

Abstract
Advances in genomic science are informing an expansion of genetic testing for neurodegenerative diseases, which can be used for diagnostic and predictive purposes and performed in both medical and consumer genomics settings. Such testing—which is often for severe and incurable conditions like Huntington’s, Alzheimer’s, and Parkinson’s diseases—raises important ethical and health communication challenges. This review addresses such challenges in the contexts of clinical, research, and direct-to-consumer genetic testing; these include informed consent, risk estimation and communication, potential benefits and psychosocial harms of genetic information (e.g., genetic discrimination), access to services, education and workforce needs, and health policies. The review also highlights future areas of likely growth in the field, including polygenic risk scores, use of genetic testing in clinical trials, and return of individual research results.
1). INTRODUCTION
Advances in genetic testing for neurodegenerative conditions have improved the ability to make accurate diagnoses, provide recurrence risk information to family members, and (in some cases) determine eligibility for clinical trials. A number of neurodegenerative conditions have an underlying genetic cause, with a 50% recurrence risk when inheritance is autosomal dominant. The diagnosis of a neurodegenerative condition can therefore have implications not only for the affected individual, but also for relatives, who may be simultaneously watching a loved one’s decline and contemplating their own at-risk status.
The identification of a specific gene change through diagnostic testing of an affected individual makes it possible to offer both diagnostic testing for symptomatic family members and predictive testing for unaffected, at-risk relatives. Prenatal genetic testing and preimplantation genetic diagnosis are also options once a pathogenic variant has been identified. Genetic testing for neurodegenerative conditions by single gene analysis was initiated beginning in the 1980s, as genes were identified for Huntington’s disease, early-onset autosomal dominant Alzheimer’s disease, and other conditions. With advances in genetic testing, use of multi-gene panels and whole exome sequencing are increasing diagnostic yield, particularly when the specific neurodegenerative condition is unknown. Diagnostic yield will likely further be increased with use of whole genome sequencing, which is becoming available. For many neurodegenerative conditions, genetic causes remain to be identified. Even when genes are known to cause disease, not all pathogenic gene changes are identifiable with today’s technology, and the significance of an identified gene change may be unknown. The American College of Medical Genetics and Genomics (ACMGG) and the Association for Molecular Pathology (AMP) established a framework whereby identified variants are classified as benign (or likely benign), pathogenic (or likely pathogenic), or as of uncertain significance (Richards et al, 2015). As an increasing number of pathogenic variants have been identified across several neurological diseases, use of—and public interest in—genetic testing has expanded considerably. Genetic risk information for several neurodegenerative disorders is now not only available through neurology and genetics clinics, but also via for-profit companies offering direct-to-consumer genetic testing services.
However testing is provided, effectively conveying genetic test results for severe, often life threatening conditions requires that sufficient attention be paid to the ethical and communication challenges involved. This review will summarize and comment on such challenges (it should be noted that pharmacogenetic testing, while highly relevant for neurodegenerative disease management, will not be addressed). In this section, we briefly review the main types of neurodegenerative diseases for which genetic testing is available (see Table 1 for a summary).
Table 1:
Overview of genetic testing for neurodegenerative diseases
| Typical age of onset | Population risks | Inheritance | Genes tested (chromosome) | DTC testing available?1 | Sources | |
|---|---|---|---|---|---|---|
| Juvenile onset: <20 yrs (5–10% of cases) Adult onset mean age: 35 to 44 yrs |
Varies across world regions European ancestry: 9.71 per 100,000 East Asia, Finland, indigenous South Africans: 0.1 to 2 per 100,000 |
Autosomal dominant | CAG trinucleotide repeat expansion in the HTT gene (4p16.3) | No | PMID: 20301482 GTR Test ID: GTR000502937.4 | |
| Early onset: age <60–65yrs (5% of cases) Late onset: age >60–65yrs (95% of cases) |
General population: 10–15% Lifetime risk to first degree family members with late onset AD: 15–25% |
25% of all AD is familial Early onset familial AD: autosomal dominant |
Early-onset autosomal dominant AD: APP (21 q21.3), PSEN1 (14q24.2), PSEN2 (1 q42.13) Risk allele for later-onset AD: APOE (19q13.32) |
Yes, for APOE | PMID: 20301340 GTR Test ID: GTR000505595.2 | |
| Lewy Body | 50–85 yrs | 3.5 per 100,000 person years | Autosomal dominant | APOE (19q13.32) GBA (1q22) SNCA (4q22.1) | No | MedGen UID: 199874 PMID: 2404291 |
| Frontotemporal Dementia | 40–60 yrs | About 20–50% of persons with FTD have a positive family history | Autosomal dominant | Multigene panel that typically includes: MAPT (17q21.31) GRN (17q21.31) C9orf72 (9p21.2) | No | PMID: 20301678 PMID: 20301545 |
| Juvenile onset: <20 years Early onset: 2050 yrs Adult onset: >50 yrs Typically, around 60 yrs |
General population: 4% First degree relatives: 8% | Autosomal dominant; Autosomal recessive | Multi-gene panels; In families with autosomal dominant inheritance: GBA (1q22), LRRK2 (12q12), SNCA, VPS35 In families with autosomal recessive inheritance: ATP13A2, DJ1, DNAJC6, FBXO7, PINK1, PODXL, PRKN, SLC6A3, SYNJ1, VPS13C |
Yes; for LRRK2 and GBA | PMID: 20301402 GTR Test ID: GTR000552609.1 | |
| Mean age males: 55 years Mean age females: mid60s Individuals with familial ALS tend to have an earlier onset of symptoms |
Males are more affected than females in 1.3/1 ratio | Autosomal dominant; Autosomal recessive; X-linked 10–15% of individuals with ALS have an inherited form |
Multigene panel that typically includes: C9orf72 (9p21.2), SOD1 (21q22.11), FUS (16p11.2), TARDBP (1p36.22) | No | PMID: 20301623 GTR Test ID: GTR000551921.12 | |
| Familial Creutzfeldt-Jakob disease: typically 30–50 yrs Germann- Staussuler- Scheinker syndrome: 30–50yrs Fatal familial insomnia: 40–50yrs |
Worldwide yearly incidence of genetic and non-genetic prion disease: 1–1.5 cases per million | Autosomal dominant | PNRP (20p13) | No. | PMID: 20301407 GTR Test ID: GT R000566062. 1 |
1.1. Huntington’s disease (HD)
As described elsewhere in this volume, HD is a severe, life-limiting, and incurable neurodegenerative disease. HD is the first genetic disease for which a polymorphic marker was used to map a gene to a chromosome; in 1983, the HD gene, now known as HTT, was mapped to chromosome number 4 (Gusella et al, 1983). With this discovery, it became possible to offer genetic testing using linkage analysis (i.e., analysis of blood samples from both affected and unaffected family members) to track inheritance of HD in a family. In 1993, the HTT gene was identified and found to be an expansion of CAG trinucleotide repeats, which opened the door to direct genetic testing for HD (MacDonald et al, 1993). CAG trinucleotide repeats for HD are categorized as follows: full-penetrance (≥40), reduced penetrance (36–39), intermediate (27–35), and normal (≤26) (Caron et al, 2018). Individuals with 36–39 CAG trinucleotide repeats may or may not develop HD, and onset is generally at a later age. Both reduced penetrance and intermediate alleles are unstable and can increase into the full-penetrance range in subsequent generations. Intermediate and reduced penetrant alleles are seen in up to 6% of the general population, making it possible in rare cases for an individual to be biallelic for HD, having inherited a pathogenic gene change from both sides of the family (Kay et al, 2016,Uhlmann et al. 2015).
1.2. Alzheimer’s disease (AD) and related dementias
Approximately 25% of AD cases are estimated to be familial, with up to 5% classified as hereditary early-onset (i.e., below age 60) AD (Bird, 2018). In the 1990s, genes causing early-onset AD were identified on chromosomes 21 (APP), 14 (PSEN1), and 1 (PSEN2) (Van Cauwenberghe et al, 2016); genetic testing is available for these pathogenic variants in families where such a mutation is known or suspected. Many other genetic risk factors for the more common later-onset form of the disease have also been identified. The APOE ε4 allele, though neither necessary nor sufficient to cause AD, is a well-established risk factor for late-onset AD. Although current practice guidelines caution against clinical use of predictive genetic testing for AD via APOE genotyping due to limitations in clinical validity and utility, the personal genomics and biotechnology company 23andMe obtained approval in 2017 from the US Food & Drug Administration for a direct-to-consumer offering of APOE testing (Goldman et al, 2011, FDA, 2017).
