

Keywords: CD36, LCFA absorption, lipid raft, LPS transport, GLP-2
Abstract
Lipopolysaccharides (LPS) are potent pro-inflammatory molecules that enter the systemic circulation from the intestinal lumen by uncertain mechanisms. We investigated these mechanisms and the effect of exogenous glucagon-like peptide-2 (GLP-2) on LPS transport in the rodent small intestine. Transmucosal LPS transport was measured in Ussing-chambered rat jejunal mucosa. In anesthetized rats, the appearance of fluorescein isothiocyanate (FITC)-LPS into the portal vein (PV) and the mesenteric lymph was simultaneously monitored after intraduodenal perfusion of FITC-LPS with oleic acid and taurocholate (OA/TCA). In vitro, luminally applied LPS rapidly appeared in the serosal solution only with luminal OA/TCA present, inhibited by the lipid raft inhibitor methyl-β-cyclodextrin (MβCD) and the CD36 inhibitor sulfosuccinimidyl oleate (SSO), or by serosal GLP-2. In vivo, perfusion of FITC-LPS with OA/TCA rapidly increased FITC-LPS appearance into the PV, followed by a gradual increase of FITC-LPS into the lymph. Rapid PV transport was inhibited by the addition of MβCD or by SSO, whereas transport into the lymph was inhibited by chylomicron synthesis inhibition. Intraveous injection of the stable GLP-2 analog teduglutide acutely inhibited FITC-LPS transport into the PV, yet accelerated FITC-LPS transport into the lymph via Nω-nitro-l-arginine methyl ester (l-NAME)- and PG97–269-sensitive mechanisms. In vivo confocal microscopy in mouse jejunum confirmed intracellular FITC-LPS uptake with no evidence of paracellular localization. This is the first direct demonstration in vivo that luminal LPS may cross the small intestinal barrier physiologically during fat absorption via lipid raft- and CD36-mediated mechanisms, followed by predominant transport into the PV, and that teduglutide inhibits LPS uptake into the PV in vivo.
NEW & NOTEWORTHY We report direct in vivo confirmation of transcellular lipopolysaccharides (LPS) uptake from the intestine into the portal vein (PV) involving CD36 and lipid rafts, with minor uptake via the canonical chylomicron pathway. The gut hormone glucagon-like peptide-2 (GLP-2) inhibited uptake into the PV. These data suggest that the bulk of LPS absorption is via the PV to the liver, helping clarify the mechanism of LPS transport into the PV as part of the “gut-liver” axis. These data do not support the paracellular transport of LPS, which has been implicated in the pathogenesis of the “leaky gut” syndrome.
INTRODUCTION
Lipopolysaccharides (LPS; endotoxin), a lipophilic pathogen-associated molecular pattern (PAMP) derived from Gram-negative bacteria, is a potent human pro-inflammatory toxin. Phosphorylated lipid A is biologically active; subsequent binding to Toll-like receptor 4 (TLR4) expressed on macrophages and other monocytes (67) initiates an intense inflammatory cascade eventuating in fever, tachycardia, and hypotension that provides the basis for referring to endotoxins as “pyrogens.” Injection of a 2 ng/kg dose of LPS into normal humans reproducibly induces fever, tachycardia and tachypnea (27) whereas higher doses induce the circulatory collapse and respiratory failure associated with septic shock and multiple organ failure (80). The gut lumen is the major reservoir of LPS, since it contains 103–1011 bacteria/ml. Of those bacteria, ~50% are Gram negative, translating to an LPS concentration of ~0.01–1,000,000 ng/ml (assuming 25 fg LPS/bacterial cell) when compared with a circulating plasma LPS concentration of 0.03 ng/ml, a ~1:3 lumen:plasma gradient in the proximal foregut, and a ~3 × 107:1 gradient in the colon. These large lumen-to-plasma gradients are consistent with the presence of multiple LPS detoxifying mechanisms and barriers to intestinal LPS uptake. Increased uptake of LPS into the portal vein (PV) with subsequent activation of TLR-4 expressed on hepatic resident macrophages (Kupffer cells) is believed to produce hepatic and systemic inflammation through activation of the “gut-liver axis” (10, 61).
The mechanism by which LPS enters the systemic circulation from the gut lumen remains speculative. Strong correlations between paracellular permeability to molecules such as lactulose, mannitol, and fluorescent dextran (mol wt 4,000) and endotoxemia have led some authors to conclude that LPS is transported by the paracellular pathway (12). Because of its molecular size and detoxification of LPS by intestinal alkaline phosphatase (IAP) (32, 49), some researchers believe that luminal LPS may enter by the paracellular pathway or via an epithelial defect only in the presence of mucosal injury (18, 64, 79, 89). Chronic mild elevations of circulating LPS are termed “metabolic endotoxemia” given their association with chronic low-grade inflammation and increased paracellular permeability, termed the “leaky gut” (12, 25), implicated in the pathogenesis of diseases such as Alzheimer’s disease and the metabolic syndrome (13, 91). Although LPS is a lipophilic molecule of ~10–15 kDa molecular size, it may cross the intestinal epithelium via a transcellular pathway. The first in vitro quantitative study of the kinetics of LPS passage across isolated everted gut sac of the rat duodeno-jejunum demonstrated the saturable LPS mucosa-to-serosa (m-to-s) transport, suggesting the transcellular LPS absorption (62). Electron microscopic studies in vitro (6) and in vivo (51) have suggested that luminal LPS is internalized by enterocytes and crosses the intestinal epithelium via clathrin- (6) and lipid raft-caveolae-mediated (53) endocytic pathways. Confocal microscopic studies also show LPS passage via goblet cell-associated pathways (54) and passive transcellular diffusion (18). Nevertheless, luminal LPS was not absorbed into the enterocytes of the rat small intestine and colon in vivo, although intravenously injected LPS was distributed into the enterocytes from the basolateral membranes (30). Lacking these direct in vivo data, the mechanism by which luminal LPS crosses the gut epithelia remains uncertain (36, 86).
Increased serum LPS concentrations are observed in humans and in experimental animals following consumption of a high-fat diet, linking such diets with increased LPS entry from small intestine into the circulation (14). A high-fat meal acutely increases circulating LPS levels in human healthy volunteers at 30 min (24), suggesting that dietary lipid facilitates LPS absorption. LPS is incorporated into chylomicron remnants, a process inhibited by the chylomicron synthesis inhibitor Pluronic-81 (PL81) (31), suggesting that LPS is absorbed by the same mechanism as are long-chain fatty acids (LCFAs). Absorbed LPS is primarily cleared from the circulation by the liver. Hepatic macrophages (Kupffer cells) appear to be the principal cells involved in the clearance of LPS (26); intraportal LPS is rapidly taken up by hepatocytes and excreted into the bile, peaking within 20 min (55). Furthermore, chylomicrons accelerate LPS clearance from the plasma by increasing the hepatic uptake of chylomicron-incorporated LPS followed by its increased excretion into the bile (71). Both chylomicrons and bile salts reversibly inhibit LPS pyrogenic activity (22, 71, 73). These results suggest that LPS uptake is in part chylomicron mediated, linked to multiple protective mechanisms that diminish LPS toxicity, preventing hepatic inflammation following Kupffer cell activation by unbound LPS.
Luminal triglycerides (TG) combine with bile acids, generating micelles that solubilize TG, which are then deesterified to LCFAs and mono-acyl glycerol (MAG) by pancreatic lipase. Digested luminal fat, LCFAs, and MAG are primarily absorbed by jejunal enterocytes via active and passive pathways. LCFAs are partially absorbed via cluster-of-differentiation (CD) 36-mediated and fatty acid transport protein (FATP)-mediated transport, followed by passive diffusion into the enterocytes (77). Lipid raft-mediated endocytosis also contributes to LCFA absorption, since CD36 localizes to cholesterol-rich lipid rafts (21).
Glucagon-like peptide-2 (GLP-2) is an intestinotrophic hormone released from enteroendocrine L cells (19). GLP-2 acutely enhances fat absorption via the chylomicron pathway (42), whereas chronic treatment with GLP-2 prevents the appearance of LPS in the circulation (14), attributed mostly to its proproliferative effects on the intestinal epithelium. Numerous studies have reported that GLP-2 reduces intestinal paracellular permeability (14, 37).
Given these data, we hypothesized that LPS is transported into the circulation via multiple pathways: the chylomicron pathway that transports LCFAs, transcellular endocytic and goblet cell-associated pathways suggested by electron and confocal microscopic studies, and a paracellular pathway suggested by associational data. We further hypothesized that GLP-2, given its barrier-strengthening properties and acceleration of lipid transport, decreases LPS uptake via transcellular and paracellular pathways while increasing uptake via the chylomicron pathway. Therefore, we studied the mechanisms of LPS transport across the lipid-absorbing small intestinal mucosa with and without exogenous GLP-2 and a stable GLP-2 analog.
MATERIALS AND METHODS
Animals.
Male Sprague-Dawley rats weighing 200–250 g and C57BL/6 mice weighing 20–25 g (Harlan, San Diego, CA) were fed a pellet diet and water ad libitum. All studies were performed with approval of the Veterans Affairs Institutional Animal Care and Use Committee, and the Animal Research Committee of University of California, Los Angeles. Rats were fasted overnight with free access to water before the experiments, whereas mice were fasted for 3 h before the experiments to empty the stomach. Animals were euthanized by terminal exsanguination under deep isoflurane anesthesia, followed by thoracotomy.
Chemicals.
Teduglutide (TDG, Shire Pharmaceuticals USA, Lexington, MA) was provided by the Pharmacy Service of the West Los Angeles VA. Rat GLP-2, NVP-728, NVP-AEW-541 and PD153035 were purchased from Tocris Bioscience (Ellisville, MO). The vasoactive intestinal peptide (VIP)/ pituitary adenylate cyclase-activating peptide (PACAP) receptor 1 (VPAC1) antagonist PG97-269; [Ac-His1, d-Phe2, Lys15, Arg16, Leu27]-VIP (1–7)-GRF (8–27) (33) was purchased from Phoenix Pharmaceuticals (Burlingame, CA) or was synthesized using solid-phase methodology according to the Fmoc-strategy using an automated peptide synthesizer (Model Pioneer, Thermo Fisher Scientific, Waltham, MA). The crude peptide was purified using reverse-phase high performance liquid chromatography (HPLC: Delta 600 HPLC System, Waters, MA) on a column of Develosil ODS-HG-5 (2 × 25 cm, Nomura Chemical, Seto, Japan). The purity of each peptide was confirmed by analytical HPLC and matrix-assisted laser desorption/ionization time of flight and mass spectrometry (MALDI-TOF MS) analysis. Seminaphthorhodafluor (SNARF)-5F 5-(and-6)-carboxylic acid (SNARF-5F) was purchased from Molecular Probes (Eugene, OR). Oleic acid (OA), taurocholic acid (TCA), LPS (from Escherichia coli O55:B5), fluorescein isothiocyanate (FITC)-conjugated LPS (FITC-LPS; from E. coli O55:B5), FITC-dextran 4000 (FD4), carbachol (CCh), glycerol phosphate (GP), methyl-β-cyclodextrin (MβCD), sulfosuccinimidyl oleate (SSO), PL81, Nω-nitro-l-arginine methyl ester (l-NAME), indomethacin (IND), and other chemicals were purchased from Sigma Chemical (St. Louis, MO). IND was dissolved in ethanol. OA was added in TCA-containing phosphate buffer saline (PBS, pH 7.4), followed by vigorous vortexing just before performing the experiments. All other chemicals were dissolved in distilled water to make a stock solution. Concentrations of OA and TCA used in this study were based on postprandial luminal concentrations of free fatty acids 4.05–75 mM (40) and of bile acids 3.7–12.2 mM (8) in humans.
Short-circuit current measurements in Ussing chambered preparations of rat jejunum.
