

Abstract
BCR transgenic mice dominate studies of B-cell tolerance; consequently, tolerance in normal mice expressing diverse sets of autoreactive B cells is poorly characterized. We have used single B-cell cultures to trace self-reactivity in BCR repertoires across the first and second tolerance checkpoints and in tolerized B-cell compartments of normal mice. This approach reveals affinity “setpoints” that define each checkpoint and a subset of tolerized, autoreactive B cells that is long-lived. In normal mice, the numbers of B cells avidly specific for DNA fall significantly as small pre-B become immature and transitional-1 (T1) B cells, revealing the first tolerance checkpoint. By contrast, DNA-reactivity does not significantly change when immature/T1 B cells become mature follicular (MF) B cells, showing that the second checkpoint does not reduce DNA-reactivity. In the spleen, autoreactivity was high in transitional 3 (T3), CD93+IgM-/loIgDhi anergic B cells and a CD93- anergic subset. Whereas splenic T3 and CD93+ anergic B cells are short-lived, CD93-IgM-/loIgDhi B cells have half-lives comparable to MF B cells. B-cell specific deletion of proapoptotic genes, Bak and Bax, resulted in increased CD93-IgM-/loIgDhi B cell but not T3 B cell numbers, suggesting that apoptosis regulates differently persistent- and ephemeral autoreactive B cells. The self-reactivity and longevity of CD93-IgM-/loIgDhi B cells, and their capacity to proliferate and differentiate into plasmacytes in response to CD40 activation in vitro lead us to propose that this persistent, self-reactive compartment may be the origin of systemic autoimmunity and a potential target for vaccines to elicit protective antibodies cross-reactive with self-antigens. 249/250
Keywords: B-cell tolerance, anergy, high-throughput screening
Introduction
Combinatorial recombination of V, D, and J gene segments of the immunoglobulin genes generates highly diverse, polyclonal sets of B-cell antigen receptors (BCRs). V(D)J recombination is essentially random, and thus generates BCRs reactive to self- as well as foreign antigens. Indeed, significant fractions (~75%) of recombinant IgG antibodies (rAbs) cloned from single early immature B cells react to self-antigens (1). Nonetheless, individuals do not normally develop pathological autoantibodies; instead, tolerance mechanisms - clonal deletion, receptor editing, and anergy - remove or inactivate self-reactive B cells (2–7).
In mice and humans, tolerance removes self-reactive B cells at two distinct checkpoints (1, 3–5, 8, 9). In the bone marrow, the first tolerance checkpoint acts to remove nascent B cells that carry self-reactive BCRs at the transition from small pre-B to immature B-cell stages by inducing BCR-mediated, apoptotic deletion (3–5). Self-reactive immature B cells can lose self-reactivity by replacing their light chains: a subset of immature B cells undergoes additional rounds of V to J gene recombination (6, 7) to generate new light chains that no longer confer self-reactivity when paired with their rearranged heavy chains. In the periphery, the second tolerance checkpoint removes self-reactive B cells that are not removed by central tolerance (1, 8, 9), or alternatively, self-reactive B cells become unresponsive to antigenic stimulations (2, 10, 11).
Studies of transgenic mice that express self-reactive BCRs (2–8, 12) have defined the mechanisms of immunological tolerance but remain artefactual models of the normal tolerization processes due to the restricted diversity of the quasi-clonal B-cell populations they support. For example, different transgenic mouse models often utilize one mode of tolerance (i.e., deletion vs. receptor editing vs. anergy) in preference – and occasionally exclusively - to others. Moreover, the phenotypes and responses of anergic B cells to activating stimuli often differ among autoreactive BCR transgenic strains (13). In contrast, studies have inferred tolerance mechanisms in humans by characterizing BCR repertoires of pre- and post-tolerance B cells by single-cell PCR and the analysis of rAbs cloned from individual B cells (1, 14–20). This approach frees investigations of immunological tolerance from any artifacts of genetically restricted BCRs but depends, obviously, on the construction and expression of rAb panels that are sufficiently large to detect reliably changes in BCR repertoires effected by tolerance.
We use here single-cell Nojima cultures (21, 22) to study the natural dynamics of the autoreactive BCR repertoire. Nojima cultures efficiently support the proliferation and eventual plasmacytic differentiation to IgG secretion of single B cells (21). Each cultured B cell generates thousands of plasmablasts that secrete sufficient quantities of IgG1 for analysis by ELISA and Luminex assays. Despite an efficient class-switch recombination, Nojima cultures do not support V(D)J mutation (21). Thus, specificity and avidity of IgG Abs in culture supernatants represent the BCRs expressed by each founder B cell; this permits efficient screening for antigen-specificity and relative avidities [Avidity Index values (21, 23)] prior to any rAb production (21, 24–26).
We find that in contrast to relevant knock-in models, absolute frequencies of DNA-reactive B cells do not significantly change during B-cell development and maturation in normal mice. Instead, the distribution of the BCR avidities for DNA changes: that fraction of DNA-reactive B cells that are most avid is lost at the transition from small pre-B to immature and transitional-1 (immature/T1) B cells in the bone marrow. The remaining B-cell sets that carry BCRs with intermediate and weak avidities for DNA are unchanged as immature/T1 B cells become mature follicular (MF) B cells, suggesting that the second tolerance checkpoint plays little, if any, role in purging DNA BCRs. In spleen, high avidity DNA-binding BCRs were enriched in the T3 and IgM−/loIgDhi anergic B cell compartments. Despite similar DNA-binding avidity, turnover-rate and regulation by apoptotic pathways readily distinguish these self-reactive B-cell compartments; T3 B and CD93+IgM−/loIgDhi B cells are short-lived, whereas CD93−IgM−/loIgDhi B cells persist with a population kinetics similar to that of the IgMintIgDhi MF B-cell compartment. CD93−IgM−/loIgDhi B cells but not T3 B cells proportionally expand in spleen in the absence of pro-apoptotic molecules, BAK and BAX. Long-term persistence of self-reactive B cells in the spleen of normal mice implies a biological importance of self-reactive B cells that can provide distinct BCR repertoire from non-self-reactive B cells (22). Learning how to control these persistent self-reactive B cells may be useful to develop strategies to elicit protective humoral responses against microbial pathogens that resemble host determinants.
Materials and Methods
Mice
C57BL/6J (B6), B6.129P2(C)-Cd19tm1(cre)Cgn/J (Cd19Cre), and B6;129-Baxtm2SjkBak1tm1Thsn/J (Bak−/−Baxfl/fl) mice were purchased from Jackson laboratory. Bak−/−Baxfl/fl mice were backcrossed with B6 mice at least ten generations before experimental use. We crossed Cd19Cre mice with Bak−/−Baxfl/fl mice to generate Cd19Cre+Bak−/−Baxfl/fl mice. All mice were maintained under specific pathogen-free conditions at the Duke University Animal Care Facility and used in experiments at 8 to 12 weeks of age. All experiments involving animals were approved by the Duke University Institutional Animal Care and Use Committee.
Single B-cell culture (Nojima culture)
On day −1, NB-21.2D9 cells were seeded into 96-well plates at 1,000 cells/well in B-cell media (BCM); RPMI-1640 medium (Invitrogen) supplemented with 10% HyClone FBS (Thermo scientific), 5.5 × 10−5 M 2-mercaptoethanol, 10 mM HEPES, 1 mM sodiumpyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1 × MEM nonessential amino acid (all Invitrogen). Next day (day 0), recombinant mouse IL-4 (Peprotech; final 2 ng/ml) was added to the cultures, and then single B cells were directly sorted into each well using cell sorters [FACSVantage or FACSAria (both BD Biosciences)]. On day 2, 50% (vol.) of the culture media were removed from cultures and 100% (vol.) of fresh BCM were added to the cultures. On days 3 to 8, two-thirds of the culture media were replaced with fresh BCM every day. On day 9, culture supernatants were harvested for ELISA determinations and culture plates were stored at −80°C for V(D)J amplifications.
Flow cytometry
Single cell suspensions, depleted of red blood cells by RBC Lysis Buffer (eBioscience), were obtained from bone marrow and spleen, and labeled with following mAbs to mouse surface molecules purchased from Biolegend or eBioscience or BD Bioscience. CD19-PE (6D5), CD21-FITC (7G8), CD23-PE or -Brilliant Violet 421 (B3B4), CD43-PE (S7), CD93-APC (AA4.1), CD138-APC (281–2), B220-eFluor 450 or -V500 or -Brilliant Violet 510 (RA3-6B2), IgM PE-Cy7 (II/41), IgD-FITC or -eFluor 450 or -Brilliant Violet 510 (11-26c), IgG1 V450 (A85-1), and IgE-FITC (RME-1). B-cell subsets were identified as described (2, 27, 28); small pre-B cells, B220loCD93+IgM−IgD−CD43−FSClo; Imm/T1 B cells, B220loCD93+IgM+IgD+/−CD23− in the bone marrow; T1 B cells, B220loCD93+IgMhiCD23−; T2 B cells, B220loCD93+IgMhiCD23+; T1/T2 B cells, B220loCD93+IgMhiIgD+/−; T3 B cells, B220loCD93+IgM−/loCD23+; MF B cells, B220+CD93−IgMintIgDhiCD21loCD23hi; MZ B cells, B220+CD93−IgMhiIgDloCD21hiCD23lo; anergic B cells, B220+IgM−/loIgDhi(CD93+ or CD93−) cells in spleen. Labeled cells were analyzed and sorted in FACSCanto, FACSCanto II, FACSAria or FACSVantage with Diva option (all BD Biosciences). Data were analyzed with FlowJo software (Tree Star). Cell doublets (determined by FSC-A/FSC-H method described elsewhere) and propidium iodide positive cells were excluded from our analyses.
ELISA determinations
Concentration of total IgG was determined by standard ELISA. Briefly, ELISA plates [(96-well plate or 384-well High binding plate (both Corning) were used in this study] were coated with anti-mouse Igκ Ab and anti-mouse Igλ Ab (2 μg/ml each; Southern Biotech) in carbonate buffer. After washing, plates were blocked with PBS containing 0.5% BSA. Samples (at 1:200 or 1:1,000 dilutions in PBS containing 0.5% BSA and 0.1% Tween-20) and serially diluted standards (described below) were then applied to the plates. After washing, HRP-conjugated anti-mouse IgG (Southern Biotech) was added as detection Ab. The HRP-activity was visualized with TMB substrate reagents (Biolegend) and OD450 was measured by spectrophotometer (Bio-Rad).
