

Abstract
Blood lactate concentrations have traditionally been utilized as an index of exercise intensity or clinical hyperlactatemia. However, more recent data suggests that fasting plasma lactate can also be indicative of the risk for subsequent metabolic disease. The hypothesis presented is that fasting blood lactate accumulation reflects impaired mitochondrial substrate utilization, which in turn influences metabolic disease risk.
Keywords: blood lactate, aerobic metabolism, skeletal muscle, TCA cycle, metabolic disease
Summary
Fasting lactate concentration and is predictive of metabolic health and disease.
INTRODUCTION
The incidence of chronic diseases such as cardiovascular disease, obesity and type 2 diabetes (T2D) is reaching epidemic proportions. In relation to intervention and prevention there is evidence indicating that cardiorespiratory fitness and health are closely related. For example, Wei et al. followed 25,714 men and reported that a low level of cardiorespiratory fitness was an independent predictor of subsequent cardiovascular disease and all-cause mortality (1). Myers et al. followed 6,213 men for 6.2 years and concluded that exercise capacity is a more powerful predictor of mortality than other established risk factors for cardiovascular disease (2). In relation to a causative factor, Zwaard et al. found a relationship between mitochondrial capacity in skeletal muscle and VO2max (r2 = 0.89, P<.001) when studying subjects ranging from chronic heart failure to professional cyclists (3). Skeletal muscle plays a substantive role in maintaining glucose homeostasis by acting as a reservoir for ~80% of insulin-mediated glucose disposal (4). The fates of glucose entering the muscle fiber includes both the glycolytic and oxidative pathways, with the mitochondria playing a fundamental role in skeletal muscle oxidative processes. Defects in skeletal muscle mitochondria are thought to precede the developmental of metabolic diseases making the mitochondria a key organelle in prevention and/or treatment (5). In support of the role of the mitochondria, there is a substantial body of evidence that mitochondrial function is impaired in the muscle of individuals with obesity and those with T2D (5–7).
A healthy metabolism appropriately adjusts the rate of substrate oxidation according to substrate availability. For example, in the fed hyperinsulinemic condition carbohydrate is the primary substrate oxidized, whereas fat is primarily oxidized in the fasting state. The ability to appropriately adjust substrate oxidation relative to substrate availability is termed metabolic flexibility. The inefficient switching of substrates in response to a metabolic challenge, or a deficit in metabolic flexibility, has been implicated in obesity and insulin resistance and linked with mitochondrial substrate utilization (8). As an example, skeletal muscle from humans with obesity minimally increased glucose oxidation with insulin exposure and preferentially partitioned carbohydrate towards net lactate release (9).
It is well established that intracellular carbohydrate and lipid metabolism are dependent on mitochondrial content and (10). In skeletal muscle during rest, glucose entering the cell can be converted to pyruvate and transported into the mitochondria as acetyl-CoA for use in the tricarboxylic (TCA) cycle. Glucose that is not completely oxidized enters glycolysis and produces lactate (11). The net release of lactate seen in skeletal muscle from obese individuals suggests that impairments in the TCA cycle could be a culprit leading to a deficit in metabolic flexibility. Together, these data suggest that an impairment in mitochondrial substrate utilization in human skeletal muscle is linked with metabolic disease and identifies a role for lactate in both glycolytic and oxidative metabolism.
Lactate is regarded as a major energy source, gluconeogenic precursor, and a signaling molecule that regulates lipid and carbohydrate metabolism even under aerobic conditions (11). Lactate is considered to be the nexus of glycolytic and oxidative metabolism as it is essentially “shuttled” between two major energy producing pathways, glycolysis and oxidative phosphorylation. In cells, the lactate shuttle is said to occur between a glycolytic producer and an oxidative consumer, which is characteristic during prolonged exercise. In healthy individuals at rest, mitochondrial oxidation supplies the majority of the energy required and lactate production through glycolysis is minimal. Lactate production is increased by either increasing energy expenditure or through a reduction in the energy produced by aerobic oxidation. Thus, elevated lactate can be an indication of increased reliance on glycolysis and used as a novel biomarker in clinical settings for predicting those at risk for metabolic disease. Therefore, we hypothesize that lactate reflects in vivo oxidative metabolism and metabolic flexibility, which in turn influences disease risk.
BLOOD LACTATE
In working muscle, lactate is often considered to be a metabolic byproduct of inadequate oxygen supply. However, lactate is now known to be produced even under aerobic conditions (11). In the exercise literature, lactate production, disposal, and clearance are higher in well trained cyclists than their healthy sedentary counterparts (12). Thus, for a similar given power output, lactate is lower in trained than sedentary individuals. It has also been demonstrated that lactate clearance is higher in skeletal muscle after endurance training (13). A recent study compared elite athletes to individuals with metabolic syndrome (14). Remarkably, at rest blood lactate concentration in individuals with the metabolic syndrome approximated that of trained cyclists exercising at a load of 300W. This dramatic difference lends credence to the notion that lactate may act as a proxy for metabolic health by functioning as an index of mitochondrial substrate utilization.