Mucosa-submucosa preparations were prepared from the midjejunum (~20 cm from the pyloric ring) by muscle stripping using fine forceps under a stereomicroscope. Two or four preparations were prepared from one animal. The tissue was then mounted between two hemisliders with an aperture = 0.3 cm2 (Physiologic Instruments, San Diego, CA). Chambers were bathed with serosal and luminal bathing solutions in a volume of 4 ml each, maintained at 37°C using a water recirculating heating system. The serosal Krebs-Ringer solution contained (in mM) 117 NaCl, 4.7 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.5 CaCl2, 25 NaHCO3, and 11 glucose and bubbled with 95% O2-5% CO2 to maintain pH at 7.4. The luminal bathing solution contained (in mM) 136 NaCl, 2.6 KCl, 1.8 CaCl2, 10 HEPES (pH 7.0), 11 mannitol, and bubbled with 100% O2. The tissues were short circuited by a voltage clamp (VCC MC6, Physiologic Instruments) at zero potential difference with automatic compensation for solution resistance. Short-circuit current (Isc) was continuously measured with tissue conductance (Gt) and transepithelial electrical resistance (TEER, Ω·cm2) determined every 20 s to assess the tissue integrity during the LPS and lipid exposure. The current was recorded by the DataQ system (Physiologic Instruments). IND (10 μM) was added to the serosal bath to eliminate the effects of endogenous prostaglandin production. The tissues were stabilized for ~30 min before the addition of test chemicals.
LPS movement from mucosal to serosal solution in rat jejunum.
LPS movement m-to-s was assessed using a limulus amebocyte lysate (LAL) test kit (Pierce Chromogenic Endotoxin Quant Kit, Pierce Biotechnology, Rockford, lL), which eliminates nonspecific detection of (1,3)-β-d-glucan, then reduces false-positive LPS measurements. After tissue stabilization, time was set as time (t)= 0 min. Fifty microliters of the serosal solution were collected at 0 min as a blank sample, followed by collection at t = 15, 30, and 45 min. At t = 0 min, LPS (10 µg/ml) was added into the mucosal solution. Vehicle (PBS alone), TCA alone (0.1 mM) in PBS, or OA (3, 10, 30 mM) with TCA (0.1 mM) in PBS was added into the mucosal solution at t = 15 min. The tissues were pretreated with drugs 10 or 15 min before the addition of LPS (i.e., at t = −10 or −15 min). The collected serosal solutions were kept at −80°C until use. LPS content in the serosal solutions was measured with the LAL test according to the manufacturer’s protocol. The value of background at t = 0 min was subtracted and the serosal LPS appearance was expressed as Δ[LPS] (EU/ml). In some experiments, FITC-LPS (10 µg/ml) was used instead of LPS. Serosal FITC-LPS content was assessed by FITC fluorescence intensity measurement using a multimode microplate reader (Synergy-2, BioTek Instruments, Winooski, VT). To detect macromolecule m-to-s movement across the mucosa, FD4 (0.1 mM) was applied into the mucosal bath and serosal FD4 content was assessed by FITC fluorescence intensity measurement.
TLR-4 reporter cell assay.
Since the LAL test may overestimate LPS levels due to nonspecific detection of monophosphoryl lipid A (MPLA) (78) and FITC-LPS method measures LPS fluorescence without regard to the functional activity of LPS, we sought to determine whether the samples contained intact, functional LPS that is a preferential TLR-4 ligand. To confirm that intact LPS was measured in the samples used for measurements in the Ussing chamber study, we further used a TLR4 reporter cell assay that consists of HEK293 cells cotransfected with murine TLR4, myeloid differentiation 2 and CD14 coreceptor genes and an inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene with nuclear factor-κ B (NF-kB) and activator protein 1 (AP-1) binding motifs (HEK-Blue mTLR-4, InvivoGen, San Diego, CA). Cells were harvested in a 96-well plate with standards and samples for 24 h. Induced SEAP activity in the medium using the HEK-Blue Detection (InvivoGen) system was used to measure LPS by measuring the absorbance at 655 nm using a Synergy-2 microplate reader 24 h after the addition of standards and samples, according to the manufacturer’s protocol. Ultrapure LPS from E. coli 055:B5 (LPS-B5 Ultrapure, InvivoGen) was used as standard at a range of 0.01–100 ng/ml.
Ussing chamber study: drug treatment.
The following drugs were pretreated by addition to the mucosal or serosal bath 10 or 15 min before LPS addition to the mucosal bath: the lipid raft inhibitor MβCD (1 mM, m), the CD36 inhibitor SSO (0.1 mM, m), the muscarinic agonist CCh (10 µM, s), a competitive substrate for IAP (IAP inhibitor) GP (10 mM, m) (57), GLP-2 (100 nM, s), the dipeptidyl peptidase-4 (DPP4) inhibitor NVP-728 (10 µM, s) (45), the stable GLP-2 analog TDG (100 nM) (20), the insulin-like growth factor-1 (IGF-1) receptor tyrosine kinase inhibitor NVP-AEW-541 (10 µM, s) (29), the epidermal growth factor (EGF) tyrosine kinase inhibitor PD153035 (1 µM, s) (9), the nitric oxide (NO) synthase (NOS) inhibitor l-NAME (0.1 mM, s), and the selective VPAC1 antagonist PG97–269 (1 µM, s) (33).
In vivo small intestinal perfusion in rats.
Intraduodenal perfusions combined with PV and mesenteric lymph cannulation were carried out according to a modification of previously reported methods (1, 58). Under isoflurane anesthesia (2%), the animal was placed supine on a recirculating heating block system (Summit Medical Systems, Bend, OR) to maintain body temperature at 36–37°C. Prewarmed saline was infused via the right femoral vein at 1.08 ml/h using a Harvard infusion pump (Harvard Apparatus, Holliston, MA). The abdomen was incised, and the PV was cannulated with a polyethylene (PE)-50 tube attached with a 23-gauge needle and fixed by methylacrylate adhesive at the insertion site. The tube was filled with heparinized saline enabling repeated blood sampling. PV samples (0.2 ml each) were collected every 15 min, followed by 0.2-ml flushes with heparinized saline. Next, the main trunk of the mesenteric lymph duct located between the celiac and superior mesenteric arteries was cannulated with another PE-50 tube attached with a 23-gauge needle and filled with saline and fixed by methylacrylate adhesive at the insertion site. The lymph solution was continuously collected into a 1.5-ml tube every 15 min. A polyethylene tube (diameter 5 mm) filled with PBS was inserted through the forestomach and tied at 0.5 cm caudal from the pyloric ring. After bolus perfusion of 2 ml PBS into the duodenum from the tube, followed by stabilization for ~30 min, the time was set as t = 0 min. FITC-LPS (50 µg/ml) in 2-ml PBS with or without OA (30 mM) and TCA (10 mM) was bolus perfused at t = 0 min. To maintain lymph flow, 2-ml PBS was perfused every 30 min. PV and lymph samples were collected every 15 min for 90 min. Blood samples were collected in a 0.5-ml tube containing 1 µl of EDTA (0.5 M) and immediately centrifuged at 5,000 g for 5 min, after which the plasma was stored at −80°C until use. The lymph solution was weighed to measure its volume and centrifuged, with the supernatant stored at −80°C until use. At 90 min, after collection of PV and lymph samples, arterial blood was taken from abdominal aorta for a reference sample, followed by euthanasia by thoracotomy.
The appearance of FITC-LPS into the PV plasma and lymph was assessed by fluorescence measurement using the Synergy-2 microplate reader. Fluorescence intensity in the t = 0 sample as background was subtracted from the fluorescence values measured in other time point samples. For lymph samples, the FITC-LPS concentration was multiplied by the lymph output to calculate total FITC-LPS appearance. FITC-LPS appearance was expressed as PV FITC-LPS content (ng/ml) or FITC-LPS transport into lymph (ng/15 min). Furthermore, total FITC-LPS transport into the PV and lymph was assessed by calculating the area under the curve (AUC) during 0–90 min of FITC-LPS appearance (AUC0–90) using the trapezoidal rule and expressed as AUC0–90 of PV FITC-LPS (µg·min/ml) and AUC0–90 of lymph FITC-LPS (µg).
Since repeated blood sampling is difficult in mice, only one PV blood sample was taken. Under isoflurane anesthesia, prewarmed saline was infused subcutaneously at 0.1 ml/h, and the abdomen was incised. A polyethylene tube (diameter 2 mm) filled with PBS was inserted through the forestomach and secured with a silk suture 0.2 cm caudal from the pyloric ring. After bolus perfusion of 0.2 ml PBS into the duodenum from the tube, followed by stabilization for ~30 min, the time was set as t = 0. FITC-LPS (50 µg/ml) in PBS with or without OA (30 mM) and TCA (10 mM) was perfused 0.2 ml at t = 0 min. At t = 15 min, PV blood and abdominal arterial blood were collected, followed by euthanasia by thoracotomy. Autofluorescence of PV plasma from the pooled nontreated fasted control mice (average 613.5 at sensitivity 80 set in Synergy-2, n = 6) was subtracted from the PV plasma samples to eliminate the background autofluorescence in plasma.
Intestinal perfusion study: drug treatment.
The lipid raft inhibitor MβCD (1 mM), the CD36 inhibitor SSO (1 mM), and PL81 (3%) were added to the initial perfusion of 2 ml PBS at t = −30 min, followed by their addition to the luminal FITC-LPS/OA/TCA solution at t = 0 min.
Effect of TDG on intestinal LPS transport in rats.
The stable, DPP4-resistant analog of GLP-2, TDG (20) was used to examine the acute effect of GLP-2 on LPS transport into PV and mesenteric lymph in vivo. TDG (50 µg/kg) was bolus injected intravenously 15 min before the luminal FITC-LPS/OA/TCA perfusion (at t = −15 min). l-NAME was added to the first perfusion of 2 ml PBS (at t = −30 min). PG97-269 (0.3 mg/kg) was bolus injected intravenously 20 min before the luminal FITC-LPS/OA/TCA perfusion (at t = −20 min).
In vivo confocal imaging of LPS transport in mouse jejunum.
In vivo confocal multiphoton microscopic imaging was performed to clarify whether LPS is transported via the transcellular or the paracellular pathway using a modification of a method originally developed in our laboratory (3, 39). Because of the limited size of the microscopic stage, mice were used for this study. Under isoflurane anesthesia (2%), the abdomen was incised and the midjejunum (~2 cm) was exposed. After the lumen was filled with 0.2 ml prewarmed PBS, the anterior wall of the jejunum was incised using an electric cautery. After the lumen was gently rinsed with prewarmed PBS, the animal was placed prone with the jejunal mucosa face down in to the incubation chamber (Warner Instrument, Hamden, CT) filled with prewarmed PBS and set on the microscopic stage of a Leica TCS-SP2 AOBS inverted confocal and multiphoton microscope (Leica, Mannheim, Germany), equipped with an argon laser and three helium-neon lasers and a Spectra-Physics MaiTai picosecond pulsed infrared laser (Mountain View, CA) set at 780 nm for two photon and infrared excitation. After stabilization for ~15 min, the mucosa was exposed to FITC-LPS (50 µg/ml) in PBS for 15 min, followed by FITC-LPS plus OA (30 mM)/TCA (10 mM) in PBS for 15 min. The mucosal images were taken by excitation at 488 nm or by two-photon excitation at 780 nm with emission at 500–550 nm. For a negative control, the cell-impermeant red fluorescence dye SNARF-5F (10 µM; Molecular Probes, Eugene, OR) was also added in the mucosal solution, and the images were taken by two-photon excitation at 780 nm and emission at 640 nm.
Statistics.
Values are expressed as means ± SE. The number of animals in each experimental group was n = 4–6. Statistical analysis was performed using GraphPad Prism 6 (La Jolla, CA) using one-way ANOVA or two-way ANOVA followed by Tukey’s multiple comparisons, or using Mann-Whitney test, if applicable. Differences were considered significant when P values were < 0.05.
RESULTS
Luminal LPS content in the gastrointestinal segments in rats.
Because most prior studies used luminally applied LPS concentration at 20–500 µg/ml in the jejunum or ileum in vitro and in vivo or in Caco-2 cell monolayers (18, 53, 62, 90) with no actual measurement of luminal LPS content in intact animals, we, therefore, measured the intraluminal content of LPS in each gastrointestinal (GI) segment of freely fed rats using the LAL test, since our experiments of LCFA perfusion in fasted animals mimic postprandial conditions. As depicted in supplemental Fig. S1 (https://doi.org/10.6084/m9.figshare.12203006.v1), LPS content in the luminal wash of each GI segment was the lowest in the duodenum [632.0 ± 88.4 EU/ml (0.06 µg/ml)] and detected in the range of 49,049.3 ± 11,225.1 (in the stomach) to 137,899.9 ± 37,400.9 EU/ml (in the cecum), equivalent to 4.9–13.8 µg/ml (n = 4). Jejunal LPS content was 8.3 µg/ml. Compared with the luminal wash, LPS content in the mucosal wash was much lower, ranging from 4.4 ± 0.9 (in the stomach) to 2,832.8 ± 381.2 EU/ml (in the proximal colon), equivalent to 0.4–283.3 ng/ml, with a longitudinal gradient (low in the stomach, duodenum, and jejunum and similarly high in the cecum, and proximal and distal colon), consistent with the predicted LPS content according to the longitudinal gradient of luminal enterobacterial density (23). Since rodents are coprophagic, the luminal wash under fed conditions might contain relatively high LPS content, except in the duodenum, where less luminal content remained in the lumen even in the fed condition, possibly due to the shortest intestinal transit time in the duodenum among intestinal segments in humans and rats (5, 11, 65, 69). This result suggests that luminal application of LPS at 10 µg/ml in the jejunum in vitro and at 50 µg/ml for intraduodenal perfusion in vivo is optimal to reflect physiological luminal LPS levels under postprandial conditions, at least in coprophagic rodents.
