

Abstract
Telomere maintenance by telomerase is essential for continuous proliferation of human cells and is vital for the survival of stem cells and 90% of cancer cells. To compensate for telomeric DNA lost during DNA replication, telomerase processively adds GGTTAG repeats to chromosome ends by copying the template region within its RNA subunit. Between repeat additions, the RNA template must be recycled. How telomerase remains associated with substrate DNA during this critical translocation step remains unknown. Using a newly developed single-molecule telomerase activity assay utilizing high-resolution optical tweezers, we demonstrate that stable substrate DNA binding at an anchor site within telomerase facilitates the processive synthesis of telomeric repeats. The product DNA synthesized by telomerase can be recaptured by the anchor site or fold into G-quadruplex structures. Our results provide detailed mechanistic insights into telomerase catalysis, a process of critical importance in aging and cancer.
Introduction
The ends of human chromosomes, termed telomeres, are composed of G-rich GGTTAG repeats, including a single stranded overhang 1. Telomeres shrink by ~50 nucleotides with each cell division, due to the end replication problem 2. A key challenge for telomere maintenance is the G-rich nature of the telomeric repeats in human cells. Specifically, the formation of G-quadruplex (GQ) structures has been shown to affect the replication of telomeres 3 and telomerase catalysis 4. GQs are nucleic acid structures that are formed by four single stranded telomeric repeats 5,6. To compensate for telomeric DNA lost during DNA replication, continuously proliferating cells such as germ cells, stem cells, and most cancer cells express telomerase 1. Telomerase is a reverse transcriptase that processively adds telomeric repeats to the single-stranded overhang at chromosome ends 7,8. Importantly, deficiencies in telomerase lead to premature aging diseases and inappropriate activation of telomerase is a hallmark of 90% of human cancers 1,9. To enhance or inhibit telomerase activity as a therapeutic approach for these diseases, it is critical to determine the molecular mechanisms underlying telomere elongation by telomerase.
Telomerase is composed of the telomerase reverse transcriptase (TERT) protein 10, telomerase RNA (TR) 11, and several accessory proteins 12. The human telomerase ribonucleoprotein (RNP) is organized into two distinct lobes. The catalytic core consists of TERT and the pseudoknot and template regions of TR, while the H/ACA lobe contains the 3’ half of TR 13. The TERT protein has four domains: the telomerase essential N-terminal (TEN) domain, the telomerase RNA binding domain (TRBD), the reverse transcriptase domain (RT), and C-terminal extension (CTE). The TRBD, RT, and CTE encircle the template region of TR, positioning it in close proximity the catalytic residues in the RT domain 13,14. The TEN-domain directly interacts with the telomeric protein TPP1 to facilitate its recruitment to telomeres 15–17, and has been suggested to contribute to processive telomerase catalysis 18. While structural and biochemical studies have revealed many molecular details of telomerase catalysis 8,13,14, how the activities of the various TERT-domains and TR are dynamically coordinated to facilitate processive telomere elongation remains poorly understood.
Cancer cells only contain ~250 assembled telomerase RNPs 19 and interactions between telomeres and telomerase are rare 20. As a consequence, telomerase is thought to elongate telomeres in a single processive lengthening event 21,22. Consistent with this model, mutations in TERT that specifically reduce telomerase processivity have been shown to have defects in telomere maintenance 23. Therefore, telomerase processivity, i.e. the total number of telomeric repeats telomerase synthesizes in a single interaction with the telomere, is the key determinant for the extent of telomere elongation.
To synthesize six nucleotide telomeric repeats, telomerase copies the template region contained in TR 8. Telomerase processivity requires repeated cycles of melting, translocation, and reannealing of substrate DNA-TR base-pairing 8. How telomerase remains associated with substrate DNA, when DNA-RNA base-pairing is disrupted, is unknown. It has been proposed that telomerase contains an anchor site to prevent substrate dissociation during TR translocation 24–26, and some reports are consistent with the presence a secondary DNA binding site within telomerase 18,27–29. But there is neither conclusive evidence to demonstrate the existence of an anchor site within telomerase nor mechanistic insight into how such an anchor site would facilitate processive telomerase catalysis.
To address how telomerase processively synthesizes telomeric DNA it is necessary to monitor telomerase catalysis and product release in real time. Bulk primer extension assays are end-point measurements and previous single-molecule approaches to study telomerase catalysis indirectly detect product formation and have very limited resolution to address product release 30,31. To overcome these challenges, we developed a single-molecule telomerase activity assay utilizing high-resolution optical tweezers. This assay allowed us to directly measure stepwise, processive telomerase activity and simultaneously monitor conformational dynamics of the product DNA. We demonstrate that telomerase tightly associates with its DNA substrate, synthesizing multiple telomeric repeats before releasing them in a single large step. The rate at which product is released from this anchor site closely corresponds to the overall rate of product dissociation from elongating telomerase 32, suggesting that it is the main substrate binding site during processive telomere elongation. In addition, we show that product DNA released from telomerase can be recaptured by the anchor site and dynamically folds and unfolds into GQs. Our results provide detailed mechanistic insight into processive telomerase catalysis, a process critical for telomere maintenance in stem cells and cancer cell survival 1,9.
Results
A Telomerase Assay Using Optical Tweezers
To investigate substrate extension by a single telomerase ribonucleoprotein (RNP), we developed a single-molecule telomerase activity assay using dual-trap high-resolution optical tweezers (Fig. 1a). Telomerase and its substrate were attached to separate polystyrene beads (Fig. 1b). The connection between the two beads was formed by the association of telomerase with its substrate DNA (Fig. 1a,b). When applying a low constant force (4.0–4.5 pN) to the tether, substrate elongation by telomerase was measured as an increase in distance between the two beads (Fig. 1a). The force was chosen because it was sufficient for high-resolution measurements while minimizing force induced perturbations of the system. To attach telomerase to the bead, we utilized a 3xFLAG-HaloTag on TERT, modified with biotin (Fig. 1b,c). Telomerase containing FLAG-TERT and 3xFLAG-HaloTag-TERT modified with biotin, co-purified with similar relative amounts of TR, TCAB1, and dyskerin (Fig. 1c–e), indicating that the tag does not affect telomerase RNP assembly. In addition, the tag had no effect on telomerase activity, stimulation by POT1/TPP1, and the presence of neutravidin did not impact primer extension (Fig. 1f, Supplementary Fig. 1a,b). Together, these results demonstrate that the 3xFLAG-HaloTag modified with biotin fused to the N-terminus TERT does not impact telomerase assembly or catalytic activity.
