

Abstract
Glucose is catabolized by two fundamental pathways, glycolysis to make ATP and the oxidative pentose phosphate pathway to make NADPH. The first step of the oxidative pentose phosphate pathway is catalyzed by the enzyme glucose-6-phosphate dehydrogenase (G6PD). Here we develop metabolite reporter and deuterium tracer assays to monitor cellular G6PD activity. Using these, we show that the most widely cited G6PD antagonist, dehydroepiandosterone (DHEA), does not robustly inhibit G6PD in cells. We then identify a small molecule (G6PDi-1) that more effectively inhibits G6PD. Across a range of cultured cells, G6PDi-1 depletes NADPH most strongly in lymphocytes. In T cells but not macrophages, G6PDi-1 markedly decreases inflammatory cytokine production. In neutrophils, it suppresses respiratory burst. Thus, we provide a cell-active small molecule tool for oxidative pentose phosphate pathway inhibition, and use it to identify G6PD as a pharmacological target for modulating immune response.
Introduction
Across all forms of life, the redox cofactor NADPH donates high-energy electrons for reductive biosynthesis and antioxidant defense 1. The critical nature of these processes requires effective maintenance of the levels of NADPH and its redox partner NADP+. In the cytosol of mammalian cells, reduction of NADP+ to NADPH mainly occurs via three routes: malic enzyme 1 (ME1), isocitrate dehydrogenase 1 (IDH1), and the oxidative pentose phosphate pathway (oxPPP) 2. While ME1 and IDH1 extract hydrides from TCA-derived metabolites, the oxPPP diverts glucose-6-phospate from glycolysis to generate two equivalents of NADPH; one by G6PD, which catalyzes the first and committed step, and one by 6-phosphogluconate dehydrogenase (PGD).
G6PD is ubiquitously expressed in mammalian tissues, with highest expression in immune cells and testes 3. It is also often upregulated in tumors 4–7. Genetically, G6PD knockout mice are inviable 8. Nevertheless, G6PD hypomorphic alleles are common in humans, affecting ~1 in 20 people world-wide 9. These mutations provide protection from malaria, but sensitize mature red blood cells (RBCs) to oxidative stressors. The vulnerability of RBCs to mutant G6PD may reflect RBC’s lack of mitochondria and thus inability to endogenously produce the substrates of ME1 or IDH1. Alternatively, it may reflect RBC’s lack of nuclei and thus inability to replace the mutant G6PD protein as the cells age. In other tissues, the function of G6PD is less investigated. Using a genetic approach, we recently showed that cancer cell lines lacking G6PD have elevated NADP+ levels, but are nevertheless able to proliferate and maintain NADPH pools through compensatory ME1 and/or IDH1 flux 10. Whether non-transformed cells are similarly flexible remains unclear.
Potent and selective small molecule inhibitors are useful tools for studying the function of metabolic enzymes. To date, several small molecule inhibitors of G6PD have been described 11–13, most notably the steroid derivative dehydroepiandosterone (1) (DHEA, Figure 1a). First reported in 1960, DHEA binds mammalian G6PD uncompetitively against both reaction substrates 14. Since then, DHEA and its derivatives have been employed as G6PD inhibitors in hundreds of studies, including a variety of in vitro and in vivo cancer settings where they display anti-proliferative activity 15–17. However, these readouts of cellular activity are indirect, and it has been proposed that the effects of DHEA may arise from alternative mechanisms other than G6PD inhibition 15,18.
Figure 1. Cellular target engagement assays reveal lack of effective G6PD inhibition by DHEA.
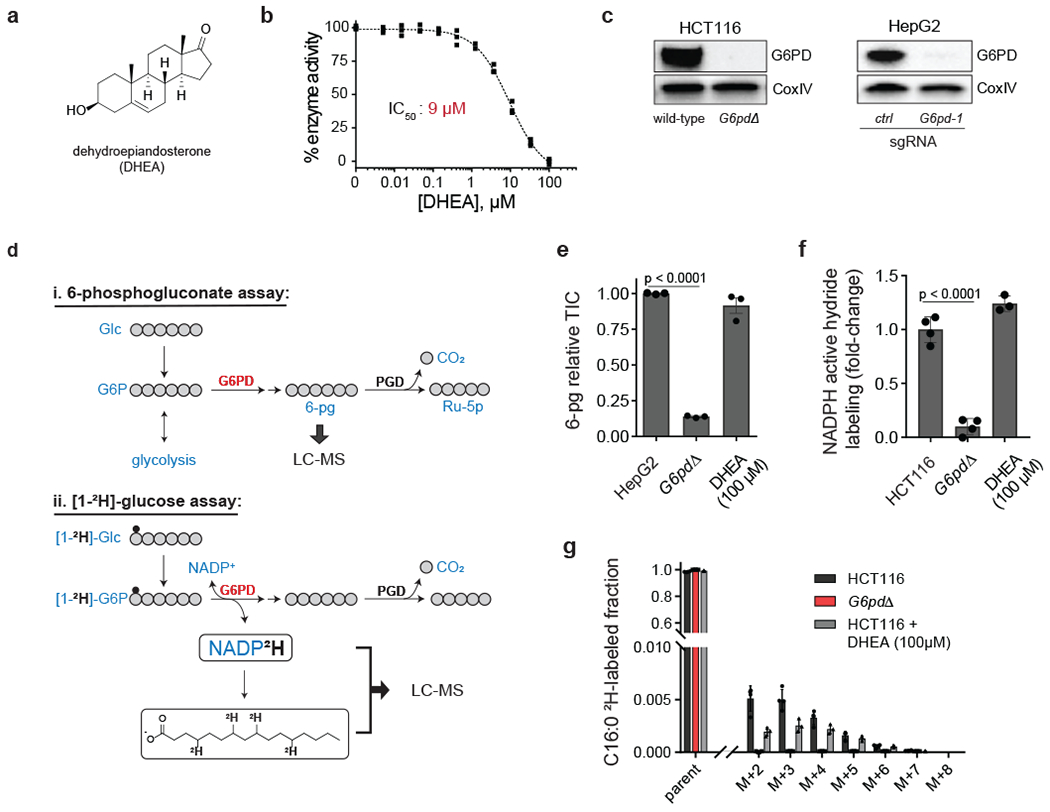
a, Chemical structure of the steroid derivative dehydroepiandosterone (DHEA). b, In vitro activity of DHEA against recombinant human G6PD (mean ± SD, n = 3). c, Western blots of G6PD knockout cells generated using CRISPR-Cas9 (HCT116 knockout is clonal; HepG2 is batch; “ctrl” represents an intergenic control). See Supplementary Figure 16 for uncropped gels. Representative results of 2 independent experiments. d, Assays for G6PD cellular activity: (i) 6-phosphogluconate (6-pg) levels in HepG2 cells, (ii) deuterium (2H, small black circle) incorporation into NADPH (active hydride) and free palmitic acid from [1-2H]-glucose in HCT116 cells. e, f, g, DHEA (100 μM, 2 h) does not phenocopy G6PD knockout (TIC, total ion count by LC-MS) (mean ± SD, n = 3). p value calculated using a two-tailed unpaired Student’s t-test.
To properly evaluate cellular target engagement, it is important to employ assays that specifically monitor the reaction of interest 19–21. However, developing assays that monitor NADPH-producing reactions can be particularly challenging, since NADPH is difficult to measure 22 and is produced by multiple pathways (where inhibition of one can be masked by compensatory production from others).
Here, we develop G6PD cellular target engagement assays and use them to show that DHEA, even at high doses, minimally inhibits G6PD in cells. We then identify a non-steroidal small molecule inhibitor of G6PD, G6PDi-1 (2), that demonstrates on-target reversible cellular activity against G6PD. Utilization of G6PDi-1 across a wide range of mammalian cells revealed that immune cells, especially T cells, are reliant on G6PD for maintaining NADPH levels and effector function.
Results
DHEA does not inhibit G6PD in cell-based assays
To examine the biochemical activity of G6PD, we established a coupled enzymatic assay using recombinant human enzyme (Supplementary figure 1a–b). Consistent with prior reports, DHEA demonstrated dose-dependent inhibition of G6PD, with a calculated half-maximal inhibitory constant (IC50) of 9 μM (Figure 1b) 23.
To assess whether DHEA effectively targets G6PD also in cells, we compared metabolomics of clonally isolated G6PD knockout cells (G6pdΔ) (Figure 1c) with parental HCT116 cells treated with high dose DHEA (100 μM). Global metabolomics showed DHEA treatment did not mirror G6PD knockout (Supplementary figure 2a). Concerningly, DHEA failed to deplete the key downstream oxPPP intermediate 6-phosphogluconate (6-pg) (Supplementary figure 2b), although measurement was challenging due to low 6-pg levels in this cell line. Through analysis of diverse cell lines, we found that the hepatocellular carcinoma line HepG2 possesses sufficient 6-pg for reliable monitoring of G6PD cellular target engagement (Figure 1d). In addition, through CRISPR manipulation of 6-phosphogluconate dehydrogenase in HCT116 cells, we identified a hypomorphic cell line (mPgd) that builds up 6-pg, facilitating assessment of G6PD target engagement (mPgd, Supplementary figure 3a–c). In mPgd HCT116 cells, DHEA (100 μM) modestly suppressed 6-pg (Supplementary figure 3d). In HepG2 cells, it had no effect (Figure 1e). Together, these observations suggest that DHEA may not consistently and effectively block cellular G6PD.
We next aimed to directly monitor G6PD mediated hydride transfer to NADPH. Specifically, we traced the transfer of deuterium from [1-2H]glucose, via glucose-6-phosphate, to the NADPH’s active hydride (Figure 1d) 24. Consistent with this labeling arising primarily from the G6PD reaction, G6PD knockout cells demonstrated a nearly complete loss of active hydride labeling (Figure 1f). The impacted step was G6PD, as no change in substrate (G6P) labeling was observed (Supplementary figure 2c). A major use of cytosolic NADPH is fat synthesis. We quantified transfer of 2H from glucose via NADPH into palmitate (C16:0), which requires two NADPH per 2-carbon unit addition during its synthesis (Figure 1d) 24. Near complete loss of labeling into C16:0 from [1-2H]-glucose (Figure 1g) was observed in G6pdΔ cells. DHEA, however, did not decrease either NADPH active hydride labeling (Figure 1f) or C16:0 labeling (Figure 1g). Thus, DHEA does not robustly inhibit cellular G6PD.