Lewy body dementia (LBD) and frontotemporal dementia (FTD) are other significant types of dementia with genetic causes. LBD accounts for 15–30% of all neurodegenerative dementias (Walker et al, 2015). At this time, genetic testing is not typically used in LBD care for diagnosis and/or susceptibility testing, although the role of genetics in LBD etiology is an area of active research. A recent systematic review of 75 articles confirmed significant associations between LBD and variants in the APOE, GBA, and SNCA genes (Sanghvi et al, 2020). The researchers suggested that other associations, particularly between Parkinson’s disease and LBD, warrant further investigation. FTD is inherited in 20–50% of cases due to a single gene change in one of the multiple genes associated with disease risk (e.g., C9orf72, MAPT, GRN) (Olszewska et al, 2016). Unless a specific familial pathogenic variant is known, genetic testing for FTD typically occurs via multi-gene panels. (Greaves & Rohrer, 2019).
1.3. Parkinson’s disease (PD)
Approximately 5–10% of PD cases are attributable to pathogenic variants in single genes that can be inherited in an autosomal dominant, autosomal recessive, or even X-linked pattern of inheritance (Shukla et al, 2019). Identified genes include GBA, LRRK2, SNCA, and VPS35. Direct-to-consumer genetic testing is now available for GBA and LRRK2, the most common genetic variants associated with PD. These genes also serve as the basis for emerging targeted therapies for PD currently being tested in clinical trial (Sardi et al, 2018).
1.4. Amyotrophic lateral sclerosis (ALS)
ALS is the most common motor neuron disease. Approximately 5–10% of ALS cases are estimated to be familial; numerous genes have been identified, including C9orf72, SOD1, FUS, and TARDBP (Peters et al, 2015). An expansion of a hexanucleotide repeat in C9orf72 is associated with up to 40% of familial ALS cases and 5% of sporadic cases, with studies actively seeking to determine what level of repeat size is pathogenic (Iacoangeli et al, 2019). Genetic test results are now being used to determine potential eligibility for emerging genotype-specific treatments (Roggenbuck et al, 2017).
1.5. Prion diseases
Genetic prion diseases are rare, with subtypes including Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker syndrome, and fatal familial insomnia. These diseases are inherited in an autosomal dominant manner and account for approximately 15% of all prion diseases (Bechtel & Geschwind, 2013). PNRP is the only gene with pathogenic variants known to cause prion disease and can be tested using next-generation sequencing or targeted analysis (Mastrianni, 2014).
2). PRACTICE ISSUES
2.1. Predictive genetic testing: The HD model and beyond
As noted above, HD was the first genetic condition for which predictive testing was offered. The HD testing protocol is described in detail here because it has become a model for offering this type of testing for other neurodegenerative conditions (e.g., early-onset autosomal dominant AD), as well as hereditary cancer syndromes (Quaid, 2017). Predictive genetic testing guidelines, developed by neurologists and geneticists with significant input from the HD patient community, have been updated periodically; the most recent guidelines in use are those developed in Europe (MacLeod et al, 2013) and by the HD Society of America (2016).
Predictive genetic testing for HD typically involves two or more in-person clinic visits with a genetic counselor and/or clinical geneticist. Evaluation by a licensed mental health professional is recommended to ensure that the patient is psychologically ready to receive the test results and that s/he will have sufficient social support in place post-disclosure. Having a serious psychiatric condition is not a contraindication for testing, although, as noted in the guidelines (MacLeod et al, 2013, HDSA 2016), a delay may be needed to establish support services for the patient. Neurological examination is offered, especially to patients concerned about possible symptoms, to confirm that the patient is truly asymptomatic; however, this evaluation is not required (MacLeod et al, 2013, HDSA 2016). For patients concerned about possible symptoms, a normal neurologic examination may be reassuring and impact decision-making if this was the only reason why they were seeking predictive genetic testing. Some neurology clinics have multidisciplinary teams to offer predictive genetic testing while others will need to refer patients to genetics clinics.
Extensive pre-test counseling covers the benefits, risks, and limitations of predictive genetic testing and explores the individual’s experience and knowledge of HD, motivations for testing, psychosocial issues, and potential impact of results. Patients should be counseled to have desired insurance coverage (e.g. health, life, long-term disability, long-term care) prior to testing, and the potential test implications for employment should be discussed. Test result disclosure is typically in-person, with the recommendation that a support person accompany the patient. For patients in geographically remote areas, telehealth approaches may be an option (Hawkins et al, 2013).
The guidelines emphasize that the decision to have predictive genetic testing should be made by the person being tested, without undue influence or coercion from others. The guidelines also contain a strong recommendation to avoid testing of minors (except in cases where juvenile onset of disease is suspected) and to avoid prenatal testing unless results would impact pregnancy management decisions (e.g., regarding termination) (Hercher et al, 2016). HD testing is not currently offered by direct-to-consumer genetic testing companies given the highly sensitive nature of test results and need for pre- and post-test education and counseling. These issues highlight just some of the many ethical implications of predictive genetic testing for neurodegenerative diseases (see Uhlmann & Roberts (2018) for a comprehensive review).
Generally, for any type of predictive genetic testing, the gene change would ideally first be identified in an affected individual before at-risk family members are tested. This may not always be possible if an affected family member is deceased or unwilling to be tested (e.g. health reasons, personal reasons, test cost). An at-risk family member may have limited to no contact with an affected family member or may not wish to request they be tested for multiple reasons (e.g. privacy, family dynamics). However, a genetic test ordered without knowing the affected family member’s specific gene change or neurodegenerative condition can be problematic. Given that different conditions can have overlapping symptoms, ordering the right test to analyze gene(s) specific for that neurodegenerative condition is critical. In addition, interpretation of a negative result will be limited since it will not be known if an at-risk family member is negative because a gene change has not been inherited (“true negative”) or because the gene change would also not have been identified (even with today’s technology) in the affected family member.
For a condition like HD with high clinical sensitivity, lack of genetic testing of an affected individual will not limit an at-risk family member’s ability to have predictive genetic testing and to be reassured if results are negative. For conditions like FTD, a multi-gene panel can be ordered for an at-risk family member; however, given lower sensitivity, a negative result will have the limitations noted above. It should also be noted that predictive testing can determine whether a gene change has been inherited but generally cannot predict age of onset, severity of symptoms, or disease course. This is particularly the case with C9orf72 hexanucleotide repeat expansions which have been seen in individuals who have ALS, motor neuron disease, frontotemporal lobar degeneration, and Parkinsonism (Cruts et al, 2015). Some conditions can have reduced penetrance, meaning that a gene change is inherited but not all individuals will become symptomatic or may become symptomatic at a later age. For example, although almost all individuals with C9orf72 hexanucleotide repeat expansions are symptomatic by age 80, nearly half live symptom-free into their late 50s. On the other hand, some individuals with C9orf72 repeats develop both ALS and FTD.
2.2. Indications for testing
The decision of whether or not to order a genetic test can be a complicated one (see Table 2 for a summary of key questions to ask prior to testing). Having symptoms and/or a positive family history of a genetic condition are common indications for testing. Genetic testing for neurodegenerative conditions is typically ordered by a neurologist or physician with expertise in clinical genetics. Ideally, this decision occurs in the context of a comprehensive review of family and medical history information and with knowledge of the benefits and limitations of the different types of genetic tests for a given condition (Uhlmann, 2009). Established frameworks for analyzing genetic tests often focus on their analytic validity, clinical validity, and clinical utility (Pitini et al, 2018). According to such frameworks, predictive genetic tests should typically only be considered for clinical use in at-risk populations if they 1) are highly accurate in determining the genotype(s) of interest (analytic validity), 2) possess suitable positive predictive value (clinical validity), and 3) inform medical care related to disease prevention or early detection (clinical utility). For those with a known diagnosis, genetic testing may lack clinical utility for that patient but could inform clinically beneficial cascade testing in at-risk family members.
Table 2.
Questions to consider regarding clinical use of genetic susceptibility testing for neurodegenerative disease
Patient Factors
|
Disease Characteristics
|
Genetic Testing and Counseling
|
Reproduced from Roberts & Uhlmann, 2013
It should be noted that individuals are often interested in genetic testing that does not meet the criteria outlined above. Although neurodegenerative diseases like HD and AD lack proven prevention options, predictive testing is an option because results might guide advanced planning related to important life decisions (e.g., marriage, childbearing, employment) and/or help those testing positive plan for future care needs. Even tests like APOE genotyping, with its modest positive predictive value, are of interest to at-risk family members for a variety of reasons that might be classified as “personal utility” (e.g., to guide advance planning, encourage monitoring of developments in the field, assist coping with uncertain risk status) rather than clinical utility (Neumann et al, 2012; Roberts et al, 2003).
