precious technologist time, and for the novice technologist they come with instructions. However, this does not mean that the theory behind ISH can be ignored. The technologist must understand ISH to order labeled probes and the ISH kits.
Probe concentration
For DNA probes the concentration of the probe will be ∼0.5–2 μg/ml. Oligonucleotide probes can be used with or without acetylation. Probes without acetylation pre-treatment of the sample will have a concentration of ∼50–200 ng/ml and may provide more intense results with minimal background. For probes with acetylation pre-treatment, a higher concentration of oligonucleotide probe may be used without incurring non-specific background staining.
Length of probe
As mentioned previously in choosing a probe, one must consider the length of the probe. Longer probes give weaker signals and they penetrate less effectively into the cross-linked (fixed) tissue. The extent of weaker signals and penetration depends also on the nature of the tissue, the choice of fixative, and whether a pretreatment has been carried out.
The length of probe can be controlled in either the synthesis reaction or the subsequent partial cleavage. In nick-translated probes the DNA length is determined by the amount of DNase in the reaction, while in random priming the length is determined by concentration of the primer. Long RNA probes may show poor tissue penetration; while chemical shortening (hydrolysis) enhances tissue penetration, it also increases the likelihood of non-selective binding to other non-targeted gene sequences.
Once a probe is prepared, the size should be checked. If the probe is too small, it may yield low signals with high background. It is necessary to know if a reduction of probe size (length) improves both the signal for the tissue and the preparation method used.
DETECTION
The choice of detection system will be principally determined by the probe label used and secondly by the ISH procedure type. One must consider the sensitivity and resolution required.
Colorimetric detection substrate systems include horseradish peroxidase with either 3-amino-9ethylcarbazole (AEC) or 3,3′-diaminobenzidine tetra-hydrochloride (DAB) substrates. AEC forms a reddish brown product which is alcohol soluble, therefore aqueous mounting media are required. Methyl green/blue has been the most often used counterstain in earlier publications but is losing popularity. DAB forms a brownish product that is compatible with solvent-based mounting solutions, and is good for permanence. Alkaline phosphatase systems can use BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitro-blue tetrazolium) or fast red. BCIP/NBT forms a purple/blue alcohol-insoluble stain. Eosin is a compatible counterstain if a nuclear target is expected, or nuclear fast red if the target is cytoplasmic. Fast red forms an intense red product which is alcohol soluble and an aqueous mounting media is required. Methyl green or blue is compatible if a nuclear target is expected, or light hematoxylin if the target is cytoplasmic.
Detection methods can be direct or indirect. Incorporation of a stable hapten to a probe is the cornerstone of non-radiographic detection. Hybridized probes can be detected by enzymatic reactions that produce a colored precipitate at the site of hybridization. The most commonly used enzymes for this application are alkaline phosphatase (AP) and horseradish peroxidase (HRPO). Although these enzymes can be conjugated directly to nucleic acid probes, such enzyme-coupled probes are often inappropriate for in situ hybridization to tissue preparations because probe penetration is hampered by the presence of the conjugated enzyme. Therefore, indirect methods are preferred (Knoll & Lichter 1995).
Biotin, fluorochromes (fluorescein), and digoxygenin (DIG) are the most common labels used. Probes that are labeled with these reporter molecules are usually detected by an AP or HRPO conjugate to avidin (for biotin) or antibodies (for DIG). Fluorescein and DIG have an advantage over biotin in that they produce lower levels of background signals in tissues that contain high amounts of endogenous biotin.
Direct detection of a fluorescent label is often employed for the demonstration of multiple chromosome targets (Nederlof et al 1989), but when single target sequences are to be identified indirect methods may be used.
To reduce non-specific staining (particularly of collagen) by indirect detection reagents it is advisable to pre-incubate preparations in a Tris (Triton-X-100 or Tween) buffer solution containing bovine serum albumin. It may also prove beneficial to use this solution as the diluent for the primary detection reagent. However, at this step it is more important to use a Fab fragment of an antibody as this may reduce background staining.
Indirect detection procedures offer increased sensitivity. The selection of the enzyme and substrate (De Jong et al 1985) should be included in weighing the benefits of different detection systems. A substrate system that employs conjugated antibodies such as anti-DIG or anti-FITC that are conjugated with AP together with the application of a colorimetric BCIP/NBT that can be cycled to produce an insoluble blue/black precipitate over a period of 24 hours is recommended. Another substrate system one could use for a more intense fluorescent signal is a fluorescent 2-hydroxy-3-naphthoic acid-2′-phenylanilide phosphate (HNPP) with fast red TR.
The main advantages of this procedure are low levels of non-specific staining, simplicity, and the use of an enzyme substrate system that can produce an insoluble blue/black precipitate.
Many commercially available probes for ISH are labeled with biotin. When used in combination with streptavidin detection systems, high sensitivity can be achieved. A disadvantage of that combination is having a widespread endogenous tissue distribution. Substantial quantities of endogenous biotin are, for example, present in the liver and kidney (Wood & Warnke 1981), and other tissues, such as pituitary, submandibular gland, thyroid, and parathyroid. Furthermore, proliferating cells may often produce enough biotin to make the discrimination between true and false-positive results difficult. However, methods of blocking endogenous biotin have greatly improved and work well to prevent false positives.