Anti-DNA IgG was measured by ELISA (29) in a hypotonic condition (hypotonic buffer: 1.5 × 10−2 M NaCl, 4.3 × 10−4 M Na2HPO4, and 1.9 × 10−3 M NaH2PO4), as the interaction between DNA and anti-DNA Ab are electrostatic and these interactions can be lost at high salt concentrations (30, 31). Calf thymus DNA (Sigma) was purified by phenol/chloroform extraction, and ELISA plates were coated with the purified DNA in 1 × SSC (10 μg/ml) and were dried up at 37°C for overnight. After blocking with hypotonic buffer containing 3% FBS and 0.5% BSA, samples (at 1:10 or 1:100 or 1:1,000 dilutions in hypotonic buffer containing 0.5% BSA and 0.1% Tween-20) and serially diluted standards (described below) were applied to the plates, and followed by addition of HRP-conjugated secondary Ab and TMB substrates as described above. The cut-off OD450 for total IgG and anti-DNA IgG was set at the point representing six-standard deviations above the mean OD450 for supernatants from mock-treated, B-cell negative samples.
DNA avidity index was obtained by taking the ratio of DNA-binding IgG (concentration) to total IgG (concentration). We used two mAbs as positive and negative controls for DNA-binding; anti-DNA mAb [HYB331–01 (Abcam)] and hapten-specific mAb [H33Lγ1 (32)]. DNA avidity index of H33Lγ1 was 0.0029, which we set as a cut-off for the DNA-binding in our assays.
Amplification of VDJ rearrangements
After Nojima cultures, VDJ rearrangements were amplified from B-cell subsets by a nested PCR as described (33) with modifications. Total RNA was extracted from selected samples after Nojima cultures using TRIzol or TRIzol LS reagents (Invitrogen) according to the manufacturer’s instruction. The extracted RNA were treated with DNase I (0.1 unit/μl; Invitrogen), and cDNA was synthesized using Superscript III with oligo (dT)20 primers (reaction volume: 20 μl) at 50°C for 50 min followed by 85°C for 5 min. One microliter of the cDNA samples was then subjected to two rounds of PCR using Herculase II fusion DNA polymerase (Agilent Technologies) with established primers (33–36). PCR conditions used in this study are as follows. IgH primary: 95°C for 4 min, followed by 2 cycles of 95°C for 30 sec, 64°C for 20 sec, and 72°C for 45 sec; 3 cycles of 95°C for 30 sec, 62°C for 20 sec, and 72°C for 45 sec; and 25 cycles of 95°C for 30 sec, 64°C for 20 sec, and 72°C for 45 sec. IgH secondary: 95°C for 4 min, followed by 40 cycles of 95°C for 30 sec, 60°C for 20 sec, and 72°C for 45 sec. VDJ amplicands were gel purified, ligated into vectors, and transformed into bacteria (37). DNA sequences were obtained at Duke DNA sequencing facility. VDJ rearrangements were identified using IMGT/V-QUEST (http://www.imgt.org/IMGT_vquest/vquest).
BrdU labeling
B6 mice were fed BrdU (Sigma) with drinking water (0.8 mg/ml) for up to 2 weeks. This BrdU water was replaced with fresh one every 2 days. Mice were sacrificed at one week and two weeks after treatment, and incorporation of BrdU in each B-cell subset was analyzed by flow cytometry using BrdU flow kit (BD Biosciences) according to manufacturer’s instruction. Dead cells were excluded using LIVE/DEAD fixable dead cell stain kit (Invitrogen).
Calcium flux assay
Splenocytes were resuspended in loading buffer (HBSS containing CaCl2 and MgCl2 (Invitrogen), supplemented with 10 mM HEPES (Invitrogen) and 1% Hyclone FCS). Cells were then incubated with the mixture of Indo-1 AM (final concentration: 2.8 μM; ThermoFisher) and Pluronic F-127 (final concentration: 22 nM; Sigma) for 30 min at room temperature in the dark. After washing, cells were labeled with mAbs against mouse surface antigens: B220-FITC (RA3–6B2), CD93-APC (AA4.1), CD23-Brilliant Violet 421 (B3B4), in the presence or absence of IgM PE-Cy7 (II/41). Cells were also labeled with PE-Cy5 conjugated mAbs to exclude non-B cells; CD4 (GK1.5), CD8α (53–6.7), CD11b (M1/70), CD11c (N418), Gr-1 (RB6–8C5), TER-119 (TER-119). Aliquots of Indo-1 loaded cells were labeled with the addition of IgD-Brilliant Violet 510 (11–26c) to check the purity of the measured B-cell subsets. After washing, cells were resuspended in loading buffer and kept in dark at room temperature until use. Labeled cells were analyzed in LSRFortessa (BD Biosciences). Baselines were established for 30 seconds, and then cells were stimulated with 1 μg/ml of F(ab’)2 fragment of goat anti-mouse kappa (Southern Biotech). Intracellular calcium concentrations were measured for 5 min. Kinetic curves were generated using Flowjo software.
Statistics
Statistical significance among samples was determined by Mann-Whitney’s U test or by Kruskal-Wallis test or by Turkey’s multiple comparison test in a mixed effects model with Geisser-Greenhouse correction. We considered P < 0.05 as statistically significant.
Results
Nojima culture efficiently supports IgG secretion by single B cells of differential developmental state
To trace self-reactivity during B-cell development in normal mice, we used single-cell, Nojima cultures, which provide representative samples of BCR diversity and repertoire (21). In the current study, we established single-cell cultures of the following B-cell subsets (Fig. S1): 1) bone marrow small pre-B cells to represent the pre-tolerance BCR repertoire; 2) bone marrow immature and transitional-1 (immature/T1) B cells to show the effects of the first tolerance checkpoint; 3) splenic mature follicular (MF) B cells to reflect the post-tolerance repertoire (1, 8, 9); 4) splenic transitional-3 (T3) B cells (27); and 5) B cells with IgM-/loIgDhi anergic phenotype (2, 38, 39) to sample the BCR repertoire that is subject to tolerization in the periphery. During the isolation of anergic phenotype B cells, we found that only a minor (~10%) fraction of splenic IgM−/loIgDhi B cells expressed CD93 (Fig. S1). These CD93+IgM−/loIgDhi B cells represented a subset of T3 B cells that were enriched for lower or negative surface IgM expression (Fig. S1).
Single small pre-B cells, immature/T1 B cells, T3 B cells, MF B cells, and CD93+ and CD93− IgM−/loIgDhi B cells proliferated in Nojima cultures to reach ≥10,000 cells after 8 days of culture (Fig. 1A). Although small pre-B cells generated 2-5-fold fewer daughter cells than did other B-cell subsets, all B-cell subsets class-switched to IgG1 uniformly (~95%), and differentiated (40% – 60%) into IgG1+CD138+ plasmablasts/-cytes (Figs. 1A–1D). Cloning efficiencies (as determined by the frequency of wells that contain IgG among total wells that were seeded with single B cells) for all B-cell subsets was about 60% (51% – 80%) with the exception of small pre-B cells (23% ± 4.1%; Table 1). The lower cloning efficiency for small pre-B cells may be due to the non-productive VJ rearrangements and resulting failure of functional BCR expression, which is required for the survival of B cells (40, 41). Nojima cultures support proliferation and differentiation of single small pre-B cells, immature/T1 B cells, T3 B cells, MF B cells, and IgM−/loIgDhi anergic B cells.
Figure 1. Nojima culture efficiently supports proliferation of single B cells, differentiation into IgG1+ plasmacytes and IgG secretion.
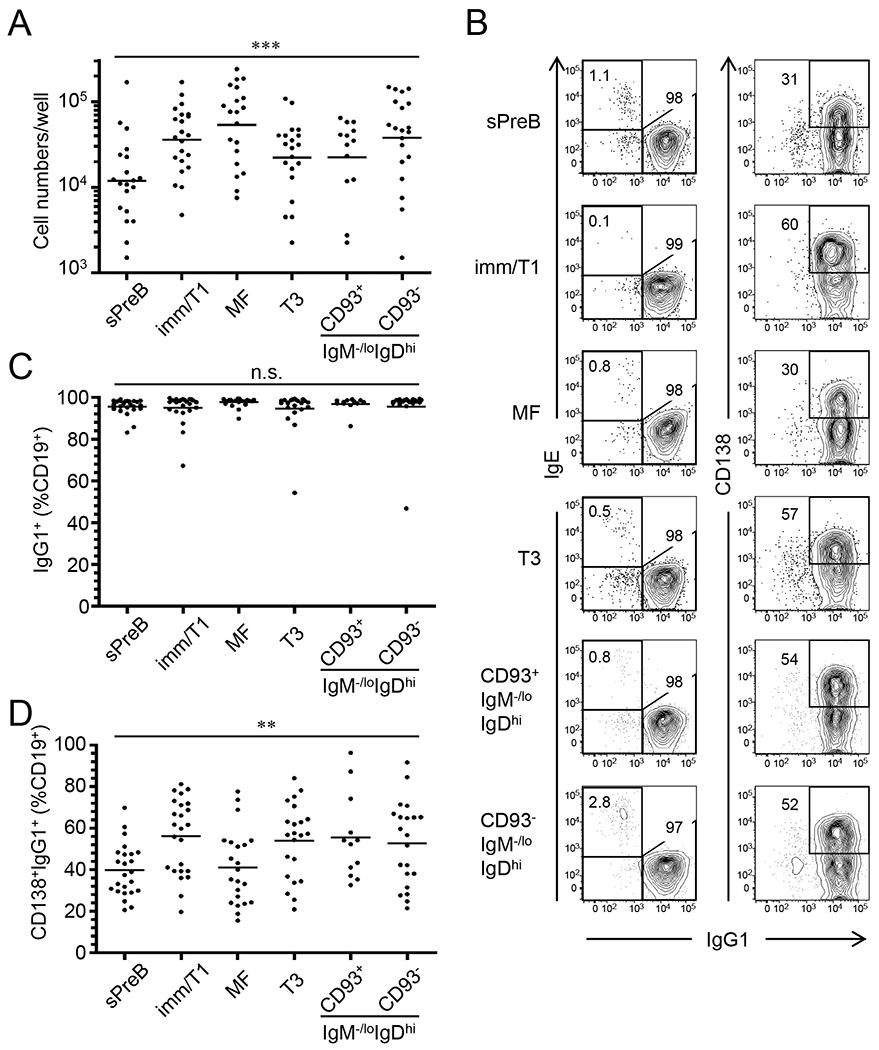
Single B cells were sorted from small pre-B (sPreB), immature/T1 (Imm/T1), mature follicular (MF), transitional-3 (T3), and CD93+ and CD93− anergic B cells of B6 mice (gatings are shown in Fig. S1), and then cultured in the presence of NB-21.2D9 feeder cells. Live B-cell numbers (A), representative FACS profiles of surface IgG1 and IgE or CD138 expression on CD19+ cells (B), and frequencies of IgG1+ cells (C) and CD138+IgG1+ cells (D) in CD19+ cells of indicated B-cell subsets after 8 days of Nojima cultures are shown. Each symbol in (A, C, and D) represents individual wells (n = 12-24) and bars indicate geometric mean of the samples. n.s., P > 0.05; **, P < 0.01; ***, P < 0.001 determined by Kruskal-Wallis test. Numbers within or close to gatings in each panel of (B) indicate the frequency of cells within gatings.
Table 1.