2.3. Informed consent
Generally, the goal of obtaining informed consent is to ensure that patients understand the benefits, risks, and limitations of genetic testing which are weighed and considered in their decision-making. Providing informed consent for genetic testing for neurodegenerative disorders can be challenging for several reasons. Depending on the condition, the following test limitations may exist: a) the specific gene(s) may not yet be known, b) all pathogenic gene changes may not yet be identifiable, c) what is known about the clinical significance of an identified variant(s) may be limited, and d) given that many tests are very new, one or more variants of uncertain significance may be identified. In these cases, providing patients with specific information about test sensitivity and significance of potential test results may not be possible. The se points need to be clearly conveyed and understood by patients. In addition to providing information about the actual test and its benefits and limitations, informed consent needs to address broader risks, including potential insurance implications (e.g., risk of genetic discrimination) if genetic test results are positive.
Many factors can influence the informed consent process; these include patients’ levels of health literacy and psychosocial stress, the complexity of information conveyed, and time limitations for clinic visits. It can be challenging to obtain informed consent in cases where patients and providers do not speak the same language and translation services may not be readily available. Obtaining informed consent can also be challenging to accomplish if patients are in the early or prodromal stages of the neurodegenerative condition for which they are being tested, as their decisional capacity may be subtly impaired. For example, they may lack the reasoning or memory skills required to make a truly informed choice regarding genetic testing. Fortunately, there are ways to address some of these challenges. In cases where a patient’s decisional capacity is in question, there are several validated instruments for use in capacity assessment (see Karlawish, 2017 for a review). In addition, there are credible and well-developed educational resources about genetic conditions and genetic testing (e.g. Genetics Home Reference, the National Society of Genetic Counselors’ “About Genetic Testing” website), and laboratories generally provide test report descriptions that are helpful to clinicians. Some national organizations for genetic conditions provide information about genetic testing for both patients and healthcare providers. Test decision aids can be helpful and save time for patients and providers. For example, an online decision aid has been developed and validated for individuals considering undergoing APOE testing (Ekstract et al, 2017).
2.4. Estimating risk
Using predictive testing to estimate risk of neurodegenerative disease is relatively straightforward for conditions with high penetrance genes (e.g. HD, autosomal dominant AD, genetic prion disease). In these cases, a positive test result means that the patient will almost inevitably develop the disease in question, assuming a normal lifespan and lack of premature death from other causes. However, even in these more certain cases the test result does not specify what the disease course or age of onset will be. A systematic review of over 1300 autosomal dominant AD cases found a mean onset of 46 years, but over 50 cases were asymptomatic into their 60s and 70s; the review concluded that family history of disease and type of mutation help explain such variation (Ryman et al, 2014). A recent case report from Colombia demonstrated how presence of a presenilin 1 mutation was seemingly counteracted by a rare APOE variant known as the Christchurch mutation that delayed disease onset by several decades (Arboleda-Velasquez et al, 2019). Disease risk is more difficult to determine if there is reduced penetrance for the condition or if genetic test results identify a novel pathogenic variant.
For conditions like late-onset Alzheimer’s disease and Parkinson’s disease, genetic factors interact with other influences on disease expression, including environmental exposures, health behaviors, comorbid disease processes, and social determinants of health. This complexity makes it questionable to quantify disease risk estimates based on genetic test results alone, and yet there are no validated risk models for neurodegenerative conditions that fully integrate relevant genetic and non-genetic factors. To complicate matters even further, studies that have attempted to quantify genetic risks of disease have often been limited by small sample sizes and/or selection biases with regard to race and socioeconomic status(Popejoy & Fullerton, 2016). As has been observed with other medical conditions, genetic risk estimates based on early studies with clinic-based recruitment tend to be significantly higher than those generated by subsequent population-based studies. For example, a recent analysis of four population-based cohort studies estimated that lifetime risk of dementia in APOE ε4 homozygotes was approximately 35–40%, significantly lower than previous studies had suggested (Qian et al, 2017). When communicating with patients and family members about predictive genetic testing results for neurodegenerative conditions, it is important to keep in mind sources of risk figures and whether the condition follows an established pattern of inheritance with full or reduced penetrance. If the neurogenerative condition is potentially impacted by other genes and non-genetic factors, this should be communicated as well.
2.5. Communicating risk
Genetic risk communication faces challenges beyond those involved in generating accurate disease risk figures. Many patients lack the health literacy and numeracy abilities required to fully grasp the probabilistic and nuanced information often conveyed in genetic susceptibility testing (Institute of Medicine, 2004). They may also be prone to cognitive biases in their interpretation of test findings. For example, they may overrate their families’ illness experiences in their perceptions of the likelihood and timing of disease, or give greater weight to genetic risk factors than is warranted due to a tendency toward genetic exceptionalism. For their part, health care providers may lack skills in effectively conveying genetic risk information and/or may face burdensome time constraints in delivering and discussing test findings. Risk communication is particularly challenging when a variant of uncertain significance (VUS) is identified. Both patients and healthcare providers may conclude that, because a change in the DNA sequence is present, a genetic cause of disease has been identified. Yet a VUS classification means that available knowledge is insufficient to classify the variant as benign or pathogenic, and the ACMGG guidelines make clear that VUS findings should not be used in medical decision-making (Richards et al, 2015). As variants may be reinterpreted over time, it is important for patients to be instructed to recontact the healthcare provider who ordered the test to determine if the variant has been reinterpreted and/or if additional genetic testing should be considered.
Despite these challenges, the literature on health risk communication in general and genetic risk communication in particular suggests some best practices to consider when conveying genetic test results for neurodegenerative diseases. Fagerlin, Zikmund-Fisher, and Ubel (2011) provide several practical recommendations for health risk communication, including the following: 1) use of plain language (vs. medical jargon), absolute (vs. relative) risks, and frequencies (vs. percentages); 2) use of visual aids to present quantitative risks, with the pictograph icon array as an especially valuable graphical tool; 3) paying special attention to the ordering of risk information presented and the time intervals being considered (e.g. lifetime risk vs. next five years); and 4) generally avoiding information that is inessential to patient decision-making (i.e., “less is more”). One of the clinical trials within the Risk Evaluation & Education for Alzheimer’s Disease (REVEAL) trials provides an instructive example here. That study used a side-by-side series of pictographs (see Figure 1) to convey to individuals with mild cognitive impairment how a given APOE genotype influenced one’s chances of progressing to clinical AD within the next three years (Lautenbach et al, 2013).
Figure 1.