Digoxygenin (Herrington et al 1989) in combination with a Fab fragment–enzyme conjugate detection system currently provides results of equal or superior sensitivity to biotin, with extremely low non-specific background staining. Another label that may be used in conjunction with a single-step detection method is fluorescein. Using this label it is possible to undertake rapid ISH methods in which target sequences of moderate to high copy number can be demonstrated in a working day.
SAMPLE PREPARATION
Fixation
Fixation is an initial step in specimen preparation or can be an intermediate step in a protocol, as in methods using cryostat sections. The duration, type, and temperature of fixation may also differ according to preparation. Together these factors will have an effect, not only on the preservation of the tissue, but also on the retention of nucleic acid and the resistance of DNA and RNA to nuclease digestion. The choice of fixative will have an influence on the conservation of nucleic acids and their availability for hybridization. Specimens that are immersion-fixed prior to paraffin embedding appear to be unaffected by ‘normal’ contamination levels of nucleases, thus indicating that only the hybridization solutions need to be scrupulously free of the enzymes.
The functional groups involved in base pairing are protected in the double helix structure of duplex DNA. RNA is fairly unreactive to cross-linking agents.
Methanol/acetic acid fixation is recommended for metaphase chromosome spreads. Cryostat sections may be fixed with 4% formaldehyde (∼30 minutes), Bouin's fixative, or paraformaldehyde vapor fixation. This fixation also helps to secure the tissue to the slide.
Proteins surround DNA and RNA target sequences and the extensive cross-linking of these proteins may mask the target nucleic acid. Therefore, permeabilization procedures may be required.
After the tissue is removed from the patient or animal, it must be fixed to prevent autolysis, inhibit bacterial/fungal growth, and make it resistant to damage from subsequent processing. There are two main groups of fixatives based on their reaction with soluble proteins. They are coagulant and non-coagulant fixatives. Coagulant fixatives are ethanol and mercuric chloride, and are not the preferred fixative for use with ISH since ethanol dehydrates, coagulates, and precipitates cellular proteins, nucleic acids, and carbohydrates. Covalent bonding does not occur with ethanol fixatives and the tissue components, so mRNA is not anchored within the tissue and is likely to be lost during post-fixation processing procedures. For ISH, non-coagulant, cross-linking aldehydes (formaldehyde, paraformaldehyde, and glutaraldehyde) are recommended.
Tissues fixed for ISH should retain mRNA within the tissue but not raise background. Both background and signal are generally higher on perfused-fixed paraffin tissue sections than on frozen sections. The signal to noise (S/N) ratio on perfused-fixed tissue sections is better.
Most commonly, tissue specimens are routinely fixed in 10% buffered formalin, processed overnight in an automatic tissue processor, and embedded in paraffin. Fixation time of 8–12 hours is optimal. Keep in mind that the longer the fixation, the more rigorous the enzyme digestion is required to optimize the signal. Alcohol-fixed tissues should be post-fixed with an aldehyde fixative to prevent the diffusion of mRNA (if you are looking at RNA).
Slide/section preparation
Sections are cut—4–6 μm on an alcohol-cleaned microtome using positively charged or hand-coated slides. Sections are drained well and then air-dried at room temperature. After deparaffinization, place slides in an alcohol-cleaned staining container of DEPC water. The staining container is then placed in the heated water bath at 23–37°C and held until the start of ISH. Gloves must be worn to prevent contamination, and all utensils, such as brushes and forceps, are cleaned with alcohol and kept within the cleaned area designated for ISH.
Proteolytic digestion
The use of formaldehyde-based fixatives prior to paraffin embedding of specimens will mask nucleic acid sequences. Digestion is a important step when performing ISH. Digestion improves probe penetration by increasing cell permeability with minimal tissue degradation. Although the nucleic acid is not directly affected by proteolytic digestion, it is important to control this step carefully as under-digestion will result in insufficient exposure of the nucleic acid while over-digestion can sufficiently weaken the protein structure surrounding the sequence to bring about its loss into subsequent solutions. mRNA sequences tend to be loosely associated with proteins while DNA targets are intimately associated with histone and other nuclear proteins. Due to these differences, the concentration of proteolytic enzyme required to unmask mRNA will be less than that necessary to expose DNA.
The selection of the proteolytic enzyme is important. This should be of molecular biology grade to ensure the absence of nuclease activity. Proteinase K and pepsin are two enzymes commonly used for digestion. The use of proteinase K has an advantage over other proteolytic enzymes because during incubation it digests any nucleases that might be present. However, higher concentrations of the enzyme may be required depending on the tissue fixation time (Fig. 26.3 ).
Fig. 26.3.