Cloning efficiency and DNA reactivity of B-cell subsets after Nojima cultures1
| Mouse strain | B-cell type | No. Exps.2 | Cloning efficiency3 | Freq. DNA-binding4 |
|---|---|---|---|---|
| B6 | Small pre-B | 6 (5) | 23 (±4.1) | 22 (±9.9) |
| Immature/T1 | 6 (4) | 56 (±9.6) | 20 (±4.8) | |
| MF | 8 (5) | 80 (±4.4) | 21 (±4.2) | |
| T3 | 2 (2) | 61 (±2.1) | 25 (±9.5) | |
| CD93+ anergic | 2 (2) | 51 (±2.2) | 22 (±3.1) | |
| CD93− anergic | 2 (2) | 68 (±6.4) | 22 (±1.2) |
Single B cells were sorted from small pre-B, immature/T1 B, MF B, T3 B, and CD93+ and CD93− anergic B cells of B6 mice, and cultured in the presence of NB-21.2D9 feeder cells for 9 days. After culture, culture supernatants were harvested for ELISA determinations.
For each B-cell subset, number of experiments for the determination of cloning efficiency (DNA reactivity) is shown. We determined DNA reactivity from most (77%; 20/26) but not all Nojima cultures.
Cloning efficiency represents frequency of IgG+ samples as a percentage of the number of samples screened. Mean values (±SD) are shown.
For each B-cell subset, frequency of DNA-binding IgG+ samples is shown as a percentage of the number of total IgG+ samples. Mean values (±SD) are shown.
Nojima cultures reveal B-cell tolerance checkpoints in normal mice
The high cloning efficiency of single B cells in Nojima cultures immediately suggested the possibility of characterizing the effects of tolerance on the B-cell repertoire in normal mice. We analyzed in total 2,021 IgG+ examples from single-cell cultures of small pre-B (n = 153), immature/T1 (n = 120), MF (n = 440), T3 (n = 447), and CD93+ (n = 362) and CD93- (n = 499) IgM−/loIgDhi B cells, respectively. All B-cell subsets produced comparable amounts of clonal IgGs in culture supernatants, 2.5 – 2.7 μg/ml, with the exception of small pre-B cells (1.3 μg/ml) (Fig. 2A).
Figure 2. DNA-reactive B cells with higher avidity are purged in bone marrow but enriched in anergic B-cell compartments in the spleen of normal mice.
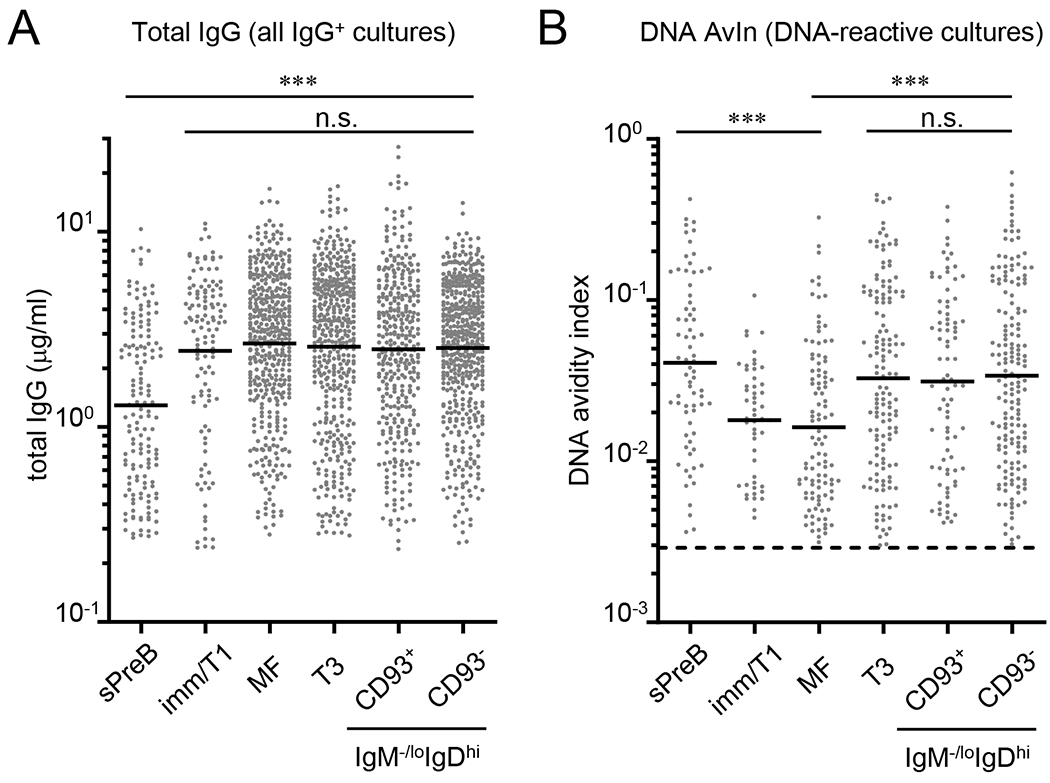
Clonal IgGs were obtained by Nojima cultures (9 days) of small pre-B (sPreB, n = 153), immature/T1 (Imm/T1, n = 120), mature follicular (MF, n = 440), transitional-3 (T3, n = 447), and CD93+ (n = 362) and CD93- (n = 499) anergic B cells from B6 mice. Concentrations of total IgG (A) and DNA AvIns (B) of each sample were determined by ELISA. Distributions of total IgG concentrations (A) and DNA AvIns (B) are shown. (B) DNA AvIns were calculated by taking the ratio of concentrations of DNA-binding IgG to total IgG (detailed in Experimental procedures). Dotted line indicates DNA AvIn (=0.0029) of non-autoreactive mAb [H33Lγ1; (32)] and therefore set as a cut-off for the determination of DNA reactivity. DNA-reactive samples among all IgG+ samples are shown. Each symbol represents individual wells and bars indicate geometric mean of the samples. Combined data from two independent experiments are shown. n.s., P > 0.05; ***, P < 0.001 determined by Kruskal-Wallis test.
By analyzing the reactivity of clonal IgG in culture supernatants, we compared the frequency of DNA-reactive cells among B-cell subsets (Table 1). In contrast to the results of studies analyzing tolerance of the human B-cell repertoire (1, 9, 22), we observed no significant differences in the frequencies of DNA-reactive B cells (i.e., clonal IgGs) among B-cell compartments: DNA-reactive, small pre-B, immature/T1, MF, T3, and CD93+ and CD93- IgM-/loIgDhi B cells were equivalently abundant in each compartment (20% – 25%; Table 1).
In contrast to unchanging frequencies of DNA-binding B cells through development (Table 1), we observed significant decreases in the mean avidity of DNA-reactive B cells during their maturation and especially across the first tolerance checkpoint. Avidities of anti-DNA IgG among B-cell subsets were estimated as the avidity index (AvIn) for each clonal IgG (21, 23) in reference to a monoclonal IgG standard, HYB331-01 (Abcam). DNA AvIn values, on average, decreased along with B-cell maturation whereas the tolerized B-cell compartments exhibited elevated DNA AvIns. Geometric mean AvIn for DNA was highest (0.041) in small pre-B cells, was significantly lower (0.018; P = 0.032) in immature/T1 B cells, and further decreased, albeit not significantly, in MF B cells (0.016; Fig. 2B). The mean DNA AvIn values were significantly higher in T3 (0.033), and CD93+ (0.031) and CD93− (0.034) IgM−/loIgDhi B cells compared to immature/T1 (P = 0.017; Fig. 2B) or MF B cells (P < 0.001; Fig. 2B). In normal mice, tolerance acts by reducing BCR avidity to DNA during B-cell maturation and those cells excluded during this process enter the T3 and IgM-/loIgDhi anergic B-cell compartments of the spleen (Table 1 and Fig. 2B).
Tolerance checkpoints set affinity thresholds for self-reactive BCRs but removal of the self-reactive BCRs is incomplete
To characterize further the distribution of BCR affinities for DNA, we binned DNA AvIns in Fig. 2 and generated histograms for each B-cell subset (21, 23). In the pre-tolerance compartment (small pre-B cells), the distribution of DNA AvIns followed a near Gaussian distribution with a median AvIn of 0.038, suggesting that both high and low DNA avidity clones are stochastically generated and present in unbiased distributions before the first tolerance checkpoint (Fig. 3). In contrast, immature/T1 B and MF B cells showed distributions of DNA AvIns highly skewed toward lower avidity DNA-binding clones; cohorts of higher avidity DNA-binding clones [groups III and IV (Gr. III+IV)] decreased by half from 48% (in small pre-B cells) to 21% (immature/T1 B) and 26% (MF B; Fig. 3) as B cells matured beyond the first checkpoint. While these results show that the first tolerance checkpoint in the bone marrow removes higher avidity, DNA-binding B cells, comparable frequencies of the higher avidity DNA-binding clones in immature/T1 and MF B cells suggest that the second checkpoint does not act to suppress DNA-reactive B cells.
Figure 3. Distributions of the avidity of DNA-binding B cells.
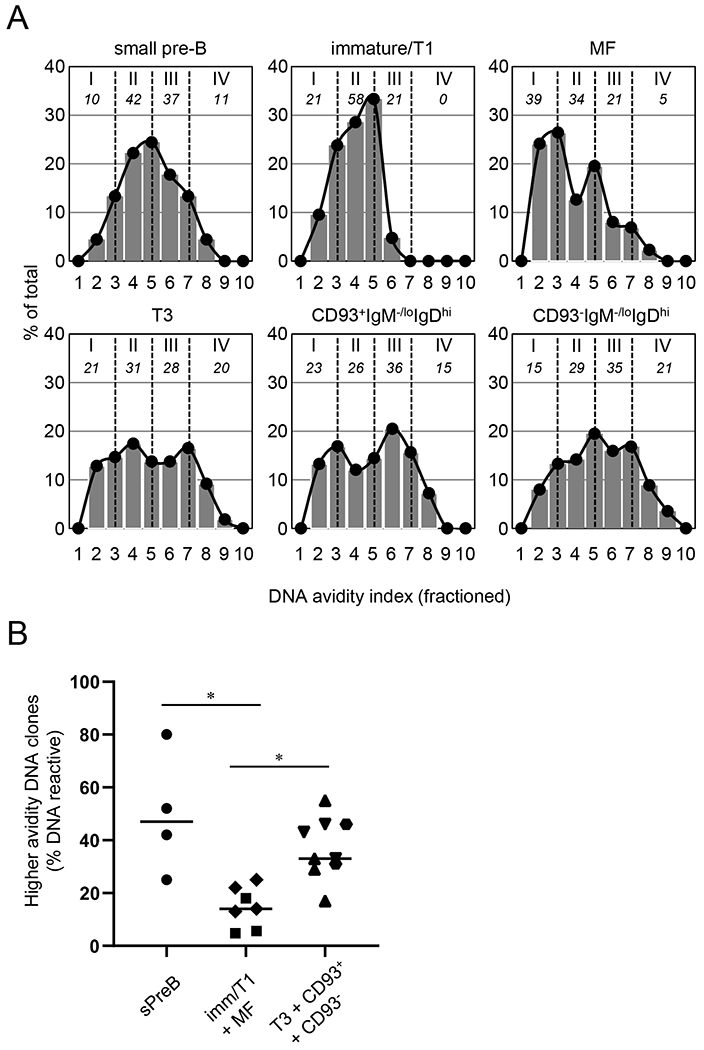
(A) DNA-binding samples in Fig. 2B were subdivided into 10 fractions (Frs.) by DNA AvIn: Fr. 1 (0-0.003), Fr. 2 (0.003-0.006), Fr. 3 (0.006-0.012), Fr. 4 (0.012-0.024), Fr. 5 (0.024-0.048), Fr. 6 (0.048-0.096), Fr. 7 (0.096-0.19), Fr. 8 (0.19-0.38), Fr. 9 (0.38-0.77), and Fr. 10 (>0.77). Histograms represent percentage of the samples in indicated fractions among total samples for each B-cell subset. Density curves were generated based on the histograms, and then subdivided into 4 groups by AvIn (I ≤ II ≤ III ≤ IV; separated by vertical dotted lines). Numbers (Italicized) underneath the group numbers represent proportion of the indicated groups (% total). (B) Frequency of higher avidity, DNA-binding samples (Frs. 6-10) among all DNA-binding samples are compared among pre-tolerance (small pre-B, ●; n = 4 mice), tolerized (immature/T1,∎, n = 3; plus MF, ◆, n = 4), and tolerizing compartments (T3, ▾, n = 3; plus CD93+IgM−/loIgDhi, ⬣, n = 2; plus CD93−IgM−/loIgDhi, ▴, n = 3). *, P < 0.05 determined by Kruskal-Wallis test.
