

Abstract

Organic–inorganic tin(II) halide perovskites have emerged as promising alternatives to lead halide perovskites in optoelectronic applications. While they suffer from considerably poorer performance and stability in comparison to their lead analogues, their performance improvements have so far largely been driven by trial and error efforts due to a critical lack of methods to probe their atomic-level microstructure. Here, we identify the challenges and devise a 119Sn solid-state NMR protocol for the determination of the local structure of mixed-cation and mixed-halide tin(II) halide perovskites as well as their degradation products and related phases. We establish that the longitudinal relaxation of 119Sn can span 6 orders of magnitude in this class of compounds, which makes judicious choice of experimental NMR parameters essential for the reliable detection of various phases. We show that Cl/Br and I/Br mixed-halide perovskites form solid alloys in any ratio, while only limited mixing is possible for I/Cl compositions. We elucidate the degradation pathways of Cs-, MA-, and FA-based tin(II) halides and show that degradation leads to highly disordered, qualitatively similar products, regardless of the A-site cation and halide. We detect the presence of metallic tin among the degradation products, which we suggest could contribute to the previously reported high conductivities in tin(II) halide perovskites. 119Sn NMR chemical shifts are a sensitive probe of the halide coordination environment as well as of the A-site cation composition. Finally, we use variable-temperature multifield relaxation measurements to quantify ion dynamics in MASnBr3 and establish activation energies for motion and show that this motion leads to spontaneous halide homogenization at room temperature whenever two different pure-halide perovskites are put in physical contact.
Introduction
Organic–inorganic halide perovskites (OIHPs) have emerged as a new class of materials for solar cells and light emission applications owing to the ease of solution processing, immunity to most defects, and long charge carrier lifetimes, which can be tuned by compositional engineering.1,2 Following the first report of perovskite-based solar cells (PSC) a decade ago,3 the field of perovskite-based photovoltaics has been developing at a very fast pace, now reaching power conversion efficiencies of over 25%.1,4,5
OIHPs are represented by the generic ABX3 formula, in which A is typically a small cation such as methylammonium (CH3NH3+, MA), formamidinium (CH3(NH2)2+, FA), and/or cesium ions. The inorganic sublattice is composed of [BX6]4– octahedra, where B is a divalent metal such as Pb2+, Sn2+, and Ge2+ or a mixture of monovalent and trivalent metals (e.g., Ag+ and In3+) and X is a halide: I–, Br–, or Cl–. Lead halide perovskites exhibit considerably higher ambient stability and optoelectronic performance6,7 in comparison to the tin- and germanium-based analogues;8,9 hence, many of the solar cells with record efficiency are based on Pb2+.4,10,11 Tin-based materials (Figure 1), while providing lower band gaps than their lead analogues, essential for tandem solar cells, suffer from easy oxidation and disproportionation which lead to self-doping, very short charge carrier lifetimes, and in turn poor power conversion efficiencies. These undesirable processes have been mitigated by introducing antioxidant additives such as SnF2,12 hydrazine,13 hydrazinium,14,15 β-tin,16 the potassium salt of hydroquinonesulfonic acid (KHQSA),17 and ascorbic acid18 and by A-/X-site compositional engineering,19−23 leading to efficiencies approaching 10%. Iodide–chloride mixing has been a widely investigated problem in the field of lead halide perovskite photovoltaics, since chloride doping leads to significantly improved thin film crystallinity and carrier diffusion lengths,24−27 and considerable improvements have also been reported for chloride doping in tin(II) halide perovskite based solar cells.28,29 However, to the best of our knowledge, there is no direct evidence for I/Cl mixing in the case of tin(II) halide perovskites.
Figure 1.
Schematic representation of the crystal structure of tin(II) halide perovskites formed by corner-sharing [SnX6]4– octahedra.
Another strategy to stabilize tin(II)-based materials is the use of mixed-metal tin(II)-lead(II) halide perovskites, which combine the advantageous optoelectronic properties of lead-based materials while providing band gaps of 1.2–1.3 eV which are close to the optimum required for all-perovskite tandem solar cells.30−34
The resulting materials are typically probed using diffraction-based methods, which provide information about long-range order, and optical spectroscopy to characterize their electronic properties. However, the atomic-level effect of various additives have not yet been evaluated, since there are currently no robust protocols for probing the local structure of multicomponent tin(II) halide perovskites. Rapid degradation of tin(II) halide perovskites has been consistently observed in device studies,35−37 and degradation mechanisms have been investigated using XRD, TGA, and UV–vis spectroscopy. However, once again, the atomic-level mechanism of degradation and the exact identity of the resulting species remain elusive.
Solid-state NMR has recently been shown to be the method of choice to determine local structure and dynamics in lead halide perovskites, which are uniquely amenable owing to the atomic-level and element-specific resolution of NMR.38 In particular, solid-state NMR can be used to evidence A-/B-site cation incorporation,39−45 halide mixing,46−49 and doping-induced phase segregation processe,40,41,43,46 and to study interfacial passivation mechanisms,50−52 cation and anion dynamics,39,53−59 and degradation processes.60 The local structure of tin halide perovskites has been previously investigated in CsSnBr3,61 MASnI3,62 and FASnI362 using pair distribution function (PDF) analysis. Given the prevalence of tin NMR studies of other groups of materials, it is surprising that it has not yet been applied to tin(II) halide perovskites. We show that this problem is not trivial. To the best of our knowledge, the only example of applying solid-state MAS NMR to tin halide perovskites to date is a 1H MAS NMR study of cation mixing in FA1–xMAxSnBr3.63 Solid-state 207Pb NMR has recently provided an abundance of atomic-level information on lead halide perovskites,46−48,60 and hence it is expected that tin NMR should be well suited to study tin analogues as well as mixed tin-lead materials.
Tin has three NMR-active isotopes, 115Sn, 117Sn, and 119Sn, with natural abundances of 0.3%, 7.7%, and 8.6%, respectively. All three isotopes have spin I = 1/2 and similar gyromagnetic ratios, which render 119Sn the most receptive of the three, with a receptivity ca. 27 times that of 13C. Solid-state tin NMR has been widely employed to study organotin compounds,64 crystalline oxides and stannates,65,66 porous networks,67−69 sulfides,70,71 nitrides,72 and all-inorganic semiconductors.73−75 Tin NMR is particularly sensitive to the difference between the +276,77 and +478 oxidation states with the corresponding chemical shift differences on the order of several hundreds of ppm, as well as to the type of atom covalently bound to the tin site. 119Sn chemical shifts span the range between 1000 and −2000 ppm for diamagnetic compounds and 7000–8000 ppm for tin metal.79 Much larger ranges of shifts are seen for paramagnetic compounds.66
Here, we probe the atomic-level microstructure of single- and mixed-halide (I, Br, Cl) tin(II) halide perovskites, single and mixed A-site cation (Cs, MA, FA) tin(II) halide perovskites, and tin(IV) non-perovskite phases using 119Sn MAS NMR spectroscopy. We show that iodide–bromide and bromide–chloride mixtures form solid solutions for any I/Br and Br/Cl ratio. On the other hand, iodide–chloride compositions, while partially miscible, yield phase-segregated mixtures of phases. We show how 119Sn MAS NMR can be applied to study degradation pathways of tin(II) halide perovskites and that degradation typically leads to highly disordered SnO2 and halostannates(IV). We have also detected traces of metallic tin in the degraded material. Three of the degradation products, FA2SnI6, MA2SnI6, and Cs2SnI6 have 119Sn chemical shifts of −4818, −4684, and −4518 ppm, respectively, values unprecedented in their magnitude for diamagnetic tin compounds. Further, we show that 119Sn longitudinal relaxation times (T1) in this class of compounds can span 6 orders of magnitude, which makes the use of optimized experimental parameters essential for the reliable detection of various phases. Finally, we use variable-temperature multi-field 119Sn MAS NMR to quantify halide dynamics in MASnBr3 and show that it leads to spontaneous halide mixing at room temperature.
Experimental Section
Materials
The following materials were used: methylammonium iodide (Sigma, 98%), formamidinium iodide (Sigma, 98%), formamidinium bromide (Sigma, 98%), formamidinium chloride (Sigma, 97%), CsI (Fischer, 99.9%), CsBr (Fischer, 99.9%), CsCl (Acros, 99.99%), SnI2 (Sigma, 99.999%), SnBr2 (Sigma), SnCl2 (Sigma, 98%), SnI4 (Sigma, 99.999%), and SnBr4 (Sigma, 99%).
Perovskite Mechanosynthesis
The materials were prepared using mechanosynthesis80,81 following recently published protocols.82−84 The precursors were stored under argon. The halostannates were synthesized by grinding the reactants in an electric ball mill (Retsch MM-400) using an agate grinding jar (10 mL) and ball (⌀ 10 mm) for 30 min at 25 Hz. XRD patterns, SEM images, and optical data of mechanochemical tin(II) halide perovskites have been previously reported82,83 and agree with those recorded on materials prepared as single crystals and thin films. The quantities of reagents used in the synthesis are given in the Supporting Information.
NMR Measurements
Solid-state MAS NMR spectra of 119Sn (74.7 MHz) were recorded on a Bruker Avance III 4.7 T spectrometer equipped with a 4 mm MAS probe using 167 kHz rf strength. About 200–250 mg of material was used for each measurement, corresponding to a full 4 mm rotor. The recycle delays were set on the basis of the measured T1 values, as described in the text. Low-temperature 1H–13C (125.8 MHz) CP MAS and room-temperature 14N (36.2 MHz) experiments were recorded on a Bruker Avance III 11.7 T spectrometer equipped with a 3.2 mm low-temperature CPMAS probe using previously optimized parameters.39 High-temperature 119Sn MAS NMR spectra were recorded on a Bruker Avance III 4.7 T spectrometer (74.7 MHz) using a 4 mm MAS Bruker probe (MgO stator) in the range between 308 and 455 K using 4 mm zirconia rotors spinning at 5 kHz with heated nitrogen. High-field data in the 308–474 K temperature range were obtained on a Bruker Avance III HD 17.6 T spectrometer (279.7 MHz) using a MAS LASER probe (Bruker) with airtight boron nitride crucibles contained in 7 mm zirconia rotors spinning at 6 kHz. The temperature was adjusted using diode laser heating.85 The sample was sandwiched between two layers of ground KBr, which allowed monitoring of the effective sample temperature through the 79Br shift of KBr.86,87 In order to prevent any interactions between KBr and the perovskite sample, a thin layer of PTFE tape was placed between the two powders. Hahn echoes of 40 μs total duration were used to mitigate ringing effects. CSA parameters were fitted using TopSpin 3.5. Further experimental details are given in the Supporting Information.
Results and Discussion
Local Structure of Mixed-Anion Tin Halostannates
Figure 2 shows 119Sn solid-state MAS NMR spectra of methylammonium mixed-halide chloro- and bromostannates(II), bromostannate(IV) as well as their tin(II) and tin(IV) halide precursors recorded at room temperature. The 119Sn chemical shift is highly sensitive to the local environment of the tin site and makes it possible to distinguish tin(II) precursors—SnCl2 (−916 ppm, Figure 2a) and SnBr2 (−640 ppm, Figure 2b)—from the corresponding perovskites—MASnCl3 (−398 ppm, Figure 2c) and MASnBr3 (−316 ppm, Figure 2g). MASnCl3 exhibits successive phase transitions at 283, 307, 331, and 463 K.88 The structure adopted by MASnCl3 under our experimental conditions (298 K) is monoclinic with slightly distorted [SnCl6]4– octahedra, which leads to the presence of chemical shift anisotropy (CSA) manifesting itself as a set of spinning sidebands (SSB) spaced by the MAS rotation frequency (Figure 2c). The fitted CSA parameters (δCSA −435 ppm, η = 0.26) are consistent with those previously reported.88 Replacing Cl– with Br– in MASnCl3 leads to solid solutions for the full range of Cl/Br ratios studied here. Low Br– concentrations, as in MASnCl2.7Br0.3, lead to a slight broadening and the appearance of two types of Sn(II) sites with similar CSA parameters (Figure 2d). The two sites correspond to different local [SnBr6–xClx]4– environments within the same phase, where the bromide content is higher for the environment at −351 ppm than it is for the environment at −393 ppm. As the concentration of Br– in the lattice is increased, the resonance broadens further and takes on a chemical shift intermediate with respect to MASnCl3 and MASnBr3 and its apparent CSA becomes smaller (δCSA −364 ppm, η = 0.1 for MASnCl2.1Br0.9) (Figure 2e). MASnBr3 is pseudocubic at room temperature; therefore, its δCSA value is ∼0 ppm and there are no SSBs associated with the main peak (Figure 2g).36 The peak is significantly broader than those of MASnCl3 and MASnCl1.5Br1.5. We attribute these line width variations to the interference between CSA and fast halide hopping, as discussed further in the text below (see also Supplementary Note 1). Note that this spectrum was acquired with no rotor synchronization in the quasi-static (νr = 600 Hz) regime (16.7 μs echo delay) due to very fast T2 relaxation. Using a rotor-synchronized echo delay (83.3 μs) leads to lower SNR but does not lead to the appearance of SSBs (Figure S1). Further, 119Sn NMR makes it possible to distinguish between bromostannates(II) and -(IV). While SnBr4 (−659 ppm) is shifted only slightly with respect to SnBr2 (−640 ppm), the difference between MASnBr3 (−316 ppm) and MA2SnBr6 (−1990 ppm) is much more pronounced.