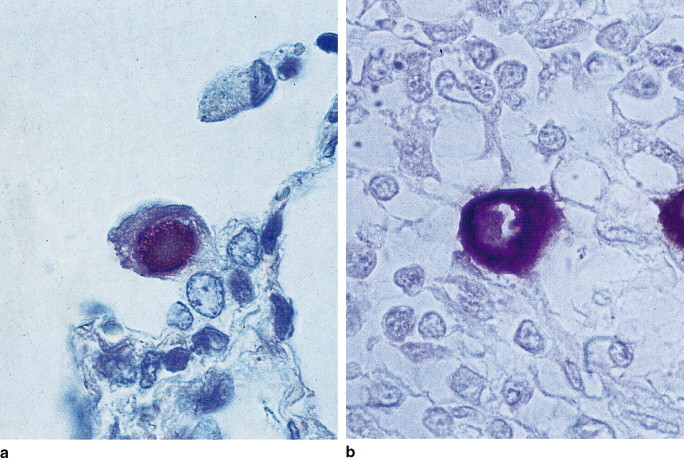
Example photographs of ISH. Human cytomegalovirus (HCMV) demonstrated by in situ hybridization in paraffin wax sections. (a) HCMV DNA in infected lung, proteinase K pretreatment 50 μg/ml for 1 hour at 37°C. (b) HCMV early gene RNA transcript in AIDS infected bowel, proteinase K pretreatment at 15 μg/ml for 30 min at 37°C.
Nuclease digestion is used as a negative control. Treatment of same tissue/patient tissue sections with RNase A will demonstrate that ISH signals are due to RNA hybridization.
Proteoglycan digestion is required for bone and cartilage. Kidney and brain may also need proteoglycan digestion if the signal is weak. Post-fixation in 4% paraformaldehyde is necessary after digestion to prevent tissue loss. In addition, post-fixation after digestion (for all digestions) prevents leaching and will inhibit RNase activity.
When in situ hybridization methods are used to demonstrate mRNA sequences, non-specific attachment of digoxygenin and fluorescein-labeled oligonucleotides to epithelial tissues can create non-specific results. To minimize this interaction, preparations can be acetylated after proteolytic digestion and before post-fixation. Acetylation decreases non-specific binding of the probe to the tissue. Positive charges on the tissue are neutralized by reducing electrostatic binding of the probe.
Prehybridization is intended to reduce non-specific binding. Sites in the tissue become saturated with the components of the prehybridization solution preventing non-specific binding. The purpose of the prehybridization solution is to equilibrate the specimen with the hybridization solution prior to the addition of the probe, and to allow anionic macromolecules to block sites of potential non-specific probe interaction. The prehybridization solution contains all the ingredients of the hybridization mixture except the probe. Non-complementary sequences such as bovine serum albumin (BSA at 1 mg/ml), Denhardt solution (Ficoll, BSA, and polyvinylpyrrolidone all at 0.02%), and tRNA are used to reduce non-specific binding. Most data indicate that blocking is required, but a separate prehybridization step may not be necessary. In some cases, adequate blocking may be accomplished during the hybridization step. Electrostatic binding of probes to the tissue and slides can be neutralized by treatment in TEA buffer containing 0.1 M triethanolamine.
Hybridization
Hybridization is the cooling after denaturization, in the presence of a complementary probe, that results in hydrogen bonding of the two strands of nucleic acids. The probe must form stable hydrogen bonds with the target with minimal hybridization with non-target sequences. The probe and target sequences must be single stranded. Simultaneously heating the probe and target to high temperatures may increase the consistency and sensitivity of detection. This can only be met if care is taken to precisely control this step of the in situ hybridization procedure. Control is achieved through balancing the various components of the hybridization solution and hybridizing at an optimal temperature for the correct length of time.
If a DNA probe is employed or a DNA target demonstrated, then it is essential that these are rendered single stranded. This is achieved by using dry heat with the hybridization solution placed over the specimen, which is then covered with a coverslip, plastic sheet, or cap. The denaturation will differ according to the percentage of guanine–cytosine base pairs within the target sequence of interest. When there is a high percentage of guanine–cytosine base pairs, the third hydrogen bond associated with the base pair will occur at a higher melting temperature (TM) than in the sequences in which adenosine and thymidine pairing predominates. Overheating, i.e. above 100°C, at this stage may compromise specimen preservation.
At the molecular level, hybridization involves an initial nucleation reaction between a few bases, followed by hydrogen bonding of the remaining sequences. The control of temperature during hybridization is crucial as variations will influence the specificity (stringency) of annealing. RNA and DNA hybrids are formed optimally at about 25°C below their TM. When lower temperatures are used, some partial homologous annealing may occur. Although this situation should be avoided, it can be usefully employed to screen for sequences with partial homology, e.g. human papillomavirus subtypes. By incorporating formamide, a helix-destabilizing reagent, annealing may be maintained using lower temperatures, e.g. 37°C, at which tissue preservation is not affected.
Stringency can also be altered by adjusting the availability of monovalent cations in the hybridization solution. These cations are usually supplied by sodium chloride and their action is to regulate the degree of natural electrostatic repulsion between the probe and target sequences. When used at high concentration their effect is to produce conditions of low stringency, while at low concentrations only sequences with complete homology can hybridize.
Anionic macromolecules are often included in the hybridization solution to reduce non-specific interactions of the probe. Sonicated and denatured salmon sperm DNA can shield non-homologous nucleic acid sequences from the probe and reduce the opportunity for cellular electrostatic interactions. Dextran sulfate will also reduce the possibility of cellular electrostatic interactions and locally concentrate the probe, enhancing the rate of hybridization. Particular attention should be taken to ensure that hybridization solutions are prepared using reagents free from nuclease contamination.