Higher avidity DNA-binding clones (Gr. III+IV), in contrast, were enriched in the splenic T3 and anergic B-cell compartments (48%-56%; Fig. 3A). Notably, distributions of the DNA AvIns were broad or bimodal, with enrichment for the highest avidity (Gr. IV) DNA-binders (15% and 21%, respectively) in these B-cell compartments (Fig. 3). Concomitant loss of higher avidity groups from the immature/T1 and MF B cells (P = 0.012, vs. small pre-B cells; Fig. 3B) and their enhancement in T3 and IgM−/loIgDhi B-cell populations (P = 0.012, vs. immature/T1 plus MF B cells; Fig. 3B) suggest that a substantial fraction of DNA-reactive small pre-B cells are not tolerized by deletion (3–5) or receptor editing (6, 7) but enter the T3 and IgM−/loIgDhi anergic B-cell compartments.
Somatic genetics of DNA reactive B cells
To assess the IgH gene features of DNA-binding and non-binding B cells, we amplified by RT-PCR VDJ rearrangements from cDNA of small pre-B, T3 B, MF B, and CD93−IgM−/loIgDhi B cells after Nojima cultures, cloned the PCR amplicands into bacteria, and sequenced (21). We selected about 20 (n = 21–26) DNA-binding and non-binding examples, respectively, from each B-cell subset (Table S1). While small sample size limited statistical assessment, there appeared to be differences in the VH usage by B-cell subsets (Fig. S2A and Table S1). For example, VH1-64, 1-26, and 1-5 were frequently recovered from small pre-B cells (29% of total VH gene segments recovered), VH1-81 and 9-3 were frequently recovered from T3 B cells (16% of total), VH1-82, 1-72, and 3-6 were frequently recovered from MF B cells (19% of total), and VH14-3 and 1-72 were frequently recovered from CD93−IgM−/loIgDhi B cells (17% of total). Among B-cell subsets, HCDR3 length, and number of positively charged amino acids in HCDR3 did not significantly differ (Figs. S2B and S2C).
To compare directly DNA-binding and non-binding B cells, we combined all sequence data and split these samples based on DNA reactivity (Table S1). DNA-binding B cells used VH1-72, 1-64, and 1-5 frequently (21% of total; Fig. S3A). By contrast, DNA non-binding B cells preferentially used VH1-26, 9-3 and 1-69 (19% of total; Fig. S3A). While HCDR3 length did not differ significantly between DNA-binding and non-binding B cells (Fig. S3B), VDJ rearrangements from DNA-binding B cells encoded a significantly (P<0.001) elevated number of positively charged amino acids in HCDR3 than did non-binding B cells (Figs. 4A and 4B); the H-chains of DNA-binding B cells carried HCDR3 regions with ≥2 positively charged amino acids twice as frequently (60% vs. 30%) as did DNA non-binding B cells (Fig. 4B).
Figure 4. Somatic genetics of DNA-reactive B cells.
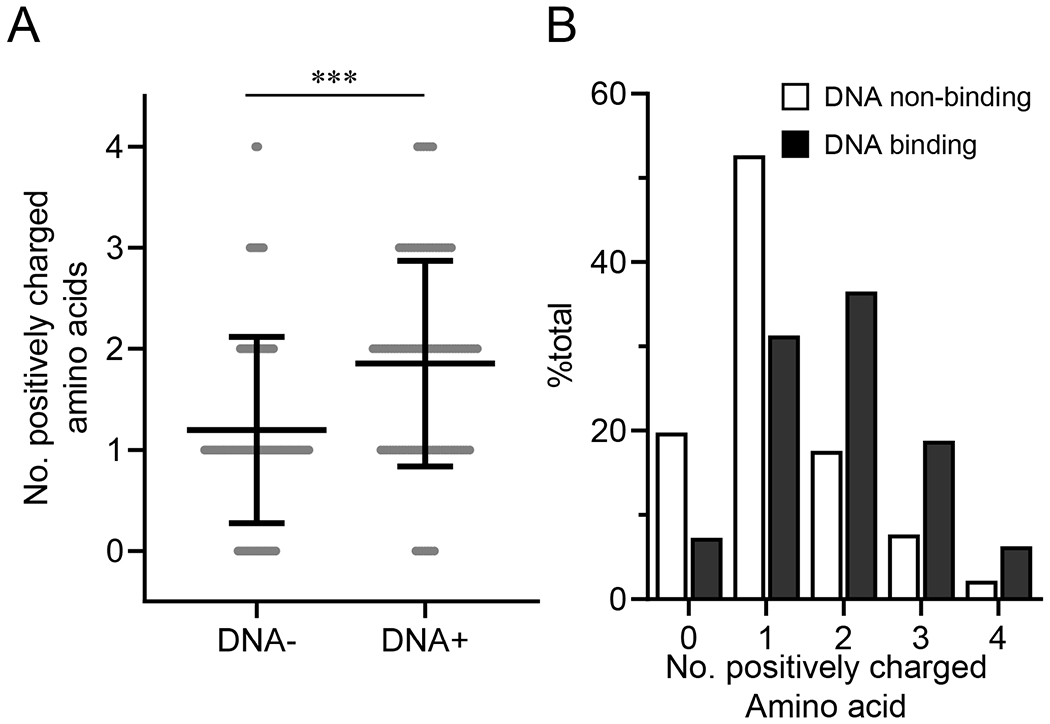
After Nojima cultures, VDJ rearrangements were amplified from cDNA samples of small pre-B (n = 45), T3 (n = 49), MF (n = 47), and CD93−IgM−/loIgD+ anergic B cells (n = 46). Data were combined and then sorted based on DNA reactivity of the corresponding culture supernatant IgGs (Table S1). (A) Number of positively charged amino acids (arginine, histidine, and lysine) within HCDR3 region was compared between DNA reactive (n = 96) and unreactive (n = 91) samples. Each symbol represents each sequence sample. Mean ± SD of samples is indicated. ***, P < 0.001 by Mann-Whitney’s U test. (B) Frequency distributions of the number of positively amino acids within HCDR3 of DNA-reactive (closed bars) and unreactive (open bars) samples described in A are shown.
Peripheral self-reactive B cells differ in persistence and in responsiveness to BCR-crosslinking
Whereas it is widely accepted that T3 and anergic B cells have half-lives of only 3 - 4 days (27, 38, 42) and that they respond poorly to stimulations through BCRs (2, 27, 39), both the lifespans and the responsiveness to BCR stimulations of anergic B cells vary widely among BCR transgenic mouse models (13), indicating that anergic B cells are heterogeneous and subsets of anergic B cells may persist in periphery and respond differentially to BCR stimulations.
To evaluate turnover in peripheral autoreactive B-cell compartments, we administered bromodeoxyuridine (BrdU) to mice in drinking water and followed the kinetics of BrdU-incorporation by T3 and IgM−/loIgDhi splenic B cells in comparison to other B-cell subsets. As expected (27), T1/T2 B cells rapidly incorporated BrdU such that frequencies of BrdU+ T1/T2 cells plateaued (>80%) within 1 week of BrdU treatment; in contrast, only about 8% of MF B cells were labeled with BrdU following 2 weeks of treatment (Figs. 5A and 5B). We also observed distinct BrdU labeling patterns within self-reactive B-cell compartments. Most (~60%) T3 and CD93+IgM−/loIgDhi B cells were labeled by BrdU incorporation after 2 weeks of treatment (Figs. 5A and 5B). Unlike, however, T3 and CD93+IgM−/loIgDhi B cells, only a minor fraction (8%) of CD93−IgM−/loIgDhi B cells became BrdU+ after 2 weeks of treatment (Figs. 5A and 5B). This slow rate of BrdU incorporation indicates that splenic CD93−IgM−/loIgDhi B cells are as persistent as MF B cells, whereas T3 B and CD93+IgM−/loIgDhi B cells are ephemeral.
Figure 5. CD93-IgM−/loIgDhi B cells are persistent in spleen.