Figure 1. A single-molecule telomerase extension assay using high-resolution dual-trap optical tweezers.

a, Experimental design to measure processive telomerase catalysis using dual-optical tweezers. b, Schematic showing the molecular details of tether formation to monitor DNA elongation by telomerase. c, Western blot of purified 3xFLAG- and 3xFLAG-HaloTag-TERT containing telomerase samples (representative example of 3 independent purifications), probed with anti-FLAG-M2-HRP (top) and poly-HRP-streptavidin (bottom). d, Northern blot of RNA extracted from purified 3xFLAG- and 3xFLAG-HaloTag-TERT containing telomerase samples, probed with three phosphorylated DNA oligonucleotides complementary to TR (representative example of 3 independent purifications). Standards are in vitro transcribed full-length TR. e, Western blot of purified 3xFLAG- and 3xFLAG-HaloTag-TERT containing telomerase samples, probed with anti-FLAG-M2-HRP (top), TCAB1 (middle), and dyskerin (bottom) antibodies (representative example of 3 independent purifications). f, Direct telomerase primer extension assay of 3xFLAG-HaloTag-TERT modified with biotin and 3xFLAG-TERT containing telomerase in reaction buffer containing 10 μM or 50 μM dNTPs and 50 mM KCl (LC = loading control). Relative activity is total lane intensity normalized to LC and the respective 3xFLAG-TERT telomerase activity (n = 3, mean ± s.d.). Specific activity is additionally normalized to the relative amount of TR (Fig. 1d). For uncropped gel images please see supplementary figure 5.
Telomerase Releases Multiple Repeats in a Single Step
Using this single-molecule telomerase assay, we set out to analyze how telomerase processively synthesizes telomeric repeats. If substrate DNA is only bound to telomerase by base-pairing to TR we would expect to observe a sequence of single nucleotide (nt) addition events as a result of RNA template movement (Supplementary Fig. 2a). In contrast, if the substrate also binds to telomerase at a secondary anchor site delaying nascent DNA strand release, extension events of one or more 6 nt telomeric repeats should be observed (Supplementary Fig. 2b). To monitor telomerase activity, tethers were formed in blank solution lacking nucleotides (Fig. 1a). Tethers were subjected to a force vs. extension measurement to confirm the expected tether length and verify stable telomerase-substrate complex formation (Supplementary Fig. 2c). After a brief control period, during which no activity was detected (Supplementary Fig. 2d), tethers were transferred into the adjacent solution containing nucleotides to initiate telomerase catalysis (Fig. 1a). We observed telomerase activity as a progression of stepwise increases in the tether length (Fig. 2a, Supplementary Fig. 2e–h). The total increase in extension ranged from approximately 20–200 nucleotides (nt, Fig. 2b). The observed elongation rates for the majority (56%) of traces ranged from 8–50 nt per minute (Fig. 2c), which closely agrees with rates measured in bulk experiments 19,33, indicating that the applied force did not alter telomerase activity. The elongation step sizes were broadly distributed, ranging from 5 nt to > 100 nt with the vast majority (86%) being greater than two telomeric repeats (12 nt, Fig. 2d). This implies that telomerase binds to the DNA substrate at a secondary site in addition to the TR-substrate base pairing, allowing multiple telomeric repeats to be synthesized before releasing them in a single large step.
Figure 2. Telomerase releases multiple telomeric repeats in a single step.
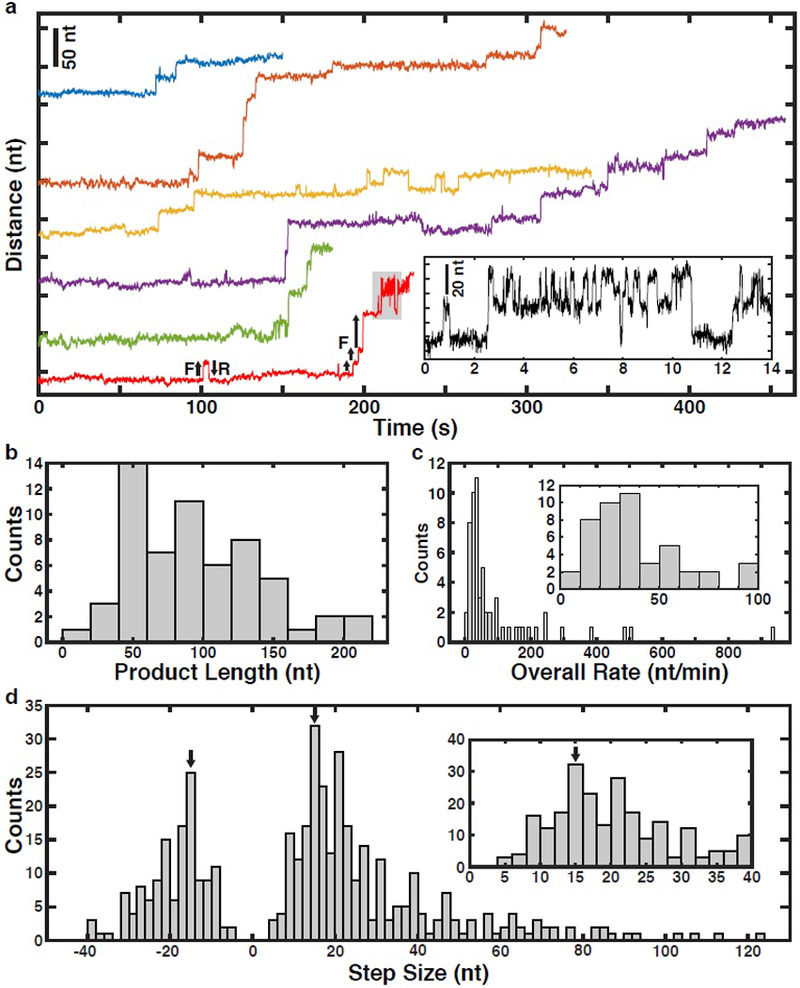
a, Representative time trajectories (120 ms per data point) of telomerase catalysis after transfer into nucleotide containing buffer (t = 0) showing processive elongation, forward (F) and reverse (R) stepping. Inset: Zoom-in of region of bottom-most example trace (in red) corresponding to the region shaded gray. b, Distribution of total tether elongation by telomerase (n = 60 traces). c, Distribution of overall elongation rate for all telomerase elongation trajectories (n = 60 traces). Inset: zoom-in around the main peak of the overall rate distribution. d, Distribution of the observed tether elongation step sizes of telomerase trajectories (n = 60 distinct tethers, 417 steps total). Inset: zoom-in around the forward step distribution peak. Arrows mark a 15 nt magnitude step size which is the expected GQ folding and unfolding step size.