To evaluate other purported inhibitors of G6PD, we obtained two recently identified small molecules, CB-83 (3) 12 and polydatin (4) 25 (Supplementary figure 4a). Like DHEA, both CB-83 and polydatin display anti-proliferative effects against transformed cells, but direct evidence of cellular G6PD inhibition is lacking 12,25. At a dose higher than that reported to impair cell growth 25, polydatin failed to decrease 6-pg levels (Supplementary figure 4b) or NADPH active hydride labeling (Supplementary figure 4c), consistent with lack of cellular target engagement of G6PD. Although individual experimental results were quite variable, CB-83 appeared to augment G6PD activity (Supplementary figures 4b–c). This could potentially reflect CB-83 activating the oxPPP by inducing oxidative stress. Despite this complexity, like DHEA, these compounds do not appear to be cell-active G6PD inhibitors.
G6PDi-1, a non-steroidal, cell active inhibitor of G6PD
We combed the literature for a compound series that could serve as a suitable chemical starting point for inhibitor discovery. Our search identified a non-steroidal aminoquinazolinone series that was recently discovered and optimized against Trypanosoma cruzi G6PD 26. Synthesis of representative compounds identified G6PDi-precursor (5) with low micromolar in vitro activity against human G6PD (Figure 2a–b). Successive rounds of optimization led to replacement of the aminophenyl ring with a cyano-thiophene and expansion of the alkyl quinazolinone region by one methylene, ultimately identifying G6PDi-1, a sub-micromolar inhibitor of human G6PD (IC50 = 0.07 μM). Additionally, we identified a structural analogue (designated G6PDi-neg-ctrl,6) that lacked any activity against G6PD to serve as a negative control compound (Figure 2a–b). In vitro activities were verified in an orthogonal, LC-MS assay that monitors 6-pg production by recombinant human G6PD (Supplementary figure 5a). Follow up in vitro dilution experiments (Supplementary figure 5b) and competition assays against both substrates (Supplementary figure 5c) showed G6PDi-1 binds to G6PD reversibly and non-competitively. Cellular thermal shift assay (CETSA) using HepG2 lysates demonstrated significant thermal stabilization of G6PD by G6PDi-1, but not DHEA up to 56 °C (Supplementary figure 6a–c). These data collectively support a reversible direct physical interaction between G6PDi-1 and G6PD at an allosteric site, with G6PDi-1 binding inhibiting enzyme catalytic activity.
Figure 2. A non-steroidal, cell active inhibitor of G6PD.
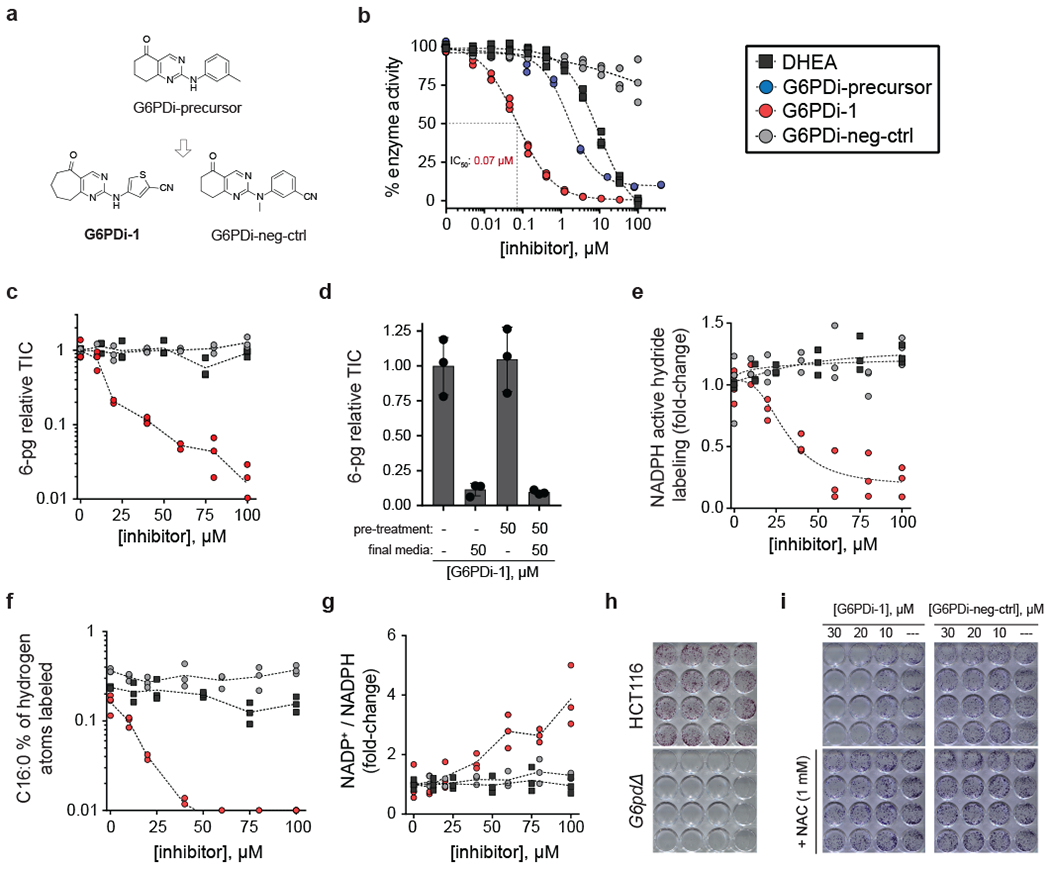
a, Chemical structures. b, In vitro dose response curves (n = 3). c, 6-pg dose response curves (HepG2 cells) (n = 3). d, Reversibility of the cellular activity of G6PDi-1. HepG2 cells were pre-treated with indicated media for two hours, followed by incubation with final media for two hours (mean ± SD, n = 3). e, NADPH active hydride 2H-labeling dose response curves (HCT116 cells, [1-2H]-glucose tracer) n = 3). f, Free palmitic acid 2H-labeling dose response curves (HCT116 cells, [1-2H]-glucose tracer) (n = 3). g, NADP+/NADPH ratio dose response curves (HCT116 cells) (n = 3). h, Representative crystal violet staining of colonies formed from HCT116 and G6PD knockout cells. i, Colony formation of HCT116 cells treated with increasing doses of G6PDi-1 and G6PDi-neg-ctrl. Representative results of 3 independent experiments. NAC = N-acetylcysteine.
To investigate cellular target engagement, G6PDi-1 was evaluated by metabolomics in wild-type and ΔG6PD HCT116 cells (Supplementary figure 7) and in our established target engagement assays. The metabolomics revealed some potential off-target effects on purine nucleosides. Nevertheless, there was clear cellular target engagement. In HepG2 cells, treatment with G6PDi-1, but not G6PDi-neg-ctrl or DHEA, led to a dose-dependent decrease in 6-pg levels (IC50 ~13 μM; Figure 2c). Similarly, 6-pg levels in HCT116-mPgd cells were much more effectively suppressed by G6PDi-1 than DHEA (Supplementary figure 8a). Consistent with this effect arising from reversible binding of G6PD, 6-pg levels completely recovered within 2 h of removing the inhibitor (Figure 2d). Additionally, treatment of HCT116 cells with G6PDi-1, but not neg-ctrl or DHEA, led to a dose-dependent decrease in 2H transfer from [1-2H]-glucose to NADPH’s active hydride (IC50 ~31 μM; Figure 2e) and downstream product C16:0 (Figure 2f). The impacted step was G6PD, as no change in G6P labeling was observed (Supplementary figure 8b). In addition, as expected for G6PD inhibition, we observed a dose-dependent increase in NADP+/NADPH (Figure 2g).
To further assess the utility of G6PDi-1 as a cellular G6PD inhibitor, we built on previous work that established epithelial cells undergoing matrix detachment are subjected to increased levels of oxidative stress, and are in turn dependent on oxPPP activity for survival 27. As expected, colony formation of G6pdΔ cells is dramatically impaired (Figure 2h). Consistent with G6PDi-1 possessing cellular G6PD activity, a dose-dependent decrease in colony formation is observed with G6PDi-1, but not G6PDi-neg-ctrl, an effect that is rescued by exogenous antioxidants (Figure 2i). Taken together, these data show that G6PDi-1 is a cell-active G6PD inhibitor.
G6PDi-1 reveals T cell dependence on oxPPP
It was recently established that transformed cells can maintain NADPH levels in the face of G6pd loss by using malic enzyme 1 (ME1) and/or isocitrate dehydrogenase 1 (1DH1) to make NADPH 10. Indeed, despite effectively penetrating HCT116 and HepG2 cells (Supplementary figure 9a) and effectively inhibiting G6PD activity, NADPH pools were largely unperturbed by G6PDi-1 treatment (Supplementary figure 9b–c). To evaluate the potential for different cells to acutely compensate for G6PD inhibition, we treated a diversity of primary and transformed cell types with G6PDi-1, reasoning that cells reliant on the oxPPP would be unable to maintain their NADPH pools. As expected, we observed that RBCs were fairly sensitive to G6PDi-1. We were surprised, however, to find that T cell lineages were substantially more strongly affected, manifesting a greater than 10-fold decrease in NADPH, accompanied by a corresponding increase in NADP+ (Figure 3a). Thus, T cells appear to be particularly dependent on the oxPPP for maintaining their NADPH pools.
Figure 3. G6PDi-1 reveals T cells depend on oxPPP for maintaining cellular NADPH.