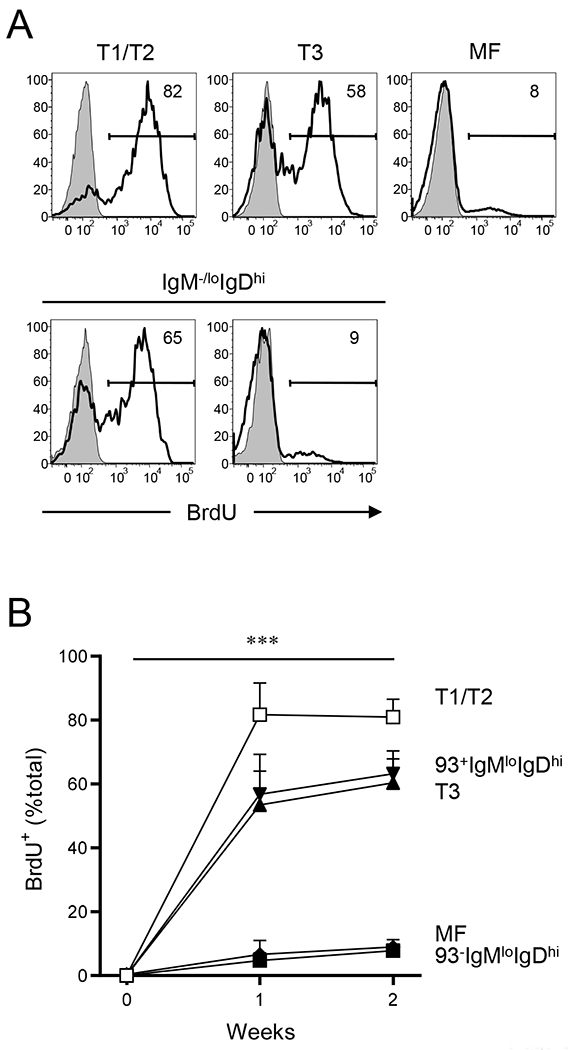
Turnover of B-cell subsets in spleen was compared by BrdU incorporation. B6 mice were fed BrdU in drinking water for up to 2 weeks, and BrdU incorporation in each B-cell subset was determined by flow cytometry. (A) Representative flow diagrams for BrdU labeling in indicated B-cell subsets are shown. Background for BrdU labeling (shaded histograms) was determined for each B-cell subset by labeling splenic B-cell compartments from untreated mice. Numbers in each flow diagram indicate percentage of BrdU+ cells. (B) Kinetics of the BrdU+ cells (% total) is shown. Data represent average ± SD (n = 5); T1/T2 (open squares), T3 (filled triangles), CD93+ (filled reverse triangles) and CD93− anergic (AN) B cells (filled diamonds), and MF B cells (filled squares). Statistical significance was determined using Turkey’s multiple comparison test in a mixed effects model with Geisser-Greenhouse correction. ***, P < 0.001. No statistical significance was observed among T1/T2, T3, and CD93+IgM−/loIgDhi B cells or between MF and CD93−IgM−/loIgDhi B cells.
We compared signaling capacity of BCRs among peripheral self-reactive B cells by measuring intracellular calcium flux after activation by BCR ligation (39, 43). Splenocytes were loaded with the calcium probe Indo-1 AM, and then labeled with B220, CD93, and CD23 mAbs in the presence or absence of anti-IgM mAb (Fig. 6A). After the baseline establishment, we added F(ab’)2 fragment of goat anti-kappa antibodies and followed the kinetics of free intracellular calcium concentrations (Fig. 6B). Consistent with previous observations and with the characteristic IgM−/loIgDhi expressions (2, 38, 39), T3 and CD93+ and CD93−IgM−/lo B cells exhibited impaired calcium flux compared with MF B cells (Fig. 6B). While impaired, CD93−IgM−/lo B cells showed better ability to flux intracellular calcium compared with their CD93+ counterpart and with T3 B cells (Fig. 6B). Thus, transient and persistent self-reactive B cells exhibit impaired, but with different degree, calcium mobilization in response to BCR stimulations.
Figure 6. Transient and persistent self-reactive B cells show different degree of impaired calcium responses in response to BCR activation.
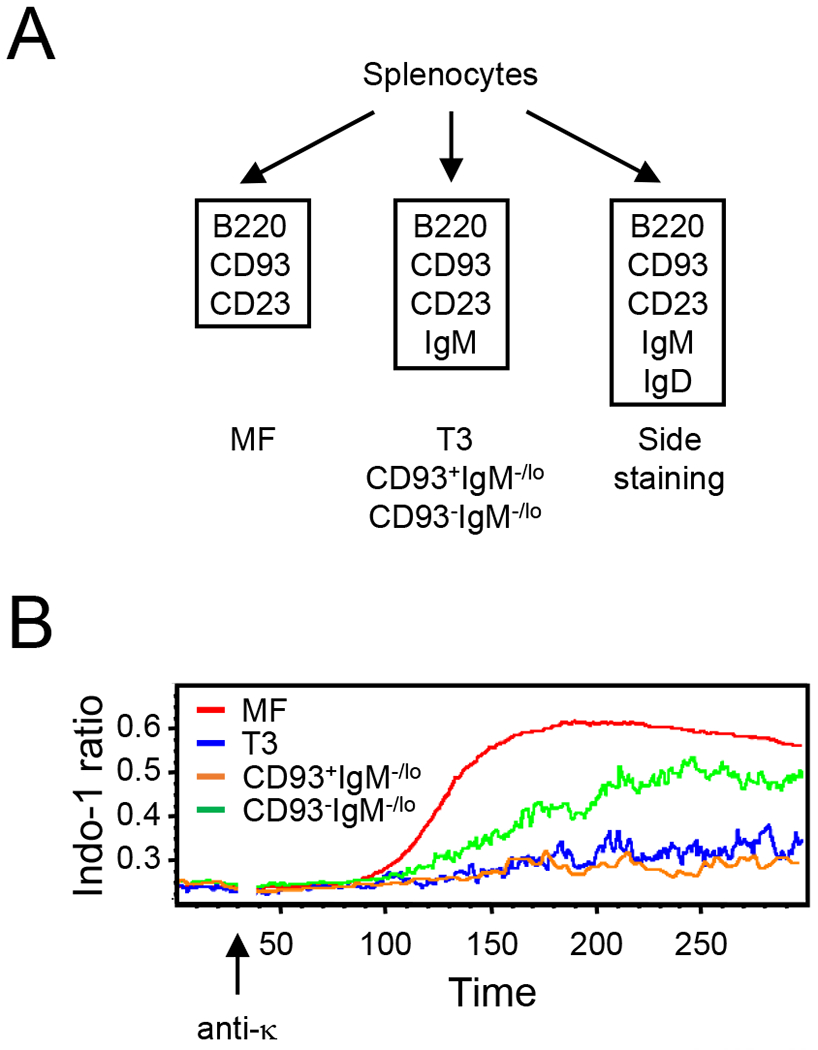
Analysis of intracellular calcium concentrations in B-cell subsets after stimulation with anti-kappa. (A) To mitigate impact of surface IgM and IgD labeling on the calcium responses, we sampled MF B cells (B220hiCD93−CD23+) that express intermediate levels of surface IgM from the staining panel without IgM (left). T3 (B220loCD93+IgM−/loCD23+), and CD93+ and CD93− anergic (B220+IgM−/loCD23+) B cells were sampled from staining panel with anti-IgM (middle). Side staining for IgD (right) revealed that MF, T3, and CD93+ and CD93− anergic B cells defined above contained IgD+ cells at 99%, 90%, 88%, and 96%, respectively (data not shown). (B) Representative kinetic graph of average MFI ratio (396 nm/496 nm) for MF (red), T3 (blue), and CD93+ (orange) and CD93− (green) anergic B cells. Anti-kappa was added to each sample at 30 s. Similar results were obtained from two independent experiments.
CD93−IgM−/loIgDhi anergic B cells expand in the absence of Bak and Bax
Apoptosis plays important roles in regulating frequency and number of anergic B cells (44–46). It is, however, unclear whether impaired apoptosis would also result in overrepresentation of anergic B cells in polyclonal mice as anergic B cells can be outcompeted for survival/maturation factors by much more abundant, MF B cells (47) and might not be able to expand in these animals (48). To determine roles for apoptosis in the regulation of anergic B cells in polyclonal mice, we followed the Korsmeyer laboratory (49) and generated mice (Cd19Cre+Bak−/−Baxfl/fl mice) in which only B cells lack expression of both Bcl-2 homologous antagonist killer (BAK) and Bcl-2-associated X protein (BAX), proapoptotic molecules that initiate apoptosis by permeabilizing mitochondrial outer membrane and by releasing cytochrome c from the mitochondria (50, 51). We then compared frequency and number of B-cell subsets in spleen among B6, (wild type control), Bak−/−Baxfl/fl (Bak-deficient, Cre-negative control), and Cd19Cre+Bak−/−Baxfl/fl mice.
Absence of BAK and BAX did not alter proportion of developing and mature B cells as frequency of CD93+ cells among all B220+ cells (4.1% ±1.3%, the mean ± SD of all mouse strains) did not significantly change between mouse strains (Fig. 7A, top panels). Within the B220+CD93+ developing B-cell compartment, IgM−/loCD23− cells doubled from 10 ±1.0% in controls (B6 and Bak−/−Baxfl/fl mice) to 23 ±6.8% in Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7A, middle panels). This increase was accompanied by a decrease of IgMhiCD23− T1 B cells from 50 ±4.0% in controls to 38 ±8.8% in Cd19Cre+Bak−/−Baxfl/fl mice but frequencies of IgMhiCD23+ T2 B cells (~30%) and IgM−/loCD23+ T3 B cells (~9%) were not significantly changed among mouse strains (Fig. 7A, middle panels). While elevated frequency and number of splenic B220loCD93+IgM−/loCD23− cells in Cd19Cre+Bak−/−Baxfl/fl mice might represent those developing B cells (pro-B, pre-B, immature B) that are mobilized from bone marrow (52–54), both anergic (T3) and non-anergic (T2) transitional B cells similarly expanded in the absence of BAK and BAX (Fig. 7A and Table 2).
Figure 7. CD93−IgM−/loIgDhi B cells overrepresented in BAK/BAX deficient mice.