In support, fasting lactate concentrations have been used as an indicator of the severity of acute illnesses or injuries such as shock, sepsis, burns, and malignancy (15). We and others suggest that fasting lactate can also be a useful predictor of metabolic disease. In the Atherosclerosis Risk in Communities (ARIC) trial the lowest quartile of fasting lactate concentrations had a 12% incidence of T2D while 30% of the highest quartile were diabetic (16). In a case-cohort analysis of 544 diabetic cases and 533 non-cases, lactate at baseline predicted incident T2D (17). Another report from the ARIC study, (8,045 subjects without T2D at baseline and median follow-up of 12 years) showed that elevated fasting lactate preceded T2D (18). These studies demonstrate an association between fasting plasma lactate and T2D and suggest that lactate may be a useful predictor of individuals at risk for this disease. However, the underlying mechanism(s) linked with the accumulation of fasting lactate is not evident; in this paper we present our hypothesis implicating an underlying deficit in mitochondrial substrate utilization of skeletal muscle due to a defect in the TCA cycle.
We have performed several longitudinal studies to validate the relationship between fasting lactate, as a proxy for in vivo oxidative metabolism, and metabolic health (19). As shown in Fig. 1 Panel A, severely obese (BMI ≥ 40 kg/m2) patients who were on an 800 kcal/day diet for a week prior to Roux-en-Y gastric bypass (RYGB) surgery displayed lactate concentrations that were lower than when consuming an ad-lib diet (obese column vs.1 week pre-RYGB). Thus, a low-calorie diet, which is known to improve metabolic health, reduces resting/fasting plasma lactate. As early as a week after RYGB (1-week post RYGB) fasting plasma lactate was further depressed and 1–9 months’ post-surgery, resting/fasting lactate decreased even more (Fig. 1, Panel B). It is important to note that at 7–9 months’ post RYGB, patients are consuming ~2,500 kcal/d and are weight-stable, indicating that energy deficit alone does not influence fasting lactate (20). As presented in Fig 1B, subjects with metabolic syndrome subjected to a 9-month exercise program reduced fasting lactate concentration. Furthermore, a study has shown that progressive weight loss (5% to16%) leads to a decrease in fasting lactate concentrations in individuals with obesity (21). The change in fasting lactate after 5% weight loss inversely correlated with the change in insulin sensitivity. Together, these data indicate that interventions which improve metabolic health reduce the accumulation of fasting plasma lactate.
Figure 1.
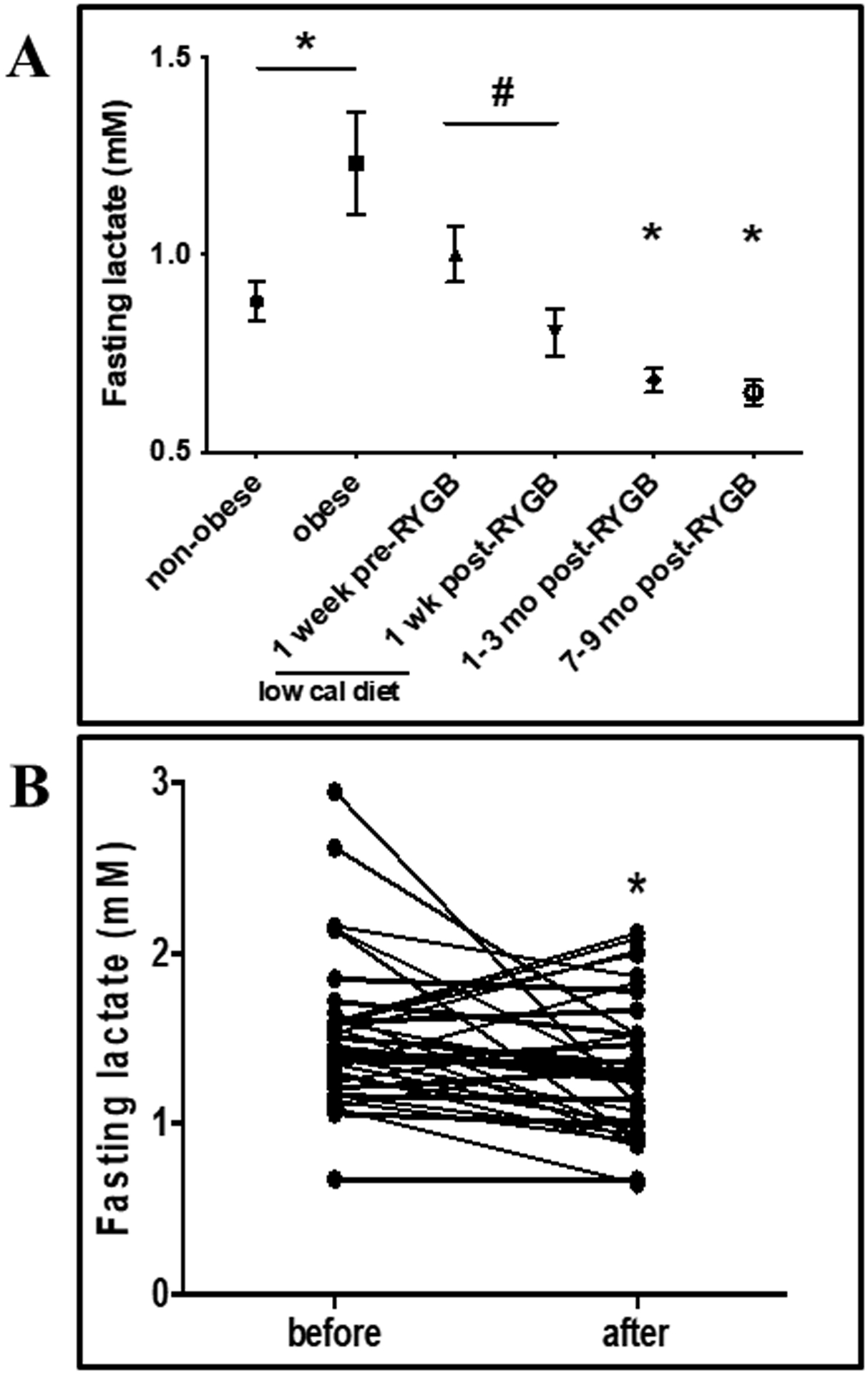
Panel A: Fasting plasma lactate (mean ± SEM) in subjects with and without obesity and after Roux-en-Y gastric bypass (RYGB) surgery. * Significantly different from non-obese at p = 0.032. # Significantly different from 1-week pre-RYGB at p = 0.02. Panel B: Fasting plasma lactate (mean ± SEM) in subjects with metabolic syndrome before and after six months of endurance exercise training. * Significantly different from before at p < 0.0001. Data from (19).
One could question how a low-calorie diet or RYGB may increase mitochondrial substrate oxidation. Studies on calorie restriction have shown improvements in mitochondrial biogenesis (22) and capacity (23). Similarly, mitochondrial capacity was also improved following RYGB (24). We believe these observations support our over-arching hypothesis that in vivo oxidative metabolism pathways are depressed in individuals at risk for metabolic disease and interventions which increase oxidative capacity reduce risk.
