

Abstract
Given that many small molecules could bind to structured regions at sites that will not affect function, approaches that trigger degradation of RNA could provide a general way to affect biology. Indeed, targeted RNA degradation is an effective strategy to selectively and potently modulate biology. We describe several approaches to endow small molecules with the power to cleave RNAs. Central to these strategies is Inforna, which designs small molecules targeting RNA from human genome sequence. Inforna deduces the uniqueness of a druggable pocket, enables generation of hypotheses about functionality of the pocket, and defines on- and off-targets to drive compound optimization. RNA-binding compounds are then converted into cleavers that degrade the target directly or recruit an endogenous nuclease to do so. Cleaving compounds have significantly contributed to understanding and manipulating biological functions. Yet, there is much to be learned about how to affect human RNA biology with small molecules.
Keywords: Targeted Degradation, Nucleic Acids, RNA, Non-coding RNA, siRNA, Antisense Oligonucleotides
Introduction.
Targeted degradation approaches have garnered much interest to study biology and for therapeutic development. Transformative forays into this area include the development of the antisense oligonucleotide (ASO) approach to target sequence [1] by Zamecnick and coworkers [2,3]. They showed that by using DNA oligonucleotides to recognize an RNA’s sequence, RNase H could be recruited to cleave the RNA strand in an RNA-DNA hybrid. In the area of degradation of RNA, much work has followed and has changed biological science and medical research. For example, small interfering RNAs (siRNAs) were discovered by Mello, Fire, and coworkers, and siRNAs have proven to be an important approach to cleave RNAs in a highly effective and sub-stoichiometric manner [4,5]. This approach led to the development of two approved FDA-approved drugs, Patisiran and Givosiran in 2018 and 2019, respectively. More recently, CRISPR-based approaches have been developed to target and cleave RNAs in cells [6–8]. Each of these tools is invaluable for bioscience research, and these approaches have facilitated easier access to cellular knock down of various targets.
Proteolysis targeting chimeras (PROTACs), developed by Deshaies and Crews, has had an accelerating and exciting impact in chemical biology and therapeutic discovery [9–11]. This approach mirrors degradation of RNA but focuses on protein targets. Small molecules that bind to a protein target are appended with a molecule that recruits an E3 ubiquitin ligase, facilitating ubiquitination and hence degradation on the targeted protein by the proteasome. Like antisense and siRNA, PROTACs not only act catalytically but also sub-stoichiometrically, where more molecules of the targeted protein are cleaved than the molecules of PROTAC present [12].
RNA decay and quality control (QC) pathways are critical processes to control transcript levels in cells [13,14]. Herein, we describe harnessing these endogenous machineries by using chimeric small molecules for targeted degradation of RNA, ribonuclease targeting chimeras (RIBOTACs) [15,16]. In addition, we describe compounds that have been designed to cleave targeted RNAs by appending small molecules that bind RNA with natural products that cleave transcripts directly such as bleomycin. This approach has proven to provide selective compounds in cells and animals [17]. Lastly, small molecules that shunt transcripts to RNA decay pathways are described. These compounds function as simple binding compounds that do not directly recruit nucleases, but rather trigger an RNA processing event to cleave toxic RNAs [18]. Factors that affect selectivity as well as methods in which the area that can be expanded to other targets and affect difficult molecular recognition pathways are also discussed.
Developing rules for targeting RNA structure with small molecules, the equivalent of sequence-targeting base pairing rules.
Much of the ability to target RNA for destruction has been enabled by the ease of design of compounds that target its sequence. The pairing rules that A pairs with T or U and G pairs with C allow for oligonucleotides to be designed to target any RNA. Decades of in-depth studies have shown that regions in an RNA structure that are not folded are the most preferred sites for oligonucleotides [19]. This information has been used in conjunction with RNA folding algorithms such as OligoWalk [20] to identify the sites with an RNA target that will be most avidly bound by an oligonucleotide. This approach considers not only the affinity of the oligonucleotide to a given RNA sequence but also if that sequence is present in a structured region [20].
One of the difficulties in targeting RNA with small molecules is the development of general and scalable methods to define bioactive ligands that target RNA. We have developed a programmatic approach in which information on the folded RNA structures that bind small molecules are integrated with bioinformatics. Named Inforna [21], our lead identification strategy allows for the sequence-based design of small molecules that target structured regions in an RNA target. Integration of this binding information with chimeric targeted degradation approaches allows for the development of small molecule functional equivalent of antisense oligonucleotides or siRNAs (Figure 1).
Figure 1. The Inforna approach to define druggable RNA targets and small molecules to target them by using human genome sequence and the RNAs that it encodes.