Figure 2.

119Sn solid-state MAS NMR spectra of mixed-anion (chloride/bromide) halostannates and their precursors at 4.7 T, 12 kHz MAS (except for SnBr4), and 298 K: (a) SnCl2; (b) SnBr2; (c) MASnCl3; (d) MASnCl2.7Br0.3; (e) MASnCl2.1Br0.9; (f) MASnCl1.5Br1.5; (g) MASnBr3; (h) SnBr4 (at 0.6 kHz MAS to prevent melting); (i) MA2SnBr6. † indicates trace unreacted SnCl2.
In turn, we investigated iodide-containing halostannate(II) and -(IV) species. SnI2 (−527 ppm Figure 3a) exhibits a partially resolved 119Sn–127I scalar coupling, 1JSn–I = 6.2 kHz, similar in magnitude to the 207Pb–127I scalar coupling in PbI2.89 The crystal structure of MASnI3 is pseudocubic at room temperature; hence, a symmetric peak with δCSA ∼0 ppm is expected. However, the material yields a very broad, slightly asymmetric resonance with T2* ≈ 10 μs (estimated from the line width), which we attribute to very efficient scalar relaxation. A similarly short T2* value has been previously observed in lead iodide perovskites.46,49,90
Figure 3.

119Sn solid-state MAS NMR spectra of mixed-anion (iodide/chloride and iodide/bromide) halostannates and their precursors at 4.7 T, 12 kHz MAS and 298 K: (a) SnI2; (b) MASnI3; (c) MASnCl2.7I0.3 (the signals at 249 and −395 ppm were detected with recycle delays of 50 ms and 50 s, respectively); (d) MASnBr0.9I2.1; (e) MASnBr1.5I1.5; (f) MASnBr2.1I0.9; (g) MASnBr2.55I0.45; (h) MASnBr2.7I0.3; (i) MASnBr3; (j) SnI4; (k) MA2SnI6. † indicates trace unreacted SnCl2.
Figure 3c shows two 119Sn spectra of MASnCl2.7I0.3, one obtained with a 50 ms recycle delay and the other with 50 s, to highlight the iodide- and chloride-rich environments, respectively. The signal corresponding to the iodide-rich phase is shifted to lower frequencies (to lower ppm values) with respect to pure MASnI3 (Figure 3b), which supports the formation of [SnI6–xClx]4– coordination environments, thereby confirming that Cl– can incorporate into the MASnI3 perovskite lattice. On the other hand, the signal corresponding to the chloride-rich phase is identical, within experimental error, with that of pure MASnCl3 (Figure 2c), which indicates that I– has not been incorporated into the perovskite lattice of MASnCl3. This result can be rationalized considering the difference in atomic radii of I– (2.2 Å) and Cl– (1.8 Å), which cause the MASnCl3 structure to be more compact in comparison to that of MASnI3.36,88
On the other hand, iodide–bromide mixing has been previously studied in polycrystalline powders using X-ray diffraction and is expected due to the smaller difference in ionic radii of I– (2.2 Å) and Br– (2.0 Å).36 As I– is replaced by Br– in the crystal structure of MASnI3 (Figure 3b), the spectrum initially broadens and shifts to higher frequencies (to higher ppm values) (MASnBr0.9I2.1, Figure 3d) and then narrows and shifts to lower frequencies as the Br/I ratio increases further (above Br/I = 1.5/1.5, Figure 3e,f). Similar spectral trends have been previously reported in 119Sn MAS NMR spectra of other disordered solids, such as stannate pyrochlores91 and in 207Pb MAS NMR spectra of mixed-cation lead halide perovskites.47,48 Here, however, we ascribe the strong line width variation to the different magnitudes of 127I and 79/81Br-induced relaxation, as described in the next section.
The difference in chemical shift between Sn(II) and Sn(IV) iodides and iodostannates in even more pronounced than for bromides and bromostannates. SnI2 (−527 ppm, Figure 3a) can be easily distinguished from SnI4 (−1746 ppm, Figure 3j), and the same is true for MASnI3 (795 ppm, Figure 3b) and MA2SnI6 (−4684 ppm, Figure 3k). The latter 119Sn chemical shift is, to the best of our knowledge, the most shielded tin environment reported to date for a diamagnetic tin compound. While on the basis of the high electronegativity of iodine one might expect strong deshielding (shift at high positive ppm values), the exact opposite is observed experimentally. This is due to the effect of spin–orbit coupling, which is important for heavy atoms, as has been previously shown by fully relativistic DFT calculations.92 The 119Sn–127I scalar coupling constant in molten SnI4 has been previously found to be 1JSn–I = 0.9 kHz and is not resolved in the solid state.93
Local Structure of FA, Cs, and Mixed A-Site Cation Tin Halostannates
The A-site cation composition, mixing, and segregation in solid lead halide perovskite has been previously explored directly using solid-state 1H, 13C and 133Cs NMR as well as indirectly using 207Pb NMR.39−41,46,48,94 Here we show that the A-site composition in tin(II) halide perovskites can be probed indirectly using 119Sn MAS NMR. Figure 4 shows 119Sn MAS NMR spectra of single- and mixed-cation cesium, methylammonium, and formamidinium tin(II) halides (I, Br, Cl). All iodides yield very broad (full width at half-maximum (fwhm) of 70–170 kHz) and largely featureless spectra due to very efficient scalar T2 relaxation (Figure 4a–c). At room temperature, MASnI3 and FASnI3 are pseudocubic,95 while CsSnI3 is orthorhombic.96 Whereas symmetrical resonances are expected for highly symmetric structures, in this case the line shapes are asymmetric, which suggests that they are not determined entirely by T2 relaxation but rather that there is another contribution to the line shape. We believe that it is caused by the well-documented effect that a fast-relaxing quadrupolar nucleus has on the line shape of a spin 1/2 nucleus which is coupled to it.97 We were able to numerically simulate the line shapes and obtained a good qualitative agreement with the experiment (Figure S7). Overall, high-sensitivity 119Sn spectra can be recorded for 3D tin iodide perovskite within minutes under the experimental conditions used here and, while they are sensitive to the halide coordination environment, their value for investigating A-site cation mixing is limited due to the lack of spectral resolution. Non-perovskite tin iodide phases (SnI2, SnI4, and MA2SnI6) do not suffer from this complication, likely owing to their different crystal structures in which the efficiency of this relaxation mechanism is reduced.
Figure 4.
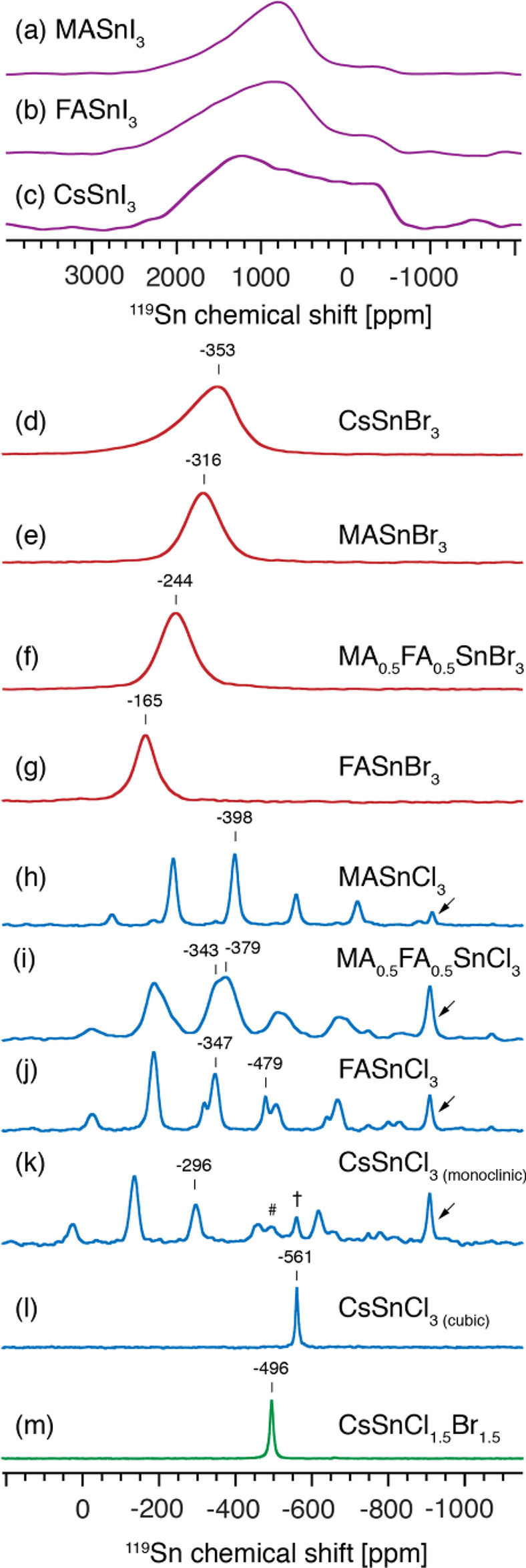
119Sn as a probe of the A-site cation (Cs, MA, FA) and A-site cation mixing in tin(II) halide perovskites. 119Sn solid-state MAS NMR spectra at 4.7 T, 12 kHz MAS (unless stated otherwise), and 298 K of iodides: (a) MASnI3; (b) FASnI3 (static, see Figure S6 for 12 kHz MAS); (c) CsSnI3; bromides (d) CsSnBr3, (e) MASnBr3, (f) MA0.5FA0.5SnBr3, and (g) FASnBr3; chlorides (h) MASnCl3, (i) MA0.5FA0.5SnCl3, (j) FASnCl3, and (k) CsSnCl3 (monoclinic) († indicates the metastable cubic phase of CsPbI3, and # is likely a second tin(II) site in the asymmetric unit cell of CsPbCl3); (l) CsSnCl3 (cubic); mixed halide (m) CsSnCl1.5Br1.5. The arrows indicate trace unreacted SnCl2.
On the other hand, tin(II) bromide perovskites yield well-resolved spectra whereby the chemical shift is a sensitive fingerprint of the A-site cation (Figure 4d–g): CsSnBr3 (−353 ppm), MASnBr3 (−316 ppm), FASnBr3 (−165 ppm). This makes it possible to probe A-site cation mixing using 119Sn NMR in the bromide systems. For example, MA0.5FA0.5SnBr3 (−244 ppm) yields a 119Sn chemical shift which is intermediate with respect to the single A-site cation species. This leads to a linear correlation between the MA/FA ratio and the 119Sn chemical shift in MAxFA1–xSnBr3: δSn (ppm) = −151x – 166. All four materials exist in the highest symmetry cubic α phase at room temperature.36,63,98 Also in this case, the line broadening was numerically simulated and is attributed to fast quadrupolar relaxation of 79/81Br bound to 119Sn (Figure S7).