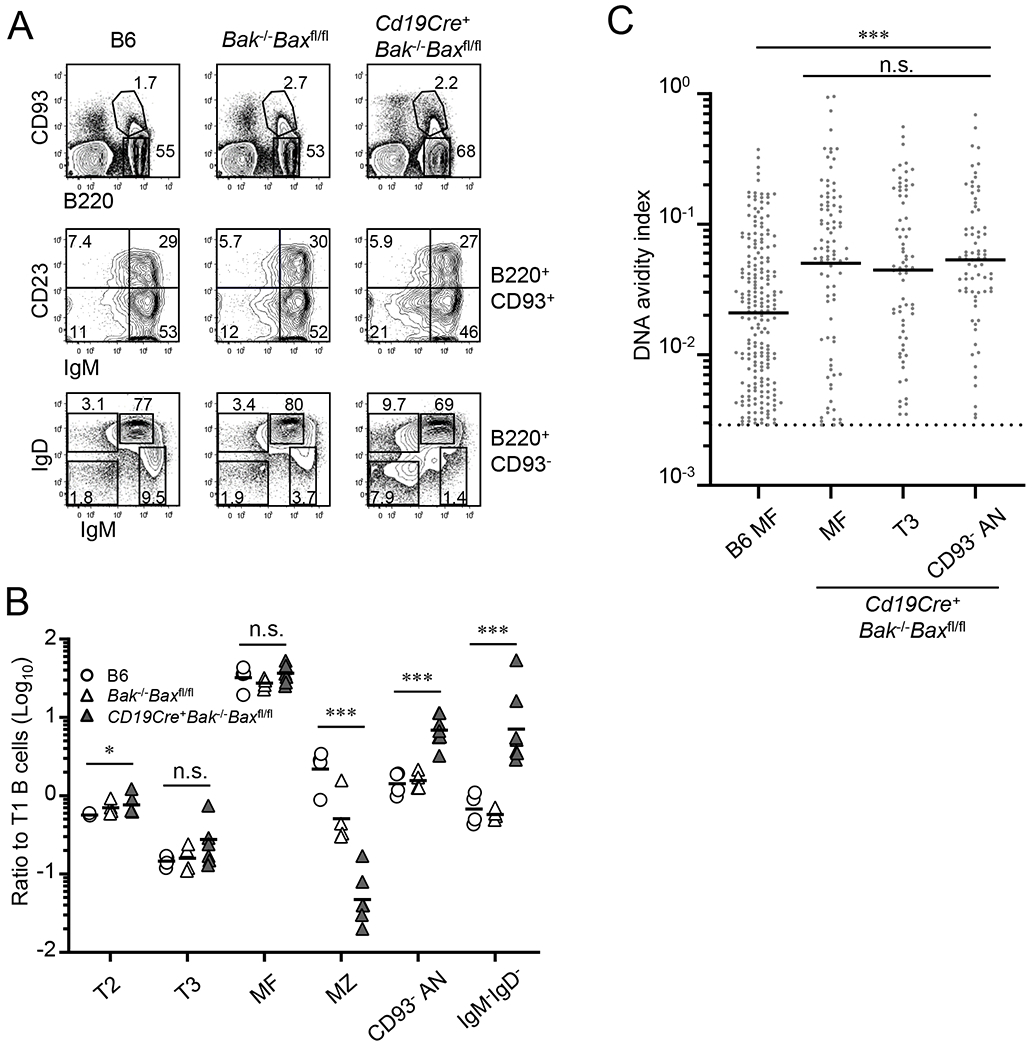
Frequency and number of B-cell subsets in spleens of B6 (n = 4), Bak−/−Baxfl/fl (n = 4), and Cd19Cre+Bak−/−BAXfl/fl mice (n = 7) were compared. (A) Representative flow diagrams of B220 and CD93 expressions on total splenocytes (top panels), IgM and CD23 expressions on B220+CD93+ developing B cells (middle panels), and IgM and IgD expressions on B220+CD93− mature B-cell compartment (bottom panels). Numbers in each panel indicate frequencies of cells in each quadrant or box. (B) Numbers of indicated B-cell subset were normalized to number of T1 B cells in individual mice. Each symbol represents individual mouse; horizontal bars indicate geometric mean. Combined data from 4 independent experiments are shown. (C) Distributions of DNA AvIns are shown for DNA-reactive, culture supernatant IgGs after Nojima cultures of B6 MF (n = 208) and MF (n = 91), T3 (n = 71), and CD93−IgM−/loIgDhi anergic (AN) B cells (n = 75) from Cd19Cre+Bak−/−BAXfl/fl mice. Dotted line indicates a cut-off for the determination of DNA reactivity (see also legend of Fig. 2B). Each symbol represents individual wells and bars indicate geometric mean of the samples. (B and C) n.s., not significant (P > 0.05); *, P < 0.05; ***, P < 0.001 by Kruskal-Wallis test.
Table 2.
Number of splenic B cells in B6, Bak−/−Baxfl/fl, and Cd19Cre+Bak−/−Baxfl/fl mice1
| Cell populations (×106) | B6 | Bak−/−Baxfl/fl | Cd19Cre+Bak−/−Baxfl/fl |
|---|---|---|---|
| Total Splenocytes | 96 (±22)2 | 120 (±23) | 280 (±97)3** |
| B220+CD93+ compartment | |||
| IgM−/loCD23− | 0.24 (±0.14) | 0.4 (±0.15) | 1.5 (±0.69)** |
| T1 | 1.3 (±0.9) | 1.8 (±0.66) | 2.6 (±1.43) |
| T2 | 0.76 (±0.51) | 1.4 (±0.81) | 1.9 (±0.93)* |
| T3 | 0.19 (±0.10) | 0.31 (±0.23) | 0.78 (±0.54)* |
| B220+CD93− compartment | |||
| MF | 38 (±9.2) | 48 (±10) | 86 (±30)** |
| MZ | 2.6 (±0.32) | 1.0 (±0.69)* | 0.12 (±0.06)** |
| IgM−/loIgDhi anergic | 1.8 (±0.94) | 2.9 (±1.3) | 19 (±12)** |
| IgM−IgD− switched | 0.86 (±0.38) | 1.0 (±0.28) | 20 (±3.2)** |
Absolute cell numbers of indicated B-cell subsets in spleens of B6 (n = 4), Bak−/−Baxfl/fl (n = 4), and Cd19Cre+Bak−/−Baxfl/fl mice (n = 7) are shown.
Mean values (±SD) are shown.
*; P < 0.05; **, P < 0.01 compared with B6 control. Statistical significance was obtained by Mann-Whitney’s U test.
In contrast to T3 B cells, frequency of IgM−/loIgDhi B cells among the B220+CD93− mature B-cell compartment increased from 3.9 ±1.2% in controls to 10 ±4.5% in Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7A, bottom panels), suggesting that these apoptotic molecules regulate differentially numbers of T3 and CD93-IgM−/loIgDhi anergic B cells in periphery. Frequency of IgMintIgDhi MF B cells moderately decreased in Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7A, bottom panels). This percentage decrease was due likely to increased representation of IgM−/loIgDhi anergic B cells and IgM−IgD− “Ig-switched” B cells but not by impaired MF B-cell maintenance as the number of MF B cells increased in Cd19Cre+Bak−/−Baxfl/fl mice compared with control animals (Table 2). By contrast, but consistent with a previous report (49), frequency (and number) of IgMhiIgD−/lo marginal zone (MZ) B cells significantly fell from 8.3 ±1.5% in B6 mice to 4.1% ±1.2% in Bak−/−Baxfl/fl mice and further declined to 1.6% ±0.2% in Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7A and Table 2).
As reported (49), number of all B-cell subsets, with the exception of MZ B cells, increased in Cd19Cre+Bak−/−Baxfl/fl mice (Table 2). To determine whether this increase can be explained simply by an increase of progenitors (T1 B cells) or BAK and BAX play roles specifically in controlling number of certain B-cell subsets, we normalized number of each B-cell subset to that of T1 B cells and compared these ratios between control mice and Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7B). This analysis revealed preferential expansion of CD93−IgM−/loIgDhi B cells in the absence of BAK and BAX in B cells. While T2, T3, and MF B cells were little (T2, <1.5-fold) or no different (T3, MF) between control mice and Cd19Cre+Bak−/−Baxfl/fl mice, CD93−IgM−/loIgDhi B cells and IgM−IgD− B cells increased 5-fold and 18-fold, respectively in Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 6B). Thus, in the absence of BAK and BAX, IgM−/loIgDhi B cells can expand in spleen comparable to or even greater than MF B cells do. Frequency and number of persistent self-reactive B cells (CD93−IgM−/loIgDhi B cells) and transient self-reactive B cells (T3 B cells) are differentially controlled by apoptosis mediated by BAK and BAX.
We next assessed DNA-reactivity of MF, T3, and CD93−IgM−/loIgDhi B cells in Cd19Cre+Bak−/−Baxfl/fl mice using Nojima culture approach. As for B6 mice (Table 1), we found no significant differences in the frequencies of DNA-reactive cells among these B-cell compartments in Cd19Cre+Bak−/−Baxfl/fl mice (21%-22%). However, in Cd19Cre+Bak−/−Baxfl/fl mice, MF B cells as well as T3 and CD93−IgM−/loIgDhi B cells exhibited significantly (P < 0.001) higher mean DNA AvIn values (0.045-0.053) compared with B6 MF B cells (0.021; Figs. 2B and 7C). Self-reactive B cells are not properly deleted in the absence of BAK and BAX.
Discussion
While tracing self-reactive B cells during B-cell development is essential to understand ontogeny of autoimmune disorders caused by aberrant activation, expansion, and differentiation of self-reactive B cells, it may also be crucial in the development of novel vaccine strategies against HIV-1 and influenza that can activate a class of BCRs reactive with both foreign antigens and self-antigens (22, 55, 56). Here we have used a single B-cell culture method, Nojima culture (21), which enables us to rapidly carry out robust screenings for BCR specificity and avidity to trace B-cell repertoires during development in normal mice. We show that the distribution of the avidity of the BCRs that bind to DNA significantly changes during B-cell development; B-cell clones that avidly bind to DNA are purged at the first tolerance checkpoint but those that escaped from the first checkpoint are retained in classic anergic compartment of IgM−/loIgDhiCD93+ T3 B cells and their CD93− counterpart in periphery.
Nojima cultures efficiently support expansion and IgG secretion of single B cells (Fig. 1). The cloning efficiency of small pre-B cells is lower (~23%) than that of other B-cell subsets (~60%). Given the dependency of B-cell proliferation on combinations of CD154-, BAFF-, and IL-21-mediated signaling in our Nojima cultures, low levels of CD40 expression (57) and little BAFF R expression (58) on small pre-B cells may explain such a low cloning efficiency. In addition, small pre-B cells need to complete IgL gene rearrangement to express surface BCRs, which generate ligand-independent ‘tonic’ signals required for B-cell survival (40, 41). As one third of the random V-J recombination generates functional light-chains, three-fold-lower cloning efficiency in small pre-B cells compared to other B-cell subsets might reflect the frequency of successful light-chain recombination. We think that combinations of these factors negatively impact the cloning efficiency of small pre-B cells.
The frequency of DNA-binding B cells did not significantly change over the course of B-cell development, from small pre-B to immature/T1 B to MF B and to anergic B-cell compartments (Table 1). Instead, we observed that B cells with high avidity for DNA were purged at the transition from small pre-B to immature/T1 B-cell stage. Clones that escaped from the first checkpoint were not purged in the MF B cells but enriched in IgM−/loIgDhi anergic B-cell compartments (Figs. 2 and 3). These results are incompatible with previous reports in humans (1, 9, 59), but this apparent ‘discrepancy’ between groups likely comes from the threshold of assays performed to determine DNA reactivity. Indeed, when we set a higher threshold for the determination of DNA reactivity and considered Gr. III+IV as DNA-reactive (Fig. 3), we found that the frequency of DNA-reactive clones decreased from 13% to 4% at the small pre-B to immature/T1 B-cell transition, and then unchanged (5%) in MF B cells (calculated from Fig. 3 and Table 1). We propose that tolerance checkpoints set an affinity threshold against self-reactive B cells and that the central tolerance checkpoint efficiently removes higher DNA avidity clones (Gr. III+IV; Fig. 3) while the peripheral checkpoint plays minor roles, if any, in purging DNA-reactive clones (Fig. 3). The removal of the high avidity, DNA-reactive BCRs by tolerance mechanisms is not deterministic but stochastic, as we observed high avidity DNA-reactive B cells in post-tolerance compartments of splenic MF B cells and anergic B cells in mice (Figs. 2 and 3). The determination of precise affinities of DNA (or any self-antigen) binding Abs will aid in estimating the ‘threshold’ of BCR avidity that is required to drive self-tolerance.