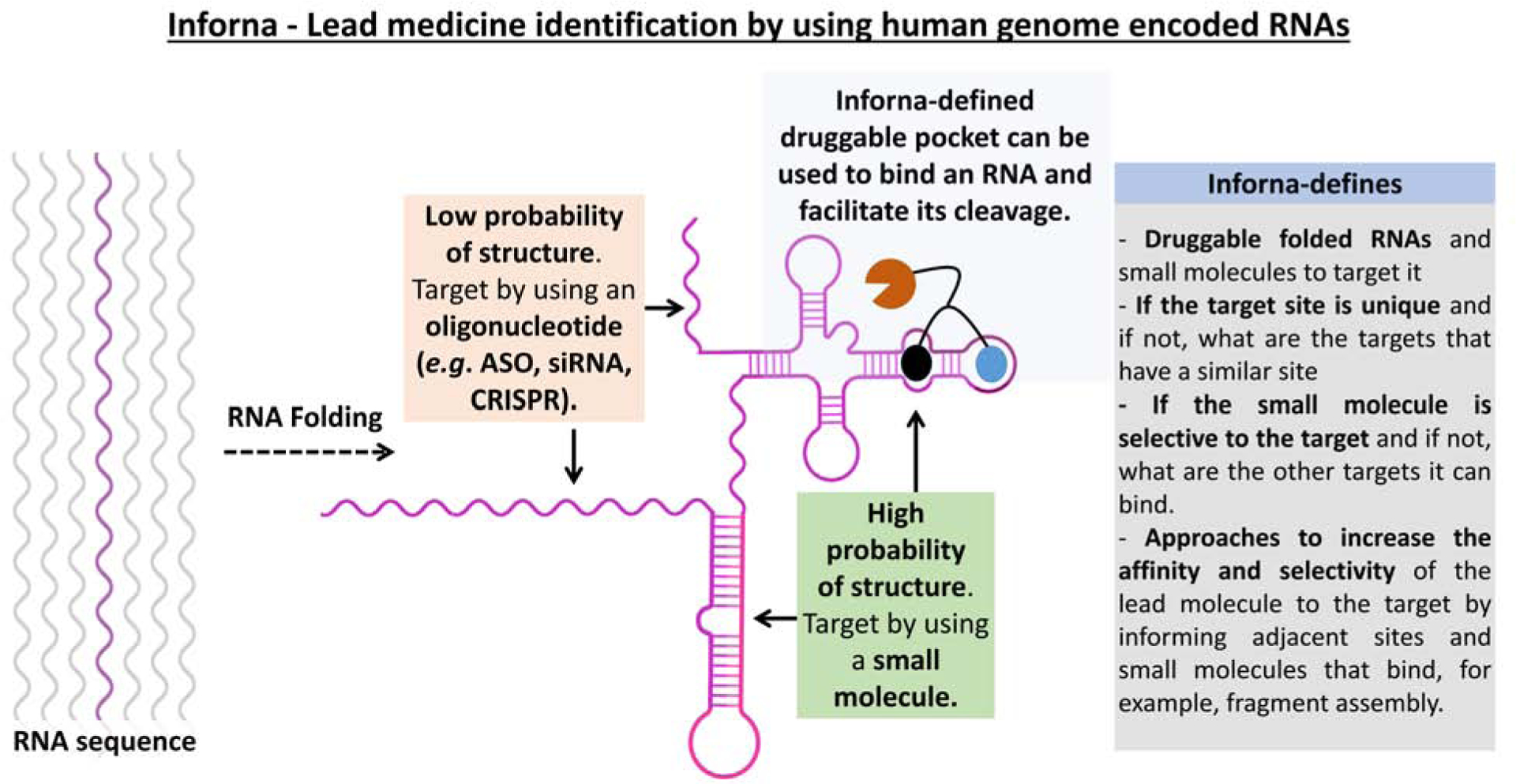
Features of Inforna include: (i) defining folded RNA structures and compounds to bind them; (ii) the uniqueness of a target site; (iii) the selectivity of the small molecule for a given target; (iv) approaches to increase selectivity and affinity of lead small molecules.
Inforna uses a set of rules for the motifs (3D structures) that are bound by small molecules to define RNAs that have sites that can be targeted by small molecules [21,22]. To decode RNA structure with small molecules, we developed two-dimensional combinatorial screening (2DCS), a massively parallel method to identify the RNA folds that bind small molecules and the small molecules that bind RNA folds [23,24]. This library-versus-library selection provides experimentally validated affinity landscapes between small molecules and folded RNA structures (Figure 2).
Figure 2: Library-versus library screening to define the folded RNA structures that are targets of small molecules named two-dimensional combinatorial screening (2DCS).
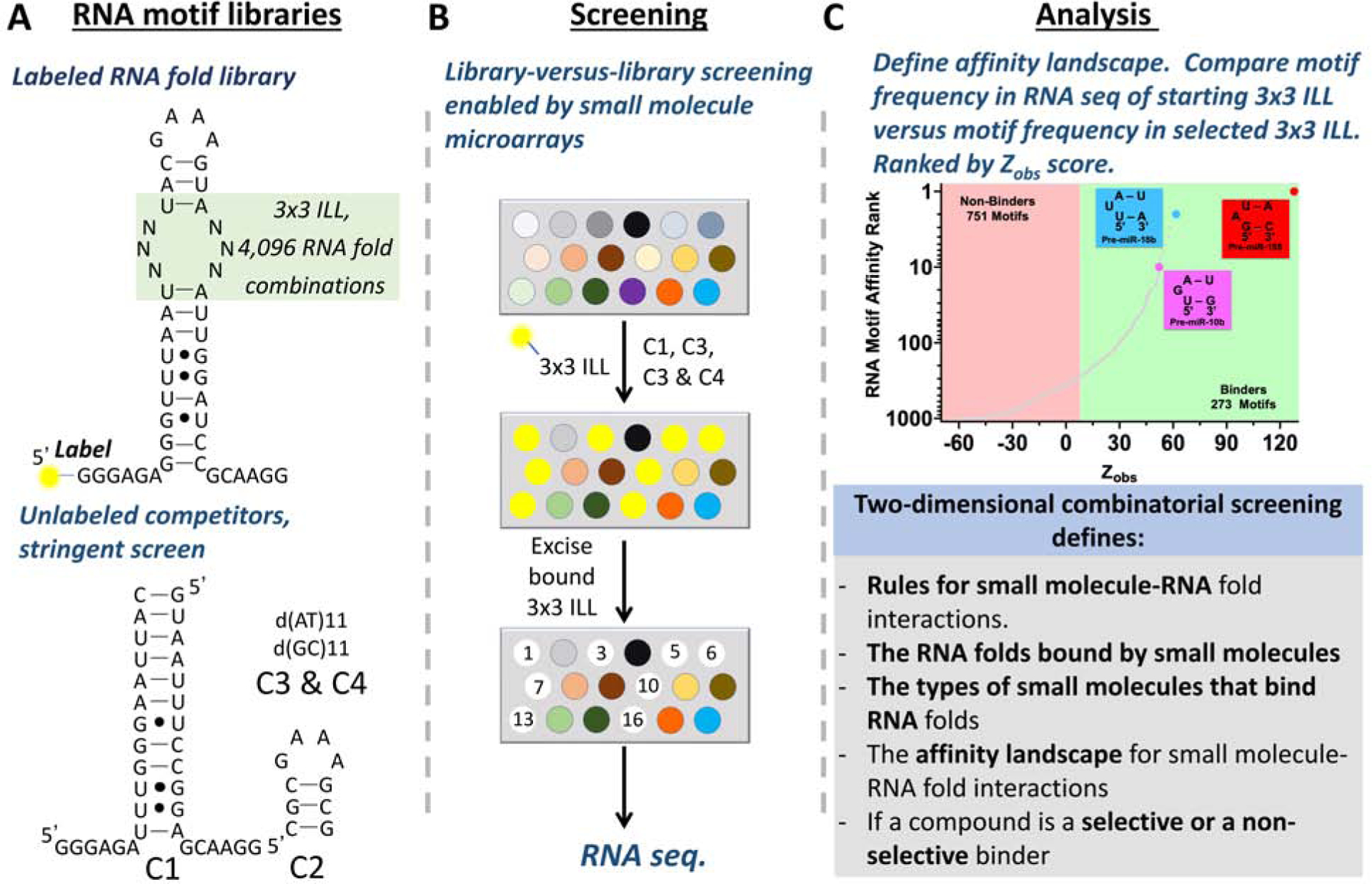
A, Base pairing rules and energy tables enable the design of oligonucleotides to target RNA sequence at unstructured regions. B, A small molecule library is immobilized onto a chip to generate the microarray and is followed by incubation with labeled RNA fold library and competitors. The RNA folds that are selectively bound to the small molecule are selected and identified via RNA-seq. C, RNA structure-recognition rules for small molecules, derived from 2DCS, enables the design of small molecules to target RNA at structured regions.