Tin(II) chloride perovskites typically exist as low-symmetry phases at room temperature (monoclinic and triclinic for CsSnCl3 and MASnCl3, respectively98,99); hence, they yield characteristic CSA patterns (Figure 4h–l). Also in this case the 119Sn chemical shift is strongly dependent on the type of the A-site cation and an additional constraint is provided by the observed (298 K) CSA parameters: MASnCl3 (δiso −398 ppm, δCSA −435 ppm, η = 0.26), FASnCl3 (δiso −347 ppm, δCSA −508 ppm, η = 0.06), CsSnCl3 (δiso −296 ppm, δCSA −568 ppm, η = 0.14). The spectrum of FASnCl3 contains a second peak at δiso −479 ppm (δCSA −401 ppm, η = 0.08), which likely corresponds to a second tin(II) site inside the asymmetric unit cell, analogous to the situation observed in the low-symmetry phase of MASnCl3.88 A-site cation mixing leads to disorder, which is exemplified by the spectrum of MA0.5FA0.5SnCl3: the resonances broaden considerably and the two broad components (FA, δiso −343, δCSA −510 ppm, η = 0.01; MA, δiso −379 ppm, δCSA −353 ppm, η = 0.12) take on values intermediate with respect to the single-cation phases. CsSnCl3 can be trapped in its high-symmetry cubic phase (δiso −561 ppm, δCSA ∼0 ppm) at room temperature if the sample is briefly heated to 380 K (Figure 4l). This phase is metastable and can be transformed back to the low-symmetry phase in the presence of humidity.98 Finally, we note that 119Sn can be used to study the halide coordination environment in tin(II) halides perovskites not only when the A site is an organic cation (Figures 2 and 3) but also when it is an inorganic cation such as cesium. Figure 4m shows that the 119Sn chemical shift of CsSnCl1.5Br1.5 (−496 ppm) is intermediate with respect to the cubic phases of CsSnCl3 (−561 ppm) and CsSnBr3 (−353 ppm). Taken together, these findings demonstrate that 119Sn MAS NMR is well-suited for probing the atomic-level microstructure of mixed-cation and mixed-anion tin(II) halide perovskites, as it is highly sensitive to both the A-site and X-site composition. The 119Sn data can be complemented by 13C, 14N, and 133Cs NMR measurements to evaluate the local structure and dynamics of the A site, as discussed further in the text.
Degradation Pathways
Having established a comprehensive database of 119Sn shifts for various relevant tin halide perovskite materials, we now explore degradation pathways in this class of compounds. Figure 5 shows a comparison between pristine and degraded MASnBr3, FASnBr3, CsSnBr3, MASnI3, FASnI3, and CsSnI3. The degradation was performed ex situ in air, and the degradation conditions (temperature and duration) were chosen phenomenologically depending on the stability of different compounds, as monitored by the disappearance of the pristine perovskite 119Sn signal.
Figure 5.
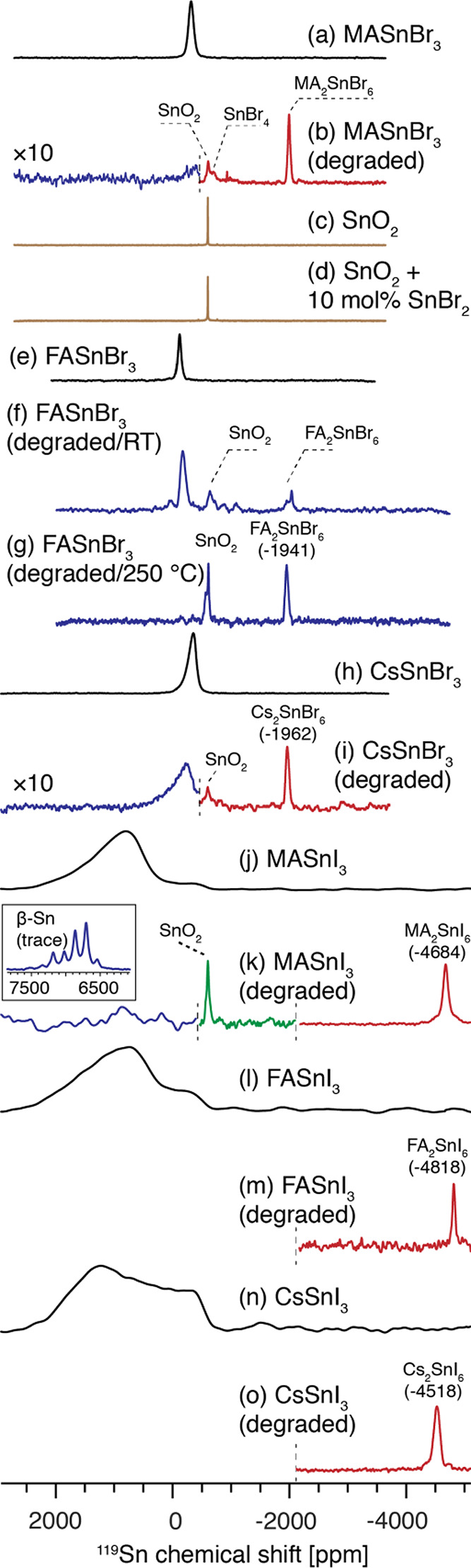
119Sn as a probe of tin(II) halide perovskite degradation. 119Sn solid-state MAS NMR spectra at 4.7 T, 12 kHz MAS.,and 298 K: (a) MASnBr3 (as prepared); (b) MASnBr3 (degraded for 1 h at 250 °C in air); (c) SnO2; (d) SnO2 + 10 mol % SnBr2 (ground and annealed at 250 °C in air); (e) FASnBr3 (as prepared); (f) FASnBr3 (degraded for 5 days at RT, in air); (g) FASnBr3 (degraded for 0.5 h at 250 °C in air); (h) CsSnBr3 (as prepared); (i) CsSnBr3 (degraded for 0.5 h at 350 °C in air); (j) MASnI3 (as prepared); (k) MASnI3 (degraded for 1 h at 150 °C in air); (l) FASnI3 (as prepared); (m) FASnI3 (degraded for 1 h at RT in air); (n) CsSnI3 (as prepared); (o) CsSnI3 (degraded for 3 h at 100 °C in air). The dashed lines indicate points at which spectra acquired at different transmitter offsets were stitched together: (b, i) two offsets; (k) three offsets. For (m) and (o) only the high-field (low ppm) part was acquired.
Thermal degradation (1 h at 250 °C in air) of MASnBr3 (Figure 5a) leads to a mixture of MA2SnBr6, SnO2, SnBr4, and trace amounts of species at −932 ppm, which we tentatively assign to an ionic product of the reaction between tin(II) and decomposition products of the organic cation. Interestingly, the SnO2 signal in the degraded perovskite is significantly broader (fwhm 3.5 kHz) in comparison to neat microcrystalline SnO2 (fwhm 0.2 kHz), which suggests that the SnO2 formed during decomposition is locally highly disordered. This could be caused by bromide doping100 or amorphization. We exclude bromide doping as the reason for the observed disorder, since a SnO2 mechanochemically doped with SnBr2 and annealed at the same temperature as the degradation process did not lead to broadening of the SnO2 resonance (Figure 5d). We therefore conclude that the SnO2 formed during the degradation of tin(II) halides perovskites is poorly crystalline or forms as nanodomains. We note that this would likely render its detection challenging by XRD.
We also observed that degradation under ambient conditions leads to products qualitatively similar to those of high-temperature degradation. However, the products formed at room temperature are considerably more locally disordered. Room-temperature degradation (5 days at RT, in air) of FASnBr3 (Figure 5e) leads to very broad peaks of SnO2 (fwhm ∼10 kHz) as well as FA2SnBr6 (two components, 3–6 kHz) (Figure 5f). After 5 days of exposure to ambient laboratory air, the sample of microcrystalline FASnBr3 still contains a large amount of the nondegraded perovskite (∼45% of the initial content). Degradation at 250 °C leads to complete disappearance of the perovskite phase and renders the peaks narrower (SnO2, two components, fwhm 2–3 kHz; FA2SnBr6, fwhm 4 kHz), presumably as a result of thermal annealing (Figure 5g). Similarly, in the case of CsSnBr3 degraded for 0.5 h at 350 °C in air, we observe the formation of SnO2 and Cs2SnBr6 (Figure 5h,i).
Analogous effects are observed during thermal degradation of MASnI3 (Figure 5j,k) as well as FASnI3 (Figure 5l,m) and CsSnI3 (Figure 5n,o), which yield FA2SnI6 (−4818 ppm) and Cs2SnI6 (−4518 ppm), respectively. We have also acquired powder XRD diffraction on the degraded materials, which show the presence of the oxidized A2SnX6 species (Figure S12). In addition, we note that we have detected metallic β-Sn in the sample of degraded MASnI3 (Figure 5k, inset), which, however, is only present as a trace impurity (see Table S4 for the necessary acquisition times). The anisotropic Knight shift of the β-Sn impurity is consistent with that of a reference β-Sn powder sample (δiso 6864 ppm or 0.68%, δaniso 486 ppm, η = 0.1) and with the values previously reported for metallic tin powder101,102 and thin films.79 Interestingly, β-Sn has been recently used as an additive to increase the stability of FASnI3.16 We suggest that the presence of metallic tin in tin(II) halide perovskites among the degradation products may contribute to the high conductivity values previously reported in the literature for tin(II) halide perovskites, an effect to date attributed uniquely to self-doping. The formation of SnO2 and SnX4 has been shown in a recent TGA study,103 which corresponds to the state in which the organic component has been fully volatilized. Solid-state 119Sn NMR carried out on materials degraded under similar conditions refines this picture by showing that the degradation proceeds through an intermediate which is the corresponding tin(IV) halostannate, A2SnX6. The conclusions of our study are therefore fully consistent with those of Leijtens et al.103 On the basis of these observations, we conclude that 119Sn MAS is well-suited for studying degradation mechanisms in tin(II) halide perovskites.
Optimal Experimental Conditions for 119Sn NMR Detection
One of the most important considerations associated with the acquisition of 119Sn MAS NMR data of tin(II) halide perovskites, their precursors, and degradation products is that the 119Sn longitudinal relaxation times (T1) can span 6 orders of magnitude (Figure 6). This makes it essential to carefully adjust the experimental parameters so as to ensure optimal sensitivity and/or quantitativeness. The physical reason behind such a large spread of T1 values is the difference in the dominating relaxation mechanism in different groups of tin compounds. T1 relaxation in tin halides has been shown to be largely due to the 119Sn–X scalar coupling, whereby the relaxation rate depends on the coupling strength.78,93 Since 1JSn–I > 1JSn–Br > 1JSn–Cl, it is expected that scalar relaxation is fastest in iodostannates, intermediate in bromostannates, and slowest in chlorostannates.78,93 This trend is clearly visible experimentally (Figure 6, blue). Beyond the coupling strength, the efficiency of scalar relaxation also depends on the rate at which the coupling is modulated (e.g., by fast relaxation of the halogen or chemical exchange). If these processes are not fast enough relative to the coupling strength, other mechanisms such as CSA or dipolar driven relaxation may prove more efficient. This is likely the case for SnBr2, SnI2, SnI4, and MA2SnBr6, since these compounds have considerably longer T1 values in comparison to the corresponding iodo- and bromostannates. Since the 1JSn–Cl values are relatively small (<0.5 kHz),93 it is possible that solid tin chlorides and chlorostannates are relaxed by these alternative processes.78,93 Relaxation in tin metal (β-Sn) is driven by the conduction electrons, as shown by Korringa.104 In the next section, we elucidate the relaxation mechanism for 119Sn in MASnBr3 and show that it is indeed determined by the scalar coupling to the halogen and driven by the motion of halides.
Figure 6.

119Sn longitudinal relaxation times (T1) at 4.7 T, 298 K, and 12 kHz MAS (except for SnBr4, which was measured at 0.6 kHz MAS to prevent melting) of the tin(II)- and tin(IV)-containing phases investigated in this work: (blue) tin(II) halide perovskites; (green) tin(II) halides; (yellow) tin(IV) halides and halostannates(IV). The numerical values are reported in Table S3.