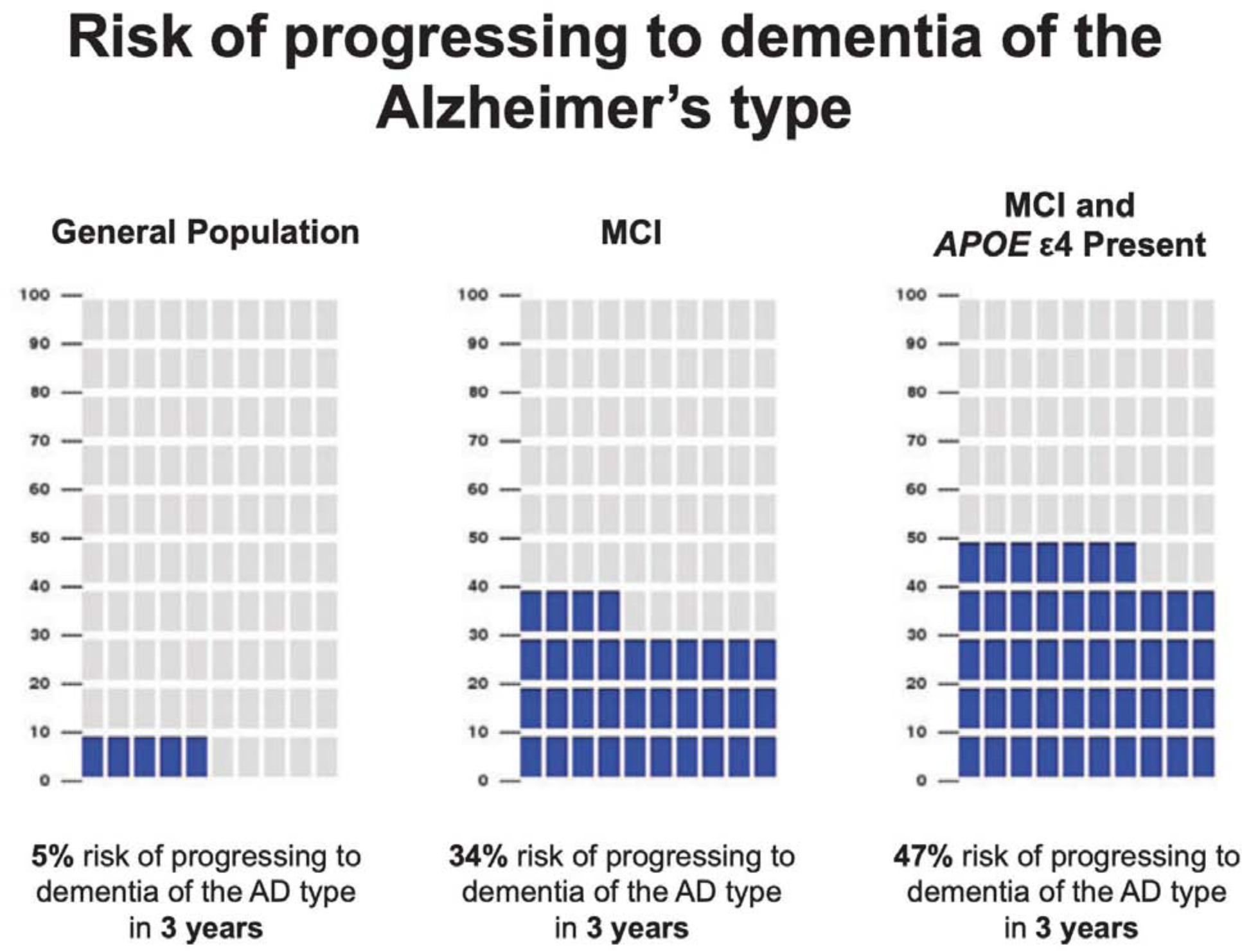
Sample use of pictographs to aid genetic risk communication for Alzheimer’s disease
Reprinted from Lautenbach et al (2013)
For inherited neurodegenerative conditions, risk communication beyond the patient to their at-risk relatives may be necessary. Because of confidentiality of medical information and HIPAA privacy concerns, health care providers cannot contact at-risk relatives without the patient’s consent; even when such consent is obtainable, providers may lack time and resources to reach out to relevant at-risk relatives. In order to assist patients with sharing information with their at-risk family members, healthcare providers can provide patients with copies of their genetic test results, a summary letter from their clinic visit, and education materials from relevant disease support groups and national organizations.
2.6. Psychological and behavioral impact of results
There are numerous potential psychological harms of genetic testing for neurodegenerative disorders, given that in some cases one is learning about the nearly certain likelihood of a feared disease. Those being tested have often watched their affected relatives suffer through many years of debilitation and decline, and test results may have profound implications for their own future care, family relationships, and key work and life decisions. Concerns about the psychological harms of testing are longstanding, dating back to the dawn of predictive genetic testing for HD in the 1980s. Given that levels of suicidal ideation and attempts are already elevated in prodromal HD, there was significant concern that positive test results could result in profound levels of psychological distress (Fiedorowicz et al, 2011). To address such concerns, the testing process itself was designed with some safeguards in place (e.g., formal pre-test psychological evaluation to assess readiness to receive results and presence of post-test support) to reduce risk for catastrophic reactions to testing. A considerable literature exists on the psychological impact of genetic testing for HD and other neurodegenerative conditions. These studies have generally found that psychological distress in response to predictive testing is generally mild and transient for those who choose to undergo testing, a finding that has been replicated in numerous HD studies and observed across multiple other disease contexts, from hereditary cancer syndromes to Alzheimer’s disease (Crozier et al, 2015, Heshka et al, 2008). Such findings suggest that the likelihood and extent of psychological harms from genetic testing—even for fatal and incurable diseases—is less than was initially feared. Yet it should be noted that this body of research has several limitations, as reviewed by Roberts (2019); these include 1) use of skewed participant samples lacking in diversity with regard to race and socioeconomic status; 2) use of traditional in-person genetic counseling models that limits generalizability of results to other formats (e.g., DTC genetic testing); and 3) an overreliance on general depression and anxiety scales as primary outcome measures. A notable exception to the latter point is a case-control study that found that disclosure of an elevated risk APOE test result had seeming adverse consequences on older adults’ perceptions of their memory skills and performance on objective memory tests (Lineweaver et al, 2014).
Genetic risk information—whether for neurodegenerative diseases or other types of conditions—is often disclosed in hopes that patients will take steps to reduce their chances of developing future disease. However, as summarized in a 2016 Cochrane systematic review of genetic susceptibility testing studies across numerous diseases, such disclosure is often insufficient to prompt risk-reducing health behavior changes (Hollands et al, 2016); this is particularly true when the recommended behaviors (e.g., dietary changes, smoking cessation, increased physical activity) require sustained patient effort and involve numerous psychological and social barriers. With many neurodegenerative diseases, health behavior recommendations are less certain than in conditions like cancer and heart disease, and individuals may undertake unproven methods of lowering their disease risk. Research on response to genetic susceptibility testing for AD has shown that individuals learning they are elevated disease risk are more likely to take additional vitamins and supplements (Vernarelli et al, 2010) and to make changes in long-term care insurance plans (Zick et al, 2005; Taylor et al, 2010). One of the randomized clinical trials from the aforementioned REVEAL Study examined the behavioral effects of disclosing APOE genotype and associated risk information for both AD and coronary artery disease (CAD). The study found that participants who received risk information about both AD and CAD engaged more frequently in health behaviors to reduce disease risk than participants who received risk information about AD alone (Christensen et al, 2016). This finding suggests that recommendations around brain health may be more successful if they highlight that certain behaviors may not only reduce risk of cognitive decline, but also preventable conditions like heart disease.
3). LEGAL & POLICY ISSUES
3.1. Access to testing
Patients’ access to genetic testing for neurodegenerative diseases can be limited by geographic and financial factors. Such testing is typically conducted in neurology or genetics clinics that are disproportionately located in large urban academic medical centers, placing needed services out of reach for the many patients who reside outside of the se sites’ catchment areas. Compounding these limitations in access to services are long wait times, lack of insurance coverage for testing and/or counseling, and lack of awareness among generalist providers on when and to whom referral is needed.
Several strategies have been proposed to help address these issues. First is the expansion of the clinical genetics workforce, including both board-certified medical geneticists and genetic counselors (Hoskovec et al, 2018). Second is the training of primary care physicians and other health care providers to know when genetic testing is appropriate and how to adequately educate and counsel patients and family members (Bartlett et al, 2014). An increasing number of continuing medical education programs are addressing this topic, guided by an established set of practice competencies in the realm of genomic medicine (Korf et al, 2014). Third is greater use of telemedicine, telephone genetic counseling services, and other technologies to expand reach and access of services. A growing number of studies, including one focused on APOE testing, have demonstrated than phone disclosure is an acceptable alternative to in-person disclosure in terms of patient comprehension of, and psychological adjustment to, genetic test results (Christensen et al, 2018). Finally, there have been calls to reconsider whether traditionally time- and labor-intensive genetic education and counseling models can be condensed or altered in certain circumstances, supported by studies where briefer models have been shown to be equivalent in terms of key genetic education and counseling outcomes (Green et al, 2015).
3.2. Consumer genomics
Another potential means of expanding access to genetic testing for neurodegenerative diseases is via consumer genomic services. 23andMe is the largest provider of direct-to-consumer testing, with a customer base that is reportedly over 10 million individuals (23andMe, 2020). As noted earlier, 23andMe provides DTC testing for risk of AD and PD, assessing for variants in the APOE, LRRK2, and GBA genotypes. However, as is the case for other types of medical conditions for which it offers testing (e.g., hereditary breast and ovarian cancer), the company does not fully test for all known pathogenic variants associated with risk of neurological disorders, and the utility of information provided may vary depending on the consumer’s racial / ethnic background. FDA approval for these tests required the company to demonstrate that their genotyping approach was analytically valid and that user comprehension of their web-delivered test results was high. Nevertheless, numerous ethical concerns have been raised about the DTC model, including the potential for negative outcomes such as the following: unnecessary or harmful medical or health decisions based on misunderstanding of test results; psychological harm from unexpected “bad news”; and unnecessary burdens on primary care providers (PCPs) given company recommendations for consumers to “ask your doctor” about genetic test results for which PCPs may not be equipped to address (McGuire & Burke, 2008). Studies of consumers of DTC genetic testing services suggest that most do not share their test results with a healthcare provider; in cases where this does occur, consumers are much more likely to discuss their results with a PCP than a genetic counselor, even though the latter is probably better suited to assist with interpretation of results and recommendations for follow-up (Koeller et al, 2017).