A 2DCS experiment is conducted by anchoring small molecules onto an agarose-coated microarray surface to allow for spatial encoding of the compounds. The small molecule microarray is then hybridized with libraries of thousands of RNA folds in the presence of competitor oligonucleotides. The oligonucleotide mixture is carefully designed to allow for highly specific interactions between small molecules and RNA motifs in the randomized region to be bound on the array surface. The small molecule-RNA fold binding interactions are individually harvested from the agarose microarray surface by simple excision. The bound RNAs are amplified and sequenced and bioinformatically analyzed. The frequency of each RNA fold member from the sequenced binders is compared to the frequency of each RNA fold member in the starting library [25] to identify the RNA motifs that are enriched or depleted in the sample. This analysis defines the encyclopedia of RNA folds that each compound interacts with and also a scoring function for the binding interactions. This scoring function assigns the relative affinity or lack of affinity to all members of the RNA fold library. 2DCS selections and its subsequent statistical analysis answers fundamental questions for each compound: (i) does the compound interact with the RNA library; that is, does it have inherent RNA-binding capacity; (ii) with which specific RNAs does it interact; and, (iii) does the compound bind many RNAs with similar affinity or a small subgroup very specifically and with high affinity.
This approach has defined the interaction landscapes for a variety of small molecules and RNA folds. For example, small molecule libraries studied include known drugs, pharmaceutical grade screening collections, and other synthesized compounds [26,27]. From a single 2DCS selection we have routinely identified tens to hundreds of binders and informed a selectivity profile [28,29].
Inforna – mining for druggable RNA folds across the human genome.
We have compiled a database of a wide variety of RNA structures in the human transcriptome including highly abundant RNAs and all human microRNA (miRNA) precursors [30]. MiRNAs are non-coding RNAs that function by translational repression of an mRNA. They are produced in the nucleus as highly structured primary transcripts (pri-miRNA) that are processed by the nuclear nuclease Drosha to liberate a precursor miRNA (pre-miRNA). Pre-miRNAs are translocated to the cytoplasm and processed by the cytoplasmic nuclease Dicer to produce mature miRNAs (miRs). The mature miRNA binds to the 3’ untranslated regions (UTRs) in complementary mRNAs to translationally repress the amount of protein made from a given mRNA. Many miRNAs are associated with various disease states and as such are important, yet underexploited, drug targets.
Our first validation of Inforna was to generate testable hypothesis about RNA structures that can be bound by small molecules and if ligands are selective. Fortuitously, the structures of miRNA precursors are very accurately predicted by using free energy minimization. Small molecule binding sites have been identified that are unique, non-unique sites [30]. This analysis of miRNA precursor structure and targetable sites within them have allowed us to uncover key factors that govern the effects of binding compounds on RNA biology. For example, we have shown that to affect the biology of an miRNA, a compound must bind to a functional site, the sites recognized and cleaved by nucleases to enable miRNA maturation [31]. Targeting Drosha and Dicer processing sites in various RNA targets – pri-miR-96, pre-miR-210 and pri-miR-515 – reduce mature miRNA levels in disease relevant systems and affect the downstream protein expression and associated phenotype. Furthermore, we discovered that compound selectivity is informed by Inforna. For example, we have found that the Drosha processing site in pri-miR-96 is a unique site thus can be targeted selectively with a small molecule [32]. In contrast, the Dicer processing site of pre-miR-210 is not unique but a compound targeting this site can be selective because off-targets are expressed at lower levels and are therefore not as highly occupied as a desired target [31]. In another example, pri-miR-515 and pri-miR-885 share a common Drosha site and are expressed at similar levels. Therefore, they are modulated similarly by a small molecule that recognizes their common structure. However, approaches have been developed to selectively target pri-miR-515 by targeting its Drosha site and an adjacent structure not found in pri-miR-885 with a single small molecule, enabled by Inforna [33].
Targeted degradation via ribonuclease targeting chimeras (RIBOTACs).
Antisense and siRNA approaches allow for targeted degradation of RNA by harnessing endogenous pathways. We sought to develop a similar approach to cleave an RNA target by using RNA QC machinery but with small molecules as the RNA recognition element. Amongst many nucleases that are expressed in cells, we also sought a nuclease whose cleavage activity could be tightly controlled. Ribonuclease L (RNase L) is latent ribonuclease that is expressed in cells in minute qualities. Upon a viral infection a 2’−5’ oligoadenylate (2’−5’An) is produced that binds to the monomeric, inactive form of RNase L, inducing its dimerization and activation [34]. Thus, we synthesized chimeric compounds in which a dimeric ligand that bind to an RNA target was appended with a short 2’−5’A4 moiety. This new strategy allows one to not only recruit but also activate RNase L only on the desired RNA target, triggering its catalytical and substoichiometric cleavage (Figure 3A) [15].