Complementarity with 13C, 14N, and 133Cs NMR
We note that the fast scalar relaxation does not affect the nuclei which are not directly bonded to the halogen. The scalar relaxation therefore has no effect on the A-site cation, which can be probed using high-resolution 1H, 13C, 133Cs, and 14N MAS NMR, as our group and others have previously shown for lead halide perovskites.39−42,45,53,55,56Figure S2 shows low-temperature 1H–13C CP spectra of methylammonium tin(II) single- and mixed-halide perovskites. The 13C resonance of MA in the mixed-halide compositions is broader in comparison to single halide compositions due to halide disorder. The 13C resonances fall within a similar chemical shift range, which makes the use of 119Sn considerably more advantageous for the elucidation of tin halide coordination environments. Figure S3 shows room-temperature 14N MAS spectra of MASnI3, FASnI3, and MA0.25FA0.75SnI3. We have previously shown that the width of the 14N SSB manifold is related to the cubooctahedral symmetry in lead halide perovskites, with narrower manifolds corresponding to cubooctahedral symmetry closer to cubic; here we show that the same considerations hold for tin(II) halide perovskites. For example, the MA and FA SSB manifolds broaden in MA0.25FA0.75SnI3 in comparison to the single-cation compositions, indicating that the overall cubooctahedral symmetry has been reduced due to A-site cation mixing, similar to the effect previously observed in mixed-cation lead halide perovskites.39 Finally, Figure S4 shows room-temperature 133Cs spectra of CsSnX3 (X = I, Br, Cl). The signals are narrow (fwhm 90–110 Hz) and well-resolved, which potentially makes 133Cs MAS NMR well suited for studying component mixing and phase segregation processes in Cs-containing tin halide perovskites, similarly to how what has previously been shown in the context of lead halide perovskites.40
Halide Dynamics in MASnBr3
NMR relaxation in solids is caused by fluctuating magnetic fields arising due to modulation of various interactions. It can therefore be used to study dynamic processes with time scales ranging from picoseconds to seconds.105 We demonstrate this by using 119Sn T1 relaxation to probe the dynamic processes in MASnBr3. The following mechanisms can in principle cause 119Sn relaxation in solids: (a) dipole–dipole interaction,106 (b) chemical shift anisotropy (CSA),106 (c) Raman process,107,108 (d) MAS-induced heteronuclear polarization exchange,109 and (e) scalar relaxation.106
In order to elucidate which mechanism is relevant in MASnBr3, we acquired variable-temperature T1 relaxation data at three magnetic field strengths, 4.7, 9.4, and 17.6 T, and found that T1 relaxation is essentially field independent (Figure 7b). The CSA mechanism has a strong field dependence and hence can be excluded. Dipole–dipole relaxation leads to a T1 minimum in the range of seconds (∼5 s); hence, this mechanism can also be excluded (see Supplementary Note 2 for the calculation). The Raman process leads to T1 values which are independent of the magnetic field strength and inversely proportional to the square of the temperature, the latter of which is the case here (Figure S5). MAS-induced heteronuclear polarization exchange arises due to crossing between energy levels of a spin 1/2 nucleus such as 119Sn, 207Pb, or 199Hg coupled to a quadrupolar spin with a very large quadrupolar coupling constant, which is the case for 127I and 79/81Br. In this mechanism, the T1 value is significantly reduced when the sample is spun. We did not observe T1 shortening between the static and spinning case (Figure S6). Finally, scalar relaxation is expected to be field independent and may be caused by modulation of the 119Sn–79/81Br scalar coupling due to either chemical exchange (scalar relaxation of the first kind) or fast quadrupolar relaxation of 79/81Br (scalar relaxation of the second kind).106 The physical origin of the process can be determined from the temperature dependence of the T2 relaxation times, which decrease with increasing temperature if they are caused by relaxation of the quadrupolar nucleus and increase with temperature if they are caused by chemical exchange, provided the system is in the extreme narrowing limit.93 However, if the system is in the slow-motion limit, both processes lead to longer 119Sn T2 values as the temperature increases (see also Supplementary Note 3). We use the fwhm of the 119Sn signal as a measure of T2 since we found that it is field independent; hence, it does not originate from a distribution of chemical environments (i.e., T2* ≈ T2). Experimentally, we observe that the 119Sn resonances become narrower as the temperature increases (Figure 7a and Table S1), which shows that T2 increases with temperature. Since determining the relaxation regime for the quadrupolar partner is not straightforward in this case due to its very large quadrupole coupling constant,110 we employ the determined activation energy as a constraint to identify the relevant relaxation mechanism. Plotting ln(119Sn T1/s) as a function of the inverse temperature yields an Arrhenius plot (Figure 7b) from which we determine the activation energy of the process driving the relaxation (Table 1 and Table S2). Averaging the results obtained at three magnetic fields and between 250 and 450 K, we obtain an average activation energy of 36 ± 6 kJ/mol or 0.37 ± 0.06 eV. This value is in fairly good agreement with those previously found for bromide diffusion in MASnBr3 using ac and dc conductivity measurements (0.30 and 0.31 eV, respectively).111,112 This value is also comparable to those previously reported for halide diffusion in α-SnI2 (0.29 eV) and MAPbI3 (0.29 ± 0.06 eV). This result suggests that T1 relaxation of 119Sn in MASnBr3 is primarily driven by scalar relaxation of the first kind: i.e., by movement of species inside the crystal lattice. Scalar relaxation of the second kind, on the other hand, would lead to activation energies corresponding to the process driving quadrupolar relaxation of 79/81Br, i.e. vibrational modes of the lattice, which are active in the far-infrared to terahertz regime (<0.03 eV).113,114 Since tin halides are ionic conductors,115 we conclude that the chemical exchange process which drives 119Sn relaxation in MASnBr3 is the diffusion of Br– ions in the crystal lattice. Ionic conductivity due to halides has been previously shown in lead halide perovskites57,116 and tin halides.115,117 These results confirm that MASnBr3 is indeed an ionic conductor. DFT calculations predict a formation energy of 0.37 eV for iodide vacancies in MASnI3 which is comparable to the experimentally measured ionic diffusion activation barrier. We note that, although these two processes are not equivalent, halide migration relies on the presence of halide vacancies.118 Since MASnBr3 starts decomposing above ∼420 K and there is no T1 minimum in the accessible temperature range, it was not possible to fit the full form of the relaxation process to access the halide diffusion rate. We note, however, that the previously calculated halide hopping rates are in the nanosecond range in lead halide perovskites.119,120
Figure 7.
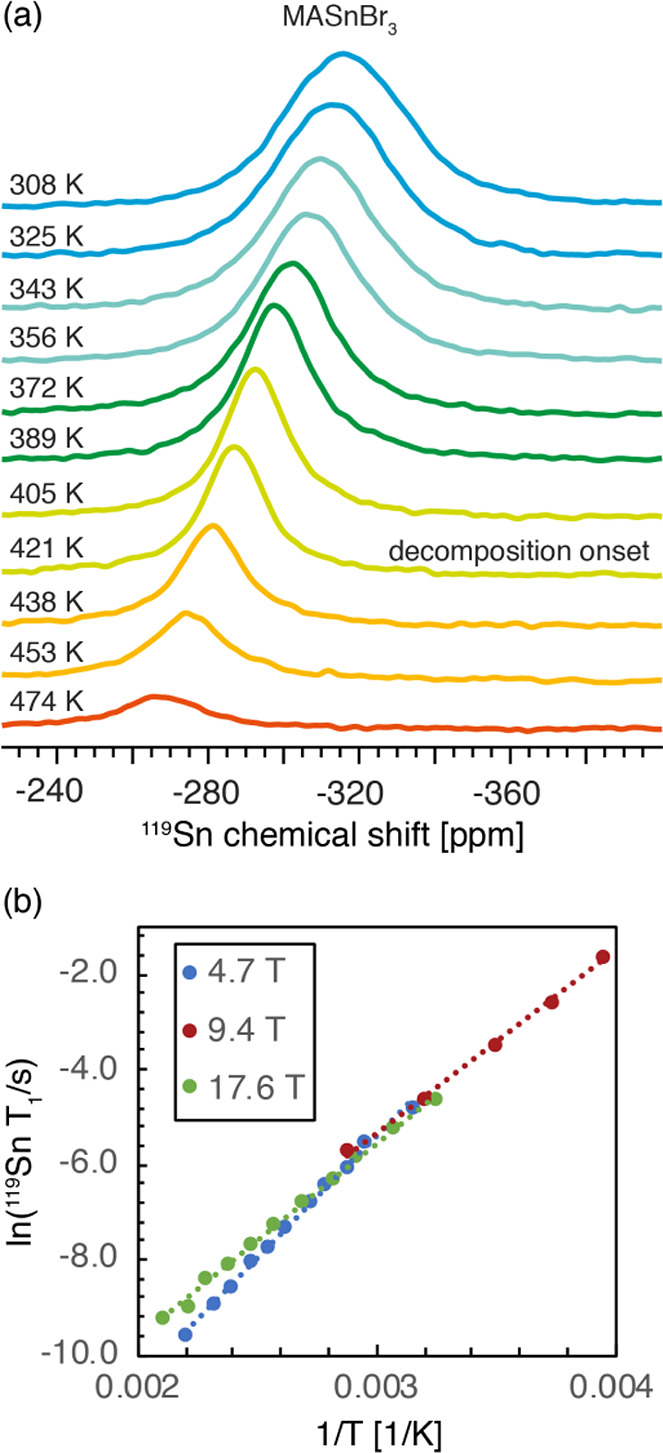
Halide dynamics in MASnBr3 from multi-field variable-temperature 119Sn solid-state MAS NMR. (a) variable–temperature (308–474 K) 119Sn spectra at 17.6 T. All spectra were acquired using the same number of scans (4096) and are quantitative. The spectrum after thermal decomposition corresponds to MA2SnBr6 (Figure 4b) with traces of SnO2 as discussed above. (b) Arrhenius plot of the 119Sn T1 relaxation data at 4.7 T (blue), 9.4 T (red), and 17.6 T (green). The linear fits are indicated by dotted lines, and the numerical values are given in Table S2.
Table 1. Activation Energies (Ea) for Halide Migration in Tin(II) Halide Perovskites and Related Phasesa.
| material | Ea (kJ/mol) | Ea (eV) | technique | ref |
|---|---|---|---|---|
| MASnBr3 (4.7 T) | 42.7 ± 0.5 | 0.44 | solid–state NMR | this work |
| MASnBr3 (9.4 T) | 31.9 ± 0.1 | 0.33 | ||
| MASnBr3 (17.6 T) | 34.1 ± 0.1 | 0.35 | ||
| MASnBr3 (average)b | 36 ± 6 | 0.37 ± 0.06 | ||
| MASnBr3 | 29.1 | 0.30 | ac conductivity | (111) |
| MASnBr3 | 30 | 0.31 | dc conductivity | (112) |
| α-SnI2 | 28 | 0.29 | ac conductivity | (115) |
| MASnI3 | 63 | 0.37 (VI)c | DFT | (118) |
| 0.65 (Ii)c | ||||
| MAPbI3 | 28 ± 6 | 0.29 ± 0.06 | transient ion-drift | (116) |
| MAPbI3 | 16 | 0.17 | 127I NQR | (57) |
The uncertainty is given as one standard deviation.
The uncertainty is calculated as the standard error of the average.
Defect formation energy at the valence band maximum: VI, iodide vacancy; Ii, iodide interstitial.
Finally, we show that the comparatively low activation energy for halide diffusion leads to spontaneous halide mixing at room temperature, which can be conveniently probed using 119Sn MAS NMR.
Spontaneous Halide Mixing
Thermally activated halide mixing has been previously demonstrated in microcrystalline46 lead halide perovskites and in polycrystalline thin films.90,121 In order to demonstrate this phenomenon in the context of tin(II) halide perovskites, we physically mixed equimolar amounts of microcrystalline MASnBr3 (Figure 8a, −316 ppm) and MASnCl3 (Figure 8b, −398 ppm) by weighing the materials into a vial and turning the vial upside down five times to provide light mixing. The spectrum recorded after 24 h of storing the mixture under argon at room temperature shows that the single-halide perovskites have fully disappeared and a new chemical species has formed (Figure 8c,d). Recording a spectrum with a short recycle delay highlights the quickly relaxing bromide-rich coordination environments (−325 ppm, Figure 8c), while using a long recycle delay accentuates the slowly relaxing chloride-rich environments (−340 ppm, Figure 8d). The resulting mixed-halide perovskite has a composition similar to that of MASnCl1.5Br1.5 (−353 ppm, Figure 8e), although the slight difference in chemical shifts demonstrates that the two materials are not identical. We expect these results to carry over to other tin(II) halide perovskite compositions and suggest that spontaneous halide mixing should occur whenever there is an intergranular halide concentration gradient.
Figure 8.