RELATIONSHIP BETWEEN METABOLIC FLEXIBILITY AND METABOLIC HEALTH.
The concept of metabolic flexibility was introduced by Kelley and Mandarino in classic experiments where they measured the respiratory quotient across a muscle bed (25). In the basal state, lean individuals oxidized predominantly fat, but when challenged with insulin switched to carbohydrate. In contrast, individuals with obesity did not adjust oxidation in response to insulin which is indicative of metabolically inflexibility. Being able to switch substrates also extends to lipid oxidation; for example, lean controls fed a high fat diet adjusted within days to a preference for fat oxidation (26). In contrast, subjects with obesity were in positive fat balance as they required a longer period to adjust oxidation; during positive fat balance, there was increased lipid deposition into adipose tissue as well as ectopic depots (muscle, liver, and pancreatic beta cells). Thus, the inability to adapt to substrate availability (metabolic flexibility) provides a physiological milieu for the development of metabolic dysfunction.
Our group has studied metabolic flexibility by imposing a high fat diet (HFD) and determining the ability of skeletal muscle to adjust lipid oxidation (27, 28). When a HFD was provided for 3 days, the ability of muscle to oxidize fatty acids increased significantly in lean individuals, but did not change in individuals with obesity. Some studies have further elucidated these mechanisms using endurance-trained subjects and lipid perfusion during a hyperinsulinemic euglycemic clamp (29, 30). Both concluded that the muscle of endurance-trained subjects can increase fatty acid oxidation (and decrease glucose oxidation) when presented with a lipid challenge. The authors also reported higher mitochondrial capacity in the muscle of the endurance-trained subjects indicating the essential role of this organelle in the control of metabolic flexibility.
The critical role of the mitochondria in metabolic flexibility was first described such that increases in fatty acids promotes lipid oxidation and suppression of glucose oxidation (31). Skeletal muscle mitochondria rely on fatty acids and glucose to produce ATP. The pyruvate dehydrogenase complex (PDH) controls entry of glucose-derived carbons into the mitochondria (32) and studies have shown that PDH plays a crucial role in metabolic flexibility in muscle (33, 34). In a recent study of isolated rodent skeletal muscle mitochondria, the authors reported decreased pyruvate oxidation when adding a fatty acid substrate to mitochondria that were already oxidizing pyruvate (35). Under conditions of either high fat or high sucrose, there is a large decrease in TCA cycle flux. Lactate can also directly supply acetyl-CoA to the TCA cycle and can act as the preferred fuel source in working muscle (13). Therefore, lactate competition can affect glucose uptake, which likely explains why both lactate and circulating glucose are higher in individuals with metabolic disease. Ultimately, this sets the stage for TCA cycle flux as a major regulator in metabolic flexibility at the cellular level since both glucose (via pyruvate) and fatty acids in addition to lactate will supply acetyl-CoA for energy production.