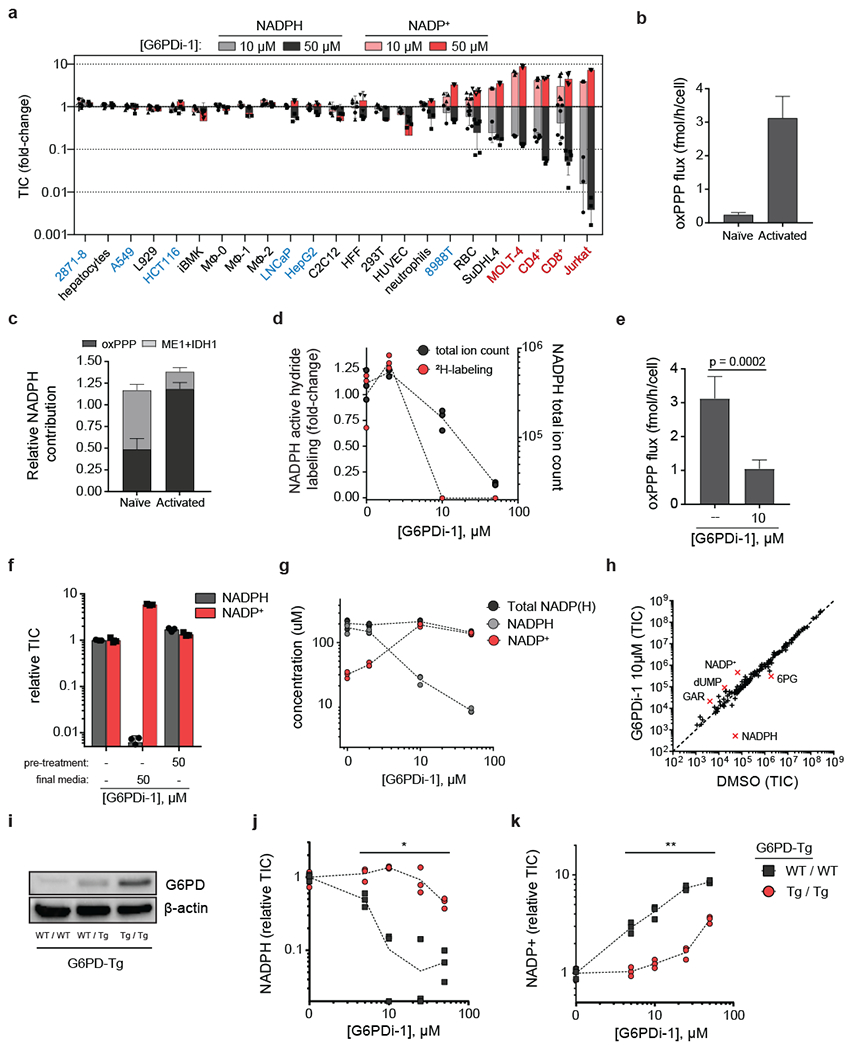
a, LC-MS quantification of NADPH and NADP+ pools across a variety of normal and transformed cell types in response to G6PDi-1 (mean ± SD, n = 6 for RBC, CD4+ and CD8+ T cells, n=3 for the rest of cells). TIC = total ion count. Cell names in red are T cell lineage, blue are cell lines derived from solid tumors. Abbreviations: 2871–8 = lung adenocarcinoma (mouse); A549 = lung adenocarcinoma (human); L929 = fibroblast (mouse); HCT116 = colorectal carcinoma (human); iBMK = immortalized baby kidney epithelial (mouse); MΦ = mouse bone-marrow derived macrophages: unstimulated (MΦ−0), stimulated with LPS+IFNγ (MΦ −1), stimulated with IL4 (MΦ −2); LNCaP = prostate adenocarcinoma (human); HepG2 = hepatocellular carcinoma (human); C2C12 = immortalized myoblasts (mouse); HFF = fibroblasts (human); 293T = immortalized embryonic kidney epithelial (human); HUVEC = umbilical vein endothelial (human); 8988T = pancreatic adenocarcinoma (human); RBC = red blood cells (mouse); SuDHL4 = B cell lympohoma (human); MOLT-4 = T cell acute lymphoblastic leukemia (human); CD4+ and CD8+ = active primary T cells (mouse); Jurkat = immortalized T lymphocyte (human). b, Total oxPPP flux as determined by 14CO2 emission in naïve mouse CD8+ T cells (unstimulated and cultured with IL-7) and activated mouse CD8+ T cells (day 4 post plate-bound αCD3/αCD28 stimulation and cultured with IL-2) (mean ± SD, n = 2 for naïve, n = 5 for active). c, Fraction cellular NADPH from the oxPPP, malic enzyme 1 (ME1) and isocitrate dehydrogenase (IDH1) in naïve and activated CD8+ T cells (mean ± SD, n = 3) (for tracers, see Supplementary figure10b–c). d, NADPH concentration and active hydride 2H-labeling dose response to G6PDi-1 after 2 h ([1-2H]-glucose tracer) (n = 3). e, G6PDi-1 blocks oxPPP flux as determined by 14CO2 emission (mean ± SD, n = 5). p value calculated using a two-tailed Student’s t-test. f, NADP+/NADPH shift in response to G6PDi-1 is rapidly reversible. Active CD8+ T cells were pre-treated with indicated media for 2 h, followed by incubation with final media for 2 h (mean ± SD, n = 3). (G) Absolute NADPH and NADP+ pools after G6PDi-1 (2 h) (n = 3). h, Water-soluble metabolite in active CD8+ T cells treated with G6PDi-1 (2 h) (mean, n = 3). Metabolites displaying a fold-change > 4 are highlighted in red. i, Western blots of G6PD (combined endogenous and transgenic) in active CD8+ T cells from G6pd overexpressing mice (G6PD-Tg mice). “WT / WT” = wild-type mice (no G6pd transgene expression); “WT / Tg” = heterozygous expression; “Tg / Tg” = homozygous expression. See Supplementary Figure 16 for uncropped gels. Representative results of 2 independent experiments. j-k, Dose response to G6PDi-1 of NADPH (j)and NADP+ (k) in active CD8+ T cells from wild-type or G6pd overexpressing mice (n = 3). * and ** denote significant differences between WT/WT and Tg/Tg mice at each of the tested doses using a a two-tailed unpaired Student’s t-test. The following p values were obtained for NADPH levels: 5 μM, p = 0.011, 10 μM, p < 0.0001, 25 μM, p = 0.019, 50 μM, p = 0.0010. The following p values were obtained for NADP+ levels: 5 μM, p = 0.0011, 10 μM, p = 0.0012, 25 μM, p < 0.0001, 50 μM, p = < 0.0001.
Since T cell activation involves substantial metabolic rewiring28, we decided to investigate whether an activation-driven metabolic program determined T cell dependency on the oxPPP. To this end, we isolated naïve CD8+ T cells from mouse spleen and either maintained them in the naïve state by culturing them with IL-7 or activated them with plate-bound αCD3/αCD28 and IL-2. Upon activation, we observed an increase in G6PD protein, which began within 8 h and became prominent by 24 h (Supplementary figure 10a). In line with this observation, absolute oxPPP flux as measured using radioactive CO2 capture increased by greater than 10-fold upon activation (Figure 3b).
Consistent with the inability of T cells to maintain NADPH using compensatory pathways, neither naïve nor activated CD8+ T cells possess substantial ME1 or IDH1 (Supplementary figure 10a). To complement these enzyme abundance measurements, we used 2H-tracing to assess the relative contribution from ME1, IDH1 and the oxPPP to cytosolic NADPH 10,24. Using a combination of five tracers (Supplementary figure 10b–c), we found that the oxPPP accounts for nearly all cytosolic NADPH production in CD8+ and CD4+ T cells activated with αCD3/αCD28 and IL-2, but not in naïve CD8+ and CD4+ T cells maintained in IL-7 supplemented media (Figure 3c and Supplementary figure 10d).
To examine further the impact of G6PDi-1, mouse CD8+ and CD4+ T cells at day 4–5 post-activation were treated with increasing G6PDi-1 in the presence of [1-2H]-glucose. G6PDi-1 (10 μM) completely blocked 2H transfer from glucose to NADPH (Figure 3d, Supplementary figure 10e) and decreased NADPH and 6-pg levels (Supplementary figure 11a). Similarly, treatment with G6PDi-1 blocked absolute oxPPP flux (Figure 3e). Consistent with their lower G6PD expression and flux and NADPH requirements for biosynthesis, naïve CD8+T cells were less sensitive to G6PDi-1 than activated CD8+T cells (Supplementary figure 10f–g). In activated CD8+ T cells, NADPH, NADP+ and 6-pg levels were restored within 2 h of removing the inhibitor (Figure 3f and Supplementary figure 11b). The effects on NADPH labeling (from [1-2H]-glucose), and NADP+, NADPH and 6-pg levels occurred within 10 min of G6PDi-1 treatment (Supplementary figure 11c). Absolute quantitation of NADP+ and NADPH revealed that the decrease in NADPH concentration induced by G6PDi-1 is matched by an increase in NADP+ concentration, with the total NADP(H) remaining around ~200 μM (Figure 3g). Collectively, these data confirm that G6PDi-1 is a rapid, reversible G6PD inhibitor that increases the NADP+/NADPH ratio in T cells.
To assess the specificity of the metabolic effects of G6PDi-1, we performed untargeted metabolomics on CD8+ T cells. The greatest metabolite concentration change occurred directly in the substrates and products of G6PD (NADPH, NADP+, 6-pg) and folate metabolites known to be perturbed by G6PD activity loss (dUMP, GAR) (Figure 3h, Supplementary figure 2a) 10. Thus, G6PDi-1 has clean on-target activity in T cells. Isotope tracing with [U-13C]-glucose and [U-13C]-glutamine revealed that G6PDi-1 decreased the glucose contribution to TCA cycle (with a corresponding increase in glutamine contribution) (Supplementary figure 12a–l). In addition, fatty acid synthesis, a major consumer of cytosolic NADPH, was nearly completely ablated (Supplementary figure 12m). Consistent with the importance of NADPH in controlling oxidative stress, G6PDi-1 treatment elevated reactive oxygen species (ROS) in both CD8+ and CD4+ T cells (Supplementary figure 11d). These effects were largely rescued by N-acetyl cysteine (Supplementary figure 11e).
Recently, a transgenic mouse strain that over-expresses human G6PD (G6PD-Tg) was reported 29 (Figure 3i). To validate the dependence of T cell NADPH pools on G6PD, CD8+ T cells from G6PD-Tg mice and littermate controls at day 4–5 post-activation were treated with increasing doses of G6PDi-1. Strikingly, G6PD overexpression markedly shifted the dose response to G6PDi-1, rescuing its effects on NADPH and NADP+ (Figure 3j–k). Thus, G6PDi-1 modulates T cell NADPH by inhibiting the catalytic activity of G6PD, with introduction of exogenous G6PD activity rescuing T cell redox state.
G6PDi-1 blocks T cell cytokine production
We next explored the functional consequences of oxPPP inhibition using G6PDi-1 in T cells. To test the effect of G6PDi-1 on activation and proliferation, naïve CD8+ T cells were isolated from spleen and activated in vitro with plate-bound αCD3/αCD28 and IL-2. Activation was evaluated by flow cytometry analysis of surface markers CD69 (levels rapidly rise upon activation) and CD25 (usually peaking at 24–48 h post-activation), and cell size, which increases over the first 24 h post-activation. To quantify proliferation, naïve cells were stained with Crystal Trace Violet (CTV) and dye dilution was measured by flow cytometry at day 4 post-activation. As expected by the late upregulation of oxPPP during activation30,31 (Supplementary Figure 10a), G6PDi-1 did not alter the normal upregulation of activation markers or activation-dependent increase in cell size (Figure 4a). More surprisingly, G6PDi-1 had a minimal effect in activation-dependent proliferation (Figure 4b and Supplementary Figure 13a) and viability (Supplementary Figure 13b). G6PDi-1 had also a minimal effect on the proliferation of CD4+ T cells (Supplementary Figure 13c).