The rate of annealing will be influenced by time and temperature as well as by the composition of the hybridization solution as discussed above. Due to steric constraints, in situ hybridization proceeds at a slower rate than in blotting methods. However, high probe concentrations can be used to compensate for this factor. Hybridization times of 1–2 hours for biotinylated and fluorescein-labeled probes are often effective but with digoxygenin-labeled probes, overnight hybridization may be required to provide high sensitivity.
Post-hybridization washes are used to adjust the stringency of hybridization. The sections must be rinsed with solutions that contain high concentrations of salt to remove the unbound probe. Subsequent washing with solutions containing decreasing salt concentrations and increasing temperature reduces mismatching of base pairs. Longer probes and those with higher G+C content are more stable and increases in temperature and formamide concentration are the destabilizing factors. By reducing the concentration of formamide in the hybridization solution, but maintaining a constant temperature, annealing conditions will become less stringent, thereby increasing the sensitivity of mRNA detection when using fluorescein-labeled oligonucleotide probes.
Controls
It is essential to include controls to verify ISH results. A positive and no-hybridization control should always be included when undertaking an ISH procedure. The positive control should contain the target sequence being demonstrated and be prepared the same as the test samples. It should receive the same solutions and go through the same procedural steps as the test samples. This will provide a gauge for the overall performance of the technique. If the same positive control is used from run to run, it will help validate the ISH staining reproducibility.
Other controls may be incorporated to test the validity of your results. The number and type of controls to be incorporated into the technique is determined at the discretion of the laboratory's personnel and standard operating procedures. As mentioned under the digestion section earlier in this chapter, nuclease digestion can be used as a negative control.
Equipment and reagent preparation
DNA and RNA can be degraded by nuclease activity. Indeed, the use of high concentrations of DNase and RNase are useful in confirming the nucleic acid type specificity of hybridization. However, at much lower concentrations, nucleases present on the skin may contaminate solutions used in hybridization methods sufficiently to degrade the quality of naked DNA and RNA. For this reason, in addition to the wearing of gloves, elaborate precautions are often taken to ensure the absence of nucleases from solutions used in hybridization techniques.
TREATMENT OF SOLUTIONS AND GLASSWARE TO DESTROY NUCLEASE ACTIVITY
DNase is destroyed by autoclaving. RNase is resistant to heat inactivation and therefore other procedures should be used as described below.
Preparation of DEPC-treated water
Add diethylpyrocarbonate (DEPC) (Sigma D5758) to pure water to a final concentration of 0.1%. This should be done in a fume hood. Shake well to dissolve and allow it to stand overnight. Autoclave the solution and container the following day.
Preparation of DEPC-treated solutions
Prepare solutions, then add DEPC to 0.1%. Shake and leave overnight, then autoclave.
Note
Adding full-strength DEPC directly to a buffer may alter the buffering properties. Solutions that require a buffer of that type should be made up in RNase-free glassware using pre-mixed autoclaved DEPC-treated water.
Preparation of glassware
Treat glassware at 200°C overnight or, if delicate:
-
1.
Wash in a mild/low suds soap or RNase Away solution.
-
2.
Rinse in double distilled nuclease-free water until detergent is removed.
-
2.
Soak in 3% aqueous hydrogen peroxide, 10 min.
-
3.
Rinse in DEPC-treated water.
-
4.
Dry and protect from dust.
Most laboratories purchase DEPC-treated water and use sterile plastic disposable containers in place of the glassware. The information is in this chapter is for those laboratories that prefer to prepare their own DEPC water and use glass bottles for buffer storage.
UNIVERSAL ISH METHOD (NO FRILLS OR SPECIALIZED EQUIPMENT)
Day 1
-
1.
Deparaffinize slides completely. Three changes of xylene and/or substitute for 4–8 min each.
Note
Incomplete deparaffinization may cause a weak reaction.
-
2.
Dehydrate through two changes 100% EtOH, 1 change 95% EtOH for 3 min each. Rinse in DEPC-treated water or use slides already in warmed DEPC-treated water. Rinse in warmed (23–37°C) Tris/saline buffer #1, pH 7.5, and drain.
-
3.
De-proteinize sections in freshly prepared proteinase K solution at 23–37°C, in a moist chamber for 15 min.
-
4.
Rinse in Tris/saline buffer #1 at room temperature for 5 min. If necessary, digest proteoglycans and/or acetylation before going to next step.
-
5.
Dehydrate slides through one change of 95% EtOH and two changes 100% EtOH for 2 min each. Air-dry for 5 min. This step is omitted if prehybridizing (next step).
-
6.
Apply the prehybridization solution by putting 1–2 drops (60–100 μl) on the sections. Incubate in a moist chamber at room temperature for 1 hour. Blot off all excess prehybridization solution before adding the probe.
-
7.
Apply hybridization fluid (probe) and cover with a heat-resistant film (microwave wrap, coverslips, or chambers). The probe is tailed with either biotin–dUTP or digoxygenin–dUTP.
-
8.