Processive Stepwise Release of Product DNA by Telomerase
In cancer cells, telomerase is thought to elongate telomeres by 50 nt in a single processive elongation event 22. Given that the majority of elongation steps (68%) are shorter than 30 nt (Fig. 2d), this would require multiple anchor site release events prior to complete product dissociation. To determine the global progression of substrate elongation by telomerase we analyzed the state transition density plot of all traces (Fig. 3a), where the x-axis indicates state extension and the y-axis the state extension of the next state (the difference is the step size). The first step of all trajectories was a forward step (trajectories begin at state extension = 0 nt), after which we observed both increases and decreases in tether length (Fig. 3a). Reverse steps (data points below the diagonal) were less frequent than forward steps (data points above the diagonal) and reverse steps never exceeded 40 nt in length (also Fig. 2d). Smaller reverse steps frequently matched the preceding forward step and occasionally repetitive forward-reverse step cycling was observed (Fig. 3a, data points mirrored across diagonal, Fig. 2a). On average, telomerase elongated substrates by approximately 72 nucleotides during the first five state transitions (Fig. 3b). The average tether length remained constant from state 6 to 10 (Fig. 3b). This coincided with an increased frequency of reverse steps and the appearance of peaks in the step size distribution at approximately 15 nucleotides in both forward and reverse directions (Fig. 3b,c). Note that overall elongation does not stall, but rather smaller, reversible steps begin to dominate the overall step distributions (Fig. 3c). Together, these observations demonstrate that telomerase can undergo multiple rounds of DNA release from the anchor site without complete product dissociation.
Figure 3. Analysis of processive DNA synthesis and product release by telomerase.

a, Kernel density plot of next state extension vs. state extension associated with each step in Fig. 2d. The diagonal dashed white line is a guide dividing forward (above line) vs. reverse (below line) steps. The upper and lower dashed yellow lines are guides indicating 15 nt steps forward and reverse, respectively. b, Dot plot of the extension for the first 10 states of all telomerase trajectories (n = 60), including the mean extension (black) and individual trajectories (color). c, Distribution of step sizes for the first 5 steps (top) and all subsequent steps (bottom) for all trajectories (n = 60 tethers, 417 steps). Arrows mark a 15 nt magnitude step size. FF and FR indicate fractions of forward and reverse steps, respectively.
Product DNA Folds into GQ and Rebinds the Anchor Site
We next sought to determine the molecular mechanisms that underlie the reverse steps observed in telomerase catalysis trajectories. Reverse steps are most likely the consequence of two distinct processes: 1) Rebinding of product DNA to the anchor site, or 2) folding of product into a G-quadruplex (GQ) structure, which has been shown to affect telomerase catalysis 4. In extended telomerase tethers, the most common reverse and forward step size is approximately 15 nt (25% and 15% of transitions, respectively, Fig. 3c bottom), which is similar to the expected change in extension for a single GQ folding event under the experimental conditions (Supplementary Fig. 3) 34,35. To test whether GQ folding contributes to the reverse steps, we conducted telomerase extension experiments in the presence of lithium chloride rather than potassium chloride. Unlike potassium, lithium cations do not support GQ formation (Supplementary Fig. 4i). In the presence of lithium, we observed similar processive telomerase extension, including reverse steps (Fig. 4a). The overall extension rate was slightly reduced, consistent with bulk experiments (Fig. 4b) 4. However, lithium eliminated steps between 14 to 17 nt in the step size distribution in both the forward and reverse directions (Fig. 2d, Fig. 4c, arrows), consistent with the interpretation that GQ dynamics are the molecular mechanism underlying these length changes. The remaining reverse steps are likely a consequence of rebinding of the product DNA to the anchor site.
Figure 4. Analysis of telomerase catalysis in the presence of LiCl.

a, Representative time trajectories (120 ms per data point) of telomerase catalysis in reaction buffer containing 50 mM LiCl after transfer into dNTP containing buffer (t = 0). b, Distribution of overall elongation rate in the presence of 50 mM LiCl for all telomerase elongation trajectories (n = 16 traces). c, Distribution of the step sizes of telomerase trajectories in reaction buffer containing 50 mM LiCl (n = 16 traces, 171 steps). d, Representative time trajectories and histograms of the folding dynamics of telomeric repeat ssDNA capable of forming a single GQ, showing simultaneous trap distance (top, 48 ms per data point) and FRET measurements (bottom, donor in green, acceptor in red, 48 ms per data point) for a single tether. e, Step size distribution of folding and unfolding events observed in single GQ tethers (n = 22 tethers, 344 steps). f, Representative time trajectory and histogram of the folding dynamics of telomeric repeat ssDNA capable of forming up to three GQs (48 ms per data point). g, Step size distribution of folding and unfolding events observed in multi GQ tethers (n = 6 tethers, 390 steps).