Transmucosal jejunal LPS transport in vitro.
First, we tested m-to-s LPS transport with or without added luminal OA in a muscle-stripped mucosa-submucosa preparation of rat jejunum using the LAL test. The jejunal mucosa was luminally exposed to LPS alone (10 µg/ml) from t = 0 min to t = 15 min, followed by the addition of vehicle (PBS), TCA (0.1 mM), or OA + TCA from t = 15 min to t = 45 min. LPS alone or LPS + TCA only in the mucosal bath had no effect on LPS concentrations in the serosal bath, whereas further addition of OA (3, 10, or 30 mM) in the mucosal bath dose-dependently increased LPS concentrations in the serosal bath at t = 30 min that were sustained at t = 45 min (Fig. 1A), suggesting that luminal LCFA exposure increases m-to-s transport of LPS. Mucosal application of LPS (10 µg/ml) with OA (30 mM) plus TCA (0.1 mM) was thus used in the following experiments. OA + TCA without LPS in the lumen had no effect on serosal LPS concentrations (Fig. 1B). LPS content in the mucosal bath at t = 0 min measured by LAL test was 14.0 ± 2.1 EU/ml (n = 6), equivalent to 2.5 ng/ml according to the conversion of 1 EU/ml = 0.18 ng/ml LPS determined by the LAL test we used. Since LPS was added at 10 µg/ml in the mucosal bath, these results suggest that endogenous LPS present in the lumen of the jejunal mucosa did not contribute to serosal LPS concentrations.
Fig. 1.

Lipopolysaccharides (LPS) transport in rat jejunal mucosa in the Ussing chamber. Muscle-stripped mucosa-submucosa preparation of rat jejunal mucosa was exposed to mucosal (m) LPS (10 µg/ml) at time (t) = 0 min, followed by the mucosal addition of vehicle (phosphate-buffered saline pH 7.4) or oleic acid (OA) and taurocholic acid (TCA) at t = 15 min. Serosal (s) LPS concentrations ([LPS]) were measured using the limulus amebocyte lysate test with colorimetric detection. Background [LPS] at t = 0 was subtracted from the value at each time point, which is expressed as Δ[LPS] (EU/ml) mucosal-to-serosal (m-to-s) (means ± SE, n = 6 rats). All data were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test. A: mucosa was exposed to mucosal LPS alone for 15 min, followed by mucosal addition of vehicle (veh), TCA (0.1 mM) alone, or OA (3–30 mM) with TCA. OA in the mucosal bath dose-dependently increased serosal [LPS] at t = 30 and 45 min, whereas LPS alone or LPS + TCA had no effect on serosal [LPS]. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + TCA group. B: vehicle was added into the mucosal bath without LPS, followed by addition of vehicle or OA/TCA. C: sulfosuccinimidyl oleate (SSO, 0.1 mM) was added into the mucosal bath 10 min before LPS application, followed by addition of vehicle or OA (30 mM)/TCA (0.1 mM). SSO inhibited OA/TCA-induced [LPS] increase, whereas SSO with LPS alone had no effect on serosal [LPS]. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. D: methyl-β-cyclodextrin (MβCD, 1 mM) was added into the mucosal bath 10 min before LPS application. MβCD abolished OA/TCA-induced [LPS] increase, whereas MβCD with LPS alone had no effect on serosal [LPS]. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. E: carbachol (CCh, 10 µM) was added into the serosal (s) bath 10 min before LPS application. CCh increased serosal [LPS], regardless the presence of mucosal OA/TCA. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. F: glycerol phosphate (GP, 10 mM) was added into the mucosal bath 10 min before LPS application. GP enhanced OA/TCA-induced [LPS] increase at t = 45 min, whereas GP with LPS alone had no effect on serosal [LPS]. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. G: effects of SSO, MβCD, CCh and GP on serosal [LPS] at t = 15 min (LPS alone exposure) and at t = 45 min (LPS+OA+TCA exposure). *P < 0.05 vs. vehicle group, †P < 0.05 vs. the corresponding LPS alone group.
Since LCFA absorption is partially mediated by the LCFA translocator CD36 (77) and by lipid raft-related endocytosis (21), we next examined the effects of the CD36 inhibitor SSO and the lipid raft inhibitor MβCD on LPS transport during OA/TCA exposure. Pretreatment with SSO (0.1 mM) had no effect on LPS transport at t = 15 min, whereas OA/TCA-augmented LPS transport (t = 30–45 min) was abolished by the addition of luminal SSO (Fig. 1C), suggesting that luminal LPS transport is mediated at least in part by CD36. MβCD (1 mM) also inhibited OA/TCA-augmented LPS transport (Fig. 1D), suggesting that lipid rafts also mediate LPS transport. These results also suggest that LPS absorption occurs during OA/TCA exposure via the transcellular pathway rather than by the paracellular pathway. In contrast, pretreatment with serosal CCh (10 µM), which empties goblet cells by stimulating mucus secretion, increased LPS transport at t = 15 min and further increased LPS transport during the OA/TCA exposure period (Fig. 1E), suggesting that emptied goblet cells may transport LPS regardless of the presence of OA/TCA in the lumen. This observation is consistent with LPS uptake through emptied goblet cells (41). GP (10 mM) in the mucosal bath further increased LPS transport at t = 45 min (Fig. 1F). Since IAP detoxifies LPS by dephosphorylating LPS (84), GP, as an inhibitor of IAP, would be predicted to increase LPS transport. This result thus suggests that IAP inhibition increases the bioavailability of LPS in the lumen. The LAL test as a functional assay detects the activity of LPS that has intact lipid A but also detects MPLA as having ~50% of the activity of intact lipid A (78). LPS transport in the Ussing chamber (LPS alone at t = 15 min and LPS+OA+TCA at t = 45 min) is summarized in Fig. 1G. Note that the value represents accumulated serosal [LPS] since the serosal bath volume was constant. SSO, MβCD, and GP had no effect on LPS transport in the absence of OA/TCA, whereas SSO and MβCD inhibited, but GP enhanced LPS transport in the presence of OA/TCA. In contrast, CCh increased LPS movement independently of luminal OA/TCA.
Since the LAL test may be confounded by interfering substances in the medium, we also examined OA/TCA-induced LPS transport using FITC-LPS with its serosal appearance measured by FITC fluorescence intensity. FITC-LPS alone in the lumen had no effect on serosal LPS content for 45 min, whereas luminal application of OA/TCA increased serosal LPS content measured by FITC fluorescence intensity, the effect abolished by the addition of SSO or MβCD (Fig. 2A), mirroring the results measured by LAL test in Fig. 1, C and D. To further validate the detection of LPS m-to-s transport, the same samples of Fig. 2A were analyzed by the TLR4 reporter cell assay, which showed that the detectable range of LPS-B5 Ultrapure was 0.1–30 ng/ml. With the use of this method, LPS m-to-s transport was increased during OA/TCA exposure and abolished by SSO or MβCD, whereas no change was observed in the FITC-LPS alone group (Fig. 2B), compatible with the FITC measurements (Fig. 2A). The detected LPS levels were ~0.3 to ~10 ng/ml and ΔLPS levels were up to ~1.5 ng/ml, much less than the FITC measurements (Fig. 2A) and relatively less than the LAL measurements (Fig. 1). By comparison, ΔLPS levels were up to ~30 EU/ml, equivalent to ~5.4 ng/ml. These results suggest that intact LPS is present in the serosal solution, although transported LPS is likely degraded. Nevertheless, all methods similarly detected ΔLPS m-to-s increase during OA/TCA exposure, which was consistently inhibited by membrane transport inhibitors, confirming that LPS transport measured by the LAL test is comparable with the fluorescence measurements of FITC-LPS or the TLR4 bioactivity assay in our in vitro experiments.
Fig. 2.

Comparison of lipopolysaccharide (LPS) measurements using fluorescein isothiocyanate (FITC)-LPS and Toll-like receptor 4 (TLR4) reporter cell assay. Rat jejunal mucosae were exposed to mucosal FITC-LPS (10 µg/ml) with or without mucosal (m) sulfosuccinimidyl oleate (SSO, 0.1 mM) or methyl-β-cyclodextrin (MβCD, 1 mM) 10 min before FITC-LPS application, followed by mucosal addition of vehicle or oleic acid (OA, 30 mM)/taurocholic acid (TCA, 0.1 mM) at time (t) = 15 min. Serosal [LPS] at each time point was detected by FITC fluorescence intensity, followed by subtraction of background [LPS] at t = 0 (A) or mTLR4-SEAP reporter cell assay (see materials and methods for detail) (B), expressed as Δ[FITC-LPS] (ng/ml) mucosal-to-serosal (m-to-s), or Δ[LPS] (ng/ml) m-to-s, respectively (means ± SE, n = 6 rats). All data were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test. *P < 0.05 vs. FITC-LPS alone group, †P < 0.05 vs. FITC-LPS + OA/TCA group.
Epithelial permeability during OA/TCA exposure in jejunal mucosa in vitro.
To examine whether paracellular permeability changes were contributed to the enhanced LPS transport observed during OA/TCA exposure, we measured transepithelial electrical resistance (TEER) in parallel with FITC-LPS or FITC-dextran 4000 (FD4) m-to-s movement in the jejunal mucosa.
Luminal addition of OA/TCA increased serosal [FITC-LPS] (Fig. 3A), whereas TEER was unchanged throughout the experiments (Fig. 3B). Luminal application of FD4 and LPS with or without further addition of OA/TCA increased serosal [FD4] (Fig. 3C) without any change in TEER (Fig. 3D), suggesting that FD4 transport in the jejunum occurs tonically regardless of the luminal presence of OA/TCA. Since OA/TCA exposure may transiently and reversibly injure villous tip cells, accompanied by increased small molecule leakage in rat jejunal perfusion study (50), we also assessed epithelial integrity histologically using brush-border F-actin staining with fluorescence-conjugated phalloidin. Fluorescent staining showed that the epithelial brush-border continuity of the villous cells was similarly conserved in FITC-LPS alone (supplemental Fig. S2, A and B) and FITC-LPS + OA/TCA (supplemental Fig. S2, C and D; https://doi.org/10.6084/m9.figshare.12203006.v1) groups, although the villous height in both groups was likely shortened with irregular villous shape. These results suggest that luminal OA/TCA exposure increases LPS transport without associated paracellular permeability changes for electrolytes and macromolecules.
Fig. 3.

Epithelial permeability during lipid exposure in rat jejunal mucosa in vitro. Rat jejunal mucosa were exposed to mucosal fluorescein isothiocyanate (FITC)-lipopolysaccharide (LPS) (10 µg/ml) or FITC-dextran 4000 (FD4, 0.1 mM) with lipopolysaccharide (LPS, 10 µg/ml) at time (t) = 0 min, followed by the mucosal (m) addition of vehicle (phosphate-buffered saline pH 7.4; PBS), or oleic acid (OA, 30 mM) and taurocholate (TCA, 0.1 mM) at t = 15 min. FITC-LPS transport [mucosal-to-serosal (m-to-s)] (A) or FD4 transport (m-to-s) (C) is expressed as Δ[FITC-LPS] (ng/ml) m-to-s, or Δ[FD4] (nM) m-to-s, respectively (means ± SE, n = 4 rats). Transepithelial electrical resistance (TEER) (Ω·cm2), recorded throughout the experiments is plotted every 15 min (B and D) for the corresponding experiments (A and C). All data were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test. A and B: luminal PBS alone or FITC-LPS alone had no effect on serosal [FITC-LPS], whereas addition of OA/TCA in the mucosal bath increased serosal [FITC-LPS] (A) with no change in TEER (B). *P < 0.05 vs. PBS alone group, †P < 0.05 vs. FITC-LPS alone group. C and D: luminal LPS alone had no effect on serosal fluorescence change. Luminal addition of FD4, with or without further addition of OA/TCA in the mucosal bath, all increased serosal [FD4] (C) with any change in TEER (D). *P < 0.05 vs. LPS alone group.