TCA CYCLE FUNCTION AND LACTATE
As described in the previous section, mitochondria govern metabolic flexibility at the cellular level. The TCA cycle is a key metabolic pathway of glucose and fatty acid oxidation that generates NADH and FADH2 for ATP production via respiration. Lactate can also drive these processes by directly providing substrate for the TCA cycle. Lactate is oxidized to pyruvate in the inner mitochondrial membrane by a lactate oxidation complex consisting of mono-carboxylate transporter-1, its chaperone CD147, mitochondrial lactate dehydrogenase, and cytochrome oxidase (11). Through this process, known as the “Intracellular Lactate Shuttle”, lactate can give rise to acetyl-CoA for use by the TCA cycle similarly to beta-oxidation of fatty acids and glucose oxidation via glycolysis. Lactate can create a competition for substrate usage between glucose and fatty acid oxidation, by regulating glucose disposal via GLUT4 and by malonyl-CoA inhibition of carnitine-palmitoyl transferase-1, respectively (36). Furthermore, lactate can directly inhibit lipolysis in white adipocytes through activation of the G-protein-coupled receptor (GPR81), further limiting fatty acid oxidation (37). This is primarily through cyclic-AMP and the cAMP response element binding (38). Activation of GPR81 has further implications for decreasing lipolysis by lactate giving rise to acetyl-CoA and subsequently malonyl-CoA, which inhibits mitochondrial FFA uptake by inhibition of CPT1 (36). However, it is not evident if lactate is a cause or consequence of these alterations in substrate oxidation and lack of metabolic flexibility.
One of our consistent observations is that individuals with severe obesity have a compromised ability to oxidize substrates, including both fat and glucose in skeletal muscle as well as the whole-body level. We have consistently reported that these individuals oxidize fatty acids and glucose at a lower rate than subjects without obesity ranging from whole body to in vitro studies using intact muscle fiber strips, muscle homogenates, and primary cultured muscle cells (39). Since fat oxidation accounts for a large proportion of energy expenditure at rest, this suggests that carbohydrate must be supplying a larger proportion of energy in individuals with obesity. Indeed, fatty acid oxidation was depressed with a compensatory increase in glycolysis in muscle from subjects with severe obesity (9). This metabolic profile in skeletal muscle is consistent with data demonstrating that resting/fasting plasma lactate is elevated in individuals with obesity (19). At rest, plasma lactate is in a steady state determined by the rates of lactate production and utilization. An increase in fasting plasma lactate could thus be due to either an increase in production or a decrease in utilization through gluconeogenesis in the liver. If glucose production via gluconeogenesis were suppressed, then we would expect that the rise in lactate would be mirrored by a decrease in fasting glucose. The opposite is observed as evidence favors increased lactate production since fasting glucose and lactate are positively correlated in groups of lean, obese, and obese/diabetic patients at 1-week and 3-months after RYGB (40).
Together, these findings suggest that in the fasting condition, glycolysis is accelerated in the skeletal muscle of subjects with obesity compared to lean subjects. This scenario has led us to the hypothesis of a “Vicious Cori Cycle” that is presented in Fig. 2, Panel B (40). Briefly, because of the impairment in pyruvate oxidation in the muscle of obese individuals, excessive lactate is exported to liver. The fact that glucose and lactate are positively correlated (Fig. 2, Panel A) suggests that pyruvate oxidation is also impaired in liver and the elevated lactate accumulation drives gluconeogenesis. The deficit in mitochondrial substrate utilization may thus be a precursor to metabolic inflexibility, glucose intolerance and subsequently T2D in individuals with overweight and obesity.
Figure 2.