Figure 4. G6PDi-1 suppresses T cell cytokine production while having a minimal effect on initial activation or proliferation.
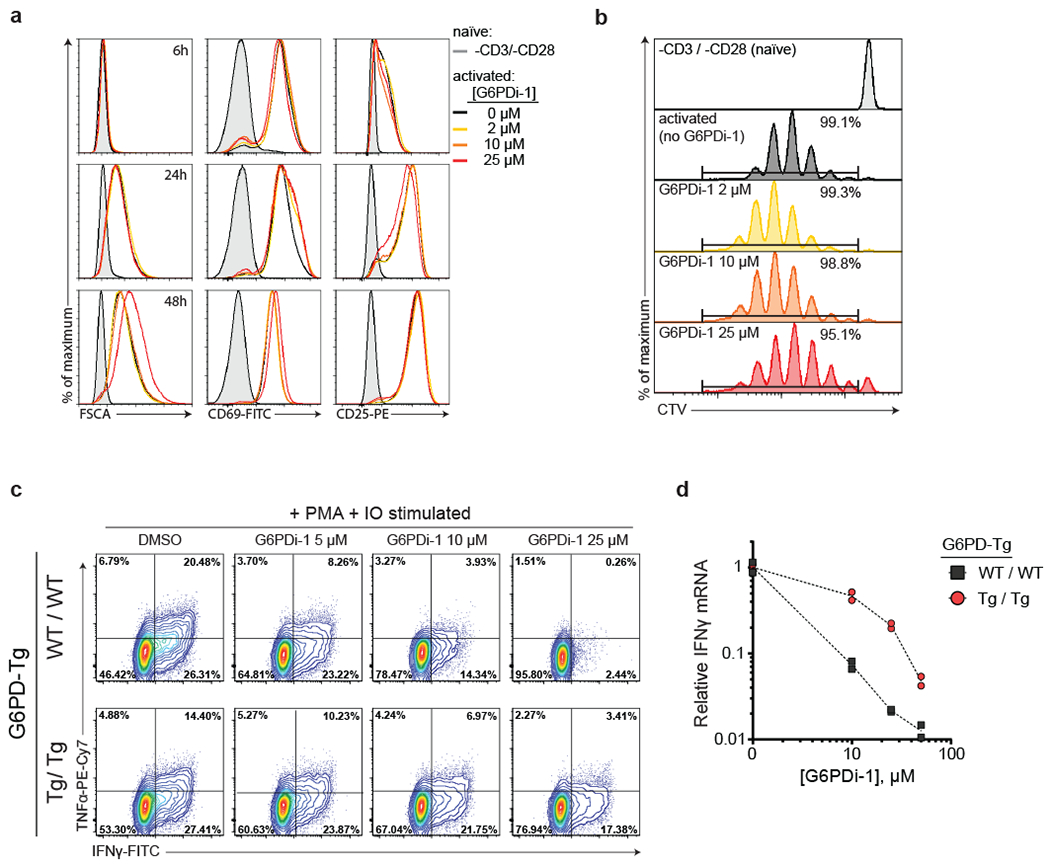
a, Flow cytometry analysis of cell size (FSCA) and activation markers (CD69 and CD25) of mouse naïve CD8+ T cells either rested in naïve state or stimulated by CD3/CD28 + IL-2 in the presence of increasing concentrations of G6PDi-1. Representative results of 2 independent experiments. b, Proliferation of CD8+ T cells either rested in naïve state or stimulated by CD3/CD28 + IL-2 in the presence of increasing concentrations of G6PDi-1 at day 4 post-activation based on Cell Trace Violet (CTV) dilution. Representative results of 2 independent experiments. c, Intracellular cytokines in active CD8+ T cells from wild-type or G6pd overexpressing mice after a 6 h stimulation with PMA and IO in the presence of the indicated dose of G6PDi-1.(c-d) Representative results of 2 independent experiments. d, Corresponding Ifng mRNA in active CD8+ T cells from wild-type or G6pd overexpression mice (normalized to Gapdh expression and no G6PDi-1 control) (n = 2).
To assess the effect of G6PD inhibition in T cell function, active CD8+ or CD4+ cells were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin (IO) in the presence of increasing doses of G6PDi-1 and cytokine production was monitored by intracellular flow cytometry. Strikingly G6PD inhibition blocked IFNγ and TNF-α production in CD8+ and IL-2 and TNF-α CD4+ T cells (Figure 4c, Supplementary Figure 13d–e).
Proper T cell activation requires ROS signaling while avoiding ROS toxicity 32–34. Accordingly, we attempted to rescue CD8+ T cell cytokine secretion with the antioxidant N-acetyl-cysteine (Supplementary figure 13f) or by providing an external source of peroxide/superoxide (Supplementary figure 13g), but neither was effective. To confirm that the defect is at the level of signaling, rather than protein synthesis, we examined IFNγ mRNA, finding that its levels were also decreased (Figure 4d). Indeed, signaling during the first hour after restimulation seems to be particularly important, as delayed addition of G6PDi-1 enabled substantial cytokine production to occur (Supplementary figure 13h). Restoration of intracellular NADPH levels by moderate overexpression of human G6PD protein decreased sensitivity to G6PDi-1 and partially normalized both protein and mRNA levels upon G6PDi-1 addition (Figure 4c–d). Thus, G6PD activity is required to maintain proper NADP+/NADPH homeostasis in activated T cells, in a manner that is not readily compensated by generic oxidant or antioxidant, and loss of such homeostasis inhibits T cell effector function.
We next sought to evaluate whether G6PD inhibition impacted the development, proliferation or suppressor function of CD4+ regulatory T cells (Treg). Stimulation with CD3/CD28 in the presence of TGF-β resulted in Foxp3+ cells, whose formation and proliferation were unaffected by G6PDi-1 (Supplementary figure 14a–c). Similarly, CD4+CD25+ Tregs proliferated and were effective in suppressing the proliferation of conventional CD4+ CD25- T cells irrespective of G6PDi-1 treatment (Supplementary figure 14d–e). Collectively, these data show that, without overtly impacting proliferation or suppressor function, G6PDi-1 inhibits pro-inflammatory cytokine production from activated T cells.
G6PDi-1 suppresses oxidative burst in neutrophils
Motivated by the key role of G6PD in effector function in CD4+ and CD8+ T cells, we decided to evaluate whether the function of other immune cells depends on G6PD activity. In macrophages, G6PDi-1 did not decrease NADPH (Figure 3a) or LPS-induced pro-inflammatory cytokine production or iNOS upregulation (Figure 5a). Thus, while in T cells G6PD activity is essential for cytokine production, it is dispensable in the case of LPS-stimulated macrophages (Figure 5b).
Figure 5. G6PDi-1 suppresses neutrophil oxidative burst.
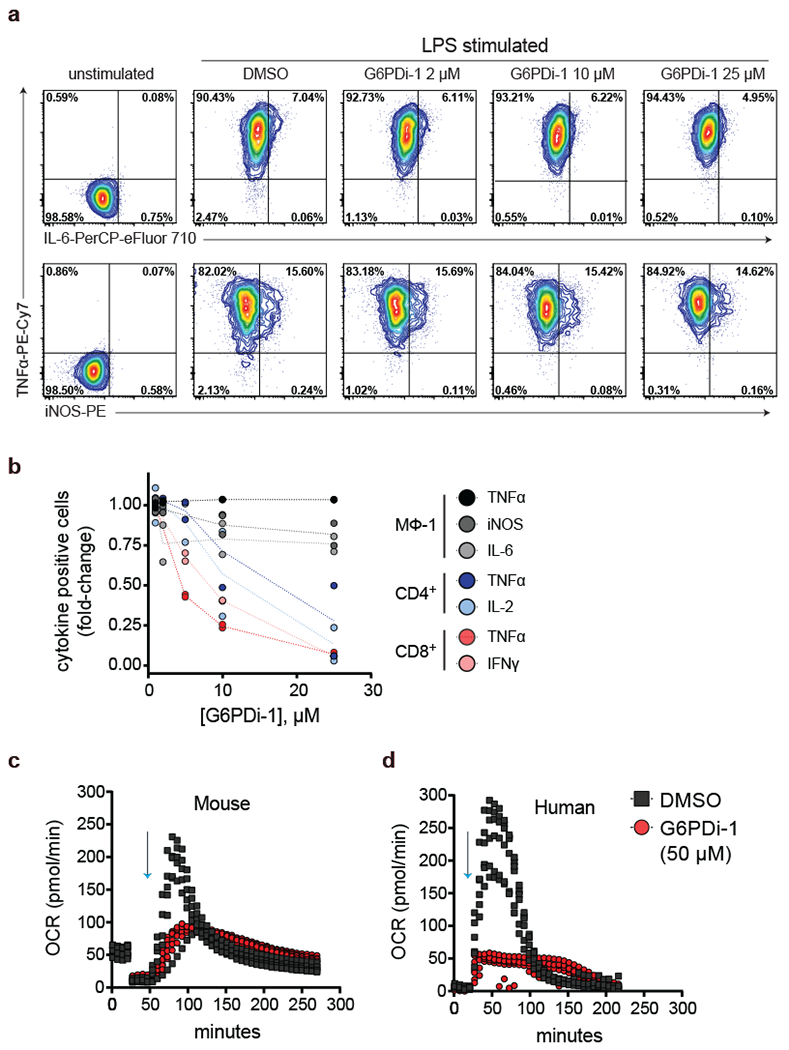
a, Intracellular cytokines in bone marrow derived macrophages after a 6 h stimulation with LPS and IFNγ in the presence of the indicated dose of G6PDi-1. Representative results of 2 independent experiments. b, Cytokine effects of G6PDi-1 across cell types (n = 2). c-d, Neutrophil oxidative burst as measured by the Seahorse Extracellular Flux Analyzer. Oxygen consumption rate (OCR) was monitored in mouse (c) and human (d) neutrophils that were activated with PMA (100 nM, indicated by blue arrows) in the presence of 50 μM G6PDi-1 or vehicle control (n = 6).