Effect of GLP-2 on jejunal LPS transport in vitro.
Next, we tested our hypothesis that GLP-2 acutely affects LPS m-to-s transport in the small intestine. Serosal application of GLP-2 (100 nM) alone at t = −15 min had no effect on OA/TCA-augmented LPS m-to-s transport, whereas further addition of the dipeptidyl peptidase-4 (DPP4) inhibitor NVP-728 (10 µM) to GLP-2 inhibited LPS transport during the OA/TCA exposure period (Fig. 4A), suggesting that prolonging the half-life of GLP-2 is necessary to acutely inhibit LPS transport during OA/TCA exposure in the Ussing chamber. This hypothesis was confirmed by using the stable GLP-2 analog teduglutide (TDG, 100 nM), which inhibited LPS transport during OA/TCA exposure in the absence of DPP4 inhibition (Fig. 4B). To examine whether endogenous GLP-2 is involved in OA/TCA-augmented LPS transport, we tested the effects of a GLP-2 receptor partial agonist/antagonist GLP-2(3–33) (81) and NVP-728 alone on LPS transport. Serosal application of GLP-2(3–33) (300 nM) or NVP-728 (10 µM) had no effect on LPS transport at t = 15 min or OA/TCA-augmented LPS transport at t = 30–45 min (Fig. 4C), suggesting that endogenous GLP-2 is not involved in OA/TCA-augmented LPS transport. To explore mechanisms underlying the inhibitory effects of GLP-2 on LPS transport during OA/TCA exposure, we investigated pathways downstream of GLP-2 receptor activation including IGF-1 and EGF and neural pathways involving NO and VIP. The IGF-1 receptor tyrosine kinase inhibitor NVP-AEW-541 (10 µM, s) abolished the effect of GLP-2 on LPS transport and further increased the amount of LPS transport (Fig. 4D). The EGF receptor tyrosine kinase inhibitor PD153035 (1 µM, s) increased the amount of LPS transport before OA/TCA exposure and further increased the amount of LPS transport after addition of OA/TCA (Fig. 4E). These results suggest that IGF-1 and EGF signaling in addition to their chronic proproliferative effects acutely regulate basal LPS permeability. In contrast, l-NAME (0.1 mM, s) (Fig. 4F) and the VPAC1 antagonist PG97-269 (1 µM, s) (Fig. 4G) had no effect on the amount of LPS transport at t = 15 min but reversed the inhibitory effect of GLP-2 on LPS transport during OA/TCA exposure. These results are summarized in Fig. 4H, demonstrating that NVP-AEW-541 and PD153035 increased the amount of LPS transport in the absence of luminal OA/TCA exposure, whereas l-NAME and PG97-269 reversed the inhibitory effect of GLP-2 on the amount of LPS transport only during OA/TCA exposure, suggesting that neural NO and VIP-VPAC1 signals are downstream of the mechanism by which GLP-2 inhibits LPS transport.
Fig. 4.

Effect of glucagon-like peptide-2 (GLP-2) on lipopolysaccharide (LPS) transport during lipid exposure in rat jejunal mucosa in vitro. Rat jejunal mucosae were exposed to mucosal LPS (10 µg/ml) at time (t) = 0 min, followed by the mucosal addition of vehicle (veh) or oleic acid (OA) and taurocholate (TCA) at t = 15 min. LPS transport [mucosal-to-serosal (m-to-s)] is expressed as Δ[LPS] (EU/ml) m-to-s (means ± SE, n = 6 rats). Each inhibitor (D–G) was added in the serosal solution 5 min before addition of GLP-2 and NVP. All data were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test. A: rat GLP-2 (100 nM) with or without NVP-728 (NVP, 10 µM) was added into the serosal (s) bath 10 min before LPS application, followed by addition of vehicle or OA (30 mM)/TCA (0.1 mM) into the mucosal (m) bath. GLP-2 with or without NVP had no effect on serosal [LPS] at t = 15 min. GLP-2 with NVP inhibited OA/TCA-induced [LPS] increase, whereas GLP-2 alone had no effect on OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. B: teduglutide (TDG, 100 nM) was added into the serosal (s) bath 10 min before LPS application, followed by addition of vehicle or OA/TCA. TDG inhibited OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group. C: GLP-2(3–33) (300 nM) or NVP (10 µM) was added into the serosal (s) bath 10 min before LPS application, followed by addition of vehicle or OA/TCA. GLP-2(3–33) or NVP had no effect on OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group. D: pretreatment with NVP-AEW-541 (AEW541, 10 µM, s) enhanced the OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group, ‡P < 0.05 vs. LPS + OA/TCA + NVP + GLP-2 group. E: pretreatment with PD153035 (1 µM, s) increased serosal [LPS] at t = 15 min and further augmented OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group, ‡P < 0.05 vs. LPS + OA/TCA + NVP + GLP-2 group. F: pretreatment with Nω-nitro-l-arginine methyl ester (l-NAME, 0.1 mM, s) reversed the inhibitory effect of GLP-2/NVP on OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group, ‡P < 0.05 vs. LPS + OA/TCA + NVP + GLP-2 group. G: pretreatment with PG97-269 (1 µM, s) reversed the inhibitory effect of GLP-2/NVP on OA/TCA-induced [LPS] increase. *P < 0.05 vs. LPS + vehicle group, †P < 0.05 vs. LPS + OA/TCA group, ‡P < 0.05 vs. LPS + OA/TCA + NVP + GLP-2 group. H: effects of NVP + GLP-2 with or without AEW541, PD153035, l-NAME, and PG97-269 on serosal [LPS] at t = 15 min (LPS alone exposure) and at t = 45 min (LPS+OA+TC exposure). *P < 0.05 vs. vehicle group, †P < 0.05 vs. NVP + GLP-2 group, ‡P < 0.05 vs. the corresponding LPS alone group.
FITC-LPS transport during LCFA absorption in vivo.
To further study the mechanism of enhanced LPS transport during lipid exposure, we measured FITC-LPS transport during LCFA absorption in vivo in anesthetized rats. First, we examined the dynamics of FITC-LPS movement from the lumen to the PV and lymph during OA/TCA exposure. Low-level FITC-LPS appearance in the PV plasma (Fig. 5A) and lymph (Fig. 5B) was present after an intraduodenal bolus perfusion of FITC-LPS alone (50 µg/ml in 2 ml PBS). In contrast, bolus perfusion of FITC-LPS with OA (30 mM)/TCA (10 mM) in 2 ml PBS rapidly increased FITC-LPS appearance in the PV at t = 15 and 30 min, which then declined to baseline, whereas FITC-LPS gradually appeared in the lymph with a peak at t = 60 min, reaching a plateau at t = 60–90 min, the latter consistent with the dynamics of chylomicron transport during physiological LCFA absorption (83). These results suggest that LPS transport during OA/TCA exposure is biphasic; rapid transport into the PV followed by a more delayed transport into the lymph.
Fig. 5.

Fluorescein isothiocyanate (FITC)-lipopolysaccharide (LPS) transport during lipid exposure in rat small intestine in vivo. FITC-LPS (50 µg/ml) in 2 ml phosphate-buffered saline (PBS) with or without oleic acid (OA, 30 mM) plus taurocholate (TCA, 10 mM) was administered by intraduodenal (id) bolus perfusion (pf) at t = 0 min, followed by 2 ml PBS perfusion every 30 min. Portal venous (PV) blood and mesenteric lymph were collected every 15 min. Fluorescence intensity was measured in PV plasma and lymph with measurement of lymph output to calculate FITC-LPS content in the samples after subtraction of background fluorescence intensity in the samples at time (t) = 0, which is expressed as PV FITC-LPS (ng/ml) and FITC-LPS transport into lymph (ng/15 min) (means ± SE, n = 6 rats). Each inhibitor was perfused in 2-ml PBS 30 min before FITC-LPS perfusion, followed by coperfusion with FITC-LPS + OA/TCA at t = 0 min. Data were analyzed by two-way ANOVA (A–F) or one-way ANOVA (G and H), followed by Tukey’s multiple comparisons test. A and B: intraduodenal perfusion of FITC-LPS alone had no effect on FITC-LPS appearance into the PV (A) and into the lymph (B). Addition of OA + TCA with FITC-LPS rapidly increased PV FITC-LPS content at t = 15 and 30 min, followed by a decline to the basal value (A), whereas FITC-LPS transport to lymph gradually increased, reaching a plateau at t = 60 min. Pretreatment and co-perfusion of SSO (1 mM) reduced OA/TCA-induced FITC-LPS transport into the PV (A), but had no effect on FITC-LPS transport into the lymph (B). *P < 0.05 vs. FITC-LPS alone group, †P < 0.05 vs. FITC-LPS + OA/TCA group. C and D: pretreatment and coperfusion of MβCD (1 mM) abolished OA/TCA-induced FITC-LPS transport into the PV (C), but had no effect on FITC-LPS transport into the lymph (D). *P < 0.05 vs. FITC-LPS alone group, †P < 0.05 vs. FITC-LPS + OA/TCA group. E and F: pretreatment and coperfusion of Pluronic-81 (PL81, 3%) delayed OA/TCA-induced FITC-LPS transport increase into the PV (E), and inhibited FITC-LPS transport into the lymph (F). *P < 0.05 vs. FITC-LPS alone group, †P < 0.05 vs. FITC-LPS + OA/TCA group. G and H: area under the curve for FITC-LPS uptake for 0–90 min (AUC0–90) into the PV (G) and lymph (H) was calculated from A, C, and E and from B, D, and F, respectively, by the trapezoidal rule. Compared with FITC-LPS alone group, OA/TCA increased AUC0–90 of PV and lymph. MβCD treatment abolished AUC0–90 of PV with no effect on AUC0–90 of lymph, whereas PL81 treatment reduced AUC0–90 of lymph with no effect on AUC0–90 of PV. SSO treatment had no effect on AUC0–90 of PV and lymph. *P < 0.05 vs. FITC-LPS alone group, †P < 0.05 vs. FITC-LPS + OA/TCA group.
Next, we examined the effects of lipid transport inhibitors on FITC-LPS transport into the PV and lymph. Pretreatment with the CD36 inhibitor SSO (1 mM), followed by intraduodenal perfusion of the OA/TCA solution inhibited rapid LPS transport to the PV at t = 15 and 30 min (Fig. 5A), whereas SSO had no effect on FITC-LPS transport into the lymph (Fig. 5B). Pretreatment and coperfusion of the lipid raft inhibitor MβCD (1 mM) abolished FITC-LPS transport into the PV (Fig. 5C) but had no effect on FITC-LPS transport into the lymph (Fig. 5D). Furthermore, pretreatment and coperfusion of the chylomicron synthesis inhibitor PL81 (3%) reduced FITC-LPS transport into the PV (Fig. 5E) and inhibited FITC-LPS transport into the lymph (Fig. 5F). These results suggest that the CD36- and lipid raft-mediated lipid absorption pathways are involved in rapid LPS transport into the PV, followed by gradual FITC-LPS transport into the lymph by LPS incorporation into chylomicrons, consistent with a previous report (31). Total LPS transport into the PV and the lymph was also assessed by calculation of AUC0–90. OA/TCA exposure increased AUC0–90 of PV FITC-LPS (Fig. 5G) and AUC0–90 of lymph FITC-LPS (Fig. 5H) compared with the FITC-LPS alone group. Increased AUC0–90 of PV FITC-LPS was abolished by MβCD treatment, whereas SSO and PL81 failed to affect overall FITC-LPS transport into the PV (Fig. 5G), although SSO and PL81 partially blunted the early rise of FITC-LPS transport into the PV at t = 15 min (Fig. 5, A and E). In contrast, OA/TCA-augmented AUC0–90 of lymph FITC-LPS was reduced only by PL81 treatment (Fig. 5H). These results also showed that total FITC-LPS transport into the lymph was ~1 µg, only ~1% of applied LPS in the lumen (100 µg). Assuming PV blood flow is ~10 ml/min for a ~250 g body weight rat [reported PV blood flow values are ~19 ml/min for a 526-g rat and 20 ml/min for a 367-g rat (75, 85)], ~60 µg (~60%) of added LPS was transported into the PV during a 90 min FITC-LPS exposure, suggesting that most of the luminal FITC-LPS was transported into the PV rather than into the lymph.