Figure 3: Methods to affect the abundance of RNAs in cells by using small molecules to bind to structured RNAs.
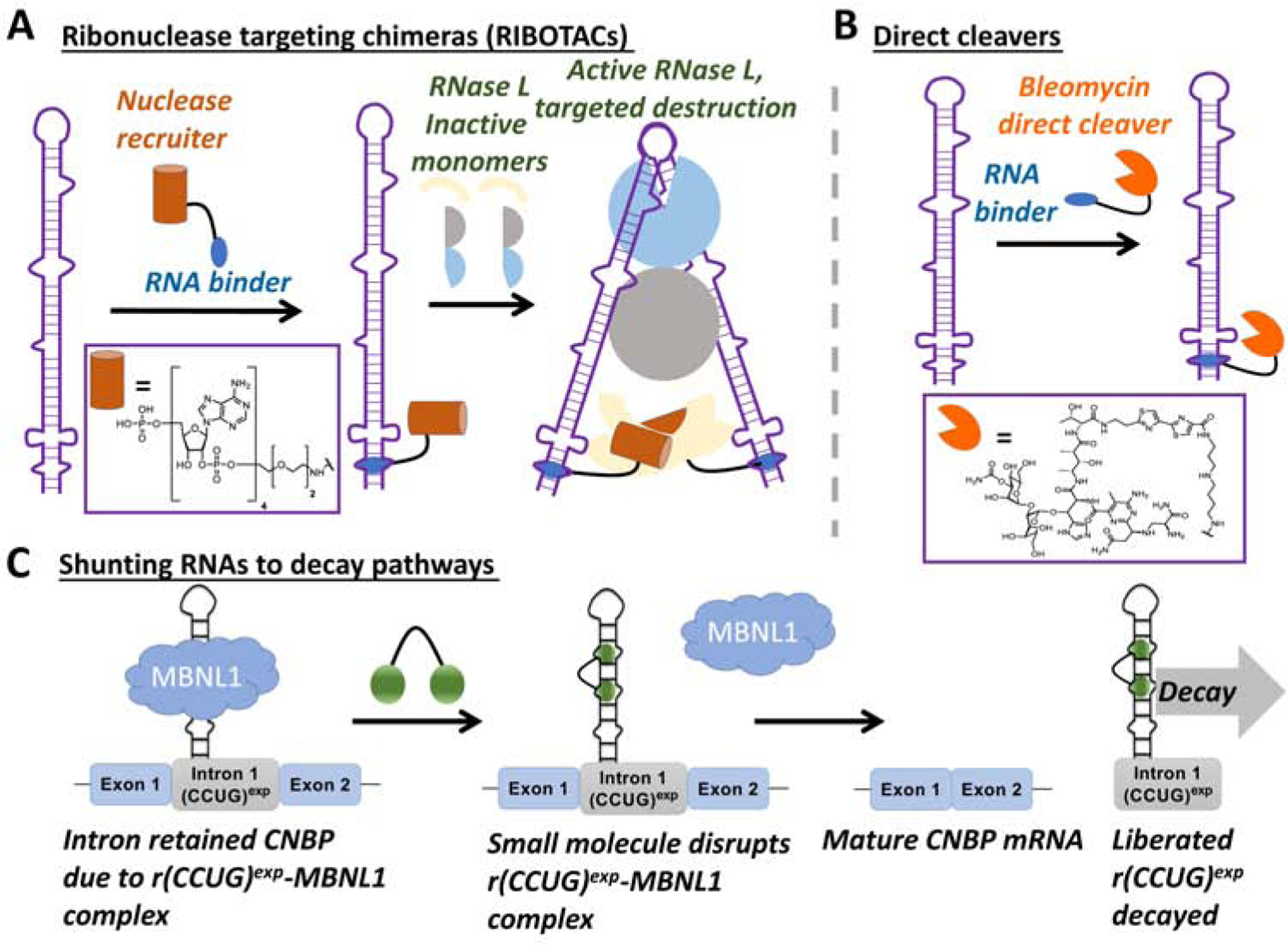
A, Ribonuclease targeting chimeras (RIBOTACs) in which a recruitment module for a nuclease is appended to a small molecule interacting with RNA, thereby facilitating cleavage of the desired RNA target. B, Coupling compounds to natural products such as bleomycin to direct cleavage of an RNA target by an oxidative mechanism. C, Toxic repeats that are present in retained introns can be subjected to decay as RNA-binding small molecules cause the RNA to be processed, liberating the intronic RNA and causing its decay.
One of the challenges with any compound, particularly those that target RNA, is achieving selectivity. Profiling of degraders in cells has shown that they have both enhanced potency and selectivity relative to their parent, simple binding compounds [16]. This observation can be attributed to several factors that includes, a lower dose of the drug required to have an effect, and the fact that the nucleases that they recruit also have substrate specificity, which further decreases affinity for off-targets. For example, a small molecule that targets pre-miR-210 also binds DNA, however, when the compound is converted into a degrader, DNA binding capacity is ablated [15,16], likely due to steric clashing or repulsion of the appended nuclease-recruitment module. Notably, to further validate direct target engagement and compound mode of action, the pre-miR-210 study showed a ternary complex formed between the RIBOTAC, pre-miR-210, and RNase L complex by immunoprecipitation (IP). Indeed, only pre-miRNA-210 was enriched in the IP fractions, while other miRNAs were not, confirming the selectivity and efficiency of the RIBOTAC approach [16].