In neutrophils, G6PDi-1 did impact NADPH, albeit to a lesser extent than in T cells (Figure 3a). A key function of neutrophils is ROS generation by NADPH oxidase 35, which requires NADPH and oxygen as substrates. To test the role for the oxPPP in this effector function, mouse and human neutrophils were stimulated with PMA in the presence of 50 μM G6PDi-1 or vehicle control, and oxygen consumption rate was used to readout oxidative burst. G6PDi-1 decreased oxidative burst in both mouse and human neutrophils (Figure 5c-d). Thus, G6PD activity is essential in providing NADPH for ROS generation by NADPH oxidase in neutrophils.
Discussion
Small molecule inhibitors with specific on-target activity are key tools for biological research. Unfortunately, however, many tool compounds fail to robustly engage their targets and/or have extensive off-target effects. Here, we show that the commonly used G6PD inhibitor DHEA, despite clear inhibition of purified enzyme, lacks robust on-target cellular activity at doses well above those needed to exhibit anti-proliferative effects. Others have previously raised doubts about DHEA’s cellular G6PD activity 18, but it has continued to be widely used as a G6PD inhibitor, in part because of evidence that it induces oxidative stress 36. This, however, is a nonspecific outcome, and in the case of DHEA (and several other recently published “G6PD inhibitors”) may occur unrelated to G6PD target engagement.
Motivated by the need for an on-target tool compound to inhibit cellular G6PD, we engaged in substantial chemistry efforts. These led to what we believe is the first cell-active on-target G6PD inhibitor, G6PDi-1. We then employed G6PDi-1 to better understand cellular NADPH homeostasis. While the oxPPP is often described as being the canonical, dominant pathway for producing cytosolic NADPH, few studies have directly tested this. As expected, RBCs, which lack mitochondria and therefore the required substrates for producing NADPH when the oxPPP is blocked, were significantly depleted of NADPH upon G6PDi-1 treatment. Many other cell lines were almost completely insensitive. Lymphocytes, however, including primary mouse active CD4+ and CD8+ T cells and human T-ALL cell lines, were yet more sensitive than RBCs. Consistent with this, we observed that activated T cells do not express substantial ME1 or IDH1, and make NADPH almost solely through the oxPPP, which is strongly upregulated during T cell activation.
If T cells are most sensitive to acute G6PD inhibition, why are clinical manifestations of G6PD deficiency most apparent in RBCs? Activated T cells, unlike mature RBCs, have intense biosynthetic requirements. Previous work has shown that biosynthesis – of proline, deoxyribonucleotides and especially fat – is a major consumer of cytosolic NADPH in proliferating mammalian cells 37. Additionally, evidence suggests RBCs in G6PD deficient patients are most often impaired through lower levels of enzyme, rather than reduced catalytic function 38. Unlike T cells, mature RBCs are enucleated, and therefore unable to express new protein. As such, G6PD levels are gradually lost over the life span of RBCs (~120 days), with older RBCs retaining <10% of their original G6PD activity 39. Mutations in G6PD accelerate this degradation 38. Indeed, patients possessing G6PD variants with the lowest enzyme stability often experience the worst clinical outcomes 40. Strikingly, variants that reduce enzyme stability and thereby deplete G6PD activity in RBCs by >95% only modestly impair G6PD activity in leukocytes, often leading to no functional deficit 41,42. This makes sense as, in their activated proliferating state, T cells are composed almost solely of freshly made protein. Interestingly, severe G6PD mutations that affect enzyme catalytic ability (rather than protein stability) can present with immune deficiency 43.
The inability of T cells to maintain NADPH homeostasis upon G6PD blockade did not prevent initial activation or growth, but profoundly inhibited pro-inflammatory cytokine production. Similar cytokine effects were not observed in macrophages, which better maintained NADPH in the face of G6PDi-1. The mechanism linking G6PD to cytokine production remains unclear, but appears to involve defects in transcriptional activation. It is tempting to speculate that previous reports linking restriction of glycolysis—via GLUT1 knockdown, glucose depletion or glucose replacement with 2-deoxyglucose or galactose—with decreased cytokine secretion may be due to oxPPP blockade 44–48.
As expected, G6PD inhibition resulted in increased total cellular ROS. The general antioxidant N-acetyl-cysteine was able to block the increased ROS but did not restore cytokine secretion. This may reflect the complex role of ROS in immune cell activation, with the right amount required in the right subcellular location. Such a precise ROS control may make T cells uniquely sensitive both to glucose availability and to G6PD inhibition. The in vivo consequences of G6PD inhibition will also reflect its impact on other immune cell types, including suppression of neutrophil oxidative burst, which requires a corresponding burst of NADPH production. Although it is unsuitable to be used in vivo, going forward, we hope that G6PDi-1 will prove to be a valuable initial tool for exploring the biological role of G6PD across diverse cellular contexts.
Materials and Methods
Cell lines, growth conditions, and reagents
HCT116, HepG2, L929, LNCaP, A549, C2C12, HFF, 293T, Molt4, Jurkat, and SuDHL4 cells were all originally obtained from ATCC (Manassas, VA). 8988T cells were obtained from DSMZ (Braunschweig, Germany). 71–8 cells and iBMK cells were a generous gift from Eileen White (Rutgers Cancer Institute of New Jersey, New Brunswick, NJ). Pooled HUVECs were obtained from ThermoFisher Scientific (#C0155C) and were maintained in EBM-2 Basal Medium (CC-3156, Lonza) supplemented with EGM-2 SingleQuots Supplements (CC-4176, Lonza). All other adherent cell lines (unless otherwise specified) were maintained in DMEM (CellGro 10–017, Mediatech Inc., Manassas, VA) supplemented with 10% fetal bovine serum (F2442, Sigma-Aldrich, St. Louis, MO). All suspension cell lines (unless otherwise specified) were maintained in RPMI-1640 media supplemented with 10% FBS, 100 U/ml penicillin, 100 ug/ml streptomycin and 50 μM 2-mercaptoethanol. All cell lines were routinely screened for mycoplasma contamination. LentiCRISPR v2 (52961) was obtained from Addgene (Cambridge, MA). All primers were synthesized by IDT (Coralville, IA). Antibodies were used according to their manufacturer’s directions. Anti-β-actin (5125) was obtained from Cell Signaling Technologies (Danvers, MA). Anti-G6PD (ab993); ME1 (ab97445) and IDH1 (EPR12296) were obtained from Abcam Inc. (Cambridge, MA). CoxIV antibody was obtained from Proteintech (11242–1-AP.). Standard laboratory chemicals were from Sigma.
Chemical compounds
Synthetic procedures are provided in the Supplementary Note
Oligonucleotides
For LentiCRISPR (Supplementary table 1).
For CRISPR-Cas9nickase (Supplementary table 2).
For RT-PCR (Supplementary table 3).
G6PD plasmid construction and expression
Partially truncated human G6PD (residues 28–515, Uniprot ID P11413) was subcloned into the pET28a vector using the Ndel and Xhol restriction enzyme sites and the following primers: 5’-agtcagcatatggtcagtcggatacacacatattcatc-3’ and 5’-agtcagctcgagtcagagcttgtgggggttcac-3’. Recombinant G6PD was expressed in Escherichia coli BL21(De3)pLysS as an N-terminal His6-tagged protein with an integrated thrombin cleavage site. Briefly, IPTG was added (final concentration of 1mM) to induce protein expression when culture density reached an OD600 of 0.6, followed by incubation at 37 °C overnight. Pellets were isolated and lysed by sonication in buffer containing 50 mM Tris (pH 8), 500 mM NaCl, 20 mM imidazole, 1mM BME, 1 mM PMSF, and 5% glycerol v/v. The lysate was centrifuged and filtered to remove insoluble debris. The resulting supernatant was fractionated twice with ammonium sulfate; first to 25% at 4 °C for 1h, with the supernatant undergoing subsequent fractionation to 50% at 4 °C for 1 h. The precipitate was collected and dissolved in binding buffer consisting of 50 mM NaH2PO4, Tris (pH 8), 500 mM NaCl, 20 mM imidazole, and 1mM BME, and was loaded onto a Ni Sepharose HisTrap HP column (GE Healthcare, 17–5248-01). The column was washed with ~10 column volumes of binding buffer. Elution of G6PD was achieved with elution buffer consisting of 50 mM NaH2PO4, Tris (pH 8), 500 mM NaCl, 250 mM imidazole, and 1mM BME. The eluted protein was desalted and concentrated to remove the imidazole before undergoing thrombin cleavage using a Thrombin CleanCleave Kit (MilliporeSigma, C974M34). The tag-less protein was purified by size-exclusion chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare) using buffer consisting of 50 mM Tris (pH 8), 150 mM NaCl, and 1mM BME. Eluted protein was concentrated using an Amicon Ultra 10 kDa MWCO filter (MilliporeSigma, UFC901008). Protein concentration was determined by Pierce BCA Assay (Thermo, 23225) and was stored in 10% glycerol at −80 °C.
G6PD enzymatic activity measurement by diaphorase-coupled assay
Inhibitor activity against recombinant human G6PD was determined using a resazurin-based diaphorase coupled kinetic assay. Increasing doses of test compound in 96-well, opaque bottom plates were treated with assay buffer containing 50 mM triethanolamine pH = 7.4, 1 mM MgCl2, 0.1 mM resazurin, 0.03 mM NADP+, 0.1 U/mL Clostridium kluyveri diaphorase (MilliporeSigma, D5540), 0.25 mg mL−1 bovine serum albumin, and ~1 nM purified G6PD. The reaction was initiated by the addition of 1 mM glucose-6-phosphate (G6P). Plates were incubated at 30 °C and read every minute by a BioTek plate reader (Synergy HT) monitoring fluorescence emission at 590 nm following excitation at 530 nm. For Michaelis-Menten experiments NADP+ and G6P concentrations were varied as described. For jump dilution experiments, inhibitor was initially incubated in assay buffer containing ~10 nM G6PD for 30 minutes at 30 °C (1x dilution), before being diluted (1:50) into an equal volume of assay buffer containing no G6PD or inhibitor (50x dilution), followed by reaction initiation of both by the addition of 1 mM G6P. G6PD inhibition was determined by calculating the change in relative fluorescence over time (RFU/min) in the presence of different doses of test compound, followed by normalization against control wells without compound. GraphPad Prism (v7.03) was used to perform a non-linear curve fit (4-parameter) to determine IC50 values. Competitive and non-competitive modes of G6PD binding by inhibitor were modeled using the built-in non-linear regression analysis packages in Graphpad (v8.2).