To confirm whether FITC-LPS transport into the PV is associated with the villous tip injury during OA/TCA exposure as previously reported (50), we assessed epithelial histology of the jejunal mucosa at t = 15 min when FITC-LPS transport into the PV reached a peak. The epithelial brush-border continuity of the villous cells with dilated subepithelial spaces was similarly observed in FITC-LPS alone (supplemental Fig. S2, E and F) and FITC-LPS + OA/TCA (supplemental Fig. S2, G and H; (https://doi.org/10.6084/m9.figshare.12203006.v1)) groups, although fluorescent staining of brush border of villous tip cells was less prominent in OA/TCA group. This result suggests that FITC-LPS transport into the PV is not associated with villous tip injury during OA/TCA exposure.
We also examined the effect of TDG on FITC-LPS transport in vivo to confirm the Ussing chamber results. Pretreatment with TDG (50 µg/kg iv) at t = −15 min abolished FITC-LPS transport into the PV (Fig. 6A), whereas FITC-LPS transport to the lymph was rapidly increased at t = 15 and 30 min (Fig. 6B). TDG-enhanced LPS transport into the lymph was accompanied by a rapid increase of lymphatic output at t = 0–30 min compared with nonpretreated controls (Fig. 6C), suggesting that TDG acutely enhances flow dynamics including mucosal blood and lymphatic flow and then increases the number of FITC-LPS-containing chylomicrons in the lymph. Pretreatment with and coperfusion of l-NAME (0.1 mM) reduced the inhibitory effect of TDG on FITC-LPS transport into the PV (Fig. 6A) and inhibited the TDG-related augmentation of FITC-LPS transport into the lymph (Fig. 6B) with reduced lymph output (Fig. 6C). Pretreatment with PG97–269 (0.3 mg/kg iv) at t = −20 min had no effect on TDG-related inhibition on FITC-LPS transport into the PV (Fig. 6D) but inhibited the TDG-related augmentation of FITC-LPS transport into the lymph (Fig. 6E) with reduced lymph output (Fig. 6F). These results suggest that TDG acutely inhibits OA/TCA-induced FITC-LPS transport into the PV via a NO-mediated pathway and enhances OA/TCA-induced FITC-LPS transport into the lymph via NO and VIP-VPAC1 pathways by increasing overall lymphatic output. TDG treatment reduced OA/TCA-augmented AUC0–90 of PV FITC-LPS (Fig. 6G). l-NAME treatment reversed the TDG-induced reduction of AUC0–90 PV FITC-LPS, whereas PG97-269 had no effect. TDG had no significant effect on OA/TCA-augmented AUC0–90 of lymph FITC-LPS (Fig. 6H), although TDG increased FITC-LPS transport into the lymph at 15–30 min (Fig. 6B, E). l-NAME and PG97-269 treatment reduced AUC0–90 of lymph FITC-LPS compared with the + TDG group (Fig. 6H). These results suggest that TDG inhibits LPS entry into the PV and initially accelerates but has a little overall effect on LPS transport into the lymph.
Fig. 6.

Effect of teduglutide (TDG on fluorescein isothiocyanate (FITC)-lipopolysaccharide (LPS) transport during lipid exposure in rat small intestine in vivo. TDG (50 µg/kg) was injected intravenously15 min before intraduodenal (id) bolus perfusion (pf) of FITC-LPS (50 µg/ml) in 2-ml PBS with oleic acid (OA, 30 mM) plus taurocholate (TCA, 10 mM). FITC-LPS appearance in the portal venous (PV) and the lymph, and lymph output are expressed as PV FITC-LPS (ng/ml), FITC-LPS transport into lymph (ng/15 min) and lymph output (µl/15 min) (means ± SE, n = 6 rats), respectively. Data were analyzed by two-way ANOVA (A–F) or one-way ANOVA (G, H), followed by Tukey’s multiple comparisons test. A–C: TDG intravenous injection abolished OA/TCA-induced FITC-LPS transport into the PV (A), but rapidly increased FITC-LPS transport into the lymph (B), accompanied by rapidly enhanced lymph output (C). Nω-nitro-l-arginine methyl ester (l-NAME, 0.1 mM) was perfused in 2-ml phosphate-buffered saline (PBS) at time (t) = −30 min, followed by TDG intravenous injection at t = −15 min. Pretreatment with and coperfusion of l-NAME reduced the inhibitory effect of TDG on OA/TCA-induced FITC-LPS transport into the PV (A) and into the lymph (B), with reduction of TDG-induced lymph output increase (C). *P < 0.05 vs. FITC-LPS + OA/TCA group, †P < 0.05 vs. + TDG group. D–F: PG97-269 (0.3 mg/kg) was intravenously injected 5 min before TDG intravenouos injection at t = −15 min. Pretreatment of PG97-269 had no effect on the inhibitory effect of TDG on OA/TCA-induced FITC-LPS transport into the PV (D), but inhibited FITC-LPS transport into the lymph (E), with reduction of TDG-induced lymph output increase (F). *P < 0.05 vs. FITC-LPS + OA/TCA group, †P < 0.05 vs. + TDG group. G, H: AUC0–90 of FITC-LPS transport into the PV (G) and lymph (H) was calculated from A and D and from B and E, respectively, by the trapezoidal rule. Compared with FITC-LPS + OA/TCA group, TDG reduced AUC0–90 of PV, which was reversed by l-NAME treatment (G). l-NAME and PG97-269 treatment reduced AUC0–90 of lymph, whereas TDG had no significant effect on AUC0–90 of lymph (H). *P < 0.05 vs. FITC-LPS + OA/TCA group, †P < 0.05 vs. + TDG group.
Direct visualization of FITC-LPS uptake into epithelial cells in vivo in mouse jejunal mucosa.
To directly distinguish intracellular uptake from paracellular diffusion of macromolecules in the small intestinal mucosa in vivo, we visualized the localization of FITC-LPS in mouse jejunal mucosa using in vivo two-photon confocal microscopy, since single-photon conventional confocal microscopy was not able to penetrate the tissue deeply enough to visualize the intracellular localization of fluorescence at the basolateral pole of the villous cells (Fig. 7, A and B). Figure 7A is single-photon confocal microscopic image of FITC-LPS incubated with OA plus TCA, demonstrating that no obvious fluorescent signal was observed in the villous cells, although the lumen had a strong FITC signal. In contrast, a two-photon confocal image of the same area showed intracellular FITC signals in the villous cells (vertical optical sections of villous cells; arrowheads) as well as FITC signals in the cytosol (red arrows) and at the basolateral pole of the villous cells contrasted with negatively stained nuclei (horizontal optical section of villous cells; white arrows) (Fig. 7B). Luminal incubation with FITC-LPS (50 µg/ml) alone in PBS over the jejunal mucosa for 15 min showed no apparent intracellular localization of fluorescent signals, with faint staining at the surface of villous cells (Fig. 7C). In contrast, the addition of OA (30 mM) plus TCA (10 mM) to luminal FITC-LPS solution for 5 min rapidly stained the intracellular space of the villous cells with negative staining of the outlines of each villous cell (Fig. 7D). Deeper scanning also revealed that FITC signals were present at the basolateral pole of the villous cells with negative staining of the nuclei (Fig. 7, B and E). Note that there were black spots in the villi with no stained structure (Fig. 7E), corresponding to the previously reported cell shedding-induced epithelial gaps (88). Furthermore, FITC-LPS was not visualized in the intercellular spaces, in contrast to in vivo confocal laser endomicroscopy of sodium fluorescein in human intestine (48).
Fig. 7.

Intracellular uptake of fluorescein isothiocyanate (FITC)-lipopolysaccharide (LPS) in mouse jejunal epithelium in vivo. Mouse jejunal mucosa was exposed to luminal FITC-LPS solution (50 µg/ml) alone for 15 min (C), followed by to FITC-LPS with oleic acid (OA, 30 mM) plus taurocholate (TCA, 10 mM) (A, B, D, E) under isoflurane anesthesia. Localization of FITC-LPS was imaged using single-photon or two-photon confocal microscope. No FITC signal was observed in the villi with single-photon confocal imaging (A), whereas two-photon confocal imaging detected intracellular FITC signals in the presence of OA plus TCA (B) in the apical cytosol of villous cells by horizontal scanning (arrowheads) and in the bottom of villous cells by vertical scanning (red arrows) with unstained nucleus (white arrows). Luminal FITC-LPS alone showed no obvious intracellular staining of FITC-LPS with faint staining on the apical surface of villous cells (C). Addition of OA plus TCA to FITC-LPS solution rapidly stained intracellular space of villous cells with a negative paracellular space (D). Deeper scanning revealed cytosolic staining in the bottom of villous cells with negative nuclei (white arrows in D and E). Exposure to luminal a cell-impermeant red fluorescence dye SNARF-5F with OA plus TCA failed to stain the intracellular space. Internal bar = 50 µm.
These results suggest that luminal FITC-LPS is rapidly absorbed via the transcellular pathway by jejunal villous cells only in the presence of OA/TCA rather than absorbed via the paracellular route. Coincubation with a cell-impermeant red fluorescent dye SNARF-5F revealed negative intracellular staining (Fig. 7F), further confirming the transcellular uptake of FITC-LPS.
FITC-LPS transport into the PV in mice.
To confirm whether acute LPS transport into the PV during LCFA exposure occurs in mice, FITC-LPS (50 µg/ml) solution (0.2 ml) with or without OA (30 mM)/TCA (10 mM) was bolus perfused from the duodenum in mice, thereafter PV blood was collected at t = 15 min. FITC-LPS levels in the PV plasma were higher in FITC-LPS + OA/TCA groups compared with the FITC-LPS alone group (Fig. 8), confirming that luminal FITC-LPS in the presence of OA/TCA is acutely transported into the PV in mice, similar to the results obtained in rats.
Fig. 8.
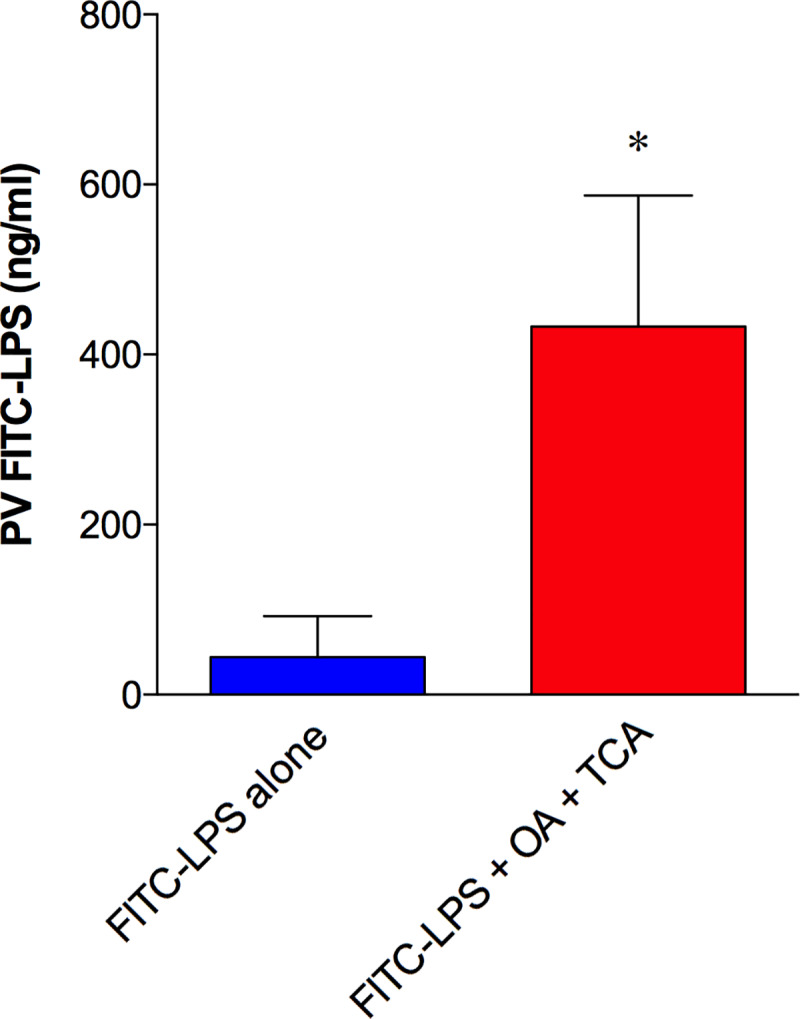
Fluorescein isothiocyanate (FITC)-lipopolysaccharide (LPS) transport during lipid exposure into the portal vein in mice. Intraduodenal perfusion of FITC-LPS with or without oleic acid (OA, 30 mM) plus taurocholate (TCA, 10 mM) was performed in anesthetized mice, followed by portal venous (PV) blood collection 15 min after perfusion. Plasma FITC-LPS levels were measured and are expressed as PV FITC-LPS (ng/ml) (means ± SE, n = 6 mice). *P < 0.05 vs. FITC-LPS alone group. Data were analyzed by the Mann-Whitney test.