A recent study showed that a promiscuous protein-binding ligand (kinase inhibitor) can be converted into a selective PROTAC [10]. It will be interesting to see if RIBOTACs and PROTACs are general strategies to enhance the selectively of RNA- and protein-binding small molecules, respectively, and if selectivity and activity are programmable. A recent study indicated programmability could indeed be possible for PROTACs, as the residence time of the ternary complex formed between the protein target, PROTAC, and ligase was correlated with cellular activity [35]. Such in-depth studies will be essential to balance activity and selectivity and therefore for translating both PROTACs and RIBOTACs into the clinic. Despite their relatively large sizes compared to traditional small molecule drugs, PROTACs have gained traction as two have favorable data from phase I clinical trials (Arvinas’ ARV-110 and ARV-417), which could bode well for RIBOTACs.
Importantly, RIBOTACs have expanded not only compound mode of action but also the number of RNAs targetable with a small molecule. That is, our previous studies targeting miRNA precursors as well as RNA repeat expansions have shown that small molecule binding to a functional site was required for bioactivity. Thus, if a functional site within an RNA had yet to be identified or if the functional site was not structured, the RNA target was not amenable to small molecule targeting. In theory, RIBOTACs could be designed for any RNA target that folds into a structure recognized by a small molecule, provided the preferred substrate for the nuclease recruited is nearby. Such a notion opens up a whole new research avenue, recruiting other nucleases with different substrate specificities to be custom matched to an RNA target and its corresponding lead small molecule.
Development of compounds that directly degrade RNA targets by coupling RNA binders to bleomycin A5.
Work by the Hecht laboratory has shown that RNA can be cleaved by the natural product bleomycin, known to cause DNA strand breaks [36–38]. Indeed, DNA cleavage is bleomycin’s believed mode of action for the treatment of cancer. Bleomycin also cleaves RNA in vitro [36], and as we later demonstrated, in situ [39–41] and in vivo [17].
We first validated our direct small molecule cleavage approach using a dimeric compound that targets the RNA triplet repeat expansion that causes myotonic dystrophy type 1 (DM1; r(CUG)exp), an incurable genetic disease that is the most common form of adult on-set muscular dystrophy. The dimer recognizes the periodic array of internal loops that form when r(CUG)exp folds. This r(CUG)exp-selective compound was appended to a bleomycin A5 cleaving module via the natural product’s primary amine (Figure 3B). The dimer-bleomycin A5 conjugate selectively cleaved the mutant allele harboring the toxic r(CUG)exp without affecting levels of the WT allele [41]. Further, the compound did not cleave DNA in vitro or in cells at the active concentration that degraded r(CUG)exp and alleviated DM1-associated molecular defects [42–44]. Specificity is due to the manner in which the bleomycin A5 is attached to the RNA binder, via conjugation of the amine that forms interactions with DNA; thus, this moiety serves two purposes –an anchor site for attachment of RN-binding module and decreasing binding to DNA.
Notably, the bleomycin conjugate selectively cleaved r(CUG)exp and broadly improved defects including a myotonic phenotype in a mouse model of DM1 [17]. Importantly, the compound had no detectable off-targets in the mouse model, as determined by RNA-seq, and improved 97% of defects observed in the transcriptome. The structure-specific molecular recognition of the cleaving compound was superior to sequence-specific binding by an oligonucleotide. The oligonucleotides triggered decay of mRNAs that contained both long and short repeats of r(CUG) while the structure-binding compound was specific for the disease-causing expanded repeat [17].
Designing small molecules to shunt toxic RNAs to native quality control pathways.
Myriad studies have shown that RNA processing pathways are affected in various diseases [45–47]. For example, in the >30 diseases caused by RNA repeat expansions, defects in RNA processing are observed. These defects are due to an RNA gain-of-function where toxic RNAs sequester proteins that are involved in RNA biogenesis, as observed in DM1 and myotonic dystrophy type 2 (DM2) and other microsatellite diseases. Transcripts harboring repeat expansions in introns in particular are improperly processed [48]. In DM2, the toxic r(CCUG)exp tetranucleotide repeat expansion is housed in intron 1 of the CCHC-type zinc finger nucleic acid binding protein gene (CNBP) [44,49], causing aberrant intron 1 retention. Intron retention appears to be a general feature of GC-rich repeats present in introns [48].
We investigated the nature of intron retention in DM2 and found that the binding of proteins to these repeats facilitated intron retention. In particular, intron retention is caused by bind the binding of muscleblind-like 1 protein (MBNL1), supported by gain- and loss-of-function studies. That is, knock-in and knock-down of MBNL1 increases and reduces intron 1 retention, respectively [18]. Thus, small molecules that bind r(CCUG)exp structure and reduce the loading of MBNL1 should decrease intron retention and cause the liberated intronic repeat to be subjected to native decay pathways. Indeed, this is the case [18]. The results of this study support that RNAs can be directed to decay pathways by using simple binding compounds (Figure 3C). Collectively, the studies described herein illustrate that there are many ways in which to affect the biology of RNA, including interfacing with RNA QC and degradation pathways.
Methods for target validation and studying cellular selectivity.
Until recently, there was a dearth of methods available to study small molecule target engagement and selectivity in situ and in vivo. Such studies are essential to confirm compound mode of action and identify off-target liabilities. However, RIBOTACs and small molecules that directly cleave the RNA target enable both to be studied simultaneously. That is, after treatment, total RNA can be analyzed for changes in expression level by comparison to untreated cells or animals; direct targets are depleted in RNA-seq data. Two additional sets of experiments are key to elucidating bona fide targets, off-targets, and downstream effects caused by inhibiting the RNA target, which can be quite complex: (i) competitive target profiling experiments; that is, co-treatment with the small molecule degrader and the parent, simple binding compound in dose response; and (ii) analogous experiments in WT cells or animals in which the target is not expressed or not aberrantly expressed [17]. Complementary target validation methods have also been developed including Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) and its competitive variant [41,50]. A Chem-CLIP probe is comprised of a small molecule conjugated to a cross-linking module (diazirine or chlorambucil) and biotin. A covalent bond is formed between the probe (small molecule-diazirine or small molecule-chlorambucil conjugate) and the targets it binds, which are then pulled down with streptavidin beads and analyzed to identify the target and its enrichment (degree of cellular occupancy).