Long and positively charged HCDR3 have been shown to correlate with autoreactivity in humans (1, 22). HCDR3 from pre-tolerance and autoreactive B-cell compartments was longer and contained more positively charged amino acids compared to those from non-autoreactive compartments. In mice, large-scale V(D)J sequencing analysis has identified that the charge, but not the length, of HCDR3s is elevated in pre-B cells compared with follicular B cells (60). Consistent with these observations, we found that DNA reactivity was associated with number of positively charged amino acids in the HCDR3 (Fig. 4) although this association was not seen between autoreactive B-cell compartments (e.g., small pre-B cells) and non-autoreactive, MF B cells (Fig. S2). Given that there are only small differences in the charge of HCDR3s between pre-B and follicular B cells in mice (60), number of samples analyzed in our study (~50 for each B-cell subset) may not be sufficient to see such small differences. Unlike in humans, HCDR3 length was very similar among mouse B-cell subsets (Fig. S2 and (60)) or between DNA-binding and DNA non-binding B cells (Fig. S3). It may be that IgH gene features of mouse B cells and human B cells are distinct, and there is little association, if any, between autoreactivity and long HCDR3, thus B cells that bear BCR with long HCDR3 are not necessarily counter-selected during B-cell development in mice. Consistent with this idea and our results, the HCDR3 length of VDJ rearrangements of the VH7183 family has actually been shown to increase during mouse B-cell development (61). We conclude that, in mouse B cells, DNA reactivity is associated with the charge of HCDR3 amino acids (62), but not HCDR3 length (Figs. 4 and S3).
Identification of self-reactive, persistent, CD93−IgM−/loIgDhi splenic B cells in normal mice demonstrates the utility of rapid determination of a BCR repertoire using Nojima cultures (Figs. 2, 3, and 5). Consistent with a previous report (39), CD93+ compartments (T3 and CD93+IgM−/loIgDhi B cells) were elevated for self-reactivity (as judged by DNA AvIn), but to our surprise, self-reactivity was also elevated in CD93−IgM−/loIgDhi B cells (Figs 2 and 3), demonstrating that CD93 expression is not a hallmark of self-reactive compartments in peripheral sites. Despite similar DNA-binding avidity, self-reactive, splenic B-cell compartments differ in expressions of surface molecules, turnover rate, and in signaling capacity in response to BCR stimulations (as judged by calcium flux); T3 and CD93+IgM−/loIgDhi B cells are short-lived and show least capacity to flux intracellular calcium, while CD93−IgM−/loIgDhi B cells exhibit long-term persistence in spleen and moderately impaired calcium responses (Figs. 5 and 6). Nojima cultures also revealed that in normal mice a minor fraction (~5%) of MF B cells bore BCRs that bind DNA with high avidity (Figs. 2 and 3), demonstrating that even B cells that avidly bind this autoantigen can mature. In the absence of B-cell BAK and BAX, this high avidity, DNA-reactive population in the MF compartment is enriched.
Our observations in polyclonal mice likely represent phenotypic heterogeneity of the self-reactive, anergic B cells among different BCR-transgenic mouse models (13). In normal mice, self-reactive B cells are heterogeneous in their specificity, avidity, surface molecule expressions and lifespan, and they co-exist in peripheral lymphoid organs. Therefore, while our strategy clearly demonstrates a utility to tracing self-reactive B cells, our results and conclusions based on tracing DNA-reactive B cells may not represent the fate of all classes of self-reactive B cells. Further study is required to uncover the determinants that lead to different surface phenotypes and persistence of self-reactive B cells.
That anergic B cells expanded equally to or greater than did MF B cells in spleen of Cd19Cre+Bak−/−Baxfl/fl mice (Fig. 7) suggests that, in the absence of BAK and BAX, anergic B cells are competent to compete with MF B cells for survival factors, such as BAFF (47) perhaps by reducing their dependency on these factors (46). Interestingly, CD93−IgM−/loIgDhi B cells but not T3 B cells proportionally expanded in Cd19Cre+Bak−/−Baxfl/fl mice despite overall increase of cell numbers for both anergic B-cell compartments (Fig. 7). One possibility is that CD93−IgM−/loIgDhi B cells depend more on the control by apoptosis mediated by BAK and BAX for their maintenance than T3 B cells do. In steady state, those CD93−IgM−/loIgDhi B cells that escape from the apoptotic regulations likely acquire persistence in spleen (Fig. 5). A non-mutually exclusive possibility is that a fraction of T3 B cells, which otherwise would die, acquire a long half-life in the absence of BAK and BAX, lose CD93 expression (39, 46), and enter into the CD93−IgM−/loIgDhi anergic B-cell compartment. Comparable DNA-reactivity (both frequency and avidity) between T3 and CD93−IgM−/loIgDhi B cells (Fig. 7) supports this possibility. Although the same scenario could occur in mice sufficient for BAK and BAX, it appears infrequent because T3 B cells that incorporated BrdU do not immediately transit to CD93−IgM−/loIgDhi anergic B cells in wild type animals (Fig. 5).
It is of interest to discuss the similarities between mouse CD93−IgM−/loIgDhi anergic B cells and human IgD+IgM− (BND) cells (43). Human BND cells are anergic cells that are present in the peripheral blood of healthy subjects. Both mouse CD93−IgM−/loIgDhi anergic B cells and human BND cells are defined as IgM-/loIgDhi and negative for cell surface markers found on developing B cells (CD93 and CD10 for mouse and human B cells, respectively). In addition, these B cells represent 3–5% of human peripheral blood B cells (BND) or mouse splenic B cells (CD93−IgM−/loIgDhi anergic) and are enriched for autoreactivity. We propose that mouse CD93−IgM−/loIgDhi anergic B cells are an analogous population to human BND cells. Our findings will provide an opportunity to further examine these mature anergic B cells in vivo as well as in vitro. Indeed, we found that mouse CD93−IgM−/loIgDhi anergic B cells were persistent in the spleen by tracing BrdU incorporation.
Self-reactive B cells demonstrated in our study seem to remain silent at steady state, as B6 mice do not normally develop autoimmune symptoms or serum autoantibodies. It is, however, possible that these self-reactive B cells can be activated through cognate help by autoreactive T cells and produce mutated, high affinity autoantibodies. Indeed, these self-reactive B cells are fully competent to proliferate, differentiate into plasmablasts/-cytes, and secrete IgG in cultures (Figs. 1 and 2). Longer persistency would raise the chance of incidental exposures to ligands (self-antigens) and subsequent activation by cognate T cells.
What is the benefit of retaining so many self-reactive B cells in periphery over the risk of developing pathogenic autoantibodies? We wish to suggest that purging the B-cell repertoire of all reactivity is a poor evolutionary strategy. Deletion of all B cells that react with self-epitopes would result in substantial “holes” in the antibody repertoire that could easily be exploited by microbial pathogens (22, 63–65). In contrast, self-reactive B-cell compartments might be beneficial to immune protection by providing BCR repertoires that are normally unavailable in non-self-reactive compartments (55, 65–67). While the association between the Guillain-Barre syndrome and an infection with C. jejuni suggests a link between the development of autoimmune diseases and the activation of self-reactive B cells with foreign antigens (68), recent reports by Goodnow (69–71) and Meffre (72) suggest an alternative fate of self-reactive B cells; in response to cross-reactive, foreign antigens, those self-reactive B cells that increased an affinity to the foreign antigens but reduced an affinity to self-antigens are selected during germinal center responses (71) and enter into memory compartment (69–72). That impaired Ca2+ responses in self-reactive B cells were rank-ordered with CD93−IgM−/loIgDhi B cells the least in the reduction, followed by T3 and then the CD93+ compartments suggests a possibility that anergy is tunable. It also suggests a possibility that CD93−IgM−/loIgDhi B cells might have a capacity to respond to antigens that may mimic self. Understanding the cellular and molecular mechanisms regulating stable, autoreactive compartments and learning how to control these populations may have substantial merit not only in the treatment or prevention of systemic autoimmunity (43), but also in the development of effective vaccines that can activate forbidden B-cell repertoires to elicit otherwise disfavored Ab responses to HIV-1 and influenza (55).
Supplementary Material
Key points.
Single-cell culture approach revealed B-cell tolerance checkpoints in normal mice.
Self-reactivity is elevated in T3 and CD93+ and CD93− anergic B cells in spleen.
Both the Lifespan and signaling capacity of BCR vary among self-reactive B cells.
Acknowledgements
We are grateful for X. Liang, S. Sanders, D. Liao, X. Nie, A. Watanabe, W. Zhang, and K. Hopper for technical assistance. We thank Dr. G. Yang and J. Finney for advice.
Funding support
This work was supported in part by grants from the US NIH (AI56363) and the Bill and Melinda Gates Foundation to G. Kelsoe.
Abbreviations:
- Ab
antibody
- BCR
B-cell antigen receptor
- CSR
class-switch recombination
- dsDNA
double-stranded DNA
- Ig
immunoglobulin
- immature/T1
immature and transitional-1
- MF
mature follicular
- MZ
marginal zone
- T1
transitional-1
- T2
transitional 2
- T3
transitional 3
Footnotes
Declaration of interests
The authors declare no competing interests.