Case reports exist both in the academic literature (Messner, 2011) and popular press (Tyrone & Sabbagh, 2019) where individuals have learned about genetic risks for neurodegenerative disease either inadvertently or without making a highly informed choice, resulting in persistent psychological distress and decisional regret. An active education and support website (www.apoe4.info), now maintained by a grassroots public charity, was launched in 2013 to share information and promote interaction among APOE ε4 carriers, many of whom learned this information in a DTC format. However, it should be noted that there is not evidence of widespread psychological or social harms resulting from DTC testing for AD, PD, or other health conditions. The largest longitudinal study of DTC service users to date found that only 2% regretted their decision to purchase services and 1% reported any kind of harm as a result. (Roberts et al, 2017)
In recent years, a modified model of DTC testing has emerged, which some have termed consumer-directed testing (Ramos & Weissman, 2018). In this approach, the consumer still initiates the request for personal genetic information (often prompted by DTC advertising), but a physician orders the test, with genetic counseling potentially available by telephone (Allyse et al, 2018). The advantages of this hybrid approach are that, at least in theory, it retains the high accessibility of the DTC model but with more health professional involvement and testing in clinically certified laboratories only. The latter elements, especially availability of genetic counseling, may be particularly important to consider if future consumer-directed testing includes genes for severe, incurable neurodegenerative diseases.
3.3. Testing of minors
Although juvenile forms of heritable neurodegenerative disease exist, the majority of genetic tests in this area are for adult-onset conditions. There is a strong consensus in the pediatrics and medical genetics communities that minors should not be tested for adult-onset conditions, given that the results are not medically actionable in childhood, pose potential psychosocial harms, and arguably violate children’s autonomy by taking away their right to an “open future” (American Academy of Pediatrics, 2013; Botkin et al, 2015). In these cases, the recommendation is to allow children to make test decisions for themselves when they reach the age of majority. However, many parents may not concur with this stance; a large British public opinion poll, for example, suggested that approximately half of the general population believes that parents should have the right to test their children for adult-onset disorders (Shkedi-Rafid et al, 2015). Because DTC companies may lack safeguards against surreptitious testing, parents who strongly desire knowing their children’s genetic risk for neurodegenerative disease could in theory bypass the health care system and submit their child’s DNA sample for analysis (Weissman et al, 2019). It remains to be seen how this potential tension between parental preferences for genomic information and standard practices in genomic medicine will play out in the coming years.
3.4. Genetic discrimination and stigma
Concerns about genetic discrimination are common among patients considering genetic testing, leading some to decline testing, pay out of pocket to avoid insurer access to information, or even undergo testing under an alias. Surveys of individuals at risk for HD in Canada and Australia (Bombard et al, 2009; Goh et al, 2013) found that approximately a third of respondents reported experiencing some type of genetic discrimination; such experiences were most common in insurance settings but were also reported in family, social, health care, and employment settings. In 2008, the Genetic Information Nondiscrimination Act (GINA) was enacted at the federal level in the US to outlaw the use of personal genetic information (including family health history) in health insurer and employer decisions (e.g., coverage and premium decisions for insurers, hiring and termination decisions for employers) (Suter, 2019). These legal protections do not extend, however, to life, disability, and long-term care (LTC) insurance, nor do they cover members of the military (Baruch & Hudson, 2008). As noted earlier, genetic testing for AD may inform some at-risk individuals’ decisions to purchase LTC insurance plans or add to existing coverage. It is not clear if or to what extent life, disability, and LTC insurers are currently using personal genetic information to inform coverage decisions, and confirmed cases of genetic discrimination in this domain are very rare. However, the rapid expansion of both the use of genetic testing and the number of older adults in the population may prompt consideration of whether GINA and related laws should be expanded to prohibit genetic discrimination in insurance domains beyond health.
4). EMERGING AREAS
4.1. Polygenic risk scores
Across numerous common diseases, the development of polygenic risk scores (PRS)—the weighted sum of the number of disease risk alleles that a given individual carries—is sparking hopes that this approach might be used clinically to inform treatment, screening, and advance planning decisions (Torkamani et al, 2018). In theory, PRS draw upon large genome-wide association studies to provide greater predictive value (and therefore clinical validity) than traditional monogenic testing approaches. Recent years have seen a dramatic rise in PRS research studies focused on Alzheimer’s disease, with a 2018 systematic review (Stocker et al, 2018) suggesting that this approach is more predictive of AD than APOE testing alone. A smaller number of PRS studies have also been conducted for Parkinson’s disease (Ibanez et al, 2019).
Some skeptics express concerns that PRS are the latest in a long line of overhyped genomic tools that ultimately do not translate into clinically useful interventions (Cecile, 2019), while others warn that increased use of PRS could inadvertently exacerbate racial health disparities (Martin et al, 2019). Yet even though PRS for neurodegenerative disease are not currently ready for clinical use, it seems likely given present trends that translational research in this area will continue to expand.
4.2. Clinical trials
Genetic testing is increasingly being used as part of clinical trials investigating the safety and efficacy of new therapies for neurodegenerative diseases. Testing can be used a) to identify high-risk individuals for prevention trials, or b) in affected patients to determine trial eligibility and suitability for a particular experimental treatment. Precision neurology approaches are being developed across a range of conditions, with SOD1 based therapies for ALS just one notable example (Capella et al, 2019). In these clinical trials, it may be advantageous or even necessary to disclose personal genetic information to prospective research participants, especially when their genetic status is a prerequisite for trial enrollment. This issue has become prominent in prevention trials for Alzheimer’s disease, where both APOE and rare familial mutations have been used to identify high-risk populations in which anti-amyloid and other therapies are being investigated (Hsu & Marshall, 2017). Ancillary studies to such trials are proving a useful means of examining the process and impact of genetic risk disclosure. For example, some trials within the Alzheimer’s Prevention Initiative’s Generation Program are using APOE ε4 homozygote status as a study inclusion criterion (Lopez et al, 2019). In these large international AD prevention trials, disclosure of APOE genotype often occurs within a genetic education and counseling protocol remotely delivered via telephone or video conferencing. The investigators have chosen this approach given the lack of genetic counseling expertise at most study sites and are formally evaluating the safety and efficacy of providing genetic risk disclosure in this format (Langlois et al, 2019).
In these investigational treatment trials, early research suggests that genetic information might help identify patients who are more likely to experience study-related adverse events. For example, APOE ε4 carriers in anti-amyloid therapy trials have been found to be at elevated risk for amyloid-related imaging abnormalities (ARIA), including vascular edema and microhemorrhages (Sperling et al, 2011). It may be appropriate in future prevention trials to use genetic testing to help inform participants’ decisions whether or not to take part in experimental treatment studies, given that the risk-benefit ratio may vary depending on a participant’s genetic profile.
4.3. Return of research results
Clinical researchers routinely collect individuals’ genetic information in studies of neurodegenerative disease, but study participants do not typically learn their own individual research results. The question of whether and when investigators should return research results has become the focus of intense debate in recent years. In general, the highest priority genetic research results to consider returning are those that are well-established pathogenic variants for clinically actionable conditions, such as the 59 genes the ACMGG has identified in its policy statement on reporting of secondary findings from genome sequencing (Kalia et al, 2017). Most genes associated with neurodegenerative disease do not meet these criteria (none are part of the aforementioned “ACMGG 59” list), but they may nevertheless be of interest to research participants and relevant to their health status and decision making. In 2018, the National Academies of Sciences, Engineering, and Medicine issued a report containing 12 recommendations based on the current evidence of the benefits, harms, and costs of returning individual research results. Some recommendations encouraged research investigators to proactively determine study policies regarding return of results and clearly communicate them to participants at the study’s outset, taking into account their needs, informational preferences, and values. Other recommendations addressed stakeholders such as research institutions and funding agencies, calling on them to provide resources and infrastructure that would ensure the high quality of laboratory results to be returned and more routinely incorporate return of results policies and budget considerations into the grants review process. An overarching recommendation was for more frequent research results disclosure than is currently taking place, in the belief that this “will demonstrate respect for participants and support transparency and the development of trust with participants, in turn bringing benefits to participants, investigators, sponsors, funding agencies, research institutions, and society.” (p.13) Although many studies lack sufficient resources (e.g., personnel with requisite clinical and communication skills; ability to retest samples in CLIA approved laboratories as appropriate) to responsibly return research results, others are responding to this call. For example, the large, NIH-led population cohort study All of Us (enrollment goal: one million participants), has begun to pilot a program for returning genomic results—including the aforementioned “ACMGG 59” list—to a subset of its participants (GenomeWeb, 2019).