Conclusions and future outlook.
The ability to design small molecules from sequence to target structured regions of RNA has provided a wide variety of chemical probes to study and manipulate RNA function. At present, there is only a limited number of ligands that have been purposefully designed to target RNA and even fewer bioactive ligands that affect RNA. RNA splicing modulators such as Risdiplam have been identified by phenotypic screens, and studies have shown that it binds an RNA-protein interface [51]. Targeting such interfaces is likely very challenging from a design perspective and as such has largely been unexplored. It remains to be seen how frequently such complexes contribute to human disease and are thus potential therapeutic targets. Ribocil, an antibacterial, riboswitch-binding ligand was also identified from phenotypic screen. Unfortunately, resistance mutations against Ribocil have emerged quickly [52]. It is also clear that known drugs can interact with RNA and as such new approaches to profile compounds emerging from phenotypic screens for binding RNA could prove to be broadly useful [22].
It is a significant challenge to design, develop, or discover bioactive ligands that target RNA and even more challenging to trace their activities to RNA binding; however, Inforna has provided such ligands. Inforna has likely been successful because of the nature of the data that is fed into the server, which is derived experimentally from 2DCS. The 2DCS approach allows one to determine if ligands are selective or non-selective and to define cellular on- and off-targets. Such information density is not acquired by using traditional high throughput screening of one target. Starting a probe discovery process with detailed knowledge of selectivity profiles is important and can enable design strategies to enhance potency and selectivity, including modular assembly.
More RNAs are likely to be targeted by small molecules as activity in the area increases [53–56]. One of the challenges with these studies will be to define selectivity profiles and target engagement in cells. Target profiling approaches such as Chem-CLIP can enable these studies and have proven to be critical in studying underlying mechanism of action [41,57]. Given the success that targeted degradation has had in only these initial forays into RNA, the potency of these ligands could be enhanced by using cleavage-based approaches, particularly nuclease and direct cleaving methods as have been described here. Also, since small molecules that bind to non-functional sites of RNA can be transformed to degraders the repertoire of small molecules that can affect RNA biology can be increased exponentially, unlocking studies of many more RNA targets and their biology.
Given the panoply of RNA QC enzymes that are present in cells, many more nucleases can be recruited to leverage their activity against a target RNA. In addition, there are many ways in which a cell rids itself of RNAs. Thus, there will likely be a great many new ways for small molecules to affect RNA lifetime. These and other developments will likely put small molecule targeting of RNA in the forefront of medicine and not as an afterthought, as it is today. We are excited to watch it unfold.
Acknowledgments.
We thank all of our laboratory co-workers over the past 15 years that have pushed the boundaries of RNA targeted small molecules. Funding for these efforts has been provided by the taxpayers of the United States of America in the form of grants from the National Institutes of Health (R01 GM97455, DP1 NS096898, P01 NS09914, and R33 NS096032 to M.D.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Angelbello AJ, Chen JL, Childs-Disney JL, Zhang P, Wang ZF, Disney MD: Using genome sequence to enable the design of medicines and chemical probes. Chem Rev (2018) 118(4):1599–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *2.Zamecnik PC, Stephenson ML: Inhibition of rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A (1978) 75(1):280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors reported a tridecamer DNA oligonucleotide, complementary to 3’ and 5’ reiterated terminal sequences of Raus sarcoma virus 35S RNA, when added to chick embryo fibroblast tissue cultures infected with the virus showed inhibition of virus production. This is the first report that showed that ASO can inhibit viral replication in-vitro.
- 3.Stephenson ML, Zamecnik PC: Inhibition of rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A (1978) 75(1):285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *4.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC: Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature (1998) 391(6669):806–811. [DOI] [PubMed] [Google Scholar]; This is the first report of dsRNA mediated interference in-vivo in C elegans to manipulate gene expression. The sense-antisense mixture produced highly effective interference with endogenous gene activity and further demonstrated target specificity of dsRNA effects and ability of interference to cross cellular boundaries.
- *5.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T: Duplexes of 21- nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature (2001) 411(6836):494–498. [DOI] [PubMed] [Google Scholar]; Here, the authors demonstrated that small interfereing RNAs (siRNAs) are capable of mediating sequence-specific RNAi in mammalian cell cultures, affecting the endogenous gene expression of target genes.
- 6.Nelles DA, Fang MY, O’Connell MR, Xu JL, Markmiller SJ, Doudna JA, Yeo GW: Programmable RNA tracking in live cells with CRISPR/Cas9. Cell (2016) 165(2):488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *7.Stojic L, Lun ATL, Mangei J, Mascalchi P, Quarantotti V, Barr AR, Bakal C, Marioni JC, Gergely F, Odom DT: Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res (2018) 46(12):5950–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors performed a quantitative study of off-target effects in widely used loss of function methods - RNAi, LNA and CRISPRi. They observed that all three methods had non negligible off target effects with CRISPRi (poly-clonal) being the cleanest. They also provided recommendations to mitigate these off target effects including use of multiple negative controls, use of log-scale threshold, using multiple target sequences etc.
- 8.Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, Aigner S et al. : Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell (2017) 170(5):899–912.e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *9.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ: Protacs: Chimeric molecules that target proteins to the skp1-cullin-f box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A (2001) 98(15):8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]; n this first report of PROTACS approach, the authors studied peptide mediated ubiquitylation and degradation of target methionine aminopeptidase 2 (MetAP2). A proteolysis targeting chimeric molecule, Protac-1 was synthesized that could bind to the target protein and ubiquitin ligase complex and the ubiquitinated MetAP2 was degraded by the endogenous ubiquitin/proteasome pathway.
- *10.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, Crews CM: Lessons in protac design from selective degradation with a promiscuous warhead. Cell Chem Biol (2018) 25(1):78–87.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors studied the target binding affinity and degradation profile of two ligase recruiters using a promiscuous kinase inhibitor and accessed global proteomic changes. The degradation profile was found to be more selective than binding profile and the extent of degradation was found to not correlate with binding affinities, suggesting that the stable ternary complex formation between ubiquitin ligase and the substrate is the determining factor for degradation.