DISCUSSION
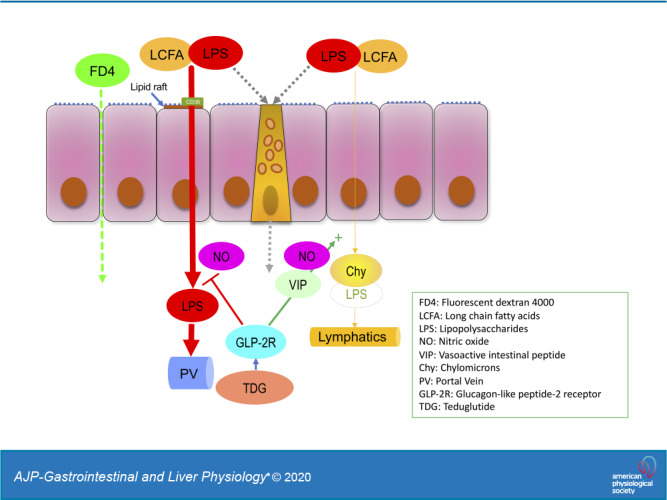
We studied the dynamics of LPS transport in rodent small intestine during lipid absorption in vitro and in vivo, observing that LPS was acutely transported from the mucosal to the serosal side via CD36- and lipid raft-mediated mechanisms during LCFA exposure. We also observed that LPS transport in vivo was biphasic with rapid transport into the PV followed by slower transport into the lymph; the former was related to lipid rafts and partially via CD36 and chylomicron formation, whereas the latter was mediated by the chylomicron-dependent LCFA uptake pathway. AUC analysis demonstrated that recovery of luminally added FITC-LPS was ~60% into the PV and only ~1% into the lymph, suggesting that most of luminal LPS is rapidly transported into the PV during lipid absorption. Furthermore, we demonstrated that exogenous GLP-2 combined with DPP4 inhibition or the stable GLP-2 analog TDG inhibited the acute phase of LPS transport during LCFA exposure in vitro and rapid LPS transport into the PV in vivo, whereas TDG accelerated LPS transport into the lymph by increasing the rate of lymphatic output early in the time course. This is the first study directly demonstrating physiological absorption of LPS from the small intestinal lumen in vivo during LCFA absorption via the chylomicron-mediated absorption pathway into the lymph.
This study also supports prior transcellular LPS transport studies (6, 18, 51) and demonstrated for the first time in vivo that luminal LPS was predominantly and rapidly transported into the PV via transcytotic pathway(s). Furthermore, we demonstrated for the first time that GLP-2 acutely inhibited the rapid uptake of LPS into the PV during LCFA absorption while supporting prior studies regarding the acute effects of GLP-2 on chylomicron transport (16, 42, 43). These results provide key insight into the mechanism 1) by which the intestinal mucosa handles LPS, a large, lipophilic, highly potent, and toxic bacterial substance present in high luminal concentrations in order to avoid activating undesired systemic inflammatory pathways; 2) how the augmentation of LPS transport by dietary fat contributes to endotoxemia; and 3) how the endogenous peptide GLP-2 modifies LPS absorption via the endocytic pathways with transport to the PV and the lymph.
We used two methods to assess LPS movement from the lumen: LPS activity measured by the LAL test in vitro and FITC-LPS uptake in vivo. There are a variety of methods used to measure LPS in biological samples that all have advantages and disadvantages: the LAL test, LPS antibody-based ELISA kit, bioassay using TLR4-expressing cells with a reporter gene, and labeled LPS molecules such as FITC-LPS. Since there is no one test that has overall superiority, we chose measurements based on the requirements of each system used. In the Ussing chamber, the serosal solution of muscle-stripped intestinal mucosa contained a substantial amount of autofluorescence, whereas blood plasma generally contains interfering factors for the LAL test or anticoagulant components that may affect the enzymatic assay on which the LAL test is based. The LAL test is probably the simplest and optimal way to detect intact lipid A activity bearing in mind that lipid A activity may be masked by the presence of chylomicrons, especially in the lymph, since chylomicrons inhibit lipid A activity (22). Therefore, we decided to use the LAL test for in vitro studies and FITC-LPS for in vivo studies. Despite these dissimilar approaches, our results demonstrated consistent transcellular LPS transport via lipid absorption-related pathways. Furthermore, we confirmed the comparability among the LAL test, FITC-LPS flux, and the TLR4 reporter cell assay in the measurement of LPS transport during OA/TCA exposure in the jejunum in vitro.
In health, LPS is limited to the intestinal lumen due to the presence of the multiple defense mechanisms including luminal antimicrobial peptides, mucins, and brush-border IAP in addition to intercellular tight junctions that are unlikely to allow passage of a 15- to 20-kDa lipophilic monomer such as LPS. These observations notwithstanding, paracellular permeability to much smaller solutes such as FITC-dextran 4000 has been used as a surrogate for gut-barrier function in endotoxin-related diseases, suggesting that the paracellular pathway may become dominant in the presence of inflammation, drugs, or other pathological states (44, 63, 74). Nevertheless, a high-fat diet increases systemic LPS concentrations in mice and humans (14, 24), chylomicrons contain LPS (31), and LPS absorption is enhanced by chylomicron formation (31), all suggesting that LPS is assimilated as part of the physiological absorption pathway of dietary LCFA. Chronic exposure to even low levels of circulating LPS due to chronic dietary intake of excessive fat, combined with increased amounts of luminal Gram-negative bacteria, may be sufficient to activate inflammatory cascades in adipocytes, pancreatic islet β cells, and hepatocytes, changes that have been related to truncal obesity, hypertension, cardiovascular disease, type 2 diabetes, hyperlipidemia, and fatty liver as components of the metabolic syndrome (23, 59).
One can assume that LPS as a lipophilic large molecule may cross the epithelium only when the paracellular spaces are widened due to dysfunction or disruption of the tight junctional complex or following epithelial cell injury. Yet, paracellular permeability markers such as TEER and FD4 movement do not always behave in parallel. We showed that FITC-LPS crossed the jejunal mucosa during OA/TCA exposure without changes in FD4 movement and TEER, suggesting that LPS is transported via the transcellular pathway rather than by the paracellular route, although FD4 crossed the jejunal mucosa somewhat nonspecifically.
Furthermore, one can question is how the colonic mucosa regulates LPS movement since the colonic lumen contains >99% of the bacterial LPS in the gut lumen. Since we focused on small intestinal LPS transport in the present study, further study is needed to examine colonic LPS movement and to clarify how sepsis is suppressed despite massive colonic luminal LPS concentrations even in the presence of mucosal injury, even though modest endotoxemia has been reported in inflammatory bowel disease (64). It thus appears that a small background of LPS entry into the PV and lymph occurs via established lipid uptake pathways in the small intestine subject to neurohormonal regulation, whereas the colon, due to its steep m-to-s LPS gradient, serves as an effective barrier to LPS entry under physiological and pathological conditions.
On the basis of in vitro data, several small intestinal transport mechanisms are implicated in LPS transport (36): endocytosis mediated by lipid raft-dependent or independent pathways, goblet cell-associated antigen passage (GAAP), chylomicron-dependent pathways, and paracellular pathways. Our in vitro results demonstrated that LPS was transported in the presence of luminal LCFA via CD36- and lipid raft-mediated endocytic mechanisms, suggesting that transport pathways used for the uptake of LCFAs also transport luminal LPS into epithelial cells. The transcellular uptake of luminal LPS was also confirmed directly by in vivo two-photon confocal microscopic imaging, wherein FITC-LPS in the presence of luminal LCFA was rapidly localized to the villous cell cytoplasm, but not to the paracellular space. Furthermore, in vivo perfusion studies documented rapid FITC-LPS appearance in the PV via CD36- and lipid raft-mediated mechanisms, followed by a gradual appearance in the lymph dependent on chylomicron synthesis. CD36 and the LPS receptors CD14 and TLR4 are present in lipid rafts (21, 66). Lipid raft disruption by MβCD decreases the LPS permeation coefficient without any change in paracellular permeability, suggesting the involvement of caveola-dependent mechanisms (53), consistent with prior in vitro studies and supporting our results.
We found that there were two major routes of LPS entry from the small intestine during lipid absorption: transport into the PV that was unanticipated, and transport into the lymph with chylomicrons that was predicted, although those pathways were previously described in inflamed models (86). Furthermore, PV LPS transport was rapid and transient compared with the gradual and sustained increase of lymphatic LPS transport. Although rapid PV LPS transport was partially inhibited by CD36 inhibition and abolished by lipid raft disruption, these interventions did not affect delayed lymphatic LPS transport, suggesting that LPS entry into the villous cells is via the physiological lipid absorption pathways that take up LCFAs from the intestinal lumen, whereas the exit pathway from the villous cells and transfer into the blood capillaries are dissimilar to chylomicron transport into the lymph. Because of their particle size, chylomicrons only enter to the central lacteals and villous lymphatic capillaries that have wide slit-like openings in the lymphatic endothelium (82). Free LPS or smaller lipid particles may exit from the villous cells and enter the blood capillaries to the PV. Another possibility is that epithelial or subepithelial lipoprotein lipase may dissociate initially transported chylomicrons with subsequent release of LPS from chylomicrons, similar to the mechanism by which lipoprotein lipase releases LCFAs from TG in chylomicrons, which then activate LCFA receptors on enteroendocrine L cells (68).
Interestingly, absorbed, circulating LPS is taken up by hepatocytes for clearance and excretion into the bile (55). Therefore, although lipid absorption is potentially harmful due to concomitant absorption of LPS, earlier studies have reported that chylomicrons inhibit lipid A activity (22) and enhance LPS uptake by hepatocytes and LPS excretion into the bile (71). Thus, LPS uptake by the chylomicron pathway has intrinsic protective mechanisms that detoxify LPS and facilitate LPS removal from the circulation. Bile salts also inhibit lipid A activity (73), suggesting that bile in the intestinal lumen and chylomicrons in the circulation prevent functional LPS lipid A from activating the immune systems in the mucosa, the circulation, and the liver. Therefore, accelerated chylomicron transport incorporating LPS, which eventually enters the systemic circulation via the thoracic duct, may induce less toxicity than does “free” LPS transport into the PV, which may directly activate TLR4 expressed on Kupffer cells and hepatic stellate cells, promoting hepatic inflammation as part of the “gut-liver axis” (76). We propose that chronic reduction of PV LPS entry may prevent low-grade inflammation in the liver, thus reducing the development of fat accumulation and inflammation (steatohepatitis), which has been linked to the activation of inflammatory cascades via TLR4 activation (15). Increased PV LPS levels present in experimental colitis augment the development of steatohepatitis (28), whereas reduction of intestinal LPS by oral antibiotics improves hepatic fibrosis with reducing intestinal permeability (17), supporting our hypothesis.