Initiate hybridization by denaturing slides for 5–10 min at 92–100°C (try not to exceed 100°C). Use a preheated ‘metal’ tray to set slides on for optimal denaturization. Cool slides to 37–42°C and incubate in humidity chamber for 18–24 hours. Agitation may enhance reaction. Since this method is not using a commercial kit, the staining continues on day 2.
Day 2
-
9.
Rinse slides twice in 2×SSC and twice in 1×SSC for 5 min each at 37°C.
-
10.
Apply a 5% blocking solution at 37°C for 10 min.
-
11.
Rinse in buffer #1 for 2 min, followed by a 10-min rinse in buffer #2.
-
12.
Add 1–2 drops of detection reagent to each slide (streptavidin–AP or anti-digoxygenin–AP). Incubate in humidity chamber for 20–30 min at 37°C. During the incubation period, prepare the substrate and warm to 37°C.
-
13.
Rinse slides in three changes of buffer #3 for 5 min each.
-
14.
Incubate in substrate for 30–60 min at 37°C.
-
15.
Stop reaction by washing in buffer at 37°C for 2 min. Rinse in two changes of distilled water (DW) for 2 min each.
-
16.
Rinse in two changes of DW for 2 min each.
-
17.
Counterstain; this is dependent on the chromogen selected. For BCIP/NBT use nuclear fast red, eosin, or methyl green. For DAB use hematoxylin or methyl green. Go on to step 18 if using BCIP/NBT or DAB. For AEC use methyl green and coverslip out of distilled water using an aqueous mounting medium. Do not go through alcohols or clearing.
-
18.
Dehydrate in increasing concentrations of alcohol.
-
19.
Clear using xylene and coverslip in permanent mounting resin (Doran & Sterchi 2000).
TROUBLESHOOTING
Tissue sections fall off
-
•
adhesive absent/insufficient poly-L-lysine or amino-alkylsilane (AS) on slide
-
•
insufficient adhesion time/temperature
-
•
over-digestion (too long/too concentrated)
-
•
overzealous coverslip removal (use pliable wrap, AS slips, or well covers)
-
•
denaturation too long or temperature too high (93–98°C is ideal)
-
•
excessive slide agitation.
Weak staining (tissue preparation)
-
•
slides incompletely deparaffinized (add an additional xylene/substitute step to insure that all the paraffin has been removed)
-
•
slides not dehydrated or drained prior to addition of probe (water in tissue will dilute probe)
-
•
over-fixation (increase digestion time)
-
•
insufficient digestion (increase time, concentration, or type of digesting reagent).
Weak staining (hybridization/detection)
-
•
probe insufficiently biotinylated
-
•
probe concentration too dilute (hybridize longer)
-
•
probe or target DNA insufficiently denatured (increase time of denaturation, check the temperature of the hot start)
-
•
incomplete hybridization (prehybridize, increase hybridization time, lower temperature if too high, or check stringency)
-
•
buffer wrong pH (should be alkaline, pH 9.5)
-
•
reagents too cold (warm reagents to 23°C).
High background
-
•
skipped blocking step
-
•
probe too concentrated
-
•
slides dried out during incubations
-
•
washes omitted or shortened
-
•
detergent wash buffer not used after label incubation
-
•
incubated substrate too long.
‘Negative’ positive control
-
•
wrong probe
-
•
reagents bad (improper storage)
-
•
digestion absent (enzymes are unstable)
-
•
denaturation absent (increase temperature)
-
•
omitted step in protocol
-
•
mixed detection reagents improperly.
Inconsistent staining (stringency conditions)
Stringency conditions are conditions that if a related but non-homologous probe binds to the target:
-
•
non-homologous sequences hybridize/some mismatch (low stringency—high salt concentration, low temperature, low formamide concentration)
-
•
complete binding only if homologous (high stringency—low salt concentration, high temperature, high formamide concentration).
RT-PISH
In situ hybridization–polymerase chain reaction overview
PCR is used to detect minute quantities of DNA or RNA in tissue sections or intact cells and to visualize, by light microscopy, the individual cells expressing or carrying specific genes. It is also used to detect low copy numbers of nucleic acid sequences in viral infections, gene mutations, gene alterations, chromosomal translocations, gene therapy, and low-level gene expression.
Conventional in situ PCR is a means to amplify small amounts of DNA or RNA down to a single copy of a gene, to an exponential accumulation of the same sequences consisting of millions or billions of identical copies for detection, sequencing, cloning or diagnosis.
There are several ways in which one can perform PISH methods. There is the direct in situ PCR, indirect in situ PCR, and reverse transcription–PCR. In all methods, tissues should be fixed to preserve both the DNA and the RNA. Tissues are cut slightly thicker for the RT-PISH method described here. Thicker sections are thought to contain more target. Tissue pre-treated with proteinase K is necessary.
Primers are usually 18–28 nucleotides long and it is important to design a pair of primers that has little or no homology to any other sequences in the tissue, to each other or within themselves. For animal tissue, it is better to select the sequence that several (but similar) species can use as the primers for PISH.
Controls are important in each step of the procedure. This is to ensure there are no false positives or negatives. Validation of the amplification is the most important reason for the controls but confirmation of the subsequent hybridization/detection is also important.