To verify that GQ folding and unfolding occurs under our experiment conditions with the predicted step size of ~15 nt and on time scales relevant for our experiments, we performed tweezers experiments tethering GQ sequences without telomerase. A single stranded DNA (ssDNA) sequence capable of forming one GQ dynamically folded and unfolded into two distinct folded extensions (F1 and F2, with extensions 18.0 ± 0.1 nt and 22.1 ± 0.2 nt, respectively, Fig. 4d,e), consistent with previous observations 36 and expectations for distinct GQ topologies (Supplementary Fig. 3). Simultaneous Förster resonance energy transfer (FRET) measurements 37 confirmed the presence of two folded extensions (Fig. 4d, Supplementary Fig. 4a,b). An extended ssDNA sequence able to form up to three GQ structures dynamically folded and unfolded with slightly shorter step sizes compared to the single GQ sequence (17.0 ± 0.5 nt and 19.8 ± 0.5 nt, Fig. 4f,g, Supplementary Fig. 4h). Importantly, the ssDNA sequence capable of forming up to three GQs only transiently formed a single GQ in lithium containing buffer (Supplemental Fig. 4i), confirming that GQ formation is suppressed under these conditions. Single GQ-tethers folded and unfolded with a mean dwell time of 11 s (Fig. 4d). The folding dwell time distribution was a single exponential as expected ( s), while unfolding distribution from both folded extensions was fit by a sum of three exponentials (Supplementary Fig. 4c,d). Separating the unfolding dwell times by folded extensions F1 and F2 revealed that both folded extensions unfolded with two distinct rate constants, yielding a total of at least four total folded states, in agreement with previous observations 5,38 (Supplementary Fig. 4e–g). The distribution of all dwell times for the multi-GQ was fit by a minimum of three exponentials with a mean dwell time of 4 s (Supplementary Fig. 4j). The folding dwell time distribution of multi GQ tethers from the completely unfolded state was a single exponential ( s) (Supplementary Fig. 4k). The unfolding dwell time distribution for multi GQ tethers was fit by a sum of three exponentials, which reflects the complexity of the underlying conformational dynamics (Supplementary Fig. 4l). These results confirm that single stranded telomeric DNA can rapidly fold and unfold into a variety of GQ structures with a range of sizes under our experimental conditions. In addition, the step sizes of GQ dynamics largely overlapped with the predicted length change for GQ formation.
Substrate DNA Dissociates Slowly from the Anchor Site
The affinity of telomerase for substrate DNA is a key contributing factor to processive telomere repeat synthesis 39. To estimate the affinity of ssDNA for the telomerase anchor site, we analyzed the dwell times prior to the first elongation step of all telomerase trajectories. Since product release is a pre-requisite for the observation of the initial tether extension, this dwell time should reflect the dissociation rate of ssDNA from the telomerase anchor site. Consistent with this hypothesis, the dwell time distribution prior to the first step was well fit by a single exponential decay with a time constant of s (Fig. 5a). After the initial extension step, telomerase could exist in at least two configurations: telomerase RNPs with released ssDNA that is either unfolded or folded into a GQ (Fig. 6). Indeed, the dwell time distributions for the second and third steps fit to the sum of two exponentials (Fig. 5b,c). The slow time constants for the second and third steps ( s, s, weights , ) were comparable to the anchor site release time. The fast time constants were approximately 30-fold faster ( s, s, weights , ), indicating that a second, more rapid, process is occurring in addition to anchor site release. Analysis of the dwell times across the first 10 transitions of all trajectories revealed a gradual decrease of the mean dwell time from approximately 95 s to 10 s from steps 1 to 5 (Fig. 5d). The mean dwell time remained constant from steps 5 to 10 (Fig. 5d). The reduction in mean dwell time to approximately 10 s is concomitant with the increased frequency of reverse steps (Fig. 3b,c) and is very close to the mean dwell time of 11 s that we measured for the single GQ sequence folding and unfolding (Fig. 4d). These observations suggest that GQ folding and unfolding is the molecular process underlying the faster rate in tether length changes.
Figure 5. Dissociation from the anchor site controls product release by telomerase.

a-c, Inverse cumulative distribution of dwell times for the a first, b second, and c third step of all telomerase trajectories (black staircase, n = 60 traces). Solid red line indicates a single (a) or double (b,c) exponential fit, dashed lines the 90% confidence intervals. d, Dot plot of individual dwell times for the first 10 steps of all telomerase trajectories (n = 60). Mean dwell time is indicated at the top of the graph and by the black horizontal lines.
Figure 6. Model for processive telomerase catalysis.

Telomerase associates with substrate DNA by base-pairing with the telomerase RNA template and at an anchor site. Multiple cycles of repeat synthesis, RNA-DNA melting, RNA translocation, and reannealing occur without product being released from the anchor site, leading to the formation of a product loop (newly synthesized DNA in red). When the product DNA dissociates from the anchor site multiple telomeric repeats are released in a single large step. Released product DNA can be recaptured by the anchor site. After and potentially before release, the product DNA can dynamically fold and unfold G-Quadruplex structures.
Discussion
Telomere maintenance by telomerase is essential for the survival of stem cell populations and the majority of cancer cells. Telomerase is thought to elongate telomeres in a single processive elongation event, but how telomerase remains bound to the chromosome end through repeated cycles of RNA-template translocation was a key unanswered question. The single-molecule analysis presented here provides detailed mechanistic insight into the critical role of an anchor site in processive telomerase catalysis (Fig. 6). In addition, we demonstrate that the product synthesized by telomerase folds into GQ structures and dynamically rebinds the anchor site (Fig. 6).
The presence of a second DNA binding site within telomerase, in addition to RNA-DNA base pairing, was proposed soon after the identification of telomerase 25,40–42. Since telomerase had been demonstrated to be processive 7, but RNA-DNA base pairing had to be disrupted to allow RNA template translocation, the presence of an anchor site was an attractive model to explain how telomerase can synthesize more than a single telomeric repeat without dissociating from its substrate. Since then, various studies have assessed the DNA binding properties of TERT from yeast 28,43, ciliates 27, and humans 18,44, but did not conclusively demonstrate the existence of an anchor site in the assembled telomerase RNP. The data presented here strongly suggests that the fully assembled human telomerase RNP actively engaged in catalysis contains a secondary DNA binding site. Our results demonstrate that multiple telomeric repeats are synthesized without release of the nascent DNA from telomerase. Since processive repeat synthesis by telomerase requires temporarily disrupting RNA-DNA base pairing, an additional anchor site must be present to prevent product dissociation (Fig. 6).
The single exponential kinetics of anchor site release suggest that a single interface between the telomerase RNP and substrate DNA is responsible for anchor site function. We cannot rule out the possibility of two distinct anchor sites. But to be consistent with our results, the affinities of these separate anchor sites would have to be very similar, which is unlikely. Reports by others have implicated the TEN-domain of TERT from various species, including human, in DNA binding 27,28,44. The TEN-domain is positioned adjacent to the catalytic ring formed by the TRBD, RT, and CTE of TERT, placing it in close proximity to the 5’-end of the DNA substrate 13. We therefore believe that the TEN-domain is a likely candidate to harbor the anchor site, but we cannot rule out that other TERT domains or telomerase RNP components contribute to anchor site function.