GLP-2 is released from enteroendocrine L cells in response to meals, likely through signaling via bacterial metabolites such as short-chain fatty acids, whose receptors are expressed on L cells (1). Although the stable GLP-2 analog TDG is approved for the treatment of short-gut syndrome due to its chronic trophic effects on the gut epithelium, it also has acute effects that are less well studied, including accelerating intestinal lipid uptake at 60–90 min (42) and enterocyte chylomicron release at 60 min (16), and reducing paracellular permeability of the mouse jejunum starting ~4 h after injection (7). Here, we demonstrated that exogenous GLP-2 combined with DPP4 inhibition or TDG acutely inhibited LCFA-facilitated LPS transport in vitro in Ussing chambered intestine. Luminal LCFA and bile acids may increase endogenous GLP-2 release via activation of the LCFA receptors free fatty acid receptor (FFA) 1 and 4 and bile acid receptor TGR5 on L cells (1, 45, 72). Basolateral LPS or systemically administered LPS increases GLP-1 release in vitro or in vivo (46, 52). Therefore, endogenous GLP-2 released by OA/TCA or transported LPS may be involved in our in vitro study. Nevertheless, a GLP-2 receptor antagonist or DPP4 inhibitor alone had no effect on OA/TCA-augmented LPS transport, suggesting that the effect of endogenous GLP-2 is minimal. The inhibitory effects of GLP-2 were mediated at the least by NO and VIP-VPAC1 pathways, details of which remain to be determined. Neuronal VIP may regulate macromolecular permeability by increased expression of the intercellular junction protein zonula occludens-1 (60), although our results suggest that LPS is transported via the transcellular pathway rather than by the paracellular pathway, at least during OA/TCA exposure. We also confirmed the acute inhibitory effects of TDG on LPS transport into the PV via the NO pathway. Our results showed that GLP-2-induced NO production suppressed LCFA-associated LPS transport in vitro, suggesting that NO directly regulates enterocyte LPS uptake from the lumen or release from the basolateral membranes. Since NO is involved in GLP-2-mediated chylomicron release from enterocytes (43), these results suggest that TDG may redirect LPS transport from the PV to the lymphatics via the NO pathway. Still, the vast majority of luminally added LPS was transported into the PV, and the TDG-induced increase of FITC-LPS transport into the lymph was transient and did not affect overall LPS transport, suggesting that TDG-NO pathway directly modifies epithelial LPS entry or intracellular/subepithelial LPS movement. The direct effects of NO on LPS movement from the lumen to the subepithelial space remain to be determined.
TDG acutely augmented LPS transport into the lymph probably by increasing lymph output via NO and VIP-VPAC1-mediated pathways, since GLP-2 increases superior mesenteric arterial blood flow possibly via NO and VIP release (35); increased intestinal microcirculatory flow enhances lymph output (56) due to increased capillary pressure that subsequently increases interstitial hydrostatic pressure (34). Although the reduction of PV LPS entry with increased chylomicron-mediated LPS absorption into the lymph in response to TDG is novel, chylomicron-bound LPS in the lymphatics is less toxic than “free” LPS in the PV due to the many detoxifying defenses inherent in the chylomicron pathway. Further study will clarify the effect of TDG on the development of high-fat diet-induced fatty liver.
LPS may also be transported through emptied goblet cells in mouse ileum (41), by the GAAP (54). We showed that CCh pretreatment, which empties goblet cells, increased LPS transport regardless of prior lipid exposure, suggesting that LPS may pass through emptied goblet cells possibly via a GAAP-related luminal lipid-independent mechanism. Another possibility is that since CCh increases mucus secretion and anion secretion, CCh-induced changes of the preepithelial mucus barrier to macromolecule diffusion and unstirred layer conditions may contribute to the increase of LPS transport. We also showed that the IAP inhibitor GP increased the transport of active LPS during lipid exposure, consistent with IAP-mediated LPS dephosphorylation of lipid A with consequent detoxification (84). Orally administered IAP inhibits metabolic changes induced by high-fat diets (47), ameliorates chemically induced colitis in a murine model (70), and attenuates alcohol-induced fatty liver in mice (38), consistent with LPS detoxification. Since IAP activity is increased at alkaline pH (4), and since the rate of secretion is maximal in the duodenum (2), the jejunum as the proximate downstream intestinal segment and primary locus of LCFA absorption has the high luminal pH and expression of brush-border IAP activity necessary to maximally detoxify LPS concurrent with LCFA absorption, serving as another protective mechanism to reduce systemic LPS toxicity. Since GLP-2 stimulates duodenal secretion via the NO and VIP pathways (87), it is possible that GLP-2 not only reduces bioactive LPS entry by reducing LPS transport into the PV, but also by increasing secretion that in turn increases the rate of lipid A dephosphorylation by IAP while augmenting chylomicron transport. Further study will clarify this possibility by monitoring bioactive lipid A transport in the PV and in chylomicrons.
In conclusion, we have demonstrated the dynamics of LPS transport during lipid exposure in the small intestine in vitro and in vivo demonstrating at least two distinct uptake pathways and finding no evidence for paracellular LPS transport or epithelial injury under the conditions used. We have also found that GLP-2 acutely impairs LPS entry into the PV, a novel finding with implications for reducing systemic inflammation via the “gut-liver axis.” Since LPS is involved in the pathogenesis of the metabolic syndrome, sepsis, and more, CD36, lipid rafts, and GLP-2 receptors may be the therapeutic targets for the prevention of LPS-related disease. Our data do not support the paracellular transport of LPS at least under physiological conditions of OA absorption and also do not support the use of LPS as a marker for intestinal paracellular permeability in rodents without inflammation.
GRANTS
This work was supported by a Department of Veterans Affairs Merit Review Award I01BX001245, National Institutes of Health Grant R01 DK54221, and an Investigator-Initiated Award from Shire Pharmaceuticals USA.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.A., I.K., A.K., and J.D.K. conceived and designed research; Y.A., K.M., T.T., K.N., H.S., and J.D.K. performed experiments; Y.A., K.M., T.T., K.N., H.S., and J.D.K. analyzed data; Y.A. and J.D.K. interpreted results of experiments; Y.A. and J.D.K. prepared figures; Y.A. and J.D.K. drafted manuscript; Y.A., I.K., A.K., and J.D.K. edited and revised manuscript; Y.A., K.M., T.T., K.N., H.S., I.K., A.K., and J.D.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Laurent Bentolila and Matthew Schibler, California NanoSystems Institute at UCLA with technical assistance for multiphoton confocal microscope.
REFERENCES
- 1.Akiba Y, Inoue T, Kaji I, Higashiyama M, Narimatsu K, Iwamoto K, Watanabe M, Guth PH, Engel E, Kuwahara A, Kaunitz JD. Short-chain fatty acid sensing in rat duodenum. J Physiol 593: 585–599, 2015. doi: 10.1113/jphysiol.2014.280792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akiba Y, Kaunitz JD. Duodenal luminal chemosensing; acid, ATP, and nutrients. Curr Pharm Des 20: 2760–2765, 2014. doi: 10.2174/13816128113199990565. [DOI] [PubMed] [Google Scholar]
- 3.Akiba Y, Kaunitz JD. Regulation of intracellular pH and blood flow in rat duodenal epithelium in vivo. Am J Physiol Gastrointest Liver Physiol 276: G293–G302, 1999. doi: 10.1152/ajpgi.1999.276.1.G293. [DOI] [PubMed] [Google Scholar]
- 4.Akiba Y, Mizumori M, Guth PH, Engel E, Kaunitz JD. Duodenal brush border intestinal alkaline phosphatase activity affects bicarbonate secretion in rats. Am J Physiol Gastrointest Liver Physiol 293: G1223–G1233, 2007. doi: 10.1152/ajpgi.00313.2007. [DOI] [PubMed] [Google Scholar]
- 5.Arhan P, Devroede G, Jehannin B, Lanza M, Faverdin C, Dornic C, Persoz B, Tétreault L, Perey B, Pellerin D. Segmental colonic transit time. Dis Colon Rectum 24: 625–629, 1981. doi: 10.1007/BF02605761. [DOI] [PubMed] [Google Scholar]
- 6.Beatty WL, Méresse S, Gounon P, Davoust J, Mounier J, Sansonetti PJ, Gorvel JP. Trafficking of Shigella lipopolysaccharide in polarized intestinal epithelial cells. J Cell Biol 145: 689–698, 1999. doi: 10.1083/jcb.145.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamin MA, McKay DM, Yang PC, Cameron H, Perdue MH. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut 47: 112–119, 2000. doi: 10.1136/gut.47.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borgström B, Dahlqvist A, Lundh G, Sjövall J. Studies of intestinal digestion and absorption in the human. J Clin Invest 36: 1521–1536, 1957. doi: 10.1172/JCI103549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bos M, Mendelsohn J, Kim YM, Albanell J, Fry DW, Baselga J. PD153035, a tyrosine kinase inhibitor, prevents epidermal growth factor receptor activation and inhibits growth of cancer cells in a receptor number-dependent manner. Clin Cancer Res 3: 2099–2106, 1997. [PubMed] [Google Scholar]
- 10.Brandl K, Kumar V, Eckmann L. Gut-liver axis at the frontier of host-microbial interactions. Am J Physiol Gastrointest Liver Physiol 312: G413–G419, 2017. doi: 10.1152/ajpgi.00361.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronner F. Mechanisms of intestinal calcium absorption. J Cell Biochem 88: 387–393, 2003. doi: 10.1002/jcb.10330. [DOI] [PubMed] [Google Scholar]
- 12.Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut 68: 1516–1526, 2019. doi: 10.1136/gutjnl-2019-318427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 14.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58: 1091–1103, 2009. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, Szabo G. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 300: G433–G441, 2011. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dash S, Xiao C, Morgantini C, Connelly PW, Patterson BW, Lewis GF. Glucagon-like peptide-2 regulates release of chylomicrons from the intestine. Gastroenterology 147: 1275–1284.e4, 2014. doi: 10.1053/j.gastro.2014.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douhara A, Moriya K, Yoshiji H, Noguchi R, Namisaki T, Kitade M, Kaji K, Aihara Y, Nishimura N, Takeda K, Okura Y, Kawaratani H, Fukui H. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol Med Rep 11: 1693–1700, 2015. doi: 10.3892/mmr.2014.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drewe J, Beglinger C, Fricker G. Effect of ischemia on intestinal permeability of lipopolysaccharides. Eur J Clin Invest 31: 138–144, 2001. doi: 10.1046/j.1365-2362.2001.00792.x. [DOI] [PubMed] [Google Scholar]
- 19.Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci USA 93: 7911–7916, 1996. doi: 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drucker DJ, Yusta B, Boushey RP, DeForest L, Brubaker PL. Human [Gly2]GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. Am J Physiol Gastrointest Liver Physiol 276: G79–G91, 1999. doi: 10.1152/ajpgi.1999.276.1.G79. [DOI] [PubMed] [Google Scholar]
- 21.Ehehalt R, Sparla R, Kulaksiz H, Herrmann T, Füllekrug J, Stremmel W. Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains (lipid rafts). BMC Cell Biol 9: 45, 2008. doi: 10.1186/1471-2121-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eichbaum EB, Harris HW, Kane JP, Rapp JH. Chylomicrons can inhibit endotoxin activity in vitro. J Surg Res 51: 413–416, 1991. doi: 10.1016/0022-4804(91)90143-A. [DOI] [PubMed] [Google Scholar]
- 23.Erridge C. Diet, commensals and the intestine as sources of pathogen-associated molecular patterns in atherosclerosis, type 2 diabetes and non-alcoholic fatty liver disease. Atherosclerosis 216: 1–6, 2011. doi: 10.1016/j.atherosclerosis.2011.02.043. [DOI] [PubMed] [Google Scholar]
- 24.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 86: 1286–1292, 2007. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 25.Fink MP. Leaky gut hypothesis: a historical perspective. Crit Care Med 18: 579–580, 1990. doi: 10.1097/00003246-199005000-00024. [DOI] [PubMed] [Google Scholar]
- 26.Freudenberg MA, Freudenberg N, Galanos C. Time course of cellular distribution of endotoxin in liver, lungs and kidneys of rats. Br J Exp Pathol 63: 56–65, 1982. [PMC free article] [PubMed] [Google Scholar]