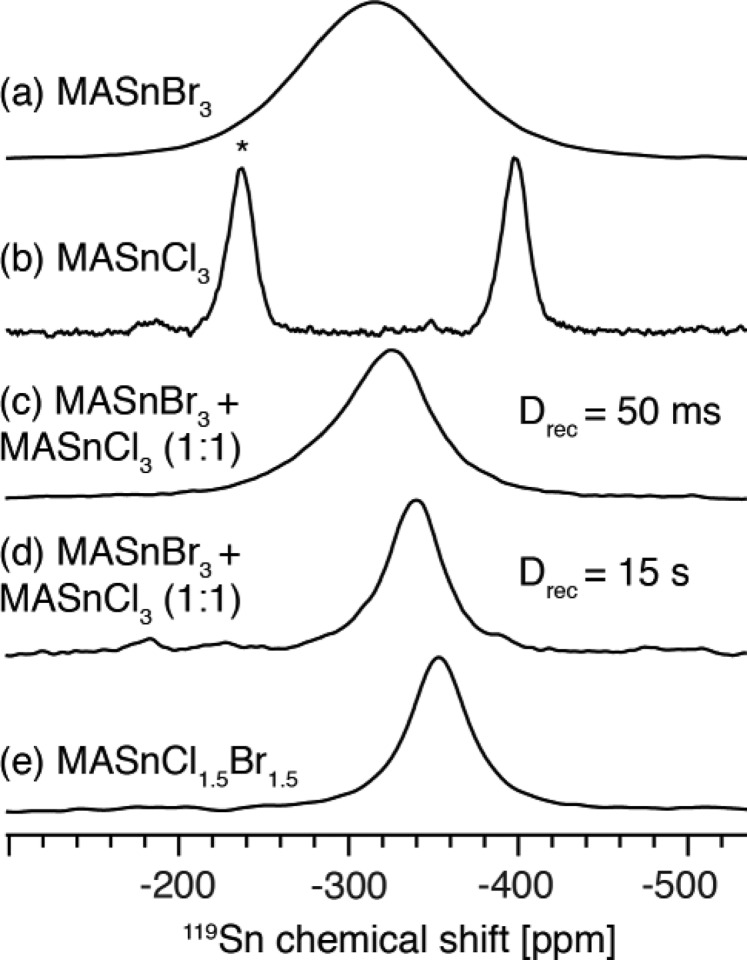
119Sn as a probe of spontaneous halide mixing. 119Sn solid-state MAS NMR spectra at 4.7 T and 12 kHz MAS: (a) MASnBr3; (b) MASnCl3 (the asterisk indicates a spinning sideband); (c) 1/1 (mol/mol) mixture of MASnBr3 and MASnCl3, lightly mixed, recorded after 24 h with a recycle delay of 50 ms; (d) same as (c) but using a recycle delay of 15 s to highlight the slowly relaxing chloride-rich environments; (e) MASnCl1.5Br1.5.
Conclusions
We have identified and overcome the challenges associated with the acquisition of solid-state 119Sn MAS NMR data, namely that the longitudinal relaxation of 119Sn in tin(II) halide perovskites and related materials spans 6 orders of magnitude, which makes it essential to judiciously choose the experimental parameters so as to obtain optimal results. We have shown that solid-state 119Sn MAS NMR can be used to characterize the local structure of tin(II) mixed-halide and mixed A-site cation perovskites and related phases as well as to distinguish between tin(II) and tin(IV) halostannate phases. This property in particular can be employed to study degradation processes in tin(II) halide perovskites, and we have exemplified it by identifying the degradation products of MASnBr3, FASnBr3, CsSnBr3, MASnI3, FASnI3, and CsSnI3. We have found that, regardless of the composition, the decomposition products include amorphous SnO2 and the corresponding tin(IV) halostannate, A2SnX6. Further, we have identified the dominant NMR relaxation mechanism of 119Sn in solid MASnBr3 as scalar relaxation of the first kind driven by bromide diffusion inside the perovskite lattice. We have quantified the activation energy of this process using variable-temperature multi-field relaxation measurements and found that the values are in excellent agreement with those extracted from previously reported electrical conductivity measurements. Finally, we have shown that spontaneous halide homogenization occurs at room temperature between microcrystalline single-halide tin(II) halide perovskites, which leads to mixed-halide materials. We expect this property of tin(II) halide perovskite to carry over to other tin(II) halide perovskite systems featuring a halide concentration gradient. Taken together, we believe that 119Sn MAS NMR is a general and versatile technique providing information on local structure and dynamics in tin(II) halide perovskites, complementary to the data obtained by diffraction techniques and optical spectroscopy.
Acknowledgments
Requests for additional data and correspondence should be addressed to C.P.G., L.E., or S.D.S. This work has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 841136. This work was supported by Swiss National Science Foundation Grant No. 200020_178860. D.P. acknowledges financial support from the HOMING programme of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund (POIR.04.04.00-00-5EE7/18-00). Financial support from the IR-RMN-THC Fr3050 CNRS for conducting the research is gratefully acknowledged. S.D.S. acknowledges the Royal Society and Tata Group (UF150033). This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC) grant EP/R023980/1.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00647.
119Sn, 13C, 133Cs , and 14N NMR spectra, numerical T1 data, and further experimental details (PDF)
The authors declare the following competing financial interest(s): Samuel D. Stranks is a co-founder of Swift Solar, Inc.
Notes
All raw data can be accessed at the following address: https://zenodo.org/record/3752869
Notes
Note added in proof: The authors acknowledge that static 119Sn NMR line shape analysis has recently been used to study halide diffusion in tin(II) halide perovskites: Yamada, K.; Fujise, K.; Hino, S.; Yamane, Y.; Nakagama, T. Characterization of Sn(II)-Based Perovskites by XRD, DTA, NQR and 119Sn NMR for Photovoltaic Applications. Chem. Lett.2019, 48, 749-752, DOI: 10.1246/cl.190262.
Supplementary Material
References
- Li W.; Wang Z.; Deschler F.; Gao S.; Friend R. H.; Cheetham A. K. Chemically Diverse and Multifunctional Hybrid Organic–Inorganic Perovskites. Nat. Rev. Mater. 2017, 2, 16099. 10.1038/natrevmats.2016.99. [DOI] [Google Scholar]
- Jena A. K.; Kulkarni A.; Miyasaka T. Halide Perovskite Photovoltaics: Background, Status, and Future Prospects. Chem. Rev. 2019, 119 (5), 3036–3103. 10.1021/acs.chemrev.8b00539. [DOI] [PubMed] [Google Scholar]
- Kojima A.; Teshima K.; Shirai Y.; Miyasaka T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. 10.1021/ja809598r. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Park B.-W.; Jung E. H.; Jeon N. J.; Kim Y. C.; Lee D. U.; Shin S. S.; Seo J.; Kim E. K.; Noh J. H.; et al. Iodide Management in Formamidinium-Lead-Halide–Based Perovskite Layers for Efficient Solar Cells. Science 2017, 356 (6345), 1376–1379. 10.1126/science.aan2301. [DOI] [PubMed] [Google Scholar]
- Https://Www.Nrel.Gov/Pv/Cell-Efficiency.Html.
- Domanski K.; Alharbi E. A.; Hagfeldt A.; Grätzel M.; Tress W. Systematic Investigation of the Impact of Operation Conditions on the Degradation Behaviour of Perovskite Solar Cells. Nat. Energy 2018, 3 (1), 61–67. 10.1038/s41560-017-0060-5. [DOI] [Google Scholar]
- Tress W.; Domanski K.; Carlsen B.; Agarwalla A.; Alharbi E. A.; Graetzel M.; Hagfeldt A. Performance of Perovskite Solar Cells under Simulated Temperature-Illumination Real-World Operating Conditions. Nat. Energy 2019, 4 (7), 568–574. 10.1038/s41560-019-0400-8. [DOI] [Google Scholar]
- Shi Z.; Guo J.; Chen Y.; Li Q.; Pan Y.; Zhang H.; Xia Y.; Huang W. Lead-Free Organic–Inorganic Hybrid Perovskites for Photovoltaic Applications: Recent Advances and Perspectives. Adv. Mater. 2017, 29 (16), 1605005. 10.1002/adma.201605005. [DOI] [PubMed] [Google Scholar]
- Leijtens T.; Eperon G. E.; Noel N. K.; Habisreutinger S. N.; Petrozza A.; Snaith H. J. Stability of Metal Halide Perovskite Solar Cells. Adv. Energy Mater. 2015, 5 (20), 1500963. 10.1002/aenm.201500963. [DOI] [Google Scholar]
- Yang W. S.; Noh J. H.; Jeon N. J.; Kim Y. C.; Ryu S.; Seo J.; Seok S. I. High-Performance Photovoltaic Perovskite Layers Fabricated through Intramolecular Exchange. Science 2015, 348 (6240), 1234–1237. 10.1126/science.aaa9272. [DOI] [PubMed] [Google Scholar]
- Abdi-Jalebi M.; Andaji-Garmaroudi Z.; Cacovich S.; Stavrakas C.; Philippe B.; Richter J. M.; Alsari M.; Booker E. P.; Hutter E. M.; Pearson A. J.; et al. Maximizing and Stabilizing Luminescence from Halide Perovskites with Potassium Passivation. Nature 2018, 555, 497. 10.1038/nature25989. [DOI] [PubMed] [Google Scholar]
- Lee S. J.; Shin S. S.; Kim Y. C.; Kim D.; Ahn T. K.; Noh J. H.; Seo J.; Seok S. I. Fabrication of Efficient Formamidinium Tin Iodide Perovskite Solar Cells through SnF2–Pyrazine Complex. J. Am. Chem. Soc. 2016, 138 (12), 3974–3977. 10.1021/jacs.6b00142. [DOI] [PubMed] [Google Scholar]
- Song T.-B.; Yokoyama T.; Stoumpos C. C.; Logsdon J.; Cao D. H.; Wasielewski M. R.; Aramaki S.; Kanatzidis M. G. Importance of Reducing Vapor Atmosphere in the Fabrication of Tin-Based Perovskite Solar Cells. J. Am. Chem. Soc. 2017, 139 (2), 836–842. 10.1021/jacs.6b10734. [DOI] [PubMed] [Google Scholar]
- Tsarev S.; Boldyreva A. G.; Luchkin S. Y.; Elshobaki M.; Afanasov M. I.; Stevenson K. J.; Troshin P. A. Hydrazinium-Assisted Stabilisation of Methylammonium Tin Iodide for Lead-Free Perovskite Solar Cells. J. Mater. Chem. A 2018, 6 (43), 21389–21395. 10.1039/C8TA07699E. [DOI] [Google Scholar]
- Li F.; Zhang C.; Huang J.-H.; Fan H.; Wang H.; Wang P.; Zhan C.; Liu C.-M.; Li X.; Yang L.-M.; et al. A Cation-Exchange Approach for the Fabrication of Efficient Methylammonium Tin Iodide Perovskite Solar Cells. Angew. Chem. 2019, 131 (20), 6760–6764. 10.1002/ange.201902418. [DOI] [PubMed] [Google Scholar]
- Gu F.; Ye S.; Zhao Z.; Rao H.; Liu Z.; Bian Z.; Huang C. Improving Performance of Lead-Free Formamidinium Tin Triiodide Perovskite Solar Cells by Tin Source Purification. Sol. RRL 2018, 2 (10), 1800136. 10.1002/solr.201800136. [DOI] [Google Scholar]
- Tai Q.; Guo X.; Tang G.; You P.; Ng T.-W.; Shen D.; Cao J.; Liu C.-K.; Wang N.; Zhu Y.; et al. Antioxidant Grain Passivation for Air-Stable Tin-Based Perovskite Solar Cells. Angew. Chem. 2019, 131 (3), 816–820. 10.1002/ange.201811539. [DOI] [PubMed] [Google Scholar]
- Xu X.; Chueh C.-C.; Yang Z.; Rajagopal A.; Xu J.; Jo S. B.; Jen A. K.-Y. Ascorbic Acid as an Effective Antioxidant Additive to Enhance the Efficiency and Stability of Pb/Sn-Based Binary Perovskite Solar Cells. Nano Energy 2017, 34, 392–398. 10.1016/j.nanoen.2017.02.040. [DOI] [Google Scholar]
- Zhao Z.; Gu F.; Li Y.; Sun W.; Ye S.; Rao H.; Liu Z.; Bian Z.; Huang C.; Sun W. Mixed-Organic-Cation Tin Iodide for Lead-Free Perovskite Solar Cells with an Efficiency of 8.12. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2017, 4, 1700204. 10.1002/advs.201700204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokar E.; Chien C.-H.; Tsai C.-M.; Fathi A.; Diau E. W.-G. Robust Tin-Based Perovskite Solar Cells with Hybrid Organic Cations to Attain Efficiency Approaching 10%. Adv. Mater. 2019, 31 (2), 1804835. 10.1002/adma.201804835. [DOI] [PubMed] [Google Scholar]