Examples of controls would be:
-
•
omission of the primers, using unrelated primers
-
•
have good positive and negative tissues
-
•
destroy the target before subjecting it to PISH
-
•
use a known cell line
-
•
verify results with conventional PCR.
See PISH Procedure 26.2
Procedure 26.1.
Oligonucleotide 3′-end labeling with DIG-ddUTP
Terminal transferase is used to add a single modified dideoxyuridine triphosphote (DIG-ddUTP, biotin-ddUTP, or fluorescein-ddUTP) to the 3′ ends of an oligonucleotide. In this method, DIG-ddUTP will be used. The method described here has been modified from the Boehringer Mannheim procedure. Now the method is described from Roche Applied Science Laboratory. The reagents in this procedure may be purchased in a kit (Roche).
Contents of labeling reagents: The amounts to prepare listed here are the amounts supplied in the kit. The kit will accommodate 25 labeling reactions. If you require other amounts, just adjust your volume calculations for less or more.
1. Reaction buffer pH 6.6 (this is a 5× concentrated solution)
1 M potassium cacodylate
0.125 M Tris-HCl
1.25 mg/ml bovine serum albumin
Prepare 50–100 μl
2. CoCl2 solution
25 mM cobalt chloride
Prepare 50–100 μl
3. DIG-ddUTP solution
1 mM digoxygenin-11-ddUTP in double distilled water
Prepare 25 μl
4a. Terminal transferase 1 (newer method)
- 25 μl terminal transferase, in the following:
- 60 mM potassium phosphate (pH 7.2 at 4°C)
- 150 mM potassium chloride
- 1 mM 2-mercaptoethanol
- 0.5% Triton X-100
- 50% glycerol
- Prepare a solution with a concentration of 400 units/μl
4b. Terminal transferase 2 (older method)
- 1 μl (50 units) terminal transferase, in the following:
- 200 mM potassium cacodylate
- 200 mM potassium chloride
- 1 mM EDTA
- 0.2 mg/ml bovine serum albumin
- 50% glycerol
- Add enough double-distilled water to make a final volume of 20 μl.
5. 0.2 M EDTA (pH 8.0) made in double distilled water.
Procedure
-
1.
Dissolve 100 pmol of the purified oligonucleotide in 10 μl of sterile double distilled water.
-
2.Add the following to a sterile microcentrifuge tube on ice:
- 4 μl of 5× concentrated reaction buffer
- 4 μl of 25 mM cobalt chloride (CoCl2)
- 1 μl DIG-ddUTP solution. For this labeling we will use DIG-ddUTP.
- 1 μl of terminal transferase (400 units/μl)
-
3.
Mix and centrifuge briefly.
-
4.
Incubate at 37°C for 15 min, then place on ice.
-
5.
Stop the reaction by adding 2 μl of 0.2 M EDTA (pH 8.0)
Procedure 26.2.
Direct PCR amplification protocol
-
1.
Fix the tissue with a cross-linking fixative.
-
2.
Cut sections at 5–7 μm on an alcohol-cleaned microtome and knife.
-
3.
Place 2–3 sections on positively charged coated slides (spaced evenly apart). Dry overnight flat on a slide warmer, 50–55°C, and then an additional 30 min before deparaffinization.
-
4.
Deparaffinize in three changes of xylene for 3–5 min each. Dehydrate in two changes 100% ethanol for 5 min. Air dry.
-
5.
Digest for 30–90 min. Do not let slides dry out. A humidity chamber is helpful for this step.*
-
6.
After digestion, rinse slides in double distilled water for 1 min.
-
7.
Dehydrate slides in two changes of 100% ethanol for 2 min each. Air dry for 5 min.
Note
Depending on the type of thermocycler used, the slides are treated differently. For example, some thermocyclers have coverwells that seal and snap tight over the individual tissue section. Other thermocyclers just have a heat block to sit the slides on. To accommodate the individual thermocycler, you may need to circle the tissue with a hydrophobic pen.
-
8.
Cover tissue with PCR mixture and cover with glass coverslip, autoclave wrap or coverwell (use what is recommended by manufacturer of thermocycler).
-
9.
Line the slide girdle of the thermocycler with aluminum foil to prevent leakage from the slides and equipment contamination. For this procedure a Perkin-Elmer thermocycler was used, which was the recommendation of the manufacturer.
-
10.
Place the slides in the aluminum foil trough. Cover each slide with 2 ml of mineral oil.
-
11.
Heat thermal block to 80°C and place on hold for 10 min while setting the thermocycler program. The amplification program used is 15 cycles of 1 min at 96°C, 1 min at 59°C, and 1 min at 72°C.
Note
Amplification program settings will vary.
-
12.
Remove slides from thermocycler and gently remove coverslips, etc. Rinse slides in two changes of xylene for 3 min each to remove mineral oil. Dehydrate and remove xylene with two changes of 100% ethanol for 3 min each. Air dry.
-
13.
Wash slides three times for 5 min each in buffer #1.
-
14.
Place slides in 150 mM NaCl with 0.2% BSA at 50°C for 10 min (this solution should be prewarmed).
-
15.