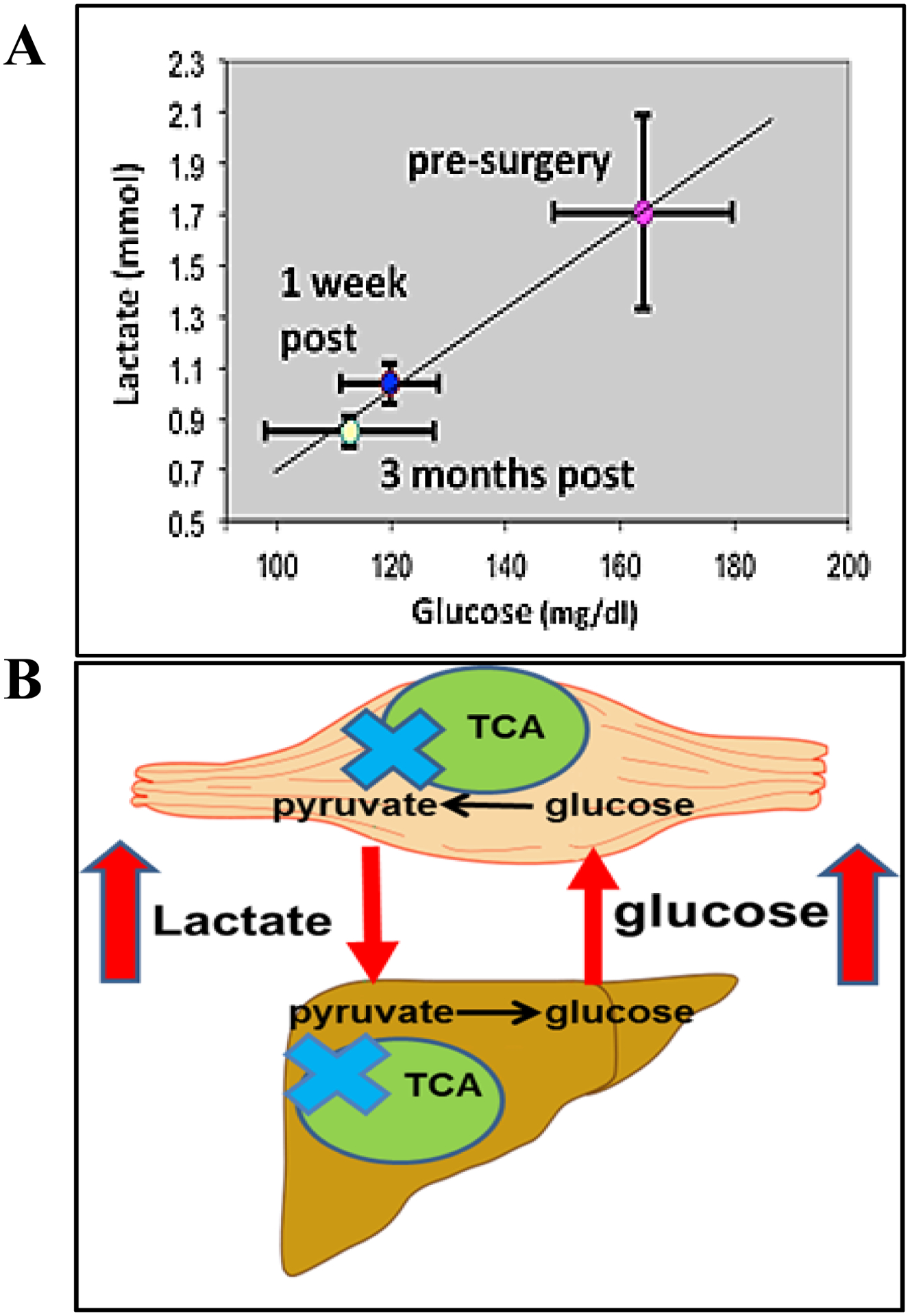
Panel A: A central tenet of our hypothesis is that complete glucose/pyruvate oxidation is impaired in severe obesity and type 2 diabetes (T2D), which increases blood lactate concentrations, drives liver gluconeogenesis, and elicits excessive hepatic glucose production which in individuals with T2D progresses to hyperglycemia. In support of this concept, when group means were plotted for patients with T2D before, 1 week after, and 3 months after Roux-en-Y gastric bypass a positive relationship between glucose and lactate was evident. Panel B: Schematic of the “Vicious Cori Cycle”. Skeletal muscle of subjects with obesity produces more lactate than lean subjects in the fasting condition and the elevated lactate drives increased glucose production. This stems from an initial lesion (blue X) in tricarboxylic acid cycle (TCA) function. The deficit in mitochondrial substrate utilization may thus be a precursor to metabolic inflexibility, glucose intolerance and subsequently T2D.