4.4. Conclusion
With the rapid expansion of genomics research in the biomedical sciences, genetic testing for neurodegenerative diseases is likely to become an increasingly important part of medical care moving forward. In this article, we have summarized the current state of this field, highlighting key ethical and health communications challenges that patients, health care providers, researchers, and policymakers face in this area. Given the anticipated growth of neurogenetic testing, the field will have to address numerous needs, including the following: greater education and training of neurologists and generalist healthcare providers; an expanded clinical genetics workforce; increased use of non-traditional genetic counseling models; patient education; insurance coverage of genetic testing; and policy changes to reduce risk of genetic discrimination and increase access to personal genomic information. Ideally, findings from research on the ethical, legal, and social implications (ELSI) of neurogenetic testing will help guide the practice and policy debates likely to be faced in the coming years.
FUNDING SOURCES
Dr. Roberts is supported by National Institute on Aging (NIA) grant P30 AG053760, and both he and Professor Uhlmann are supported by NIA grant RF1 AG047866.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Contributor Information
J. Scott Roberts, Department of Health Behavior & Health Education, University of Michigan School of Public Health.
Anne Patterson, University of Michigan School of Public Health.
Wendy Uhlmann, Department of Internal Medicine, Division of Genetic Medicine, Department of Human Genetics, University of Michigan School of Medicine.
REFERENCES
- Allyse MA, Robinson DH, Ferber MJ, Sharp RR, n.d. Direct-to-Consumer Testing 2.0: Emerging Models of Direct-to-Consumer Genetic Testing 10.1016/j.mayocp.2017.11.001 [DOI] [PubMed]
- ApoE4.info, n.d. ApoE4.Info – The Alzheimer’s Gene Resource https://www.apoe4.info/wp/ (accessed January 2020).
- Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT, 2019. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med 25, 1680–1683. 10.1038/s41591-019-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Academy of Pediatrics, 2013. Ethical and Policy Issues in Genetic Testing and Screening of Children. Pediatrics 131, 620 LP– 622. 10.1542/peds.2012-3680 [DOI] [PubMed] [Google Scholar]
- Bartlett G, Rahimzadeh V, Longo C, Orlando LA, Dawes M, Lachaine J, Bochud M, Paccaud F, Bergman H, Crimi L, Issa AM, 2014. The future of genomic testing in primary care: The changing face of personalized medicine. Per. Med 10.2217/pme.14.36 [DOI] [PubMed] [Google Scholar]
- Baruch S, Hudson K, 2008. Civilian and Military Genetics: Nondiscrimination Policy in a Post-GINA World. Am. J. Hum. Genet 10.1016/j.ajhg.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel K, Geschwind MD, 2013. Ethics in prion disease. Prog. Neurobiol 10.1016/j.pneurobio.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TD., 1998. [Updated 2018]. Alzheimer Disease Overview In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. https://www.ncbi.nlm.nih.gov/books/NBK1161/ [PubMed] [Google Scholar]
- Bombard Y, Veenstra G, Friedman JM, Creighton S, Currie L, Paulsen JS, Bottorff JL, Hayden MR, Guttman M, Giambattista C, Ludman M, Murphy J, Babineau-Sturk T, Macleod P, Rice J, Martin W, Wieler M, Meschino W, Gibbons C, Raymond L, Decolongon J, Suchowersky O, Klimek M Lou, 2009. Perceptions of genetic discrimination among people at risk for Huntington’s disease: A cross sectional survey. BMJ 338 10.1136/bmj.b2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botkin JR, Belmont JW Berg JS, Berkman BE, Bombard Y, Holm IA, Levy HP, Ormond KE, Saal HM, Spinner NB, Wilfond BS, McInerney JD, 2015. Points to consider: Ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet, 97(1), 6–21. doi: 10.1016/j.ajhg.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG, 2019. Gene Therapy for ALS—A Perspective. Int. J. Mol. Sci 20, 4388 10.3390/ijms20184388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron NS, Wright GEB, Hayden MR., 1998. [Updated 2018 Jul 5]. Huntington Disease In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1305/ [PubMed] [Google Scholar]
- Cecile A, Janssens JW, Joyner MJ, 2019. Polygenic Risk Scores That Predict Common Diseases Using Millions of Single Nucleotide Polymorphisms: Is More, Better?, Clinical Chemistry, Volume 65, Issue 5, Pages 609–611, 10.1373/clinchem.2018.296103 [DOI] [PubMed] [Google Scholar]
- Christensen KD, Roberts JS, Whitehouse PJ, Royal CDM, Obisesan TO, Cupples LA, Vernarelli JA, Bhatt DL, Linnenbringer E, Butson MB, Fasaye GA, Uhlmann WR, Hiraki S, Wang N, Cook-Deegan R, Green RC, 2016. Disclosing pleiotropic effects during genetic risk assessment for Alzheimer disease. Ann. Intern. Med 164, 155–163. 10.7326/M15-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KD, Uhlmann WR, Roberts JS, Linnenbringer E, Whitehouse PJ, Royal CDM, Obisesan TO, Cupples LA, Butson MB, Fasaye GA, Hiraki S, Chen CA, Siebert U, Cook-Deegan R, Green RC, 2018. A randomized controlled trial of disclosing genetic risk information for Alzheimer disease via telephone. Genet. Med 20, 132–141. 10.1038/gim.2017.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook Shukla L, Schulze J, Farlow J, Pankratz N, Wojcieszek J, Foroud T Parkinson Disease Overview 2004. May 25 [Updated 2019 Jul 25]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. https://www.ncbi.nlm.nih.gov/books/NBK1223/ (accessed January 2020). [Google Scholar]
- Crozier S, Robertson N, Dale M, 2015. The Psychological Impact of Predictive Genetic Testing for Huntington’s Disease: A Systematic Review of the Literature. J. Genet. Couns 24, 29–39. 10.1007/s10897-014-9755-y [DOI] [PubMed] [Google Scholar]
- Cruts M, Engelborghs S, van der Zee J, van Broeckhoven C, 2015. C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. https://www.ncbi.nlm.nih.gov/books/NBK268647/ [Google Scholar]
- Ekstract M, Holtzman GI, Kim KY, Willis SM, Zallen DT, 2017. Evaluation of a Web-based decision aid for people considering the APOE genetic test for Alzheimer risk. Genet. Med 19, 676–682. 10.1038/gim.2016.170 [DOI] [PubMed] [Google Scholar]
- Fagerlin A, Zikmund-Fisher BJ, Ubel PA, 2011. Helping Patients Decide: Ten Steps to Better Risk Communication . JNCI J. Natl. Cancer Inst 103, 1436–1443. 10.1093/jnci/djr318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow J, Pankratz ND, Wojcieszek J, Foroud T, 1993. Parkinson Disease Overview, GeneReviews®. [Google Scholar]
- Fiedorowicz J,G, Mills J,A, Ruggle, Langbehn D, Paulsen J,S: Suicidal Behavior in Prodromal Huntington Disease. Neurodegener Dis 2011;8:483–490. doi: 10.1159/000327754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Test Registry, 2018. Alzheimer dementia and dementia panel https://www.ncbi.nlm.nih.gov/gtr/tests/505595/ (February 2020).
- Genetic Test Registry, 2018. Huntington Disease. https://www.ncbi.nlm.nih.gov/gtr/tests/502937/ (accessed February 2020).
- Genetic Test Registry, 2018. Parkinsons disease panel. https://www.ncbi.nlm.nih.gov/gtr/tests/552609/ (accessed February 2020).
- Genetic Test Registry, 2019. Amyotrophic Lateral Sclerosis Panel https://www.ncbi.nlm.nih.gov/gtr/tests/551921/ (accessed February 2020).
- Genetic Test Registry, 2019. Prion Disease (PRNP Single Gene Test) https://www.ncbi.nlm.nih.gov/gtr/tests/566062/ (accessed February 2020).
- GenomeWeb, 2019. All of Us Research Program to Test Options for Returning Results https://www.genomeweb.com/sequencing/allus-research-program-test-options-returning-results#.XjR0ty3MyL5 (accessed February 2020).