Generation of G6PD-null and hypomorphic PGD cell lines
Generation of clonal G6PD-null line in the HCT116 background have been previously described 10. Using a similar approach, a clonal hypomorphic PGD cell line in the HCT116 background was generated. Briefly, paired guide RNAs (mPgd-1 and mPgd-2) against exon 3 of human Pgd were cloned into plasmids containing Cas9 nickase expression vector and puromycin-resistant genes. Cells were transiently transfected with these plasmids using Lipofectamine 3000 (Life Technologies) and selected for 48 h with 2 μg/mL puromycin. After selection, cells were grown to confluence before single-cell plating in 96-well plates. Hypomorphic cell lines showed decreased but not ablated protein expression by western blotting.
Generation of batch G6PD-null cells in the HepG2 background was achieved using the lentiviral CRISPR–Cas9 vector system LentiCRISPR v2 (Addgene #52961). Briefly, sgRNA sequences targeting exon-5 of human G6pd were designed using the crispr.mit.edu design tool. The identified PAM sequences (see table above) were subcloned into the LentiCRISPR v2 using the BsmBI restriction endonuclease (NEB R0580S). Virus was produced through PEI (MilliporeSigma, 408727) transfection of vectors and lentiviral packaging plasmids psPax2 and VSVG in 293T cells. Medium containing lentivirus was collected after two days and filtered through a PES filter (0.22 μm, MilliporeSigma). HepG2 cells transfected with virus targeting non-coding control or G6pd and Polybrene (8 μg/mL, Invitrogen). Cells were split after 48 h into RPMI-media (10% FBS) containing puromycin (2 μg/mL) and cultured for 3 days. G6PD knockout was confirmed by western blotting.
Cellular thermal shift assay (CETSA)
Lysates from HepG2 cells at 75% confluence were isolated with 0.5% Triton in TBS (20 mM, Tris pH 7.4, 150 mM NaCl) for 30 min on ice, pre-cleared by centrifugation and used for thermal shift assay as described 49. Briefly, inhibitor or DMSO control was added to lysates at indicated concentration and incubated for 30min on ice followed by 3 min heating at 47 °C, 50 °C, 53 °C, and 56 °C in a thermal cycler. After heating, tubes were cooled at room temperature for 3 min and insoluble fraction removed by centrifugation at 17,000 g for 20 min. The soluble fraction was separated by SDS-PAGE, transferred to PVDF membrane, and immunoblotted using indicated antibodies at a dilution of 1:2000. Blots were developed by chemiluminescence and imaged using LI-COR C-DiGit Western Blot Scanner. Two independent experiments were performed. Signal intensity of proteins from immunoblots was quantified using Image Studio version 5.2 for C-DiGit Scanner, and bands were normalized to signal intensity of the 47 °C treated samples. Relative signal intensities were plotted as bar graph relative to the DMSO treated control.
G6PD enzymatic activity measurement by LC-MS
Inhibitor activity against recombinant human G6PD was determined by direct product monitoring by LC-MS. Test compounds at indicated doses were treated with assay buffer containing 50 mM triethanolamine pH = 7.4, 1 mM MgCl2, 0.30 mM NADP+, 0.25 mg/mL bovine serum albumin, and ~1 nM purified G6PD. The reaction was initiated by the addition of 1 mM G6P (or water for negative control condition). Aliquots of reaction mixture were collected at indicated time points and rapidly quenched by diluting (1:5) into methanol precooled on dry ice. The mixtures were centrifuged at 13,000 g for 20 min at 4 °C, and the resulting supernatants were diluted (1:20) into 40:40:20 methanol/ acetonitrile/ water and analyzed by LC-MS.
Mice
Seven- to twelve-week-old mice were used for all experiments. Wild-type C57BL/6 were purchased from Charles River or Harlan laboratories. The mice were on normal light cycle (8 AM – 8 PM) and had free access to water and standard chow diet.The G6PD-Tg mouse line was generated at the Spanish National Cancer Research Center (CNIO) at the Transgenic Mice core facility using a 20Kb human genomic DNA construct containing the entire G6PD gene, including 2.5 Kb of upstream flanking sequence and 2.0 Kb of downstream flanking sequence 29. G6PD-Tg mice were bred and maintained in the facilities of the University of Valencia.
Isolation, culture and stimulation of naïve CD8+ or CD4+ T cells
To isolate naïve CD8+ or CD4+ T cells, spleens were harvested and single cell suspensions prepared by manual disruption and passage through a 70 µm cell strainer in PBS supplemented with 0.5% BSA and 2 mM EDTA. After red blood cell lysis, naïve CD8+ or CD4+ T cells were purified by magnetic bead separation using commercially available kits following vendor instructions (Naive CD8a+ T Cell Isolation Kit, mouse or Naive CD4+ T Cell Isolation Kit, mouse, Miltenyi Biotec Inc).
Cells were cultured in complete RPMI media (RPMI 1640, supplemented with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 55 μM 2-mercaptoethanol). Naïve T cells were either rested in complete RPMI media supplemented with recombinant IL-7 (50U/mL) or stimulated for 48 h with plate-bound anti-CD3 (10 µg/ml) and anti-CD28 (5 µg/ml) in complete RPMI media supplemented with 100 μM alanine (to ensure proper activation50) and recombinant IL-2 (100 U/mL). Activated T cells were maintained in complete RPMI media supplemented with recombinant IL-2 (100 U/mL). All experiments on “active” T cells were performed at day 4–5 post-activation.
Isolation, culture and stimulation of bone marrow-derived macrophages
Mouse bone marrow monocyte/macrophage progenitors were isolated from femur and tibia and cultured in BMM media (DMEM supplemented with 10% FBS, 20% L929-conditioned media, 100 U/ml penicillin, and 100 µg/ml streptomycin). Expression of CD11b and F4/80 was checked by flow cytometry after 6 days in culture. Mature macrophages were either maintained in BMM media (MΦ−0 macrophages) or stimulated overnight with LPS (100 ng/mL) + IFNγ (20 ng/mL) for MΦ−1 activation or IL-4 (20 ng/mL) for MΦ−2 activation.
Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from C57Bl/6 mice by perfusion of the liver with liver perfusion medium (1x) (Thermo Fisher 17701038) followed by digestion with one bottle of collagenase/elastase (Worthington Biochemical LK002066) and DNase1 (Worthington Biochemical, LK003170) in Krebs Ringer Buffer with HEPES and 0.5mM CaCl2. Digested liver was minced in hepatocyte wash medium (Thermo Fisher 17704024), passed through a 70 μm strainer, and centrifuged at 50 g. Dead cells were removed by adding a 25% percoll solution, centrifuging at 120 g, and aspirating the supernatant. Primary hepatocytes were plated at 1.2 M cells/well in collagen coated 6 well plates in pre-warmed DMEM with 100 nM insulin, 100 nM dexamethasone, and 1% Glutamax.
Isolation and culture of erythrocytes from mouse spleen
To isolate red blood cells, mice were euthanized by cervical dislocation followed by collection of ~200 µL whole blood via cardiac puncture into tubes containing 7.5 µL heparin (1000USP/mL, H3393, Sigma Aldrich). The cells were incubated on ice for ~5 min, then centrifuged (5 min, 500 rpm, 4 °C) followed by aspiration of the serum and buffy coat layer. Cells were gently resuspended in PBS and then pelleted (5 min, 500 rpm, 4 °C) three times. Cells were then resuspended in RPMI media and used immediately for experiments.
Isolation, culture, and stimulation of neutrophils
Murine neutrophils were isolated from 8–12 week old C56BL/6 mice. Mice were euthanized by cervical dislocation, and bone marrow cells were harvested from femur and tibia within 30 min. Cell suspensions were passed through 70 µm cell strainer. Neutrophils were prepared using a negative selection kit (EasySep Mouse Neutrophil Enrichment Kit, Stem Cell Technologies), following manufacturer instruction. Cells were cultured in RPMI 1640 media supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 5 mM HEPES, and 2 mM EDTA. To stimulate neutrophils, 100 nM phorbol 12-myristate 13-acetate (PMA) (Cayman Chemical) was added to the media.
Human neutrophils were isolated from 8 ml of blood collected from healthy donors. Neutrophils were purified using the MACSxpress Whole Blood Neutrophil Isolation Kit (Miltenyi Biotec 130–104-434) followed by erythrocyte depletion (Miltenyi Biotec 130–098-196) according to manufacturer’s instructions.
Neutrophil Oxidative Burst Assay
Suspended neutrophils were plated in culture wells pre-coated with Cell-Tak™ (Corning), spun at the 200 x g for 1 min. with minimal acceleration/deceleration, and then incubated for 1h at 37 °C. Murine neutrophils were plated at 2 × 105 cells/ well, in RPMI 1640 media without sodium bicarbonate. Human neutrophils were plated at 5 × 104 cells/ well, in RPMI 1640 media supplemented with 0.1% human serum albumin. Inhibitor (G6PDi-1, 50µM) or vehicle control were added just prior to starting assay. Mouse neutrophils were also treated with rotenone (0.5µM, BioVision) + antimycin A (0.5 µM, BioVision) at t = 20 min. Oxidative burst was stimulated with PMA (100 nM, Cayman Chemical); oxygen consumption rate using the XF-96e extracellular flux analyzer (Seahorse Bioscience).
Flow cytometry analysis
To analyze cell surface markers expression, cells were collected, washed with PBS and stained with the viability dye Live/Dead Aqua. Cells were then washed with staining buffer and stained for surface markers: CD4 (APC-Cy7, clone RM4–5,), CD8a (PerCP-Cy5.5, clone 53–6.7), CD25 (APC, clone PC61), CD44 (PE-Cy7, clone IM7), CD62L (PE, clone MEL-14) CD69 (FITC, clone H1.2F3), for T cells; CD11b (APC, clone M1/70 and F4/80 (FITC, clone BM8) for macrophages.
To analyze proliferation, naïve CD8+ T cells were stained with CTV dye and either maintained in a naïve state with IL7 or stimulated with αCD3/αCD28 + recombinant IL-2 in the presence of increasing concentrations of G6PDi-1. Cells were refed at day 2 and 3 post stimulation and proliferation measured at day 4 post activation. Cells were collected, washed with staining buffered and stained with the viability dye propidium iodide.