Increased reliance on muscle glycolysis under resting conditions is a puzzle as there is clearly sufficient oxygen to supply the muscle with ATP, as indicated by the ability to substantially increase ATP production to perform exercise. It is plausible that a decrease in metabolic clearance rate may give rise to lactatemia. One explanation could be “accelerated glycolysis”, which leads to an increase in net lactate release (41). However, we speculate that there is a defect in the TCA cycle in the skeletal muscle of individuals with metabolic disease that likely drive this accumulation of blood lactate. In support, we observed that primary skeletal muscle cell cultures from individuals with severe obesity retained alterations in glucose partitioning and displayed a depressed ability to stimulate glucose oxidation and glycogen storage with insulin (42). This appeared to be due, at least in part, to a reduction in TCA cycle flux. Together, these data suggest that the TCA cycle is altered in human skeletal muscle with severe obesity in a manner which shunts glucose towards the production of non-oxidized glycolytic end products (e.g. lactate). This was also reported in data from individuals with T2D (43). Furthermore, we reported that TCA cycle intermediates were lower in addition to a lower concentration of citrate with severe obesity (42). This suggests that either the step of conversion from acetyl-CoA to citrate (citrate synthase) is reduced and/or the amount of acetyl-CoA available for entry into the TCA cycle is lowered with obesity. Direct measures of acetyl-CoA are needed to disentangle these two conclusions. It has been shown that a mismatch between the early stages of TCA flux can lead to mitochondrial accumulation of acetyl-CoA (44). Citrate synthase is the main entry into the TCA cycle and does not bind acetyl-CoA without first binding oxaloacetate. We have shown previously that citrate synthase is lower in skeletal muscle of individuals with obesity (45). However, we did not see any differences in oxaloacetate nor pyruvate oxidation rates, which suggests no defects in acetyl-CoA production (42).
CONCLUSIONS
Blood lactate concentrations can provide valuable insights into metabolic flexibility and overall metabolic health. Herein, we provide a schematic (Fig. 3) that proposes the interactions leading to accumulation of blood lactate. Defects in mitochondrial substrate utilization and impaired TCA cycle flux leads to reduced aerobic oxidation. This reduction in aerobic oxidation leads to a compensatory increase in glycolysis and a shift in metabolism towards glycolytic products such as lactate. Impairments of the TCA at the level of the muscle cell may be a key determinant in substrate partitioning and metabolic health. Overall, these alterations in substrate handling at the cellular level contribute to metabolic inflexibility and the advancement toward metabolic disease. This scenario thus provides a rationale by which elevated resting and/or fasting lactate can be used as a biomarker for intrinsic metabolic health. Lactate is already in wide use in acute care medicine and trauma as a reliable prognostic indicator of recovery and survival. Lactate infusion has been shown to improve myocardial function in patients with septic shock, after cardiac surgery, and with acute heart failure (46). In addition, lactate has the potential to be used for treatment of tumors (47).
Figure 3.
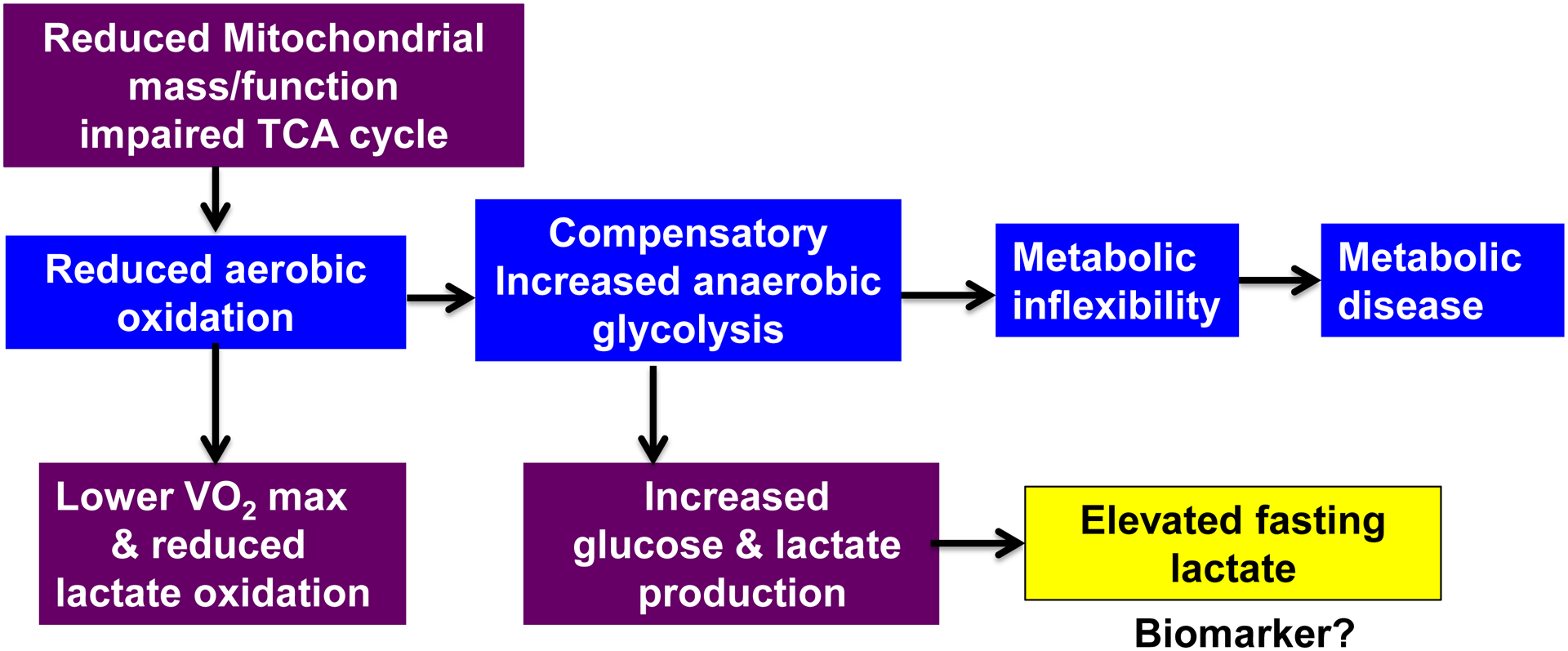
Overall summary schematic of proposed mechanisms. We hypothesize that reduced aerobic oxidation elicits a compensatory increase in glycolysis, which leads to metabolic inflexibility and subsequent metabolic disease. Therefore, accumulation of blood lactate can be used as a proxy biomarker for metabolic health.