- Kopacic I.; Friesenbichler B.; Hoefler S. F.; Kunert B.; Plank H.; Rath T.; Trimmel G. Enhanced Performance of Germanium Halide Perovskite Solar Cells through Compositional Engineering. ACS Appl. Energy Mater. 2018, 1 (2), 343–347. 10.1021/acsaem.8b00007. [DOI] [Google Scholar]
- Liu J.; Ozaki M.; Yakumaru S.; Handa T.; Nishikubo R.; Kanemitsu Y.; Saeki A.; Murata Y.; Murdey R.; Wakamiya A. Lead-Free Solar Cells Based on Tin Halide Perovskite Films with High Coverage and Improved Aggregation. Angew. Chem. 2018, 130 (40), 13405–13409. 10.1002/ange.201808385. [DOI] [PubMed] [Google Scholar]
- Shao S.; Liu J.; Portale G.; Fang H.-H.; Blake G. R.; ten Brink G. H.; Koster L. J. A.; Loi M. A. Highly Reproducible Sn-Based Hybrid Perovskite Solar Cells with 9% Efficiency. Adv. Energy Mater. 2018, 8 (4), 1702019. 10.1002/aenm.201702019. [DOI] [Google Scholar]
- Stranks S. D.; Eperon G. E.; Grancini G.; Menelaou C.; Alcocer M. J. P.; Leijtens T.; Herz L. M.; Petrozza A.; Snaith H. J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342 (6156), 341–344. 10.1126/science.1243982. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Yuan Y.; Shao Y.; Fang Y.; Wang Q.; Huang J. Abnormal Crystal Growth in CH3NH3PbI3–xClx Using a Multi-Cycle Solution Coating Process. Energy Environ. Sci. 2015, 8 (8), 2464–2470. 10.1039/C5EE01179E. [DOI] [Google Scholar]
- Yang B.; Keum J.; Ovchinnikova O. S.; Belianinov A.; Chen S.; Du M.-H.; Ivanov I. N.; Rouleau C. M.; Geohegan D. B.; Xiao K. Deciphering Halogen Competition in Organometallic Halide Perovskite Growth. J. Am. Chem. Soc. 2016, 138 (15), 5028–5035. 10.1021/jacs.5b13254. [DOI] [PubMed] [Google Scholar]
- Yang B.; Brown C. C.; Huang J.; Collins L.; Sang X.; Unocic R. R.; Jesse S.; Kalinin S. V.; Belianinov A.; Jakowski J.; et al. Enhancing Ion Migration in Grain Boundaries of Hybrid Organic–Inorganic Perovskites by Chlorine. Adv. Funct. Mater. 2017, 27 (26), 1700749. 10.1002/adfm.201700749. [DOI] [Google Scholar]
- Marshall K. P.; Walker M.; Walton R. I.; Hatton R. A. Enhanced Stability and Efficiency in Hole-Transport-Layer-Free CsSnI3 Perovskite Photovoltaics. Nat. Energy 2016, 1, 16178. 10.1038/nenergy.2016.178. [DOI] [Google Scholar]
- Marshall K. P.; Walton R. I.; Hatton R. A. Tin Perovskite/Fullerene Planar Layer Photovoltaics: Improving the Efficiency and Stability of Lead-Free Devices. J. Mater. Chem. A 2015, 3 (21), 11631–11640. 10.1039/C5TA02950C. [DOI] [Google Scholar]
- Eperon G. E.; Leijtens T.; Bush K. A.; Prasanna R.; Green T.; Wang J. T.-W.; McMeekin D. P.; Volonakis G.; Milot R. L.; May R.; et al. Perovskite-Perovskite Tandem Photovoltaics with Optimized Band Gaps. Science 2016, 354 (6314), 861–865. 10.1126/science.aaf9717. [DOI] [PubMed] [Google Scholar]
- Prasanna R.; Gold-Parker A.; Leijtens T.; Conings B.; Babayigit A.; Boyen H.-G.; Toney M. F.; McGehee M. D. Band Gap Tuning via Lattice Contraction and Octahedral Tilting in Perovskite Materials for Photovoltaics. J. Am. Chem. Soc. 2017, 139 (32), 11117–11124. 10.1021/jacs.7b04981. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Abdi-Jalebi M.; Tabachnyk M.; Glass H.; Kamboj V. S.; Nie W.; Pearson A. J.; Puttisong Y.; Gödel K. C.; Beere H. E.; et al. High Open-Circuit Voltages in Tin-Rich Low-Bandgap Perovskite-Based Planar Heterojunction Photovoltaics. Adv. Mater. 2017, 29 (2), 1604744. 10.1002/adma.201604744. [DOI] [PubMed] [Google Scholar]
- Rajagopal A.; Yang Z.; Jo S. B.; Braly I. L.; Liang P.-W.; Hillhouse H. W.; Jen A. K.-Y. Highly Efficient Perovskite–Perovskite Tandem Solar Cells Reaching 80% of the Theoretical Limit in Photovoltage. Adv. Mater. 2017, 29 (34), 1702140. 10.1002/adma.201702140. [DOI] [PubMed] [Google Scholar]
- Leijtens T.; Prasanna R.; Bush K. A.; Eperon G. E.; Raiford J. A.; Gold-Parker A.; Wolf E. J.; Swifter S. A.; Boyd C. C.; Wang H.-P.; et al. Tin–Lead Halide Perovskites with Improved Thermal and Air Stability for Efficient All-Perovskite Tandem Solar Cells. Sustain. Energy Fuels 2018, 2 (11), 2450–2459. 10.1039/C8SE00314A. [DOI] [Google Scholar]
- Noel N. K.; Stranks S. D.; Abate A.; Wehrenfennig C.; Guarnera S.; Haghighirad A.-A.; Sadhanala A.; Eperon G. E.; Pathak S. K.; Johnston M. B.; et al. Lead-Free Organic-Inorganic Tin Halide Perovskites for Photovoltaic Applications. Energy Environ. Sci. 2014, 7 (9), 3061–3068. 10.1039/C4EE01076K. [DOI] [Google Scholar]
- Hao F.; Stoumpos C. C.; Cao D. H.; Chang R. P. H.; Kanatzidis M. G. Lead-Free Solid-State Organic-Inorganic Halide Perovskite Solar Cells. Nat. Photonics 2014, 8 (6), 489–494. 10.1038/nphoton.2014.82. [DOI] [Google Scholar]
- Boyd C. C.; Cheacharoen R.; Leijtens T.; McGehee M. D. Understanding Degradation Mechanisms and Improving Stability of Perovskite Photovoltaics. Chem. Rev. 2019, 119 (5), 3418–3451. 10.1021/acs.chemrev.8b00336. [DOI] [PubMed] [Google Scholar]
- Franssen W. M. J.; Kentgens A. P. M. Solid–State NMR of Hybrid Halide Perovskites. Solid State Nucl. Magn. Reson. 2019, 100, 36–44. 10.1016/j.ssnmr.2019.03.005. [DOI] [PubMed] [Google Scholar]
- Kubicki D. J.; Prochowicz D.; Hofstetter A.; Péchy P.; Zakeeruddin S. M.; Grätzel M.; Emsley L. Cation Dynamics in Mixed-Cation (MA)x(FA)1–XPbI3 Hybrid Perovskites from Solid-State NMR. J. Am. Chem. Soc. 2017, 139 (29), 10055–10061. 10.1021/jacs.7b04930. [DOI] [PubMed] [Google Scholar]
- Kubicki D. J.; Prochowicz D.; Hofstetter A.; Zakeeruddin S. M.; Grätzel M.; Emsley L. Phase Segregation in Cs-, Rb- and K-Doped Mixed-Cation (MA)x(FA)1–XPbI3 Hybrid Perovskites from Solid-State NMR. J. Am. Chem. Soc. 2017, 139 (40), 14173–14180. 10.1021/jacs.7b07223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicki D. J.; Prochowicz D.; Hofstetter A.; Saski M.; Yadav P.; Bi D.; Pellet N.; Lewiński J.; Zakeeruddin S. M.; Grätzel M.; et al. Formation of Stable Mixed Guanidinium–Methylammonium Phases with Exceptionally Long Carrier Lifetimes for High-Efficiency Lead Iodide-Based Perovskite Photovoltaics. J. Am. Chem. Soc. 2018, 140 (9), 3345–3351. 10.1021/jacs.7b12860. [DOI] [PubMed] [Google Scholar]
- Kubicki D. J.; Prochowicz D.; Hofstetter A.; Zakeeruddin S. M.; Grätzel M.; Emsley L. Phase Segregation in Potassium-Doped Lead Halide Perovskites from 39K Solid-State NMR at 21.1 T. J. Am. Chem. Soc. 2018, 140 (23), 7232–7238. 10.1021/jacs.8b03191. [DOI] [PubMed] [Google Scholar]
- Kubicki D. J.; Prochowicz D.; Pinon A.; Stevanato G.; Hofstetter A.; Zakeeruddin S. M.; Grätzel M.; Emsley L. Doping and Phase Segregation in Mn2+- and Co2+-Doped Lead Halide Perovskites from 133Cs and 1H NMR Relaxation Enhancement. J. Mater. Chem. A 2019, 7, 2326–2333. 10.1039/C8TA11457A. [DOI] [Google Scholar]
- Xiang W.; Wang Z.; Kubicki D. J.; Tress W.; Luo J.; Prochowicz D.; Akin S.; Emsley L.; Zhou J.; Dietler G.; et al. Europium-Doped CsPbI2Br for Stable and Highly Efficient Inorganic Perovskite Solar Cells. Joule 2019, 3 (1), 205–214. 10.1016/j.joule.2018.10.008. [DOI] [Google Scholar]
- Franssen W. M. J.; Bruijnaers B. J.; Portengen V. H. L.; Kentgens A. P. M. Dimethylammonium Incorporation in Lead Acetate Based MAPbI3 Perovskite Solar Cells. ChemPhysChem 2018, 19 (22), 3107–3115. 10.1002/cphc.201800732. [DOI] [PubMed] [Google Scholar]
- Rosales B. A.; Men L.; Cady S. D.; Hanrahan M. P.; Rossini A. J.; Vela J. Persistent Dopants and Phase Segregation in Organolead Mixed-Halide Perovskites. Chem. Mater. 2016, 28, 6848–6859. 10.1021/acs.chemmater.6b01874. [DOI] [Google Scholar]
- Karmakar A.; Askar A. M.; Bernard G. M.; Terskikh V. V.; Ha M.; Patel S.; Shankar K.; Michaelis V. K. Mechanochemical Synthesis of Methylammonium Lead Mixed–Halide Perovskites: Unraveling the Solid-Solution Behavior Using Solid-State NMR. Chem. Mater. 2018, 30, 2309–2321. 10.1021/acs.chemmater.7b05209. [DOI] [Google Scholar]
- Askar A. M.; Karmakar A.; Bernard G. M.; Ha M.; Terskikh V. V.; Wiltshire B. D.; Patel S.; Fleet J.; Shankar K.; Michaelis V. K. Composition-Tunable Formamidinium Lead Mixed Halide Perovskites via Solvent-Free Mechanochemical Synthesis: Decoding the Pb Environments Using Solid-State NMR Spectroscopy. J. Phys. Chem. Lett. 2018, 9, 2671–2677. 10.1021/acs.jpclett.8b01084. [DOI] [PubMed] [Google Scholar]
- Hanrahan M. P.; Men L.; Rosales B. A.; Vela J.; Rossini A. J. Sensitivity-Enhanced 207Pb Solid-State NMR Spectroscopy for the Rapid, Non-Destructive Characterization of Organolead Halide Perovskites. Chem. Mater. 2018, 30 (20), 7005–7015. 10.1021/acs.chemmater.8b01899. [DOI] [Google Scholar]