References
- 1.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC. 2003. Predominant autoantibody production by early human B cell precursors. Science 301: 1374–1377. [DOI] [PubMed] [Google Scholar]
- 2.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, and et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334: 676–682. [DOI] [PubMed] [Google Scholar]
- 3.Nemazee DA, and Burki K. 1989. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 337: 562–566. [DOI] [PubMed] [Google Scholar]
- 4.Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, and Weigert M. 1991. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature 349: 331–334. [DOI] [PubMed] [Google Scholar]
- 5.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, and Goodnow CC. 1991. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 353: 765–769. [DOI] [PubMed] [Google Scholar]
- 6.Gay D, Saunders T, Camper S, and Weigert M. 1993. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med 177: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiegs SL, Russell DM, and Nemazee D. 1993. Receptor editing in self-reactive bone marrow B cells. J Exp Med 177: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell DM, Dembic Z, Morahan G, Miller JF, Burki K, and Nemazee D. 1991. Peripheral deletion of self-reactive B cells. Nature 354: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meffre E, and Wardemann H. 2008. B-cell tolerance checkpoints in health and autoimmunity. Current opinion in immunology 20: 632–638. [DOI] [PubMed] [Google Scholar]
- 10.Nossal GJ, and Pike BL. 1980. Clonal anergy: persistence in tolerant mice of antigen-binding B lymphocytes incapable of responding to antigen or mitogen. Proc Natl Acad Sci U S A 77: 1602–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodnow CC, Crosbie J, Jorgensen H, Brink RA, and Basten A. 1989. Induction of self-tolerance in mature peripheral B lymphocytes. Nature 342: 385–391. [DOI] [PubMed] [Google Scholar]
- 12.Taylor JJ, Martinez RJ, Titcombe PJ, Barsness LO, Thomas SR, Zhang N, Katzman SD, Jenkins MK, and Mueller DL. 2012. Deletion and anergy of polyclonal B cells specific for ubiquitous membrane-bound self-antigen. J Exp Med 209: 2065–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cambier JC, Gauld SB, Merrell KT, and Vilen BJ. 2007. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol 7: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, and Meffre E. 2004. Bruton’s tyrosine kinase is essential for human B cell tolerance. J Exp Med 200: 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, and Wardemann H. 2007. Autoreactivity in human IgG+ memory B cells. Immunity 26: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herve M, Isnardi I, Ng YS, Bussel JB, Ochs HD, Cunningham-Rundles C, and Meffre E. 2007. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med 204: 1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, Zhang SY, Puel A, Jouanguy E, Picard C, Garty BZ, Camcioglu Y, Doffinger R, Kumararatne D, Davies G, Gallin JI, Haraguchi S, Day NK, Casanova JL, and Meffre E. 2008. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 29: 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, Kilic SS, Aksu G, Debre M, Rieux-Laucat F, Conley ME, Cunningham-Rundles C, Durandy A, and Meffre E. 2011. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH, Gregersen PK, and Meffre E. 2011. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest 121: 3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, Virdee M, Menard L, Cantaert T, Morbach H, Rachid R, Martinez-Pomar N, Matamoros N, Geha R, Grimbacher B, Cerutti A, Cunningham-Rundles C, and Meffre E. 2013. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest 123: 4283–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, and Kelsoe G. 2016. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity 44: 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe A, Su KY, Kuraoka M, Yang G, Reynolds AE, Schmidt AG, Harrison SC, Haynes BF, St Clair EW, and Kelsoe G. 2019. Self-tolerance curtails the B cell repertoire to microbial epitopes. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuraoka M, Snowden PB, Nojima T, Verkoczy L, Haynes BF, Kitamura D, and Kelsoe G. 2017. BCR and Endosomal TLR Signals Synergize to Increase AID Expression and Establish Central B Cell Tolerance. Cell Rep 18: 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy KR, Watanabe A, Kuraoka M, Do KT, McGee CE, Sempowski GD, Kepler TB, Schmidt AG, Kelsoe G, and Harrison SC. 2018. Memory B Cells that Cross-React with Group 1 and Group 2 Influenza A Viruses Are Abundant in Adult Human Repertoires. Immunity 48: 174–184 e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe A, McCarthy KR, Kuraoka M, Schmidt AG, Adachi Y, Onodera T, Tonouchi K, Caradonna TM, Bajic G, Song S, McGee CE, Sempowski GD, Feng F, Urick P, Kepler TB, Takahashi Y, Harrison SC, and Kelsoe G. 2019. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell 177: 1124–1135 e1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bajic G, Maron MJ, Adachi Y, Onodera T, McCarthy KR, McGee CE, Sempowski GD, Takahashi Y, Kelsoe G, Kuraoka M, and Schmidt AG. 2019. Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, and Hardy RR. 2001. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol 167: 6834–6840. [DOI] [PubMed] [Google Scholar]
- 28.Kuraoka M, Holl TM, Liao D, Womble M, Cain DW, Reynolds AE, and Kelsoe G. 2011. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Koralov SB, and Kelsoe G. 2000. Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2. J Exp Med 192: 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smeenk R, van der Lelij G, and Aarden L. 1982. Avidity of antibodies to dsDNA: comparison of IFT on Crithidia luciliae, Farr assay, and PEG assay. J Immunol 128: 73–78. [PubMed] [Google Scholar]
- 31.Smeenk RJ, Brinkman K, van den Brink HG, and Westgeest AA. 1988. Reaction patterns of monoclonal antibodies to DNA. J Immunol 140: 3786–3792. [PubMed] [Google Scholar]
- 32.Dal Porto JM, Haberman AM, Shlomchik MJ, and Kelsoe G. 1998. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol 161: 5373–5381. [PubMed] [Google Scholar]
- 33.Tiller T, Busse CE, and Wardemann H. 2009. Cloning and expression of murine Ig genes from single B cells. Journal of immunological methods 350: 183–193. [DOI] [PubMed] [Google Scholar]
- 34.Kantor AB, Merrill CE, Herzenberg LA, and Hillson JL. 1997. An unbiased analysis of V(H)-D-J(H) sequences from B-1a, B-1b, and conventional B cells. J Immunol 158: 1175–1186. [PubMed] [Google Scholar]
- 35.Ehlers M, Fukuyama H, McGaha TL, Aderem A, and Ravetch JV. 2006. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203: 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohatgi S, Ganju P, and Sehgal D. 2008. Systematic design and testing of nested (RT-)PCR primers for specific amplification of mouse rearranged/expressed immunoglobulin variable region genes from small number of B cells. Journal of immunological methods 339: 205–219. [DOI] [PubMed] [Google Scholar]
- 37.McWilliams L, Su KY, Liang X, Liao D, Floyd S, Amos J, Moody MA, Kelsoe G, and Kuraoka M. 2013. The human fetal lymphocyte lineage: identification by CD27 and LIN28B expression in B cell progenitors. J Leukoc Biol 94: 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fulcher DA, and Basten A. 1994. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med 179: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, and Cambier JC. 2006. Identification of anergic B cells within a wild-type repertoire. Immunity 25: 953–962. [DOI] [PubMed] [Google Scholar]
- 40.Lam KP, Kuhn R, and Rajewsky K. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death [see comments]. Cell 90: 1073–1083. [DOI] [PubMed] [Google Scholar]
- 41.Kraus M, Alimzhanov MB, Rajewsky N, and Rajewsky K. 2004. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117: 787–800. [DOI] [PubMed] [Google Scholar]
- 42.Cyster JG, and Goodnow CC. 1995. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity 3: 691–701. [DOI] [PubMed] [Google Scholar]
- 43.Duty JA, Szodoray P, Zheng NY, Koelsch KA, Zhang Q, Swiatkowski M, Mathias M, Garman L, Helms C, Nakken B, Smith K, Farris AD, and Wilson PC. 2009. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 206: 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang W, Weintraub BC, Dunlap B, Garside P, Pape KA, Jenkins MK, Goodnow CC, Mueller DL, and Behrens TW. 1998. Self-reactive B lymphocytes overexpressing Bcl-xL escape negative selection and are tolerized by clonal anergy and receptor editing. Immunity 9: 35–45. [DOI] [PubMed] [Google Scholar]
- 45.Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, and Strasser A. 2003. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med 198: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver PM, Vass T, Kappler J, and Marrack P. 2006. Loss of the proapoptotic protein, Bim, breaks B cell anergy. J Exp Med 203: 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, and Cyster JG. 2004. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20: 441–453. [DOI] [PubMed] [Google Scholar]
- 48.Cyster JG, Hartley SB, and Goodnow CC. 1994. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature 371: 389–395. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, and Korsmeyer SJ. 2005. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A 102: 11272–11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, and Cheng EH. 2009. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36: 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Renault TT, and Chipuk JE. 2014. Death upon a kiss: mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX-dependent apoptosis. Chem Biol 21: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monroe RJ, Seidl KJ, Gaertner F, Han S, Chen F, Sekiguchi J, Wang J, Ferrini R, Davidson L, Kelsoe G, and Alt FW. 1999. RAG2:GFP knockin mice reveal novel aspects of RAG2 expression in primary and peripheral lymphoid tissues. Immunity 11: 201–212. [DOI] [PubMed] [Google Scholar]
- 53.Gartner F, Alt FW, Monroe RJ, and Seidl KJ. 2000. Antigen-independent appearance of recombination activating gene (RAG)-positive bone marrow B cells in the spleens of immunized mice. J Exp Med 192: 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ueda Y, Yang K, Foster SJ, Kondo M, and Kelsoe G. 2004. Inflammation Controls B Lymphopoiesis by Regulating Chemokine CXCL12 Expression. J Exp Med 199: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haynes BF, Kelsoe G, Harrison SC, and Kepler TB. 2012. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol 30: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bajic G, van der Poel CE, Kuraoka M, Schmidt AG, Carroll MC, Kelsoe G, and Harrison SC. 2019. Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Sci Rep 9: 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castigli E, Young F, Carossino AM, Alt FW, and Geha RS. 1996. CD40 expression and function in murine B cell ontogeny. Int Immunol 8: 405–411. [DOI] [PubMed] [Google Scholar]
- 58.Gorelik L, Cutler AH, Thill G, Miklasz SD, Shea DE, Ambrose C, Bixler SA, Su L, Scott ML, and Kalled SL. 2004. Cutting edge: BAFF regulates CD21/35 and CD23 expression independent of its B cell survival function. J Immunol 172: 762–766. [DOI] [PubMed] [Google Scholar]
- 59.Meffre E 2011. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci 1246: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaplinsky J, Li A, Sun A, Coffre M, Koralov SB, and Arnaout R. 2014. Antibody repertoire deep sequencing reveals antigen-independent selection in maturing B cells. Proc Natl Acad Sci U S A 111: E2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ivanov II, Schelonka RL, Zhuang Y, Gartland GL, Zemlin M, and Schroeder HW Jr. 2005. Development of the expressed Ig CDR-H3 repertoire is marked by focusing of constraints in length, amino acid use, and charge that are first established in early B cell progenitors. J Immunol 174: 7773–7780. [DOI] [PubMed] [Google Scholar]
- 62.Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, and Weigert M. 1993. Residues that mediate DNA binding of autoimmune antibodies. J Immunol 150: 4966–4977. [PubMed] [Google Scholar]
- 63.Goodnow CC 1996. Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci U S A 93: 2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelsoe G, Verkoczy L, and Haynes BF. 2014. Immune System Regulation in the Induction of Broadly Neutralizing HIV-1 Antibodies. Vaccines (Basel) 2: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finney J, Watanabe A, Kelsoe G, and Kuraoka M. 2019. Minding the gap: The impact of B-cell tolerance on the microbial antibody repertoire. Immunol Rev 292: 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, Ortel TL, Liao HX, and Alam SM. 2005. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308: 1906–1908. [DOI] [PubMed] [Google Scholar]
- 67.Finney J, Yang G, Kuraoka M, Song S, Nojima T, Verkoczy L, Kitamura D, Haynes BF, and Kelsoe G. 2019. Cross-Reactivity to Kynureninase Tolerizes B Cells That Express the HIV-1 Broadly Neutralizing Antibody 2F5. J Immunol 203: 3268–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jacobs BC, Rothbarth PH, van der Meche FG, Herbrink P, Schmitz PI, de Klerk MA, and van Doorn PA. 1998. The spectrum of antecedent infections in Guillain-Barre syndrome: a case-control study. Neurology 51: 1110–1115. [DOI] [PubMed] [Google Scholar]
- 69.Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, Langley D, Roome B, Vazquez-Lombardi R, Rouet R, Hermes J, Chan TD, Brink R, Dunn-Walters DK, Christ D, and Goodnow CC. 2014. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc Natl Acad Sci U S A 111: E2567–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reed JH, Jackson J, Christ D, and Goodnow CC. 2016. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J Exp Med 213: 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, Bourne K, Reed JH, Patterson K, Porebski BT, Brink R, Christ D, and Goodnow CC. 2018. Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science 360: 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schickel JN, Glauzy S, Ng YS, Chamberlain N, Massad C, Isnardi I, Katz N, Uzel G, Holland SM, Picard C, Puel A, Casanova JL, and Meffre E. 2017. Self-reactive VH4–34-expressing IgG B cells recognize commensal bacteria. J Exp Med 214: 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.