Fasting levels may, according to our studies, also prove to be a tool for a rapid and inexpensive evaluation of the staging of the metabolic syndrome. Future studies should use tracers at either the whole-body or skeletal muscle levels to determine if TCA cycle flux is indeed compromised or if some other mechanism is involved. This tracer approach may be particularly useful in primary myotubes acquired from donors with varying health statuses. The primary myotube model offers the capacity to specifically isolate skeletal muscle metabolism since these primary cells retain the metabolic characteristics of the donors (28), and is thus particularly relevant to the study of human skeletal muscle. The role of lactate metabolism and the regulation of these processes in human metabolic disease remains to be fully elucidated.
Key Points.
Lactate accumulation reflects the gap between lactate production and lactate clearance.
Lactate may be a useful marker for the assessment and staging of severe obesity, type 2 diabetes and other expressions of the metabolic syndrome.
The biological mechanisms linked with an accumulation of blood lactate remain to be determined.
Acknowledgments
This work was funded by NIH DK56112 (JAH) and DK120296 (JAH and GLD) and supported by the American Heart Association Postdoctoral Fellowship (15POST25080003 KZ).
Footnotes
Conflicts of Interest: None
References
- 1.Wei M, Kampert JB, Barlow CE et al. Relationship between low cardiorespiratory fitness and mortality in normal-weight, overweight, and obese men. JAMA. 1999;282(16):1547–53. [DOI] [PubMed] [Google Scholar]
- 2.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346(11):793–801. [DOI] [PubMed] [Google Scholar]
- 3.van der Zwaard S, de Ruiter CJ, Noordhof DA et al. Maximal oxygen uptake is proportional to muscle fiber oxidative capacity, from chronic heart failure patients to professional cyclists. J Appl Physiol (1985). 2016;121(3):636–45. [DOI] [PubMed] [Google Scholar]
- 4.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322(4):223–8. [DOI] [PubMed] [Google Scholar]
- 5.Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol. 2016;12(11):633–45. [DOI] [PubMed] [Google Scholar]
- 6.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54(1):8–14. [DOI] [PubMed] [Google Scholar]
- 7.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49(5):677–83. [DOI] [PubMed] [Google Scholar]
- 9.Friedman JE, Caro JF, Pories WJ, Azevedo JL Jr., Dohm GL. Glucose metabolism in incubated human muscle: effect of obesity and non-insulin-dependent diabetes mellitus. Metabolism. 1994;43(8):1047–54. [DOI] [PubMed] [Google Scholar]
- 10.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–50. [DOI] [PubMed] [Google Scholar]
- 11.Brooks GA. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018;27(4):757–85. [DOI] [PubMed] [Google Scholar]
- 12.Messonnier LA, Emhoff CA, Fattor JA, Horning MA, Carlson TJ, Brooks GA. Lactate kinetics at the lactate threshold in trained and untrained men. J Appl Physiol (1985). 2013;114(11):1593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergman BC, Wolfel EE, Butterfield GE et al. Active muscle and whole body lactate kinetics after endurance training in men. J Appl Physiol (1985). 1999;87(5):1684–96. [DOI] [PubMed] [Google Scholar]
- 14.San-Millan I, Brooks GA. Assessment of Metabolic Flexibility by Means of Measuring Blood Lactate, Fat, and Carbohydrate Oxidation Responses to Exercise in Professional Endurance Athletes and Less-Fit Individuals. Sports Med. 2018;48(2):467–79. [DOI] [PubMed] [Google Scholar]
- 15.Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc. 2013;88(10):1127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford SO, Hoogeveen RC, Brancati FL et al. Association of blood lactate with type 2 diabetes: the Atherosclerosis Risk in Communities Carotid MRI Study. Int J Epidemiol. 2010;39(6):1647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juraschek SP, Shantha GP, Chu AY et al. Lactate and risk of incident diabetes in a case-cohort of the atherosclerosis risk in communities (ARIC) study. PLoS One. 2013;8(1):e55113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juraschek SP, Selvin E, Miller ER, Brancati FL, Young JH. Plasma lactate and diabetes risk in 8045 participants of the atherosclerosis risk in communities study. Ann Epidemiol. 2013;23(12):791–6 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones TE, Pories WJ, Houmard JA et al. Plasma lactate as a marker of metabolic health: Implications of elevated lactate for impairment of aerobic metabolism in the metabolic syndrome. Surgery. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guesbeck NR, Hickey MS, MacDonald KG et al. Substrate utilization during exercise in formerly morbidly obese women. J Appl Physiol (1985). 2001;90(3):1007–12. [DOI] [PubMed] [Google Scholar]