- Bi D.; Li X.; Milić J. V.; Kubicki D. J.; Pellet N.; Luo J.; LaGrange T.; Mettraux P.; Emsley L.; Zakeeruddin S. M.; et al. Multifunctional Molecular Modulators for Perovskite Solar Cells with over 20% Efficiency and High Operational Stability. Nat. Commun. 2018, 9 (1), 4482. 10.1038/s41467-018-06709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alharbi E. A.; Alyamani A. Y.; Kubicki D. J.; Uhl A. R.; Walder B. J.; Alanazi A. Q.; Luo J.; Burgos-Caminal A.; Albadri A.; Albrithen H.; et al. Atomic-Level Passivation Mechanism of Ammonium Salts Enabling Highly Efficient Perovskite Solar Cells. Nat. Commun. 2019, 10 (1), 1–9. 10.1038/s41467-019-10985-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakoli M. M.; Tress W.; Milić J. V.; Kubicki D.; Emsley L.; Grätzel M. Addition of Adamantylammonium Iodide to Hole Transport Layers Enables Highly Efficient and Electroluminescent Perovskite Solar Cells. Energy Environ. Sci. 2018, 11 (11), 3310–3320. 10.1039/C8EE02404A. [DOI] [Google Scholar]
- Wasylishen R. E.; Knop O.; Macdonald J. B. Cation Rotation in Methylammonium Lead Halides. Solid State Commun. 1985, 56 (7), 581–582. 10.1016/0038-1098(85)90959-7. [DOI] [Google Scholar]
- Knop O.; Wasylishen R. E.; White M. A.; Cameron T. S.; Van Oort M. J. M. Alkylammonium Lead Halides. Part 2. CH3NH3PbX3 (X = Chlorine, Bromine, Iodine) Perovskites: Cuboctahedral Halide Cages with Isotropic Cation Reorientation. Can. J. Chem. 1990, 68, 412–422. 10.1139/v90-063. [DOI] [Google Scholar]
- Franssen W. M. J.; van Es S. G. D.; Dervisoglu R.; de Wijs G. A.; Kentgens A. P. M. Symmetry, Dynamics, and Defects in Methylammonium Lead Halide Perovskites. J. Phys. Chem. Lett. 2017, 8, 61–66. 10.1021/acs.jpclett.6b02542. [DOI] [PubMed] [Google Scholar]
- Bernard G. M.; Wasylishen R. E.; Ratcliffe C. I.; Terskikh V.; Wu Q.; Buriak J. M.; Hauger T. Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective. J. Phys. Chem. A 2018, 122 (6), 1560–1573. 10.1021/acs.jpca.7b11558. [DOI] [PubMed] [Google Scholar]
- Senocrate A.; Moudrakovski I.; Kim G. Y.; Yang T.-Y.; Gregori G.; Gratzel M.; Maier J. The Nature of Ion Conduction in Methylammonium Lead Iodide: A Multimethod Approach. Angew. Chem., Int. Ed. 2017, 56, 7755–7759. 10.1002/anie.201701724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senocrate A.; Moudrakovski I.; Maier J. Short-Range Ion Dynamics in Methylammonium Lead Iodide by Multinuclear Solid State NMR and 127I NQR. Phys. Chem. Chem. Phys. 2018, 20 (30), 20043–20055. 10.1039/C8CP01535J. [DOI] [PubMed] [Google Scholar]
- Senocrate A.; Moudrakovski I.; Acartürk T.; Merkle R.; Kim G. Y.; Starke U.; Grätzel M.; Maier J. Slow CH3NH3+ Diffusion in CH3NH3PbI3 under Light Measured by Solid-State NMR and Tracer Diffusion. J. Phys. Chem. C 2018, 122 (38), 21803–21806. 10.1021/acs.jpcc.8b06814. [DOI] [Google Scholar]
- Askar A. M.; Bernard G. M.; Wiltshire B.; Shankar K.; Michaelis V. K. Multinuclear Magnetic Resonance Tracking of Hydro, Thermal, and Hydrothermal Decomposition of CH3NH3PbI3. J. Phys. Chem. C 2017, 121 (2), 1013–1024. 10.1021/acs.jpcc.6b10865. [DOI] [Google Scholar]
- Fabini D. H.; Laurita G.; Bechtel J. S.; Stoumpos C. C.; Evans H. A.; Kontos A. G.; Raptis Y. S.; Falaras P.; Van der Ven A.; Kanatzidis M. G.; et al. Dynamic Stereochemical Activity of the Sn2+ Lone Pair in Perovskite CsSnBr3. J. Am. Chem. Soc. 2016, 138 (36), 11820–11832. 10.1021/jacs.6b06287. [DOI] [PubMed] [Google Scholar]
- Laurita G.; Fabini D. H.; Stoumpos C. C.; Kanatzidis M. G.; Seshadri R. Chemical Tuning of Dynamic Cation Off-Centering in the Cubic Phases of Hybrid Tin and Lead Halide Perovskites. Chem. Sci. 2017, 8 (8), 5628–5635. 10.1039/C7SC01429E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara C.; Patrini M.; Pisanu A.; Quadrelli P.; Milanese C.; Tealdi C.; Malavasi L. Wide Band-Gap Tuning in Sn-Based Hybrid Perovskites through Cation Replacement: The FA(1-x)MA(x)SnBr(3) Mixed System. J. Mater. Chem. A 2017, 5 (19), 9391–9395. 10.1039/C7TA01668A. [DOI] [Google Scholar]
- Wrackmeyer B.; Webb G. A. Application of 119Sn NMR Parameters. Annu. Rep. NMR Spectrosc. 1999, 38, 203–264. 10.1016/S0066-4103(08)60038-1. [DOI] [Google Scholar]
- Clayden N. J.; Dobson C. M.; Fern A. High-Resolution Solid-State Tin-119 Nuclear Magnetic Resonance Spectroscopy of Ternary Tin Oxides. J. Chem. Soc., Dalton Trans. 1989, (5), 843–847. 10.1039/dt9890000843. [DOI] [Google Scholar]
- Grey C. P.; Dobson C. M.; Cheetham A. K.; Jakeman R. J. B. Studies of Rare-Earth Stannates by Tin-119 MAS NMR. The Use of Paramagnetic Shift Probes in the Solid State. J. Am. Chem. Soc. 1989, 111 (2), 505–511. 10.1021/ja00184a017. [DOI] [Google Scholar]
- Lin Z.; Rocha J.; Jesus J. D. P. de; Ferreira A. Synthesis and Structure of a Novel Microporous Framework Stannosilicate. J. Mater. Chem. 2000, 10 (6), 1353–1356. 10.1039/b000102n. [DOI] [Google Scholar]
- Ferreira A.; Lin Z.; Rocha J.; Morais C. M.; Lopes M.; Fernandez C. Ab Initio Structure Determination of a Small-Pore Framework Sodium Stannosilicate. Inorg. Chem. 2001, 40 (14), 3330–3335. 10.1021/ic0012571. [DOI] [PubMed] [Google Scholar]
- Wolf P.; Valla M.; Rossini A. J.; Comas-Vives A.; Nunez-Zarur F.; Malaman B.; Lesage A.; Emsley L.; Coperet C.; Hermans I. NMR Signatures of the Active Sites in Sn-β Zeolite. Angew. Chem., Int. Ed. 2014, 53 (38), 10179–10183. 10.1002/anie.201403905. [DOI] [PubMed] [Google Scholar]
- Mundus C.; Taillades G.; Pradel A.; Ribes M. A 119Sn Solid-State Nuclear Magnetic Resonance Study of Crystalline Tin Sulphides. Solid State Nucl. Magn. Reson. 1996, 7 (2), 141–146. 10.1016/S0926-2040(96)01243-X. [DOI] [PubMed] [Google Scholar]
- Pietrass T.; Taulelle F. 119Sn Solid-State NMR of Tin Sulfides. Evidence of Polytypism in SnS2. Magn. Reson. Chem. 1997, 35 (6), 363–366. . [DOI] [Google Scholar]
- Scotti N.; Kockelmann W.; Senker J.; Traßel S.; Jacobs H. Sn3N4, ein Zinn(IV)-nitrid – Synthese und erste Strukturbestimmung einer binären Zinn–Stickstoff-Verbindung. Z. Anorg. Allg. Chem. 1999, 625 (9), 1435–1439. . [DOI] [Google Scholar]
- Grykałowska A.; Nowak B. High-Resolution Solid-State 119Sn and 195Pt NMR Studies of MPtSn Semiconductors (M = Ti, Zr, Hf, Th). Solid State Nucl. Magn. Reson. 2005, 27 (4), 223–227. 10.1016/j.ssnmr.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lock H.; Xiong J.; Wen Y.-C.; Parkinson B. A.; Maciel G. E. Solid-State 29Si, 113Cd, 119Sn, and 31P NMR Studies of II-IV-P2 Semiconductors. Solid State Nucl. Magn. Reson. 2001, 20 (3), 118–129. 10.1006/snmr.2001.0036. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Rossini A. J.; Kriegner D.; Valla M.; De Kergommeaux A.; Walter M.; Kravchyk K. V.; Nachtegaal M.; Stangl J.; Malaman B.; et al. Unraveling the Core-Shell Structure of Ligand-Capped Sn/SnOx Nanoparticles by Surface-Enhanced Nuclear Magnetic Resonance, Mössbauer, and X-Ray Absorption Spectroscopies. ACS Nano 2014, 8 (3), 2639–2648. 10.1021/nn406344n. [DOI] [PubMed] [Google Scholar]
- Amornsakchai P.; Apperley D. C.; Harris R. K.; Hodgkinson P.; Waterfield P. C. Solid-State NMR Studies of Some Tin(II) Compounds. Solid State Nucl. Magn. Reson. 2004, 26 (3), 160–171. 10.1016/j.ssnmr.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Yeh H.-M. M.; Geanangel R. A. 119Sn NMR Spectra of Tin(II) Halides. Inorg. Chim. Acta 1981, 52, 113–118. 10.1016/S0020-1693(00)88583-7. [DOI] [Google Scholar]
- Sharp R. R. Field Dependence of Nuclear Magnetic Relaxation of 119Sn in SnCl4, SnBr4, and SnI4. J. Chem. Phys. 1974, 60 (3), 1149–1157. 10.1063/1.1681126. [DOI] [Google Scholar]
- Nagashima T.; Tajima S.. On the Anisotropic Knight Shift of Nuclear Magnetic Resonance in Metallic Tin. Bulletin of Osaka Prefectural College of Technology 1968, 1, 95−100. https://core.ac.uk/display/67693624. [Google Scholar]
- Prochowicz D.; Franckevicius M.; Cieslak A. M.; Zakeeruddin S. M.; Gratzel M.; Lewinski J. Mechanosynthesis of the Hybrid Perovskite CH3NH3PbI3: Characterization and the Corresponding Solar Cell Efficiency. J. Mater. Chem. A 2015, 3, 20772–20777. 10.1039/C5TA04904K. [DOI] [Google Scholar]
- Prochowicz D.; Yadav P.; Saliba M.; Saski M.; Zakeeruddin S. M.; Lewinski J.; Gratzel M. Mechanosynthesis of Pure Phase Mixed-Cation MAxFA1-XPbI3 Hybrid Perovskites: Photovoltaic Performance and Electrochemical Properties. Sustain. Energy Fuels 2017, 1, 689–693. 10.1039/C7SE00094D. [DOI] [Google Scholar]
- Saski M.; Prochowicz D.; Marynowski W.; Lewiński J. Mechanosynthesis, Optical, and Morphological Properties of MA, FA, Cs-SnX3 (X = I, Br) and Phase-Pure Mixed-Halide MASnIxBr3–x Perovskites. Eur. J. Inorg. Chem. 2019, 2019 (22), 2680–2684. 10.1002/ejic.201801506. [DOI] [Google Scholar]
- Hong Z.; Tan D.; John R. A.; Tay Y. K. E.; Ho Y. K. T.; Zhao X.; Sum T. C.; Mathews N.; García F.; Soo H. S. Completely Solvent-Free Protocols to Access Phase-Pure, Metastable Metal Halide Perovskites and Functional Photodetectors from the Precursor Salts. iScience 2019, 16, 312–325. 10.1016/j.isci.2019.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ajjouri Y.; Locardi F.; Gelvez-Rueda M. C.; Prato M.; Sessolo M.; Ferretti M.; Grozema F. C.; Palazon F.; Bolink H. J. Mechanochemical Synthesis of Sn(II) and Sn(IV) Iodide Perovskites and Study of Their Structural, Chemical, Thermal, Optical, and Electrical Properties. Energy Technol. 2020, 8, 1900788. 10.1002/ente.201900788. [DOI] [Google Scholar]