- 11.Schneekloth JS Jr., Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM: Chemical genetic control of protein levels: Selective in vivo targeted degradation. J Am Chem Soc (2004) 126(12):3748–3754. [DOI] [PubMed] [Google Scholar]
- 12.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N et al. : Catalytic in vivo protein knockdown by small-molecule protacs. Nature Chem Biol (2015) 11(8):611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doma MK, Parker R: RNA quality control in eukaryotes. Cell (2007) 131(4):660–668. [DOI] [PubMed] [Google Scholar]
- 14.Harigaya Y, Parker R: No-go decay: A quality control mechanism for RNA in translation. Wiley Interdiscip Rev RNA (2010) 1(1):132–141. [DOI] [PubMed] [Google Scholar]
- **15.Costales MG, Matsumoto Y, Velagapudi SP, Disney MD: Small molecule targeted recruitment of a nuclease to RNA. J Am Chem Soc (2018) 140(22):6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors report the development of Ribonuclease-targeting chimeras (RIBOTACs) to recruit endogenous cellular nucleases to catalytically and substoichiometrically cleave a non-coding RNA.
- 16.Costales MG, Suresh B, Vishnu K, Disney MD: Targeted degradation of a hypoxia- associated non-coding RNA enhances the selectivity of a small molecule interacting with rna. Cell Chem Biol (2019) 26(8):1180–1186.e1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Angelbello AJ, Rzuczek SG, McKee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, Disney MD: Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc Natl Acad Sci U S A (2019) 116(16):7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors have reported the design of a small molecule, cugamycin, that selectively cleaved r(CUG) repeat expansion associated with mytotic dystrophy type 1. The small molecule was demonstrated to cleave target RNA in disease relevant mouse model, improve the splicing effects and rescue other disease associated phenotype. Furthermore, in-vivo transcriptome-wide analysis of on and off target effects were perfomed vis RNA-seq of RNA isolated from mouse.
- **18.Benhamou RI, Angelbello AJ, Wang ET, Disney MD: A toxic RNA catalyzes the cellular synthesis of its own inhibitor, shunting it to endogenous decay pathways. bioRxiv (2019) 741926. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors reported a designer compound that targets the strcutured regions of r(CCUG) repeats, associated with mytotic dystrophy type 2. The compound was further appended with bioorthogonal alkyne and azide moieties that transformed the compound to a very potent compound which on binding to RNA repeats templated synthesis of their own inhibitor via in-situ click reaction.
- 19.Lima WF, Monia BP, Ecker DJ, Freier SM: Implication of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry (1992) 31(48):12055–12061. [DOI] [PubMed] [Google Scholar]
- *20.Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH: Predicting oligonucleotide affinity to nucleic acid targets. RNA (1999) 5(11):1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors reported a computer program, oligowalk, that predicts the duplex stability of oligonucleotide and target nucleic acid while taking into account the duplex structure and self structure of both oligonucleotide and target. This predicted measurement is particularly useful in design of oligonucleotide and is part of RNA structure package developed by their lab.
- *21.Velagapudi SP, Gallo SM, Disney MD: Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol (2014) 10(4):291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]; Here, the authors have reported a computational approach named inforna, which identifies lead molecules targeting structured RNA regions. This approach informs a sequence-based design of bioactive ligands against any RNA target of interest.
- 22.Disney MD: Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J Am Chem Soc (2019) 141(17):6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD: A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem Biol (2007) 2(11):745–754. [DOI] [PubMed] [Google Scholar]
- 24.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL: Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J Am Chem Soc (2008) 130(33):11185–11194. [DOI] [PubMed] [Google Scholar]
- 25.Velagapudi SP, Luo Y, Tran T, Haniff HS, Nakai Y, Fallahi M, Martinez GJ, Childs-Disney JL, Disney MD: Defining RNA -small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent Sci (2017) 3(3):205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velagapudi SP, Costales MG, Vummidi BR, Nakai Y, Angelbello AJ, Tran T, Haniff HS, Matsumoto Y, Wang ZF, Chatterjee AK, Childs-Disney JL et al. : Approved anti-cancer drugs target oncogenic non-coding RNA s. Cell Chem Biol (2018) 25(9):1086–1094.e1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velagapudi SP, Disney MD: Two-dimensional combinatorial screening enables the bottom-up design of a microRNA-10b inhibitor. Chem Commun (2014) 50(23):3027–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childs-Disney JL, Tran T, Vummidi BR, Velagapudi SP, Haniff HS, Matsumoto Y, Crynen G, Southern MR, Biswas A, Wang ZF, Tellinghuisen TL et al. : A massively parallel selection of small molecule-RNA motif binding partners informs design of an antiviral from sequence. Chem (2018) 4(10):2384–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haniff H, Knerr L, Liu X, Crynen G, Boström J, Abegg D, Adibekian A, Lemurell M, Disney MD: Design of a small molecule that stimulates VEGFA informed from an expanded encyclopedia of RNA fold-small molecule interactions. bioRxiv (2019) DOI: 10.26434/chemrxiv.7967291.v1. [DOI] [Google Scholar]
- *30.Liu B, Childs-Disney JL, Znosko BM, Wang D, Fallahi M, Gallo SM, Disney MD: Analysis of secondary structural elements in human microRNA hairpin precursors. BMC Bioinformatics (2016) 17, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]; Herein, the authors reported a database of microRNA (miRNA) motifs present in human. The sequences obtained from miRbase were used in secondary structure prediction by RNAstructure, a software that uses free energy minimization. Quantitative analysis gave insights into the preferred non canonical base pairings and structural elements that are prevalent in miRNA motifs. In particular, the authors identified that single nucleotide bulges and 1×1 internal loops are highly represented in these motifs. This database is widely used in informing design of small molecule for miRNA targets.