Product release by the anchor site could have two distinct mechanisms. The anchor site could bind to a single portion of ssDNA and product release would result in the complete dissociation of the anchor site from the ssDNA followed by product rebinding. In our tweezers assay, such ssDNA release and rebinding steps would be clearly observable as increases and decreases respectively in tether extension. However, we frequently observe consecutive product release steps without observing any intervening anchor site rebinding (reverse) steps (Supplementary Fig. 2e–h). We also observed partial product release (Supplementary Fig. 2e, it is unlikely that telomerase produced 40 nt of ssDNA in the ~1.5 s intervening release steps). These results suggest an alternate anchor site mechanism, where dissociation from one portion of ssDNA occurs simultaneously with binding to another portion of ssDNA. In the case of telomerase this mechanism would increase processivity by minimizing the amount of time that the anchor site is not bound to the substrate.
The anchor site tightly associates with substrate DNA. But, the half-life of anchor site binding (t1/2 = 1.1 min) is substantially shorter than the half-life of primer binding to the telomerase RNP not engaged in catalysis (t1/2 = 15 – 1200 min) 45. This is consistent with the anchor site and RNA-DNA base pairing contributing to the affinity of telomerase for ssDNA when it is idle. Our measured anchor site off-rate (koff = 0.01±0.001 s−1) together with a conservative estimation of the on-rate (kon = 106 M−1s−1) results in a dissociation constant of 10 nM. This dissociation constant is comparable to the Michaelis-Menten constant for telomerase relative to the DNA substrate (Km = 10–20 nM) 46,47. Considering that DNA substrate turnover by telomerase is fairly slow, this Km should be comparable to the dissociation constant of primer DNA binding to telomerase that is actively synthesizing telomeric repeats. Therefore, the close correspondence of the anchor site dissociation constant and the Michaelis-Menten constant of telomerase and substrate DNA suggests that the anchor site is the main DNA binding site when telomerase is engaged in catalysis. Consistent with this model, the half-life of substrate bound to the anchor site (t1/2 = 1.1 min) is similar to the half-life of primer bound to telomerase that is actively synthesizing telomeric repeats (t1/2 = 1.0 min) 32. Importantly, both the Km and half-life of primer binding to actively elongating telomerase were determined under conditions with low levels of dGTP. When dGTP is limiting, the initiation of repeat synthesis after translocation is slow (Fig. 6, inset) 33. Thus, telomerase is “stuck” with minimal base-pairing and the anchor site is especially important to prevent substrate dissociation (Fig. 6, inset).
In addition to directly visualizing product release, we were also able to analyze conformational dynamics of the telomeric DNA synthesized by telomerase. Several lines of evidence suggest that product DNA released from telomerase forms GQs. First, the frequency of GQ folding (shortening in tether length), increases with product length, because longer stretches of telomeric repeats have more opportunities to form a GQ (Fig. 3). Second, this increased frequency in tether length reductions coincides with the appearance of peaks in the step size distribution (~14–17 nt) in both forward and reverse direction that match the predicted step size of GQ formation (Fig. 3). Third, in the presence of lithium cations, which do not support GQ formation, precisely these peaks centered around ±15 nt in length are completely absent (Fig. 4). In addition, in the absence of telomerase, telomeric repeat DNA folded and unfolded with similar step sizes and dwell times as length changes observed in extended telomerase products. It is also possible that GQs can form in the product ssDNA prior to release from the anchor site, as has been reported by others 4. Future studies, combining our ability to monitor anchor site release with FRET probes reporting on product folding will be required to address this question.
Importantly, not all reverse steps can be explained by folding of the product ssDNA into a GQ. Reverse step sizes both shorter and longer than GQ folding events were observed (Fig. 2), and we also observed reverse steps in the presence of lithium, where GQ formation is suppressed (Fig. 4). These reverse steps likely reflect recapture of the product DNA by the anchor site. Although, some tether length changes could be the result of force induced conformational changes, we believe this is unlikely because no length changes were observed in the absence of nucleotides. Overall, release steps have a broader size distribution (up to 120 nt) than reverse steps (up to 40 nt). The shorter length in the distribution of reverse steps is likely a consequence of the bias imposed by the applied force. Since the product DNA is partially extended by the force, segments closer to the RNP are more likely to rebind. The frequency of reverse steps is comparable (~40%) in potassium and lithium containing buffer. Since GQ folding is suppressed in the presence of lithium, this suggests that the frequency of rebinding is higher under these conditions. It is possible that anchor site binding of product DNA and GQ formation are mutually exclusive, which would increase the chances of product recapture when telomerase elongates DNA in lithium containing buffer. Consistent with this interpretation a recent study demonstrated that GQ formation increases the dissociation rate of product DNA from telomerase 4, potentially by interfering with anchor site binding.
The results presented in this study address a long-standing question: How does telomerase remain bound to its DNA substrate when RNA-DNA base pairing is disrupted during the translocation step of the telomerase catalytic cycle? The anchor site facilitates synthesis of multiple telomeric repeats, which could loop out from the catalytic core, prior to product release in a single large step (Fig. 6). This suggests that telomerase can synthesize a sufficient amount of telomeric repeats to compensate for the ~50 nucleotides lost during DNA replication without releasing the chromosome end from the anchor site. Furthermore, since telomerase can bind a large loop of single stranded DNA it is also possible that the anchor site captures the chromosome end at an internal site rather than directly at the 3’ end.
In total our results provide a comprehensive picture of the molecular mechanisms underlying processive telomerase catalysis, which represent an important advance in our understanding of telomere maintenance, a cellular process that plays a central role in human aging, as well as cancer formation and survival 1.