- Goh AMY, Chiu E, Yastrubetskaya O, Erwin C, Williams JK, Juhl AR, Paulsen JS, 2013. Perception, experience, and response to genetic discrimination in Huntington’s disease: The Australian results of the international RESPOND-HD study. Genet. Test. Mol. Biomarkers 17, 115–121. 10.1089/gtmb.2012.0288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JS, Hahn SE, Catania JW, Larusse-Eckert S, Butson MB, Rumbaugh M, Strecker MN, Roberts JS, Burke W, Mayeux R, Bird T, 2011. Genetic counseling and testing for Alzheimer disease: Joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet. Med 13, 597–605. 10.1097/GIM.0b013e31821d69b8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves CV, Rohrer JD, 2019. An update on genetic frontotemporal dementia. J. Neurol 10.1007/s00415-019-09363-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Christensen KD, Cupples LA, Relkin NR, Whitehouse PJ, Royal CDM, Obisesan TO, Cook-Deegan R, Linnenbringer E, Butson MB, Fasaye GA, Levinson E, Roberts JS, Bhatt D, Biesecker B, Blacker D, Chen C, Cox E, Davis JG, Farrer L, Griffith P, Harkins K, Hiraki S, Johnson M, Johnson S, Juengst E, Karlawish J, Le L, McCarty Wood E, Obisesan T, Post S, Quaid K, Ravdin L, Roter D, Stern R, Sadovnick A, Sami S, Sankar P, Topol E, Uhlmann W, Waterston L, Wright L, 2015. A randomized noninferiority trial of condensed protocols for genetic risk disclosure of Alzheimer’s disease. Alzheimer’s Dement. 11, 1222–1230. 10.1016/j.jalz.2014.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shoulson I, Bonilla E, Martin JB, 1983. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238. 10.1038/306234a0 [DOI] [PubMed] [Google Scholar]
- Hawkins A, Creighton S, Ho A, McManus B, Hayden M, 2013. Providing predictive testing for Huntington disease via telehealth: results of a pilot study in British Columbia, Canada. Clin. Genet 84, 60–64. 10.1111/cge.12033 [DOI] [PubMed] [Google Scholar]
- Hercher L, Uhlmann WR, Hoffman EP, Gustafson S, Chen KM, 2016. Prenatal Testing for Adult-Onset Conditions: the Position of the National Society of Genetic Counselors. J. Genet. Couns 25, 1139–1145. 10.1007/s10897-016-9992-3 [DOI] [PubMed] [Google Scholar]
- Heshka JT, Palleschi C, Howley H, Wilson B, Wells PS, 2008. A systematic review of perceived risks, psychological and behavioral impacts of genetic testing. Genet. Med 10.1097/GIM.0b013e31815f524f [DOI] [PubMed] [Google Scholar]
- Hollands GJ, French DP, Griffin SJ, Prevost AT, Sutton S, King S, Marteau TM, 2016. The impact of communicating genetic risks of disease on risk-reducing health behaviour: Systematic review with meta-analysis. BMJ 352 10.1136/bmj.i1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskovec JM, Bennett RL, Carey ME, DaVanzo JE, Dougherty M, Hahn SE, LeRoy BS, O’Neal S, Richardson JG, Wicklund CA, 2018. Projecting the Supply and Demand for Certified Genetic Counselors: a Workforce Study. J. Genet. Couns 27, 16–20. 10.1007/s10897-017-0158-8 [DOI] [PubMed] [Google Scholar]
- Hsiung G-YR, Feldman HH, 1993. GRN-Related Frontotemporal Dementia, GeneReviews®. [Google Scholar]
- Hsu D, Marshall G, 2017. Primary and Secondary Prevention Trials in Alzheimer Disease: Looking Back, Moving Forward. Curr. Alzheimer Res. 13, 1–1. 10.2174/1567205013666160930112125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington’s Disease Society of America, 2016. Genetic Testing Protocol for Huntington’s Disease. http://hdsa.org/wp-content/uploads/2015/02/HDSA-Gen-Testing-Protocol-for-HD.pdf
- Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, Morrison KE, Shaw PJ, Shaw CE, Fogh I, Dobson RJ, Newhouse SJ, Al-Chalabi A, 2019. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun 7 10.1186/s40478-019-0724-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez L, Farias FHG, Dube U, Mihindukulasuriya KA, Harari O, 2019. Polygenic Risk Scores in Neurodegenerative Diseases: a Review. Curr. Genet. Med. Rep 7, 22–29. 10.1007/s40142-019-0158-0 [DOI] [Google Scholar]
- Institute of Medicine; Board on Neuroscience and Behavioral Health; Committee on Health Literacy; Lynn Nielsen-Bohlman, Panzer Allison M., and David A Kindig E, 2004. Health literacy: a prescription to end confusion. Choice Rev. Online 42, 42-4059-42-4059. 10.5860/choice.42-4059 [DOI] [Google Scholar]
- Janssens AC, Joyner MJ, 2019. Polygenic risk scores that predict common diseases using millions of single nucleotide polymorphisms: Is more, better? Clin. Chem 10.1373/clinchem.2018.296103 [DOI] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT, 2017. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med 19, 249–255. 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Karlawish J, 2007. Measuring decision-making capacity in cognitively impaired individuals. NeuroSignals 10.1159/000109763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay C, Collins JA, Miedzybrodzka Z, Madore SJ, Gordon ES, Gerry N, Davidson M, Slama RA, Hayden MR, 2016. Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology 87, 282–288. 10.1212/WNL.0000000000002858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeller DR, Uhlmann WR, Carere DA, Green RC, Roberts JS, 2017. Utilization of Genetic Counseling after Direct-to-Consumer Genetic Testing: Findings from the Impact of Personal Genomics (PGen) Study. J. Genet. Couns 26, 1270–1279. 10.1007/s10897-017-0106-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf BR, Berry AB, Limson M, Marian AJ, Murray MF, O’Rourke PP, Passamani ER, Relling MV, Tooker J, Tsongalis GJ, Rodriguez LL, 2014. Framework for development of physician competencies in genomic medicine: Report of the competencies working group of the inter-society coordinating committee for physician education in genomics. Genet. Med 16, 804–809. 10.1038/gim.2014.35 [DOI] [PubMed] [Google Scholar]
- Langlois CM, Bradbury A, Wood EM, Roberts JS, Kim SYH, Riviere M-E, Liu F, Reiman EM, Tariot PN, Karlawish J, Langbaum JB, 2019. Alzheimer’s Prevention Initiative Generation Program: Development of an APOE genetic counseling and disclosure process in the context of clinical trials. Alzheimer’s Dement. Transl. Res. Clin. Interv 5, 705–716. 10.1016/j.trci.2019.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenbach DM, Christensen KD, Sparks JA, Green RC, 2013. Communicating Genetic Risk Information for Common Disorders in the Era of Genomic Medicine. Annu. Rev. Genomics Hum. Genet 14, 491–513. 10.1146/annurevgenom-092010-110722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lineweaver TT, Bondi MW, Galasko D, Salmon DP, 2014. Effect of Knowledge of APOE Genotype on Subjective and Objective Memory Performance in Healthy Older Adults. Am. J. Psychiatry 171, 201–208. 10.1176/appi.ajp.2013.12121590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C, Tariot PN, Caputo A, Langbaum JB, Liu F, Riviere M-E, Langlois C, Rouzade-Dominguez M-L, Zalesak M, Hendrix S, Thomas RG, Viglietta V, Lenz R, Ryan JM, Graf A, Reiman EM, 2019. The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv 5, 216–227. 10.1016/j.trci.2019.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucette-Stamm L, O’Donovan MC, Riba-Ramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS, 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- MacLeod R, Tibben A, Frontali M, Evers-Kiebooms G, Jones A, Martinez-Descales A, Roos R, 2013. Recommendations for the predictive genetic test in Huntington’s disease. Clin. Genet 83, 221–231. 10.1111/j.1399-0004.2012.01900.x [DOI] [PubMed] [Google Scholar]
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ, 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591. 10.1038/s41588-019-0379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrianni JA. Genetic Prion Diseases. 2003. March 27 [Updated 2014 Jan 2]. In: Adam MP, Ardinger HH, Pagon, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1229/ (accessed January 2020). [Google Scholar]
- McGuire AL, Burke W, 2008. An unwelcome side effect of direct-to-consumer personal genome testing: Raiding the medical commons. JAMA - J. Am. Med. Assoc 10.1001/jama.2008.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MedGen n.d. Lewy body dementia (Concept Id: C0752347). https://www.ncbi.nlm.nih.gov/medgen/C0752347 (accessed February 2020).