- Xu J.; Wang J.; Rakhmatullin A.; Ory S.; Fernández-Carrión A. J.; Yi H.; Kuang X.; Allix M. Interstitial Oxide Ion Migration Mechanism in Aluminate Melilite La1+xCa1–XAl3O7 + 0.5x Ceramics Synthesized by Glass Crystallization. ACS Appl. Energy Mater. 2019, 2 (4), 2878–2888. 10.1021/acsaem.9b00224. [DOI] [Google Scholar]
- Thurber K. R.; Tycko R. Measurement of Sample Temperatures under Magic-Angle Spinning from the Chemical Shift and Spin-Lattice Relaxation Rate of 79Br in KBr Powder. J. Magn. Reson. 2009, 196 (1), 84–87. 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam S.; Schröder C.; Wegner S.; van Wüllen L. The Structure of a Borosilicate and Phosphosilicate Glasses and Its Evolution at Temperatures above the Glass Transition Temperature: Lessons from in Situ MAS NMR. Phys. Chem. Glas. - Eur. J. Glass Sci. Technology Part B 2014, 55 (6), 280–287. [Google Scholar]
- Yamada K.; Kuranaga Y.; Ueda K.; Goto S.; Okuda T.; Furukawa Y. Phase Transition and Electric Conductivity of ASnCl3 (A = Cs and CH3NH3). Bull. Chem. Soc. Jpn. 1998, 71 (1), 127–134. 10.1246/bcsj.71.127. [DOI] [Google Scholar]
- Taylor R. E.; Beckmann P. A.; Bai S.; Dybowski C. 127I and 207Pb Solid-State NMR Spectroscopy and Nuclear Spin Relaxation in PbI2: A Preliminary Study. J. Phys. Chem. C 2014, 118 (17), 9143–9153. 10.1021/jp5023423. [DOI] [Google Scholar]
- Rosales B. A.; Hanrahan M. P.; Boote B. W.; Rossini A. J.; Smith E. A.; Vela J. Lead Halide Perovskites: Challenges and Opportunities in Advanced Synthesis and Spectroscopy. ACS Energy Lett. 2017, 2 (4), 906–914. 10.1021/acsenergylett.6b00674. [DOI] [Google Scholar]
- Mitchell M. R.; Reader S. W.; Johnston K. E.; Pickard C. J.; Whittle K. R.; Ashbrook S. E. 119Sn MAS NMR and First-Principles Calculations for the Investigation of Disorder in Stannate Pyrochlores. Phys. Chem. Chem. Phys. 2011, 13 (2), 488–497. 10.1039/C0CP01274B. [DOI] [PubMed] [Google Scholar]
- Bagno A.; Casella G.; Saielli G. Relativistic DFT Calculation of 119Sn Chemical Shifts and Coupling Constants in Tin Compounds. J. Chem. Theory Comput. 2006, 2 (1), 37–46. 10.1021/ct050173k. [DOI] [PubMed] [Google Scholar]
- Sharp R. R. Rotational Diffusion and Magnetic Relaxation of 119Sn in Liquid SnCl4 and SnI4. J. Chem. Phys. 1972, 57 (12), 5321–5330. 10.1063/1.1678224. [DOI] [Google Scholar]
- Van Gompel W. T. M.; Herckens R.; Reekmans G.; Ruttens B.; D’Haen J.; Adriaensens P.; Lutsen L.; Vanderzande D. Degradation of the Formamidinium Cation and the Quantification of the Formamidinium-Methylammonium Ratio in Lead Iodide Hybrid Perovskites by Nuclear Magnetic Resonance Spectroscopy. J. Phys. Chem. C 2018, 122, 4117–4124. 10.1021/acs.jpcc.7b09805. [DOI] [Google Scholar]
- Dang Y.; Zhou Y.; Liu X.; Ju D.; Xia S.; Xia H.; Tao X. Formation of Hybrid Perovskite Tin Iodide Single Crystals by Top-Seeded Solution Growth. Angew. Chem., Int. Ed. 2016, 55 (10), 3447–3450. 10.1002/anie.201511792. [DOI] [PubMed] [Google Scholar]
- Chung I.; Song J.-H.; Im J.; Androulakis J.; Malliakas C. D.; Li H.; Freeman A. J.; Kenney J. T.; Kanatzidis M. G. CsSnI3: Semiconductor or Metal? High Electrical Conductivity and Strong Near-Infrared Photoluminescence from a Single Material. High Hole Mobility and Phase-Transitions. J. Am. Chem. Soc. 2012, 134 (20), 8579–8587. 10.1021/ja301539s. [DOI] [PubMed] [Google Scholar]
- Kofod P. Lineshapes of a Spin-12 Nucleus with Scalar Coupling to a Quadrupolar Nucleus Subject to Random Field Relaxation. J. Magn. Reson., Ser. A 1996, 119 (2), 219–224. 10.1006/jmra.1996.0076. [DOI] [Google Scholar]
- Sharma S.; Weiden N.; Weiss A. Phase Transitions in CsSnCl3 and CsPbBr3 An NMR and NQR Study. Z. Naturforsch. A 2014, 46, 329. 10.1515/zna-1991-0406. [DOI] [Google Scholar]
- Mosca R.; Ferro P.; Besagni T.; Calestani D.; Chiarella F.; Licci F. Effect of Humidity on the a.c. Impedance of CH3NH3SnCl3 Hybrid Films. Appl. Phys. A: Mater. Sci. Process. 2011, 104 (4), 1181–1187. 10.1007/s00339-011-6407-z. [DOI] [Google Scholar]
- Agashe C.; Major S. S. Effect of F, Cl and Br Doping on Electrical Properties of Sprayed SnO2 Films. J. Mater. Sci. Lett. 1996, 15 (6), 497–499. 10.1007/BF00275412. [DOI] [Google Scholar]
- Bloembergen N.; Rowland T. J. On the Nuclear Magnetic Resonance in Metals and Alloys. Acta Metall. 1953, 1 (6), 731–746. 10.1016/0001-6160(53)90033-9. [DOI] [Google Scholar]
- Borsa F.; Barnes R. G. Temperature Dependence of the Isotropic and Anisotropic Knight Shift in Polycrystalline Cadmium and β-Tin. J. Phys. Chem. Solids 1966, 27 (3), 567–573. 10.1016/0022-3697(66)90200-9. [DOI] [Google Scholar]
- Leijtens T.; Prasanna R.; Gold-Parker A.; Toney M. F.; McGehee M. D. Mechanism of Tin Oxidation and Stabilization by Lead Substitution in Tin Halide Perovskites. ACS Energy Lett. 2017, 2 (9), 2159–2165. 10.1021/acsenergylett.7b00636. [DOI] [Google Scholar]
- Korringa J. Nuclear Magnetic Relaxation and Resonance Line Shift in Metals. Physica 1950, 16 (7), 601–610. 10.1016/0031-8914(50)90105-4. [DOI] [Google Scholar]
- Kowalewski J.; Maler L.. Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications, 2nd ed.; CRC Press: 2019.
- Abragam A.The Principles of Nuclear Magnetism; Clarendon Press: 1961. [Google Scholar]
- Grutzner J. B.; Stewart K. W.; Wasylishen R. E.; Lumsden M. D.; Dybowski C.; Beckmann P. A. A New Mechanism for Spin–Lattice Relaxation of Heavy Nuclei in the Solid State: 207Pb Relaxation in Lead Nitrate. J. Am. Chem. Soc. 2001, 123 (29), 7094–7100. 10.1021/ja0040924. [DOI] [PubMed] [Google Scholar]
- Neue G.; Bai S.; Taylor R. E.; Beckmann P. A.; Vega A. J.; Dybowski C. 119Sn Spin-Lattice Relaxation in Alpha-SnF2. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 79 (21), 214302. 10.1103/PhysRevB.79.214302. [DOI] [Google Scholar]
- Shmyreva A. A.; Safdari M.; Furó I.; Dvinskikh S. V. NMR Longitudinal Relaxation Enhancement in Metal Halides by Heteronuclear Polarization Exchange during Magic-Angle Spinning. J. Chem. Phys. 2016, 144 (22), 224201. 10.1063/1.4953540. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Nose S.; Umehara T.; Okuda T.; Ichiba S. 81Br NQR and 119Sn Mössbauer Study for MSnBr3 (M = Cs and CH3NH3). Bull. Chem. Soc. Jpn. 1988, 61 (12), 4265–4268. 10.1246/bcsj.61.4265. [DOI] [Google Scholar]
- Onoda-Yamamuro N.; Matsuo T.; Suga H. Thermal, Electric, and Dielectric Properties of CH3NH3SnBr3 at Low Temperatures. J. Chem. Thermodyn. 1991, 23 (10), 987–999. 10.1016/S0021-9614(05)80179-X. [DOI] [Google Scholar]
- Yamada K.; Kawaguchi H.; Matsui T.; Okuda T.; Ichiba S. Structural Phase Transition and Electrical Conductivity of the Perovskite CH3NH3Sn1-XPbxBr3 and CsSnBr3. Bull. Chem. Soc. Jpn. 1990, 63 (9), 2521–2525. 10.1246/bcsj.63.2521. [DOI] [Google Scholar]
- Van Kranendonk J.; Walker M. Theory of Quadrupolar Nuclear Spin-Lattice Relaxation Due to Anharmonic Raman Phonon Processes. Phys. Rev. Lett. 1967, 18 (17), 701–703. 10.1103/PhysRevLett.18.701. [DOI] [Google Scholar]
- Pérez-Osorio M. A.; Lin Q.; Phillips R. T.; Milot R. L.; Herz L. M.; Johnston M. B.; Giustino F. Raman Spectrum of the Organic–Inorganic Halide Perovskite CH3NH3PbI3 from First Principles and High-Resolution Low-Temperature Raman Measurements. J. Phys. Chem. C 2018, 122 (38), 21703–21717. 10.1021/acs.jpcc.8b04669. [DOI] [Google Scholar]
- Kuku T. A. Ionic Conductivity of SnI2. Solid State Ionics 1986, 20 (3), 217–222. 10.1016/0167-2738(86)90218-3. [DOI] [Google Scholar]
- Futscher M. H.; Lee J. M.; McGovern L.; Muscarella L. A.; Wang T.; Haider M. I.; Fakharuddin A.; Schmidt-Mende L.; Ehrler B. Quantification of Ion Migration in CH3NH3PbI3 Perovskite Solar Cells by Transient Capacitance Measurements. Mater. Horiz. 2019, 6, 1497. 10.1039/C9MH00445A. [DOI] [Google Scholar]
- Ansel D.; Debuigne J.; Denes G.; Pannetier J.; Lucas J. About SnF2 Stannous Fluoride V.: Conduction Characteristics. Berichte Bunsenges. Für Phys. Chem. 1978, 82 (4), 376–380. 10.1002/bbpc.197800067. [DOI] [Google Scholar]
- Shi T.; Zhang H.-S.; Meng W.; Teng Q.; Liu M.; Yang X.; Yan Y.; Yip H.-L.; Zhao Y.-J. Effects of Organic Cations on the Defect Physics of Tin Halide Perovskites. J. Mater. Chem. A 2017, 5 (29), 15124–15129. 10.1039/C7TA02662E. [DOI] [Google Scholar]
- Yang D.; Ming W.; Shi H.; Zhang L.; Du M.-H. Fast Diffusion of Native Defects and Impurities in Perovskite Solar Cell Material CH3NH3PbI3. Chem. Mater. 2016, 28 (12), 4349–4357. 10.1021/acs.chemmater.6b01348. [DOI] [Google Scholar]
- Lai M.; Obliger A.; Lu D.; Kley C. S.; Bischak C. G.; Kong Q.; Lei T.; Dou L.; Ginsberg N. S.; Limmer D. T.; et al. Intrinsic Anion Diffusivity in Lead Halide Perovskites Is Facilitated by a Soft Lattice. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (47), 11929–11934. 10.1073/pnas.1812718115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmelund T.; Scheidt R. A.; Seger B.; Kamat P. V. Bidirectional Halide Ion Exchange in Paired Lead Halide Perovskite Films with Thermal Activation. ACS Energy Lett. 2019, 4 (8), 1961–1969. 10.1021/acsenergylett.9b01280. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.