- 31.Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, Disney MD: Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J Am Chem Soc (2017) 139(9):3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velagapudi SP, Cameron MD, Haga CL, Rosenberg LH, Lafitte M, Duckett DR, Phinney DG, Disney MD: Design of a small molecule against an oncogenic noncoding RNA. Proc Natl Acad Sci U S A (2016) 113(21):5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costales MG, Hoch DG, Abegg D, Childs-Disney JL, Velagapudi SP, Adibekian A, Disney MD: A designed small molecule inhibitor of a non-coding RNA sensitizes her2 negative cancers to herceptin. J Am Chem Soc (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman RH: A scientific journey through the 2–5A/Rnase L system. Cytokine Growth Factor Rev (2007) 18(5–6):381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy MJ, Winkler S, Hughes SJ, Whitworth C, Galant M, Farnaby W, Rumpel K, Ciulli A: SPR-measured dissociation kinetics of protac ternary complexes influence target degradation rate. ACS Chem Biol (2019) 14(3):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *36.Carter BJ, de Vroom E, Long EC, van der Marel GA, van Boom JH, Hecht SM: Site-specific cleavage of RNA by Fe(ii).Bleomycin. Proc Natl Acad Sci U S A (1990) 87(23):9373–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors reported for the first time cleavage of RNA by bleomycin, an anti tumor agent which degrades DNA. They found that Fe (II) coordinated bleomycin was able to cleave RNA with ten fold better selectivity than DNA and this site-specific oxidative cleavage activity was attributed to its ability to reognize RNA structured regions.
- 37.Hecht SM: RNA degradation by bleomycin, a naturally occurring bioconjugate. Bioconj Chem (1994) 5(6):513–526. [DOI] [PubMed] [Google Scholar]
- 38.Hecht SM: Bleomycin: New perspectives on the mechanism of action. J Nat Prod (2000) 63(1):158–168. [DOI] [PubMed] [Google Scholar]
- 39.Angelbello AJ, Disney MD: Bleomycin can cleave an oncogenic noncoding RNA. Chembiochem (2018) 19(1):43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Disney MD: Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem Biol (2018) 13(3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, Disney MD: Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat Chem Biol (2017) 13(2):188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.La Spada AR, Paulson HL, Fischbeck KH: Trinucleotide repeat expansion in neurological disease. Ann Neurol (1994) 36(6):814–822. [DOI] [PubMed] [Google Scholar]
- 43.Ranum LP, Cooper TA: RNA-mediated neuromuscular disorders. Annu Rev Neurosci (2006) 29(259–277. [DOI] [PubMed] [Google Scholar]
- 44.Day JW, Ranum LP: RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord (2005) 15(1):5–16. [DOI] [PubMed] [Google Scholar]
- 45.Baralle D, Buratti E: RNA splicing in human disease and in the clinic. Clin Sci (2017) 131(5):355–368. [DOI] [PubMed] [Google Scholar]
- 46.Scotti MM, Swanson MS: RNA mis-splicing in disease. Nat Rev Genet (2016) 17(1):19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montes M, Sanford BL, Comiskey DF, Chandler DS: RNA splicing and disease: Animal models to therapies. Trends Genet (2019) 35(1):68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *48.Sznajder LJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, Oliveira R, Nutter CA, Bhatt K, Sobczak K, Ashizawa T et al. : Intron retention induced by microsatellite expansions as a disease biomarker. Proc Natl Acad Sci U S A (2018) 115(16):4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study the authors queried GC and A/AT rich repeats present in the intron region of mRNAs associated with diseases including DM2, FECD and C9-ALS/FTD as examples. The study analyzed RNA-seq data obtained from disease cells or tissues and compared it to healthy control and found that GC rich microsatellites are present in close proximity to splice sites and form highly structured regions that impairs proper spliceosome assembly, leading to intron retention. Furthermore, they demonstrated with r(CCUG) repeats as an example that the repeats can be used as a disease biomarker.
- 49.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP: Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293(5531):864–867. [DOI] [PubMed] [Google Scholar]
- 50.Guan L, Disney MD: Covalent small-molecule-RNA complex formation enables cellular profiling of small-molecule-RNA interactions. Angew Chem Int Ed Engl (2013) 52(38):10010–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *51.Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, Jablonski P et al. : Discovery of risdiplam, a selective survival of motor neuron-2 (smn2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem (2018) 61(15):6501–6517. [DOI] [PubMed] [Google Scholar]; The authors described the discovery and development of a small molecule risdiplam, orally bioavailable and brain penetrant therapeutic drug candidate currently in clinical trials to treat spinal muscular atropy (SMA). It functions by modulating SMN2 gene splicing to produce full SMN protein. The authors have described chemical optimization pathway and extensive DMPK and toxicity studies.
- 52.Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinverni JC, Mayhood T, Villafania A, Nahvi A, Murgolo N et al. : Selective small-molecule inhibition of an RNA structural element. Nature (2015) 526(7575):672–677. [DOI] [PubMed] [Google Scholar]
- 53.Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr, Hargrove AE: Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew. Chem Int. Ed. Engl (2018) 57(40):13242–13247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorenz DA, Song JM, Garner AL: High-throughput platform assay technology for the discovery of pre-microRNA-selective small molecule probes. Bioconj Chem (2015) 26(1):19–23. [DOI] [PubMed] [Google Scholar]
- 55.Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, Ranken R, Massire C, Sampath R, Ecker DJ, Swayze EE et al. : Sar by ms: Discovery of a new class of RNA-binding small molecules for the hepatitis C virus: Internal ribosome entry site iia subdomain. J Med Chem (2005) 48(23):7099–7102. [DOI] [PubMed] [Google Scholar]
- 56.Abulwerdi FA, Shortridge MD, Sztuba-Solinska J, Wilson R, Le Grice SFJ, Varani G, Schneekloth JS: Development of small molecules with a noncanonical binding mode to HIV-1 trans activation response (TAR) RNA. J Med Chem (2016) 59(24):11148–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang WY, Wilson HD, Velagapudi SP, Disney MD: Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J Am Chem Soc (2015) 137(16):5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]