- Messner DA, 2011. Informed choice in direct-to-consumer genetic testing for Alzheimer and other diseases: Lessons from two cases. New Genet. Soc 10.1080/14636778.2011.552300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique Nailah, RN,MSN and Siddique Teepu, MD, Ds. (hc)., n.d. Amyotrophic Lateral Sclerosis Overview – GeneReviews™ - NCBI Bookshelf.
- National Academies of Sciences, Engineering, and Medicine, 2018. Returning Individual Research Results to Participants: Guidance for a New Research Paradigm. 10.17226/25094 [DOI] [PubMed]
- National Society of Genetic Counselors, n.d. About Genetic Testing [WWW Document] URL http://www.aboutgeneticcounselors.com/Genetic-Testing (accessed February 2020).
- Neumann PJ, Cohen JT, Hammitt JK, Concannon TW, Auerbach HR, Fang C, Kent DM, 2012. Willingness-to-pay for predictive tests with no immediate treatment implications: A survey of US residents. Health Econ. 21, 238–251. 10.1002/hec.1704 [DOI] [PubMed] [Google Scholar]
- Nguyen HHP, Weydt P, 2018. Huntington disease. Medizinische Genet 10.1007/s11825-018-0190-6 [DOI]
- Olszewska DA, Lonergan R, Fallon EM, Lynch T, 2016. Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep 10.1007/s11910-016-0707-9 [DOI] [PubMed] [Google Scholar]
- Peters OM, Ghasemi M, Brown RH, 2015. Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest 10.1172/JCI71601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitini E, De Vito C, Marzuillo C, D’Andrea E, Rosso A, Federici A, Di Maria E, Villari P, 2018. How is genetic testing evaluated? A systematic review of the literature. Eur. J. Hum. Genet 10.1038/s41431-018-0095-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popejoy AB, Fullerton SM, 2016. Genomics is failing on diversity. Nature 10.1038/538161a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, Langbaum JB, Neuhaus JM, Reiman EM, Roberts JS, Seshadri S, Tariot PN, Woods BM, Betensky RA, Blacker D, 2017. APOE-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts. PLOS Med. 14, e1002254 10.1371/journal.pmed.1002254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaid KA, 2017. Genetic testing for Huntington disease, in: Handbook of Clinical Neurology. Elsevier B.V., pp. 113–126. 10.1016/B978-0-12-801893-4.00010-9 [DOI] [PubMed] [Google Scholar]
- Ramos E, Weissman SM, 2018. The dawn of consumer‐directed testing. Am. J. Med. Genet. Part C Semin. Med. Genet 178, 89–97. 10.1002/ajmg.c.31603 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, 2015. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JS, 2019. Assessing the Psychological Impact of Genetic Susceptibility Testing. Hastings Cent. Rep 49, S38–S43. 10.1002/hast.1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JS, Gornick MC, Carere DA, Uhlmann WR, Ruffin MT, Green RC, 2017. Direct-to-consumer genetic testing: User motivations, decision making, and perceived utility of results. Public Health Genomics 20, 36–45. 10.1159/000455006 [DOI] [PubMed] [Google Scholar]
- Roberts JS, LaRusse SA, Katzen H, Whitehouse PJ, Barber M, Post SG, Relkin N, Quaid K, Pietrzak RH, Cupples LA, Farrer LA, Brown T, Green RC, 2003. Reasons for seeking genetic susceptibility testing among first-degree relatives of people with Alzheimer disease. Alzheimer Dis. Assoc. Disord 17, 86–93. 10.1097/00002093-200304000-00006 [DOI] [PubMed] [Google Scholar]
- Roggenbuck J, Quick A, Kolb SJ, 2017. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: An update for clinicians. Genet. Med 10.1038/gim.2016.107 [DOI] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JBS, Lopera F, Martins R, Masters CL, Mayeux RP, McDade E, Moreno S, Reiman EM, Ringman JM, Salloway S, Schofield PR, Sperling R, Tariot PN, Xiong C, Morris JC, Bateman RJ, 2014. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 83, 253–260. 10.1212/WNL.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanghvi H, Singh R, Morrin H, Rajkumar AP, 2020. Systematic review of genetic association studies in people with Lewy body dementia. Int. J. Geriatr. Psychiatry 10.1002/gps.5260 [DOI] [PubMed] [Google Scholar]
- Sardi SP, Cedarbaum JM, Brundin P, 2018. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord 10.1002/mds.27414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savica R, Grossardt BR, Bower JH, Boeve BF, Ahlskog JE, Rocca WA, 2013. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol. 70, 1396–1402. 10.1001/jamaneurol.2013.3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkedi-Rafid S, Fenwick A, Dheensa S, Lucassen AM, 2015. Genetic testing of children for adult-onset conditions: Opinions of the British adult population and implications for clinical practice. Eur. J. Hum. Genet 23, 1281–1285. 10.1038/ejhg.2014.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ, 2011. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 7, 367–385. 10.1016/j.jalz.2011.05.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundhedsstyrelsen, 2009. Health literacy: a prescription to end confusion. The National Academies Press; 10.17226/10883 [DOI] [PubMed] [Google Scholar]
- Suter SM, 2019. GINA at 10 years: The battle over “genetic information” continues in court. J. Law Biosci 5, 495–526. 10.1093/jlb/lsz002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DH, Cook-Deegan RM, Hiraki S, Roberts JS, Blazer DG, Green RC, 2010. Genetic testing for Alzheimer’s and long-term care insurance. Health Aff. 29, 102–108. 10.1377/hlthaff.2009.0525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, Wineinger NE, Topol EJ, 2018. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet 10.1038/s41576-018-0018-x [DOI] [PubMed] [Google Scholar]
- 23andMe, 2020. About Us - 23andMe. https://mediacenter.23andme.com/company/about-us/ (accessed February 2020).
- Tyrone JT, Sabbagh MN, Hanc J, n.d. Fighting for my life : how to thrive in the shadow of Alzheimer’s
- U.S. Food and Drug Administration, 2017. FDA allows marketing of first direct-to-consumer tests that provide genetic risk information for certain conditions. https://www.fda.gov/news-events/press-announcements/fda-allows-marketing-first-direct-consumer-tests-provide-genetic-risk-information-certain-conditions (accessed January 2020).
- Uhlmann WR, 2009. Thinking It All Through: Case Preparation and Management, in: Uhlmann WR; Schuette JL; Yashar B (Ed.), A Guide to Genetic Counseling. Elsevier BV, New York, NY, pp. 93–121. 10.1016/j.ajhg.2010.07.024 [DOI] [Google Scholar]
- Uhlmann WR, Peñaherrera MS, Robinson WP, Milunsky JM, Nicholson JM, Albin RL, 2015. Biallelic mutations in huntington disease: A new case with just one affected parent, review of the literature and terminology. Am. J. Med. Genet. Part A 167, 1152–1160. 10.1002/ajmg.a.37009 [DOI] [PubMed] [Google Scholar]
- Uhlmann WR, Roberts JS, 2018. Ethical issues in neurogenetics, in: Handbook of Clinical Neurology. Elsevier B.V., pp. 23–36. 10.1016/B978-0-444-63233-3.00003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K, 2016. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med 18, 421–430. 10.1038/gim.2015.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Rosso SM, Heutink P. MAPT-Related Disorders. 2000. November 7 [Updated 2010 Oct 26]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1505/ [Google Scholar]
- Vernarelli JA, Roberts JS, Hiraki S, Chen CA, Cupples LA, Green RC, 2010. Effect of Alzheimer disease genetic risk disclosure on dietary supplement use. Am. J. Clin. Nutr 91, 1402–1407. 10.3945/ajcn.2009.28981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker Z, Possin KL, Boeve BF, Aarsland D, 2015. Lewy body dementias. Lancet. 386, 1683–1697. 10.1016/S0140-6736(15)00462-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman SM, Kirkpatrick B, Ramos E, 2019. At-home genetic testing in pediatrics. Curr. Opin. Pediatr 10.1097/MOP.0000000000000824 [DOI] [PubMed] [Google Scholar]
- Zick CD, Mathews CJ, Roberts JS, Cook-Deegan R, Pokorski RJ, Green RC, 2005. Genetic testing for Alzheimer’s disease and its impact on insurance purchasing behavior. Health Aff 24, 483–490. 10.1377/hlthaff.24.2.483 [DOI] [PMC free article] [PubMed] [Google Scholar]