Drain NaCl from slides and cover with 100 μl alkaline phosphatase-conjugated anti-digoxygenin (1:50 dilution in 0.1 M Tris pH 7.5 with 0.1 M NaCl) for 30 min at 37°C in a humidity chamber.
-
16.
Rinse slides in 0.1 M Tris pH 9.5 with 0.1 M NaCl for 1 min.
-
17.
Incubate slides in NBT/BCIP solution (10 μl of NBT/BCIP in 2.0 ml Tris pH 9.5 with 0.1 M NaCl) for 5–15 min. Check the proper end-point under the microscope.
-
18.
Wash slides in distilled water for 2 min. Counterstain with nuclear fast red for 5 min. Rinse in distilled water for 1 min, dehydrate, clear, and mount.
Reagent list for the RT-PISH procedure 26.2.
Digestion reagents
1. Pepsin or trypsin at 2 mg/ml (mild digestion)
Note
Stock solution is 20 mg pepsin + 9.5 ml sterile water + 0.5 ml 2 N HCl.
2. Protease K (harsh)
Dilute 1.0 ml proteinase K at 1 mg/ml in 150 ml of PBS.
Use at 55°C.
3. Buffer #1
4. 20× SSC
5. 2× SSC
6. Formamide SSC
| Formamide | 50 ml |
| 2× SSC | 50 ml |
7. PCR mixture
00.25 μM primers
10 μM dATP
10 μM dCTP
10 μM GTP
3.5 μM dTTP
TROUBLESHOOTING DIGESTION
If there is no or a weak signal, you need to increase digestion by 10 minutes.
Suggested digestion times are indicated in Table 26.2 .
Table 26.2.
Suggested digestion times adjusted to fixation time
| Fixation time (hours) | Enzyme | Digestion time (minutes) |
|---|---|---|
| 4 | Pepsin or trypsin | 10 |
| 15 | Pepsin or trypsin | 90 |
| 24 | Pepsin or trypsin | 120 |
Acknowledgments
The author would like to acknowledge Tony Warford and Lamar Jones for writing the first two versions of this chapter. Some of their writing remains in this chapter. Special thanks go to Maureen Doran for her technical sharing, to Dr Bruce Gitter for his scientific review, and to Eli Lilly and Company for the support and time to write this chapter.
Footnotes
Quick test for sufficient digestion is: Place slide under a microscope; using a 40× objective look at one cell. When you can count 20 dots in nucleus, digestion is complete.
REFERENCES
- Davis M., Malcolm S., Rabbitts T.H. Chromosome translocation can occur on either side of the c-myc oncogene in Burkitt lymphoma cells. Nature. 1984;308:286–288. doi: 10.1038/308286a0. [DOI] [PubMed] [Google Scholar]
- De Jong A.S.H., Van Kessel-Van Vark M., Raap A.K. Sensitivity of various visualization methods for peroxidase and alkaline phosphatase activity in immu-noenzyme histochemistry. Histochemical Journal. 1985;17:1119–1130. doi: 10.1007/BF01002537. [DOI] [PubMed] [Google Scholar]
- Doran M., Sterchi D.L. Let's do in situ (workshop no. 67) National Society for Histotechnology; Milwaukee, WI: 2000. [Google Scholar]
- Gall J.G., Pardue M.L. Formation and detection of RNA–DNA hybrid molecules in cytological preparations. Proceedings of the National Academy of Sciences USA. 1969;63:378–383. doi: 10.1073/pnas.63.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith C.S., Tatti K.M., Ksiazek T.G. Ultrastructural characterization of SARS coronavirus. Emerging Infectious Diseases. 2004;10(2):320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland R.P., Spence M.T.Z., editors. The handbook: a guide to fluorescent probes and labeling technologies. 10th edn. Invitrogen; Paisley: 2005. [Google Scholar]
- Herrington C.S., Burns J., Graham A.K. Interphase cytogenetics using biotin and digoxigenin labeled probes II: Simultaneous differential detection of human and papilloma virus nucleic acids in individual nuclei. Journal of Clinical Pathology. 1989;41:601–606. doi: 10.1136/jcp.42.6.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis M.A., Gelfand D.H., Sminsky J.J., editors. PCR protocols: a guide to methods and applications. Academic Press; New York: 1990. [Google Scholar]
- Janneke C., Alers P-J.K., Kees J. Effect of bone decalcification procedures on DNA in situ hybridization and comparative genomic hybridization: EDTA is highly preferable to a routinely used acid decalcifier. Journal of Histochemistry and Cytochemistry. 1999;47(5):703–709. doi: 10.1177/002215549904700512. [DOI] [PubMed] [Google Scholar]
- John H.A., Birnstiel M.L., Jones K.W. RNA–DNA hybrids at the cytological level. Nature. 1969;223:582–587. doi: 10.1038/223582a0. [DOI] [PubMed] [Google Scholar]
- Hopman A.H., Ramaekers F.C., Raap A.K. In situ hybridization as a tool to study numerical chromosome aberrations in solid bladder tumors. Histochemistry. 1988;89:307–316. doi: 10.1007/BF00500631. [DOI] [PubMed] [Google Scholar]
- Kendall C.H., Roberts P.A., Pringle J.H. The expression of parathyroid hormone messenger RNA in normal and abnormal parathyroid tissue. Journal of Pathology. 1991;165:111–118. doi: 10.1002/path.1711650205. [DOI] [PubMed] [Google Scholar]