Online Methods
Telomerase expression and purification
Telomerase was expressed in HEK293T cells as previously described 48. 30×106 cells were transfected with 15 μg of TERT plasmid (ProA-3xFLAG- or 3xFLAG-HaloTag-TERT) and 60 μg of pSUPER-hTR using 150 μl of Lipofectamine 2000 (Invitrogen) in a total volume of 7.5 ml of OPTI-MEM (Gibco). Cells were harvested and lysed with CHAPS buffer (10 mM TRIS-HCl pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.5% CHAPS, 10% glycerol, 5 mM beta-mercapto-ethanol) 48 hours after transfection (100×106 cells). Telomerase was bound to Anti-FLAG-M2 affinity gel (Sigma-Aldrich, A2220, 100 μl of resin), washed three times with washing buffer (20 mM HEPES pH 7.9, 300 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 0,1% Triton X-100, 10% glycerol), and eluted with 200 μl of wash buffer supplemented with 0.15 mg/ml 3xFLAG peptide (Sigma-Aldrich). Telomerase quantity, composition, assembly, and biotin modification was assessed with western and northern blots as previously described 46, using TR probes 1, 2, and 3 (IDT, Supplementary Table 1), and antibodies against FLAG (Sigma-Aldrich, anti-FLAG-M2), TCAB1 (Proteintech, 14761–1-AP), dyskerin (Santa Cruz, sc-373956), and poly-HRP-streptavidin (Thermo Scientific. #21140). TR standards for northern blots were in vitro transcribed from PCR products using the HiScribe™ T7 high yield RNA synthesis kit (New England Biolabs). Band intensities were quantified using ImageQuant TL 8.2 (GE Healthcare Life Sciences).
Direct telomerase primer elongation assay
Telomerase activity assays were carried out for 1 hour at 30 °C as previously described in reaction buffer (50 mM TRIS-HCl pH 8.0, 50 mM or 150 mM KCl, 1 mM MgCl2) 49. Assays with telomerase alone were carried out with 50 nM primer A, 50 mM KCl, 10 μM or 50 μM dATP, dTTP, and dGTP, and 0.33 μM dGTP [α−32P] (Perkin Elmer). Telomerase assays to assess POT1/TPP1 stimulation were carried out with 50 nM primer A5, 150 mM KCl, 100 nM POT1/TPP1, 500 μM dATP, dTTP, 3.3 μM dGTP, and 0.33 μM dGTP [α−32P] (Perkin Elmer). POT1/TPP1 was purified as previously described 15. Telomerase products were separated on 12% poly-acrylamide, 1xTBE, 7 M Urea, sequencing gels and detected using storage phosphor screens (GE Healthcare Life Sciences, BAS-IP MS 3543) and an Amersham Typhoon IP (GE Healthcare Life Sciences). Phosphorylated primer A-2 was used as a loading control. Activity was quantified as total radioactive incorporated per lane normalized to the loading control. To compare activity between 3xFLAG- and 3xFLAG-HaloTag containing telomerase, specific activity was calculated by dividing the activity measured by telomerase assays by the TR levels determined by northern blot. Processivity was quantified as the intensity of products longer than 6 repeats divided by the total signal per lane.
Optical trapping measurements
All optical trapping measurements were carried out at 23 °C. Optical trapping was carried out using a home-built timeshared dual optical trap combined with confocal single-molecule fluorescence microscope instrument as previously described in detail 37,50,51. Briefly, an acousto-optic modulator (AOM) deflected a 1,064 nm laser (IPG) between two positions to create two independent optical traps (66.7 kHz modulation rate). A PID (proportional-integral-derivative) feedback loop stabilized the trap laser intensity to maintain constant trap stiffness (0.2–0.3 pN/nm) during trap positioning. Bead positions were measured at 66.7 kHz sampling rate using the transmitted trap laser light by infrared-enhanced quadrant photodiodes (Pacific Silicon Devices) via the standard back focal plane interferometry method. Bead position and trap stiffness calibrations were performed via standard fitting of the bead Brownian motion power spectra. A 532 nm fluorescence excitation laser was coaligned between the dual traps at the center of the tether and interlaced with a second AOM to be on only during trap laser off intervals. Fluorescence emission was collected confocally and split by color into standard donor (Cy3) and acceptor (Cy5) emission channels and detected by a pair of single photon counting avalanche photodiodes (APD). Instrument control and data acquisition were performed using a field programmable gate array-based PC card system (National Instruments PCIe-7852R). Instrument control software was written in LabVIEW version 2012 (National Instruments).
The substrate handle (SH) was generated by annealing the substrate handle oligo- nucleotides 1 and 2 (IDT, Supplementary Table 1). The biotin-digoxigenin handle (BDH) was generate by PCR using biotin and digoxygenin modified oligos 1 and 2 (IDT, Supplementary Table 1) and pBR322 as template. To establish tethers, the telomerase-substrate complex was formed by incubating 3xFLAG-HaloTag(biotin)-TERT containing telomerase with the SH for 30 minutes at room temperature, prior to binding of the complex to 1 μM diameter polystyrene beads (Spherotech) functionalized with an anti-digoxigenin antibody (Roche) for 10 minutes at room temperature. The BDH was incubated with a 50-fold excess of neutravidin (Thermo Scientific) for 10 minutes at room temperature, prior to binding to anti-digoxigenin antibody modified polystyrene beads 52. Tethers were formed by holding the beads close together in telomerase reaction buffer lacking nucleotides (50 mM TRIS-HCl pH 8.0, 50 mM KCl, 1 mM MgCl2, 1% glucose, 1 mg/ml glucose oxidase, 0.13 mg/ml catalase, 1 mg/ml Trolox) under continuous laminar flow in a multi-channel home-made chamber 53 to establish the linkage between the neutravidin on the BDH and the biotin-modified 3xFLAG-HaloTag-TERT telomerase. Stable tethers were subjected to force-extension measurements at a ramp rate of 100 nm s−1, which were fit to worm-like chain model DNA polymer modelling parameters. Measurements of telomerase catalysis were initiated in blank buffer. After a control period of approximately 30 seconds, tethers were transferred into reaction buffer supplemented with 50 μM dATP, dTTP, and dGTP. To determine the contribution of G-quadruplex folding and unfolding on product extension by telomerase, trapping experiments were carried out in buffers containing 50 mM LiCl instead of KCl.
To analyze GQ folding and unfolding G-quadruplex forming oligos GGG(TTAGGG)3 and (TTAGGG)12 were purchased from IDT (Supplementary Table 1). GQ constructs were generated by ligating the oligos to DNA handles amplified from Lambda DNA and digested with TspRI (right handle, RH) and pBR322 digested with HindIII or PspGI to make the left handle (LH). The RH and LH were amplified using primers modified with digoxygenin and biotin, respectively (Supplementary Table 1). FRET donor and acceptor fluorophores flanking the single GQ sequence were added by annealing and ligating Cy3 and Cy5 labeled 9 nt long oligos to complimentary sequences flanking the GQ sequence. The GQ construct was incubated with the streptavidin coated beads (Spherotech) for 1 hour and tethers were formed in situ by establishing a digoxigenin-anti-digoxigenin linkage of the RH with anti-digoxygenin antibody coated beads. Stable tethers were first subjected to force extension at 100 nm s−1 spanning a force range from 0–30 pN to unfold any pre-existing structures and to assess tether quality. The trap distance was then set to apply a constant force of 4–4.5 pN and measurements were carried in force-feedback mode.