To analyze intracellular cytokines, active T cells were re-stimulated with PMA (20 ng/ml) and ionomycin (1 ug/ml) in the presence of GolgiStop and increasing concentrations of G6PDi-1. After a 6h incubation period, cells were collected, washed with PBS and stained with the viability dye Live/Dead Aqua. Cells were then washed with staining buffer and stained for surface markers: CD4 (APC-Cy7, clone RM4–5,), CD8a (PerCP-Cy5.5, clone 53–6.7). Cells were then fixed and permeabilized and stained for intracellular cytokines: IFNγ (FITC, clone XMG1.2), TNFα (PE-Cyanine7, clone MP6-XT22) and IL-2 (PE, clone JES6–5H4). To analyze intracellular cytokines, mature macrophages were stimulated with LPS (100 ng/mL) + IFNγ (20ng/mL) in the presence of GolgiStop and increasing concentrations of G6PDi-1. After a 6 h incubation period, cells were collected, washed with PBS and stained with the viability dye Live/Dead Aqua. Cells were then fixed and permeabilized and stained for intracellular cytokines: TNFα (PE-Cyanine7, clone MP6-XT22) and IL-6 (PerCP-eFluor 710, clone MP5–20F3) and for iNOS (PE, clone CXNFT).
For measuring intracellular ROS levels, cells were incubated for 2 h in the presence of increasing concentrations of G6PDi-1 and then stained for ROS and viability using CellROX Green Flow Cytometry Assay Kit following manufacturer instructions. In some experiments, n-acetyl cysteine (1 mM), galactose oxidase (0.045 U/mL) + galactose (500 µM), or potassium superoxide (0.5 µg/mL) were added to the culture media at the same time the drug and/or PMA and ionomycin were added. All flow cytometry was analyzed with an LSR II (BDbiosciences) using standard filter sets and FCS express 7.01 flow cytometry software (De Novo Software). Gating strategies for flow cytometry analyses are shown in Supplementary Figure 15.
Treg assays
Spleen and peripheral lymph nodes were harvested and processed to single cell suspensions of lymphocytes. Red blood cells were removed with hypotonic lysis. We used magnetic beads (Miltenyi Biotec, San Diego, CA) for isolation of Tconv (CD4+CD25–), Treg (CD4+CD25+), and antigen presenting cells (CD90.2–). For cell culture medium, we used RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 µg/mL), and 55 μM β-mercaptoethanol Treg suppression and iTreg polarization were conducted as previously reported51. For Treg suppression assays, Tconv were purified and stimulated with irradiated antigen presenting cells plus CD3ε mAb (1 µg/mL, BD Pharmingen). To assess proliferation, Tconv cells were labeled with carboxyfluorescein succinimidyl ester (CFSE), and Treg cells with CellTrace Violet. After 72 h, proliferation of Tconv and Treg cells was determined by flow cytometric analysis of CFSE and CellTrace Violet dilution, respectively. For conversion to Foxp3+ Tregs, Tconv cells were incubated for 3–5 days with CD3ε/CD28 mAb beads, plus TGF-β (3 ng/mL) and IL-2 (25 U/mL), and analyzed by flow cytometry for Foxp3+ iTreg. Flow cytometry data was captured using Cytoflex (Beckman Coulter, Brea, CA) and analyzed using the FlowJo 10.2 software.
Absolute quantification of oxPPP flux using [1-14C]-glucose and [6-14C]-glucose
Glucose oxidation flux through oxPPP was determined from difference in the rate of 14CO2 released from [1-14C]-glucose and [6-14C]-glucose, as previously described with some modification 52,53. RPMI 1640 media without sodium bicarbonate was supplemented with 0.74 g/L of NaHCO3, 2.5 mM HEPES pH 7.4, 10% dFBS, and 1% of either [1-14C]-glucose or [6-14C]-glucose. Three million primary mouse naïve (rested in IL-7) or active CD8+ T cells (in the presence of IL-2 and 0.1% DMSO or 10 μM G6PDi-1) were incubated for 4 h in a sealed 12.5 cm2 flask. To facilitate the collection of 14CO2, 100 uL of a 10 M KOH solution was introduced into the sealed flask using a center well for incubation flask (8823200000, DWK Life Sciences). The assay was stopped by injection of 1 mL 1M HCL and the KOH solution then transferred to scintillation vials containing 10 mL scintillation solution for counting. The signal was corrected for the percentage of radioactive tracer in the medium. OxPPP flux is calculated as follow:
Tracer experiments
For all experiments involving stable isotope tracers (e.g. [1-2H]-glucose), the isotope tracer nutrient was substituted for unlabeled nutrient at the same concentration normally found in the base media for a given cell type (e.g. DMEM for HCT116, RPMI for T cells, etc). In addition, dialyzed FBS was used as a supplement in place of FBS.
Cytosolic NADPH sources were traced, and redox active hydride labeling was calculated, using a previously described strategy 10,24. [1-2H]-glucose (which directly traces G6PD) or [3-2H]-glucose (which directly traces PGD) were used for tracing oxPPP contribution to NADPH; [4-2H]glucose for ME1; [2,3,3,4,4-2H5]-glutamine, for both ME1 and IDH1; and D2O, to account for solvent exchange.
The mass difference between 13C1 and 2H1 NADPH and NADP+ cannot be resolved using the Q Exactive Plus. Therefore, the natural 13C abundance was corrected from the raw data. The labeling of the redox-active hydrogen of NADPH ([Active-H]) and correction for solvent exchange were done as previously described 24. [1-2H]-glucose and [3-2H]-glucose contribution were corrected by glucose-6-phosphate labeling, [4-2H]-glucose by malate labeling and [2,3,3,4,4-2H5]-glutamine by the average of citrate and malate labeling. OxPPP contribution was calculated as the sum of the normalized active H labeling for [1-2H]-glucose and [3-2H]-glucose, and ME1 plus IDH1 as the sum of the normalized active H labeling for [4-2H]-glucose and [2,3,3,4,4-2H5]-glutamine.
Metabolite extraction
For analysis of intracellular metabolites by LC-MS, adherent cell lines were plated and grown to 80% confluency in 6-well plates. At the start of an experiment, the appropriate media was added to cells, which included isotope tracers and/or chemical inhibitors as described. Cells were incubated at 37 ºC at 5% CO2 for 2 h (unless otherwise noted). For all experiments involving small molecule agents, DMSO concentrations were <0.2%. After 2 h, media was removed by aspiration and metabolome extraction was performed (without any wash steps) by the addition of 800 µL of ice cold solvent (40:40:20 acetonitrile:methanol:water + 0.5% formic acid). After a 1-min incubation on ice, the extract was neutralized by the addition of NH4HCO3 (15% w/v). The samples were incubated at −20°C for ~30 min, at which point the wells were scraped and the extract transferred to Eppendorf tubes and centrifuged (15 min, 16,000 rpm, 4 °C). The resulting supernatant was frozen on dry ice and kept at −80 °C until LC-MS analysis.
For suspension cells (including T cells and red blood cells), 2×106 cells were seeded in 1 mL of media in 12-well plates and incubated with appropriate media, which included isotope tracers and/or chemical inhibitors and/or cytokines as described. For all experiments involving small molecule agents, DMSO concentrations were <0.2%. After 2 h, cell suspensions were transferred to 1.5 mL tubes and pelleted (30 s, 6,000 rpm, room temperature). Media was removed by aspiration and metabolome extraction was performed by the addition of 75 µL of ice cold solvent (40:40:20 ACN:MeOH:H2O + 0.5% formic acid). After a 5 min incubation on ice, acid was neutralized by the addition of NH4HCO3. After centrifugation (15 min, 16,000 rpm, 4°C), the resulting supernatant was transferred to a clean tube, frozen on dry ice and kept at −80°C until LC-MS analysis. For neutrophil experiments, metabolites were extracted after 30 min of stimulation with PMA using similar extraction conditions.
LC-MS analysis
Unless otherwise noted, metabolites were analyzed using a quadrupole-orbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific, Waltham, MA), coupled to hydrophilic interaction chromatography (HILIC) with LC separation on a XBridge BEH Amide column (Waters), or a stand-alone orbitrap (Thermo-Fisher Exactive) coupled to reversed-phase ion-pairing chromatography with LC separation on a HSS-T3 column (Waters).Both mass spectrometers were operating in negative ion mode and were coupled to their respective liquid chromatography methods via electrospray-ionization. Detailed analytical conditions have been previously described 24,54. Metabolites from neutrophils were analyzed using similar methods that have been previously described55.
Adherent cell metabolite abundances were normalized by packed cell volume; suspension cells to cell count. Unless otherwise indicated, isotopic labeling of metabolites arising from incubation with 13C or 2H labeled nutrients were corrected for natural abundance, as previously described 56. Data were analyzed using the ElMaven software (v 0.2.4, Elucidata), with compounds identified based on exact mass and retention time match to commercial standards. For metabolomics analysis, metabolites data were normalized to control condition and clustered using Cluster 3.0 software. Heatmaps were plotted using Java Treeview.
Absolute quantification of NADP+ and NADPH in active CD8+ T cells
Active CD8+ T cells were cultured and metabolome extraction was performed as previously described. Packed cell volume was measured using Midwest Scientific PCV cell counting tubes and estimated to be 1.5 µL per 2 × 106 cells. Cell extracts were spiked with 1.5 uL of NADP+ 2.5 or 25 μM or NADPH 20 or 200 μM. Absolute concentration was calculated based on the increase in NADP+ or NADPH signal in the spiked samples.
Statistics
Samples sizes are defined in each figure legend. Results for technical replicates are presented as mean ± SD or SEM. Statistical significance between conditions was calculated using Student’s t-test (two-tailed) when comparing two groups, and one-way ANOVA followed by Dunnett’s post hoc analysis when comparing more than two. All statistical calculations were performed using the software package GraphPad Prism (v7.03 and v8.2).
Ethics
We have complied with all relevant ethical regulations regarding the use of research animals and Human research participants.
Unless otherwise specified, animal studies followed protocols approved by the Princeton University Institutional Animal Care and Use Committee (protocol number 2032–17). Mouse neutrophil studies followed protocols approved by the University of Wisconsin Institutional Animal Care and Use Committee (protocol number M006219). Mouse Treg studies followed protocols approved by the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committees (protocol number 19–000561). G6PD-Tg mouse studies followed protocols approved by the University of Valencia Institutional Animal Care and Use Committee (protocol number A1444079171882).