- 21.Chondronikola M, Magkos F, Yoshino J et al. Effect of Progressive Weight Loss on Lactate Metabolism: A Randomized Controlled Trial. Obesity (Silver Spring). 2018;26(4):683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Civitarese AE, Carling S, Heilbronn LK et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4(3):e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menshikova EV, Ritov VB, Dube JJ et al. Calorie Restriction-induced Weight Loss and Exercise Have Differential Effects on Skeletal Muscle Mitochondria Despite Similar Effects on Insulin Sensitivity. J Gerontol A Biol Sci Med Sci. 2017;73(1):81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernstrom M, Bakkman L, Loogna P et al. Improved Muscle Mitochondrial Capacity Following Gastric Bypass Surgery in Obese Subjects. Obes Surg. 2016;26(7):1391–7. [DOI] [PubMed] [Google Scholar]
- 25.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol. 1999;277(6):E1130–41. [DOI] [PubMed] [Google Scholar]
- 26.Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295(5):E1009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Battaglia GM, Zheng D, Hickner RC, Houmard JA. Effect of exercise training on metabolic flexibility in response to a high-fat diet in obese individuals. Am J Physiol Endocrinol Metab. 2012;303(12):E1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyle KE, Zheng D, Anderson EJ, Neufer PD, Houmard JA. Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. Int J Obes (Lond). 2012;36(8):1025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dube JJ, Coen PM, DiStefano G et al. Effects of acute lipid overload on skeletal muscle insulin resistance, metabolic flexibility, and mitochondrial performance. Am J Physiol Endocrinol Metab. 2014;307(12):E1117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phielix E, Meex R, Ouwens DM et al. High oxidative capacity due to chronic exercise training attenuates lipid-induced insulin resistance. Diabetes. 2012;61(10):2472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–9. [DOI] [PubMed] [Google Scholar]
- 32.Mandarino LJ, Wright KS, Verity LS et al. Effects of insulin infusion on human skeletal muscle pyruvate dehydrogenase, phosphofructokinase, and glycogen synthase. Evidence for their role in oxidative and nonoxidative glucose metabolism. J Clin Invest. 1987;80(3):655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahimi Y, Camporez JP, Petersen MC et al. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. Proc Natl Acad Sci U S A. 2014;111(46):16508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond). 2014;11(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorgensen W, Rud KA, Mortensen OH, Frandsen L, Grunnet N, Quistorff B. Your mitochondria are what you eat: a high-fat or a high-sucrose diet eliminates metabolic flexibility in isolated mitochondria from rat skeletal muscle. Physiol Rep. 2017;5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem. 1993;268(34):25836–45. [PubMed] [Google Scholar]
- 37.Liu C, Wu J, Zhu J et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. 2009;284(5):2811–22. [DOI] [PubMed] [Google Scholar]
- 38.Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146(7):1763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houmard JA, Pories WJ, Dohm GL. Is there a metabolic program in the skeletal muscle of obese individuals? J Obes. 2011;2011:250496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pories WJ, Dohm GL. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care. 2012;35(12):2438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z, Gladden LB. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol. 2018;118(4):691–728. [DOI] [PubMed] [Google Scholar]
- 42.Zou K, Hinkley JM, Park S et al. Altered tricarboxylic acid cycle flux in primary myotubes from severely obese humans. Int J Obes (Lond). 2018. [DOI] [PubMed] [Google Scholar]
- 43.Gaster M Reduced TCA flux in diabetic myotubes: A governing influence on the diabetic phenotype? Biochem Biophys Res Commun. 2009;387(4):651–5. [DOI] [PubMed] [Google Scholar]
- 44.Koves TR, Ussher JR, Noland RC et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. [DOI] [PubMed] [Google Scholar]
- 45.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279(5):E1039–44. [DOI] [PubMed] [Google Scholar]
- 46.Nalos M, Leverve X, Huang S et al. Half-molar sodium lactate infusion improves cardiac performance in acute heart failure: a pilot randomised controlled clinical trial. Crit Care. 2014;18(2):R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nijsten MW, van Dam GM. Hypothesis: using the Warburg effect against cancer by reducing glucose and providing lactate. Med Hypotheses. 2009;73(1):48–51. [DOI] [PubMed] [Google Scholar]