Template elongation and GQ-dynamics were monitored as a change extension in the tether length by tracking the bead position within the movable trap. The optical trap was operated under active force feedback, where bead positions within the traps were monitored and kept constant by adjusting the location of one trap, thereby applying a constant force of 4 – 4.5 pN to the tether. The recorded motion of the trap tracked the changing extension of the tether e.g., the elongation activity of telomerase, release and rebinding of product, folding and unfolding of GQ structures etc. The change in location of the trap was calibrated to the equivalent change in the number of tether ssDNA nucleotides (nt, e.g., due to the catalytic elongation of the tethered DNA substrate) via standard ssDNA polymer modeling (via the extensible freely jointed chain model, persistence length = 0.75 nm, contour length = 0.56 nm/nt, stretch modulus = 800 pN). Force extension were carried out at 100 nm s-1. The models for tether extension vs force are produced by the sum of the standard polymer models of dsDNA representing the handles (extensible worm-like chain model, persistence length = 53 nm, contour length = 0.34 nm/bp, stretch modulus = 1200 pN) and the relevant quantity of ssDNA 54–56.
Data analysis
Offline data analysis was performed using standard methods implemented via custom codes programmed in MATLAB version 2018b (Mathworks). Data collected at 66.7 kHz was boxcar averaged to a final lower rate, as specified, but generally sufficiently fast to accurately determine telomerase template elongation states and lifetimes, between 1.5 and 24 ms per data point. Automatic state and step finding in telomerase trajectories was carried out using the code XL-ICON which is a MATLAB based implementation of the infinite Hidden Markov Modelling (iHMM) method which is a Bayesian nonparametric extension of the very commonly used HMM method and allows for state discovery with simultaneous correction for high-resolution tweezers measurement drift and finite response time 57. For the iHMM analysis of telomerase activity vs time using the XL-ICON code, the following priors and settings were used. Note: The value of priors affected only the efficiency of sampling and analysis convergence, not the final results. The key priors are: The mean number of active states prior was set to two (even though the number of states was generally > 2), the mean dwell time prior was set to 10 s, and the mode dwell time prior was set to 3.3 s. To give priors on the tweezers measurement emission, ‘noise’ was sampled from the specific data file from a region lacking transitions and for a duration of 5 to 10 s. The related priors were set to noise_a = 2 (Gaussian shape for noise) and noise_s = the standard deviation of the noise (scale for noise). The drift node density was fixed at one drift node every 2 s and splines were used to interpolate the drift between nodes. Additional priors: M = 50 (weak limit approximation), K = 3 (trap response autoregressive order), resp_m = 0 (trap response coefficient mean), resp_s = 1/3 (trap response coefficient standard deviation), and drift_a = 2 (beta prior for drift node placement). Regions of a small number of traces (4 out of 60) were excluded from contributing to the state and step size distributions because they had a very large number of reversing states that would have obscured the contributions from the remainder of traces. The overall extension rate was calculated by dividing the maximal extension of each trace by the time point at which it was first observed. Step finding for GQ trajectories was carried out in a simpler manner using standard HMM methods because the total length changes were limited to the folded and unfolded extensions unlike the highly processive telomerase elongation data. All trajectories were manually inspected for accuracy of the automated state and step finding. Step sizes in nm were converted to nucleotides using the force dependent extension based on the freely jointed chain model using the same parameters as above. For dwell time analysis, fitting was never performed on binned data. For dwell time distributions well fit by single exponentials, the mean dwell time, which is the maximum likelihood estimator for the time constant, is reported along with the standard error of the mean to estimate uncertainty. For dwell time distributions that were well fit by the sum of two or three exponentials (e.g., the unfolding of the single GQ tether and the distribution of all multi-GQ state dwell times), fitting was performed by minimizing the least squares error in the dwell time cumulative distribution. Error was estimated by finding fit parameter one sigma (68%) confidence bounds by standard bootstrapping of the fitting using resampled data with replacement.
Supplementary Material
Acknowledgements
We thank Jayakrishnan Nandakumar and Iain Cheeseman for comments on the manuscript. This work was support by grants from the NSF (MCB-1919439) to M.J.C. and the NIH (R00 GM120386) to J.C.S. J.C.S. is a Damon Runyon Dale F. Frey Scientist supported (in part) by the Damon Runyon Cancer Research Foundation (DFS-24-17).
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
Data availability
All data generated or analyzed during this study are included in this manuscript (and its supplemental information). Reagents and raw data are available from J.C.S. and M.J.C. upon request.
Computer Code
Custom MATLAB code used to analyze the data generated in this study is available from M.J.C. upon request.