Collection of blood from healthy donors followed the protocol approved by the University of Wisconsin Institutional Review Board (Protocol number 2019–1031-CP001). Informed consent was obtained from all participants
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank C. DeCoste of the Princeton University flow cytometry resource facility and the Cytomics Unit of the IIS-La Fe for experimental set-up and design; R. S. O’Connor of University of Pennsylvania for assistance in setting up T cell experiments and for comments and suggestions on the figures; J. Jiao of the Children’s Hospital of Philadelphia for technical assistance with the Treg experiments; I. Babic of Nerdbio for assistance with the CETSA experiments; Y. Huang of Peking University for helpful suggestions pertaining to structure–activity relationship (SAR) analysis; C. Bartman and the rest of members of the Rabinowitz laboratory for comments and suggestions. This work was supported by NIH grants 1DP1DK113643 and R01 CA163591. J.C.G.C. is supported by funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 751423.
Footnotes
Competing interests
Princeton University has filed a patent relating to the novel G6PD inhibitors and their uses. J.D.R. is a co-founder of Raze Therapeutics, advisor and stock owner in Kadmon, Agios, C.R.P., LEAF, and Bantam Pharmaceuticals and consultant to Pfizer. No competing interests were disclosed by the other authors.
References
- 1.Voet D, Voet JG & Pratt CW Fundamentals of biochemistry: life at the molecular level. (2016). [Google Scholar]
- 2.Stanton RC Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB life 64, 362–369, doi: 10.1002/iub.1017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science (New York, N.Y.) 347, 1260419, doi: 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Kowalik MA, Columbano A & Perra A Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Frontiers in Oncology 7, doi: 10.3389/fonc.2017.00087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q et al. Overexpression of G6PD Represents a Potential Prognostic Factor in Clear Cell Renal Cell Carcinoma. Journal of Cancer 8, 665–673, doi: 10.7150/jca.16858 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagashio R et al. Prognostic significance of G6PD expression and localization in lung adenocarcinoma. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1867, 38–46, doi: 10.1016/j.bbapap.2018.05.005 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Pu H et al. Overexpression of G6PD is associated with high risks of recurrent metastasis and poor progression-free survival in primary breast carcinoma. World journal of surgical oncology 13, 323–323, doi: 10.1186/s12957-015-0733-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longo L et al. Maternally transmitted severe glucose 6‐phosphate dehydrogenase deficiency is an embryonic lethal. The EMBO journal 21, 4229–4239, doi: 10.1093/emboj/cdf426 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cappellini MD & Fiorelli G Glucose-6-phosphate dehydrogenase deficiency. The Lancet 371, 64–74, doi: 10.1016/S0140-6736(08)60073-2 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Chen L et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nature Metabolism 1, 404–415, doi: 10.1038/s42255-019-0043-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamilton NM et al. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. Journal of medicinal chemistry 55, 4431–4445, doi: 10.1021/jm300317k (2012). [DOI] [PubMed] [Google Scholar]
- 12.Preuss J et al. Identification and Characterization of Novel Human Glucose-6-Phosphate Dehydrogenase Inhibitors. Journal of biomolecular screening 18, 286–297, doi: 10.1177/1087057112462131 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Mele L et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis 9, 572, doi: 10.1038/s41419-018-0635-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marks PA & Banks J INHIBITION OF MAMMALIAN GLUCOSE-6-PHOSPHATE DEHYDROGENASE BY STEROIDS. Proceedings of the National Academy of Sciences of the United States of America 46, 447–452 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Monaco M et al. Role of glucose-6-phosphate dehydrogenase inhibition in the antiproliferative effects of dehydroepiandrosterone on human breast cancer cells. British journal of cancer 75, 589–592 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pashko LL, Lewbart ML & Schwartz AG Inhibition of 12-O-tetradecanoylphorbol-13-acetate-promoted skin tumor formation in mice by 16α-fluoro-5-androsten-17-one and its reversal by deoxyribonucleosides. Carcinogenesis 12, 2189–2192, doi: 10.1093/carcin/12.11.2189 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Girón RA, Montaño LF, Escobar ML & López-Marure R Dehydroepiandrosterone inhibits the proliferation and induces the death of HPV-positive and HPV-negative cervical cancer cells through an androgen- and estrogen-receptor independent mechanism. The FEBS journal 276, 5598–5609, doi:doi: 10.1111/j.1742-4658.2009.07253.x (2009). [DOI] [PubMed] [Google Scholar]
- 18.Ho HY, Cheng ML, Chiu HY, Weng SF & Chiu DT Dehydroepiandrosterone induces growth arrest of hepatoma cells via alteration of mitochondrial gene expression and function. Int J Oncol 33, 969–977 (2008). [PubMed] [Google Scholar]
- 19.Pacold ME et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nature Chemical Biology 12, 452, doi: 10.1038/nchembio.2070. https://www.nature.com/articles/nchembio.2070#supplementary-information (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mullarky E et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proceedings of the National Academy of Sciences 113, 1778, doi: 10.1073/pnas.1521548113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ducker GS et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 114, 11404–11409, doi: 10.1073/pnas.1706617114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu W, Wang L, Chen L, Hui S & Rabinowitz JD Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxidants & redox signaling 28, 167–179, doi: 10.1089/ars.2017.7014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon G, Mackow MC & Levy HR On the mechanism of interaction of steroids with human glucose 6-phosphate dehydrogenase. Arch Biochem Biophys 318, 25–29, doi: 10.1006/abbi.1995.1199 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Chen L, Liu L, Su X & Rabinowitz JD Chemical Basis for Deuterium Labeling of Fat and NADPH. Journal of the American Chemical Society 139, 14368–14371, doi: 10.1021/jacs.7b08012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mele L et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death & Disease 9, 572, doi: 10.1038/s41419-018-0635-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercaldi GF, Ranzani AT & Cordeiro AT Discovery of new uncompetitive inhibitors of glucose-6-phosphate dehydrogenase. Journal of biomolecular screening 19, 1362–1371, doi: 10.1177/1087057114546896 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Schafer ZT et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113, doi: 10.1038/nature08268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buck MD, O’Sullivan D & Pearce EL T cell metabolism drives immunity. The Journal of experimental medicine 212, 1345–1360, doi: 10.1084/jem.20151159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nobrega-Pereira S et al. G6PD protects from oxidative damage and improves healthspan in mice. Nature communications 7, doi: 10.1038/ncomms10894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ron-Harel N et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell metabolism 24, 104–117, doi: 10.1016/j.cmet.2016.06.007.(2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geiger R et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842.e813, doi: 10.1016/j.cell.2016.09.031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sena LA et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236, doi: 10.1016/j.immuni.2012.10.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padgett LE & Tse HM NADPH Oxidase-Derived Superoxide Provides a Third Signal for CD4 T Cell Effector Responses. Journal of immunology (Baltimore, Md. : 1950) 197, 1733–1742, doi: 10.4049/jimmunol.1502581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mak TW et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 46, 675–689, doi: 10.1016/j.immuni.2017.03.019 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Nguyen GT, Green ER & Mecsas J Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol 7, 373, doi: 10.3389/fcimb.2017.00373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldfarb AH, McIntosh MK & Boyer BT Vitamin E attenuates myocardial oxidative stress induced by DHEA in rested and exercised rats. Journal of Applied Physiology 80, 486–490, doi: 10.1152/jappl.1996.80.2.486 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Fan J et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302, doi: 10.1038/nature13236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morelli A, Benatti U, Gaetani GF & De Flora A Biochemical mechanisms of glucose-6-phosphate dehydrogenase deficiency. Proceedings of the National Academy of Sciences of the United States of America 75, 1979–1983, doi: 10.1073/pnas.75.4.1979 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marks PA, Johnson AB & Hirschberg E EFFECT OF AGE ON THE ENZYME ACTIVITY IN ERYTHROCYTES. Proceedings of the National Academy of Sciences of the United States of America 44, 529–536, doi: 10.1073/pnas.44.6.529 (1958). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham AD, Colavin A, Huang KC & Mochly-Rosen D Coupling between Protein Stability and Catalytic Activity Determines Pathogenicity of G6PD Variants. Cell reports 18, 2592–2599, doi: 10.1016/j.celrep.2017.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morellini M, Colonna-Romano S, Meloni T, Battistuzzi G & Gandini E Glucose-6-phosphate dehydrogenase of leukocyte subpopulations in normal and enzyme deficient individuals. Haematologica 70, 390–395 (1985). [PubMed] [Google Scholar]
- 42.Ardati KO, Bajakian KM & Tabbara KS Effect of Glucose-6-Phosphate Dehydrogenase Deficiency on Neutrophil Function. Acta Haematologica 97, 211–215, doi: 10.1159/000203685 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Vives Corrons JL et al. Severe-glucose-6-phosphate dehydrogenase (G6PD) deficiency associated with chronic hemolytic anemia, granulocyte dysfunction, and increased susceptibility to infections: description of a new molecular variant (G6PD Barcelona). Blood 59, 428–434 (1982). [PubMed] [Google Scholar]
- 44.Macintyre AN et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell metabolism 20, 61–72, doi: 10.1016/j.cmet.2014.05.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho PC et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228, doi: 10.1016/j.cell.2015.08.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cham CM & Gajewski TF Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. Journal of immunology (Baltimore, Md. : 1950) 174, 4670–4677, doi: 10.4049/jimmunol.174.8.4670 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Shi LZ et al. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of experimental medicine 208, 1367–1376, doi: 10.1084/jem.20110278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang CH et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251, doi: 10.1016/j.cell.2013.05.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez Molina D et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science (New York, N.Y.) 341, 84–87, doi: 10.1126/science.1233606 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Ron-Harel N et al. T Cell Activation Depends on Extracellular Alanine. Cell reports 28, 3011–3021.e3014, doi: 10.1016/j.celrep.2019.08.034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao H et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. International journal of cancer 138, 2477–2486, doi: 10.1002/ijc.29979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang R et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882, doi: 10.1016/j.immuni.2011.09.021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katz J & Wood HG The Use of C14O2 Yields from Glucose-1- and −6-C14 for the Evaluation of the Pathways of Glucose Metabolism. Journal of Biological Chemistry 238, 517–523 (1963). [PubMed] [Google Scholar]
- 54.Lu W et al. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Analytical chemistry 82, 3212–3221, doi: 10.1021/ac902837x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seim GL et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nature Metabolism 1, 731–742, doi: 10.1038/s42255-019-0083-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su X, Lu W & Rabinowitz JD Metabolite Spectral Accuracy on Orbitraps. Analytical chemistry 89, 5940–5948, doi: 10.1021/acs.analchem.7b00396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.