- Knoll J.H.M., Lichter P. Current protocols in molecular biology. In situ hybridization and detection using nonisotopic probes. Wiley; New York: 1995. [DOI] [PubMed] [Google Scholar]
- Lux S.E., Tse W.T., Menninger J.C. Hereditary spherocytosis associated with deletion of human erythrocyte ankyrin gene on chromosome 8. Nature. 1990;345:736–739. doi: 10.1038/345736a0. [DOI] [PubMed] [Google Scholar]
- Madrid M.A., Lo R.W. Chromogenic in situ hybridization (CISH): a novel alternative in screening archival breat cancer tissue samples for HER-2/neu. Breast Cancer Research. 2004;6(5):R593–R600. doi: 10.1186/bcr915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell B.S., Dhami D., Schumacher U. In situ hybridization: a review of methodologies and applications in the biomedical sciences. Medical Laboratory Sciences. 1992;49:107–118. [PubMed] [Google Scholar]
- Nederlof P.M., Robinson D., Abuknesha R. Three-color fluorescence in situ hybridization for the simultaneous detection of multiple nucleic acid sequences. Cytometry. 1989;10:20–27. doi: 10.1002/cyto.990100105. [DOI] [PubMed] [Google Scholar]
- Netterwald J. Molecular testing? Emerging technologies show promise for helping to prevent spread of the epidemic disease. Advance for Medical Laboratory Professionals. 2006;April:17–18. [Google Scholar]
- Poddighe P.J., Ramaekers F.C.S., Hopman A.H.N. Interphase cytogenetics of tumors. Journal of Pathology. 1992;166:215–224. doi: 10.1002/path.1711660303. [DOI] [PubMed] [Google Scholar]
- Pringle J.H., Ruprai A.K., Primrose L. In situ hybridization of immunoglobulin light chain mRNA in paraffin sections using biotinylated or hapten-labeled oligonucleotide probes. Journal of Pathology. 1990;162:197–207. doi: 10.1002/path.1711620305. [DOI] [PubMed] [Google Scholar]
- Pringle J.H., Barker S. Demonstration of Epstein-Barr virus in tissue sections by in situ hybridization for viral RNA. Journal of Pathology. 1992;167(Suppl):133A. [Google Scholar]
- Pringle J.H., Baker J., Colloby P.S. The detection of cell proliferation in normal and malignant formalin-fixed paraffin-embedded tissue sections using in situ hybridization for histone mRNA. Journal of Pathology. 1993;169(Suppl):144A. [Google Scholar]
- Ruprai A.K., Pringle J.H., Angel C.A. Localization of immunoglobulin light chain mRNA expression in Hodgkin's disease by in situ hybridization. Journal of Pathology. 1991;164:37–40. doi: 10.1002/path.1711640107. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular cloning—a laboratory manual. 2nd edn. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- Van den Berg F., Schipper M., Jiwa M. Implausibility of an aetiological association between cytomegalovirus and Kaposi's sarcoma shown by four techniques. Journal of Clinical Pathology. 1989;42:128–131. doi: 10.1136/jcp.42.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu N., King D.J., Seal B.S. Detection of Newcastle disease virus RNA by reverse transcription polymerase chain reaction using formalin-fixed, paraffin-embedded tissue and comparison with immunohistochemistry and in situ hybridization. [abstract] American Association of Veterinary Laboratory Diagnosticians. 2005;48:166. doi: 10.1177/104063870701900410. [DOI] [PubMed] [Google Scholar]
- Warford A., Lauder I. In situ hybridization in perspective. Journal of Clinical Pathology. 1991;44:177–181. doi: 10.1136/jcp.44.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. An introduction to chromogenic in situ hybridization. Journal of Histotechnology. 2005;28:229–234. [Google Scholar]
- Wood G.S., Warnke R. Suppression of endogenous avidin binding activity in tissues and its relevance to biotin–avidin detection systems. Journal of Histochemistry and Cytochemistry. 1981;29:1196–1204. doi: 10.1177/29.10.7028859. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Sato S., Oguchi T. High sensitivity of PCR in situ hybridization for the detection of human papillomavirus infection in uterine cervical neoplasias. Gynecologic Oncology. 2001;82(2):350–354. doi: 10.1006/gyno.2001.6261. [DOI] [PubMed] [Google Scholar]
- Yan S.J., Blomme E.A.G. In situ zymography: a molecular pathology technique to localize endogenous protease activity in tissue sections. Veterinary Pathology. 2003;40:227–236. doi: 10.1354/vp.40-3-227. [DOI] [PubMed] [Google Scholar]
FURTHER READING
- Darby I.A., editor. In situ hybridization protocols. 2nd edn. Humana Press; Totowa, NJ: 2000. [Google Scholar]
- Wilkinson D.G., editor. In situ hybridization: a practical approach. 2nd edn. Oxford University Press; New York: 1999. [Google Scholar]