References
- 1.Stewart SA & Weinberg RA Telomeres: cancer to human aging. Annu Rev Cell Dev Biol 22, 531–557 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Harley CB, Futcher AB & Greider CW Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Sfeir A et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansson LI et al. Telomere DNA G-quadruplex folding within actively extending human telomerase. Proceedings of the National Academy of Sciences 116, 9350–9359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra J et al. Extreme mechanical diversity of human telomeric DNA revealed by fluorescence-force spectroscopy. Proceedings of the National Academy of Sciences 116, 8350–8359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abraham Punnoose J et al. Random Formation of G-Quadruplexes in the Full-Length Human Telomere Overhangs Leads to a Kinetic Folding Pattern with Targetable Vacant G-Tracts. Biochemistry 57, 6946–6955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greider CW Telomerase is processive. Mol. Cell. Biol. 11, 4572–4580 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu RA, Upton HE, Vogan JM & Collins K Telomerase Mechanism of Telomere Synthesis. Annu. Rev. Biochem 86, 439–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armanios M & Blackburn EH The telomere syndromes. Nat Rev Genet 13, 693–704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura TM et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Feng J et al. The RNA component of human telomerase. Science 269, 1236–1241 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Blackburn EH & Collins K Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol 3, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen THD et al. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557, 190–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J et al. Structure of Telomerase with Telomeric DNA. Cell 173, 1179–1190.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt JC, Dalby AB & Cech TR Identification of human TERT elements necessary for telomerase recruitment to telomeres. Elife 3, e03563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nandakumar J et al. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 492, 285–289 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong FL et al. TPP1 OB-Fold Domain Controls Telomere Maintenance by Recruiting Telomerase to Chromosome Ends. Cell 150, 481–494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robart AR & Collins K Human telomerase domain interactions capture DNA for TEN domain-dependent processive elongation. Mol Cell 42, 308–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xi L & Cech TR Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res 42, 8565–8577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt JC, Zaug AJ & Cech TR Live Cell Imaging Reveals the Dynamics of Telomerase Recruitment to Telomeres. Cell 166, 1188–1197.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y et al. Processive and distributive extension of human telomeres by telomerase under homeostatic and nonequilibrium conditions. Mol Cell 42, 297–307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y et al. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell 138, 463–475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu RA, Tam J & Collins K DNA-binding determinants and cellular thresholds for human telomerase repeat addition processivity. EMBO J 36, 1908–1927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greider CW & Blackburn EH Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405–413 (1985). [DOI] [PubMed] [Google Scholar]
- 25.Morin GB Recognition of a chromosome truncation site associated with alpha-thalassaemia by human telomerase. Nature 353, 454–456 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Collins K & Greider CW Tetrahymena telomerase catalyzes nucleolytic cleavage and nonprocessive elongation. Genes Dev 7, 1364–1376 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Jacobs SA, Podell ER & Cech TR Crystal structure of the essential N-terminal domain of telomerase reverse transcriptase. Nat Struct Mol Biol 13, 218–225 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Lue NF A Physical and Functional Constituent of Telomerase Anchor Site. J Biol Chem 280, 26586–26591 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiyama BM, Parks JW & Stone MD The telomerase essential N-terminal domain promotes DNA synthesis by stabilizing short RNA–DNA hybrids. Nucleic Acids Res 43, 5537–5549 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang H, Opresko P & Myong S Single-molecule real-time detection of telomerase extension activity. Sci Rep 4, 6391–6391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parks JW & Stone MD Coordinated DNA dynamics during the human telomerase catalytic cycle. Nature Communications 5, 4146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalby AB, Hofr C & Cech TR Contributions of the TEL-patch Amino Acid Cluster on TPP1 to Telomeric DNA Synthesis by Human Telomerase. J Mol Biol 427, 1291–1303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Podlevsky JD, Logeswaran D & Chen JJ-L A single nucleotide incorporation step limits human telomerase repeat addition activity. EMBO J 37, e97953 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai J, Carver M, Punchihewa C, Jones RA & Yang D Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res 35, 4927–4940 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phan AT Human telomeric G-quadruplex: structures of DNA and RNA sequences. FEBS J. 277, 1107–1117 (2010). [DOI] [PubMed] [Google Scholar]
- 36.You H et al. Dynamics and stability of polymorphic human telomeric G-quadruplex under tension. Nucleic Acids Res 42, 8789–8795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comstock MJ, Ha T & Chemla YR Ultrahigh-resolution optical trap with single-fluorophore sensitivity. Nat Meth 8, 335–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JY, Okumus B, Kim DS & Ha T Extreme conformational diversity in human telomeric DNA. Proc Natl Acad Sci USA 102, 18938–18943 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latrick CM & Cech TR POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J 29, 924–933 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morin GB The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529 (1989). [DOI] [PubMed] [Google Scholar]
- 41.Harrington LA & Greider CW Telomerase primer specificity and chromosome healing. Nature 353, 451–454 (1991). [DOI] [PubMed] [Google Scholar]
- 42.Blackburn EH et al. Recognition and elongation of telomeres by telomerase. Genome 31, 553–560 (1989). [DOI] [PubMed] [Google Scholar]
- 43.Shastry S, Steinberg-Neifach O, Lue N & Stone MD Direct observation of nucleic acid binding dynamics by the telomerase essential N-terminal domain. Nucleic Acids Res 46, 3088–3102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sealey DCF et al. The N-terminus of hTERT contains a DNA-binding domain and is required for telomerase activity and cellular immortalization. Nucleic Acids Res 38, 2019–2035 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallweber G, Gryaznov S, Pongracz K & Pruzan R Interaction of human telomerase with its primer substrate. Biochemistry 42, 589–600 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Schmidt JC, Zaug AJ, Kufer R & Cech TR Dynamics of human telomerase recruitment depend on template-telomere base pairing. 29, 869–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jurczyluk J et al. Direct involvement of the TEN domain at the active site of human telomerase. Nucleic Acids Res 39, 1774–1788 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 48.Sauerwald A et al. Structure of active dimeric human telomerase. Nat Struct Mol Biol 20, 454–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaug AJ, Crary SM, Jesse Fioravanti M, Campbell K & Cech TR Many disease-associated variants of hTERT retain high telomerase enzymatic activity. Nucleic Acids Res 41, 8969–8978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitley KD, Comstock MJ & Chemla YR High-Resolution ‘Fleezers’: Dual-Trap Optical Tweezers Combined with Single-Molecule Fluorescence Detection. Methods Mol. Biol 1486, 183–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitley KD, Comstock MJ & Chemla YR High-Resolution Optical Tweezers Combined With Single-Molecule Confocal Microscopy. Meth. Enzymol 582, 137–169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Righini M et al. Full molecular trajectories of RNA polymerase at single base-pair resolution. Proceedings of the National Academy of Sciences 115, 1286–1291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patrick EM, Srinivasan S, Jankowsky E & Comstock MJ The RNA helicase Mtr4p is a duplex-sensing translocase. Nat Chem Biol 13, 99–104 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Bustamante C, Marko JF, Siggia ED & Smith S Entropic elasticity of lambda-phage DNA. Science 265, 1599–1600 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Smith SB, Cui Y & Bustamante C Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science 271, 795–799 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Cluzel P et al. DNA: an extensible molecule. Science 271, 792–794 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Sgouralis I, Whitmore M, Lapidus L, Comstock MJ & Pressé S Single molecule force spectroscopy at high data acquisition: A Bayesian nonparametric analysis. J Chem Phys 148, 123320 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.