- 27.Fullerton JN, Segre E, De Maeyer RP, Maini AA, Gilroy DW. Intravenous endotoxin challenge in healthy humans: an experimental platform to investigate and modulate systemic inflammation. J Vis Exp 111: e53913, 2016. doi: 10.3791/53913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C, Obermeier F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol 55: 1391–1399, 2011. doi: 10.1016/j.jhep.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 29.García-Echeverría C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Furet P, Porta DG, Liebetanz J, Martiny-Baron G, Ruetz S, Hofmann F. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 5: 231–239, 2004. doi: 10.1016/S1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 30.Ge Y, Ezzell RM, Warren HS. Localization of endotoxin in the rat intestinal epithelium. J Infect Dis 182: 873–881, 2000. doi: 10.1086/315784. [DOI] [PubMed] [Google Scholar]
- 31.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 50: 90–97, 2009. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millán JL, Hodin RA. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci USA 105: 3551–3556, 2008. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gourlet P, De Neef P, Cnudde J, Waelbroeck M, Robberecht P. In vitro properties of a high affinity selective antagonist of the VIP1 receptor. Peptides 18: 1555–1560, 1997. doi: 10.1016/S0196-9781(97)00230-1. [DOI] [PubMed] [Google Scholar]
- 34.Granger DN, Perry MA, Kvietys PR, Taylor AE. Capillary and interstitial forces during fluid absorption in the cat small intestine. Gastroenterology 86: 267–273, 1984. doi: 10.1016/0016-5085(84)90410-4. [DOI] [PubMed] [Google Scholar]
- 35.Guan X, Karpen HE, Stephens J, Bukowski JT, Niu S, Zhang G, Stoll B, Finegold MJ, Holst JJ, Hadsell D, Nichols BL, Burrin DG. GLP-2 receptor localizes to enteric neurons and endocrine cells expressing vasoactive peptides and mediates increased blood flow. Gastroenterology 130: 150–164, 2006. [Erratum in Gastroenterology 130: 1019–1021, 2006] doi: 10.1053/j.gastro.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Guerville M, Boudry G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointest Liver Physiol 311: G1–G15, 2016. doi: 10.1152/ajpgi.00098.2016. [DOI] [PubMed] [Google Scholar]
- 37.Hadjiyanni I, Li KK, Drucker DJ. Glucagon-like peptide-2 reduces intestinal permeability but does not modify the onset of type 1 diabetes in the nonobese diabetic mouse. Endocrinology 150: 592–599, 2009. doi: 10.1210/en.2008-1228. [DOI] [PubMed] [Google Scholar]
- 38.Hamarneh SR, Kim BM, Kaliannan K, Morrison SA, Tantillo TJ, Tao Q, Mohamed MMR, Ramirez JM, Karas A, Liu W, Hu D, Teshager A, Gul SS, Economopoulos KP, Bhan AK, Malo MS, Choi MY, Hodin RA. Intestinal alkaline phosphatase attenuates alcohol-induced hepatosteatosis in mice. Dig Dis Sci 62: 2021–2034, 2017. doi: 10.1007/s10620-017-4576-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirokawa M, Takeuchi T, Chu S, Akiba Y, Wu V, Guth PH, Engel E, Montrose MH, Kaunitz JD. Cystic fibrosis gene mutation reduces epithelial cell acidification and injury in acid-perfused mouse duodenum. Gastroenterology 127: 1162–1173, 2004. doi: 10.1053/j.gastro.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 40.Hofmann AF, Borgström B. The intraluminal phase of fat digestion in man: the lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J Clin Invest 43: 247–257, 1964. doi: 10.1172/JCI104909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howe SE, Lickteig DJ, Plunkett KN, Ryerse JS, Konjufca V. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS One 9: e86656, 2014. doi: 10.1371/journal.pone.0086656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsieh J, Longuet C, Maida A, Bahrami J, Xu E, Baker CL, Brubaker PL, Drucker DJ, and Adeli K. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology 137: 997–1005.e1-4, 2009. doi: 10.1053/j.gastro.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh J, Trajcevski KE, Farr SL, Baker CL, Lake EJ, Taher J, Iqbal J, Hussain MM, Adeli K. Glucagon-like peptide 2 (GLP-2) stimulates postprandial chylomicron production and postabsorptive release of intestinal triglyceride storage pools via induction of nitric oxide signaling in male hamsters and mice. Endocrinology 156: 3538–3547, 2015. doi: 10.1210/EN.2015-1110. [DOI] [PubMed] [Google Scholar]
- 44.Hurni MA, Noach AB, Blom-Roosemalen MC, de Boer AG, Nagelkerke JF, Breimer DD. Permeability enhancement in Caco-2 cell monolayers by sodium salicylate and sodium taurodihydrofusidate: assessment of effect-reversibility and imaging of transepithelial transport routes by confocal laser scanning microscopy. J Pharmacol Exp Ther 267: 942–950, 1993. [PubMed] [Google Scholar]
- 45.Inoue T, Wang JH, Higashiyama M, Rudenkyy S, Higuchi K, Guth PH, Engel E, Kaunitz JD, Akiba Y. Dipeptidyl peptidase IV inhibition potentiates amino acid- and bile acid-induced bicarbonate secretion in rat duodenum. Am J Physiol Gastrointest Liver Physiol 303: G810–G816, 2012. doi: 10.1152/ajpgi.00195.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahles F, Meyer C, Möllmann J, Diebold S, Findeisen HM, Lebherz C, Trautwein C, Koch A, Tacke F, Marx N, Lehrke M. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes 63: 3221–3229, 2014. doi: 10.2337/db14-0100. [DOI] [PubMed] [Google Scholar]
- 47.Kaliannan K, Hamarneh SR, Economopoulos KP, Nasrin Alam S, Moaven O, Patel P, Malo NS, Ray M, Abtahi SM, Muhammad N, Raychowdhury A, Teshager A, Mohamed MM, Moss AK, Ahmed R, Hakimian S, Narisawa S, Millán JL, Hohmann E, Warren HS, Bhan AK, Malo MS, Hodin RA. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci USA 110: 7003–7008, 2013. doi: 10.1073/pnas.1220180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiesslich R, Duckworth CA, Moussata D, Gloeckner A, Lim LG, Goetz M, Pritchard DM, Galle PR, Neurath MF, Watson AJ. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 61: 1146–1153, 2012. doi: 10.1136/gutjnl-2011-300695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyama I, Matsunaga T, Harada T, Hokari S, Komoda T. Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin Biochem 35: 455–461, 2002. doi: 10.1016/S0009-9120(02)00330-2. [DOI] [PubMed] [Google Scholar]
- 50.Kvietys PR, Specian RD, Grisham MB, Tso P. Jejunal mucosal injury and restitution: role of hydrolytic products of food digestion. Am J Physiol Gastrointest Liver Physiol 261: G384–G391, 1991. doi: 10.1152/ajpgi.1991.261.3.G384. [DOI] [PubMed] [Google Scholar]
- 51.Laugerette F, Furet JP, Debard C, Daira P, Loizon E, Géloën A, Soulage CO, Simonet C, Lefils-Lacourtablaise J, Bernoud-Hubac N, Bodennec J, Peretti N, Vidal H, Michalski MC. Oil composition of high-fat diet affects metabolic inflammation differently in connection with endotoxin receptors in mice. Am J Physiol Endocrinol Metab 302: E374–E386, 2012. doi: 10.1152/ajpendo.00314.2011. [DOI] [PubMed] [Google Scholar]
- 52.Lebrun LJ, Lenaerts K, Kiers D, Pais de Barros JP, Le Guern N, Plesnik J, Thomas C, Bourgeois T, Dejong CHC, Kox M, Hundscheid IHR, Khan NA, Mandard S, Deckert V, Pickkers P, Drucker DJ, Lagrost L, Grober J. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Reports 21: 1160–1168, 2017. doi: 10.1016/j.celrep.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 53.Mani V, Hollis JH, Gabler NK. Dietary oil composition differentially modulates intestinal endotoxin transport and postprandial endotoxemia. Nutr Metab (Lond) 10: 6, 2013. doi: 10.1186/1743-7075-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, Newberry RD, Miller MJ. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483: 345–349, 2012. doi: 10.1038/nature10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology 109: 1969–1976, 1995. doi: 10.1016/0016-5085(95)90765-3. [DOI] [PubMed] [Google Scholar]
- 56.Miura S, Tashiro H, Kurose I, Suematsu M, Serizawa H, Sekizuka E, Nagata H, Yoshioka M, Tsuchiya M. Intestinal microcirculatory changes during fat absorption and the effect of cholecystokinin inhibitor. Am J Physiol Gastrointest Liver Physiol 262: G399–G404, 1992. doi: 10.1152/ajpgi.1992.262.3.G399. [DOI] [PubMed] [Google Scholar]
- 57.Mizumori M, Ham M, Guth PH, Engel E, Kaunitz JD, Akiba Y. Intestinal alkaline phosphatase regulates protective surface microclimate pH in rat duodenum. J Physiol 587: 3651–3663, 2009. doi: 10.1113/jphysiol.2009.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizumori M, Meyerowitz J, Takeuchi T, Lim S, Lee P, Supuran CT, Guth PH, Engel E, Kaunitz JD, Akiba Y. Epithelial carbonic anhydrases facilitate PCO2 and pH regulation in rat duodenal mucosa. J Physiol 573: 827–842, 2006. doi: 10.1113/jphysiol.2006.107581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreira AP, Texeira TFS, Ferreira AB, Peluzio MC, Alfenas RC. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 108: 801–809, 2012. doi: 10.1017/S0007114512001213. [DOI] [PubMed] [Google Scholar]
- 60.Neunlist M, Toumi F, Oreschkova T, Denis M, Leborgne J, Laboisse CL, Galmiche JP, Jarry A. Human ENS regulates the intestinal epithelial barrier permeability and a tight junction-associated protein ZO-1 via VIPergic pathways. Am J Physiol Gastrointest Liver Physiol 285: G1028–G1036, 2003. doi: 10.1152/ajpgi.00066.2003. [DOI] [PubMed] [Google Scholar]
- 61.Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology 52: 1829–1835, 2010. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 62.Nolan JP, Hare DK, McDevitt JJ, Ali MV. In vitro studies of intestinal endotoxin absorption. I. Kinetics of absorption in the isolated everted gut sac. Gastroenterology 72: 434–439, 1977. doi: 10.1016/S0016-5085(77)80253-9. [DOI] [PubMed] [Google Scholar]
- 63.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 32: 742–747, 2000. doi: 10.1016/S0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 64.Pastor Rojo O, López San Román A, Albéniz Arbizu E, de la Hera Martínez A, Ripoll Sevillano E, Albillos Martínez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis 13: 269–277, 2007. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]
- 65.Penagini R, Spiller RC, Misiewicz JJ, Frost PG. Effect of ileal infusion of glycochenodeoxycholic acid on segmental transit, motility, and flow in the human jejunum and ileum. Gut 30: 609–617, 1989. doi: 10.1136/gut.30.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 72: 557–581, 2015. doi: 10.1007/s00018-014-1762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088, 1998. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 68.Psichas A, Larraufie PF, Goldspink DA, Gribble FM, Reimann F. Chylomicrons stimulate incretin secretion in mouse and human cells. Diabetologia 60: 2475–2485, 2017. doi: 10.1007/s00125-017-4420-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quon MG, Mena I, Valenzuela JE. Abnormalities in the duodenal transit and motility in duodenal ulcer patients: studies with a new isotopic technique. Gut 30: 579–585, 1989. doi: 10.1136/gut.30.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramasamy S, Nguyen DD, Eston MA, Alam SN, Moss AK, Ebrahimi F, Biswas B, Mostafa G, Chen KT, Kaliannan K, Yammine H, Narisawa S, Millán JL, Warren HS, Hohmann EL, Mizoguchi E, Reinecker HC, Bhan AK, Snapper SB, Malo MS, Hodin RA. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis 17: 532–542, 2011. doi: 10.1002/ibd.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Read TE, Harris HW, Grunfeld C, Feingold KR, Calhoun MC, Kane JP, Rapp JH. Chylomicrons enhance endotoxin excretion in bile. Infect Immun 61: 3496–3502, 1993. doi: 10.1128/IAI.61.8.3496-3502.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reimann F, Tolhurst G, Gribble FM. G-protein-coupled receptors in intestinal chemosensation. Cell Metab 15: 421–431, 2012. doi: 10.1016/j.cmet.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 73.Rudbach JA, Anacker RL, Haskins WT, Johnson AG, Milner KC, Ribi E. Physical aspects of reversible inactivation of endotoxin. Ann NY Acad Sci 133: 629–643, 1966. doi: 10.1111/j.1749-6632.1966.tb52394.x. [DOI] [PubMed] [Google Scholar]
- 74.Salzman AL, Wang H, Wollert PS, Vandermeer TJ, Compton CC, Denenberg AG, Fink MP. Endotoxin-induced ileal mucosal hyperpermeability in pigs: role of tissue acidosis. Am J Physiol Gastrointest Liver Physiol 266: G633–G646, 1994. doi: 10.1152/ajpgi.1994.266.4.G633. [DOI] [PubMed] [Google Scholar]
- 75.Schaffner D, von Elverfeldt D, Deibert P, Lazaro A, Merfort I, Lutz L, Neubauer J, Baumstark MW, Kreisel W, Reichardt W. Phase-contrast MR flow imaging: A tool to determine hepatic hemodynamics in rats with a healthy, fibrotic, or cirrhotic liver. J Magn Res Imaging 46: 1526–1534, 2017. doi: 10.1002/jmri.25677. [DOI] [PubMed] [Google Scholar]
- 76.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13: 1324–1332, 2007. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 77.Stahl A, Hirsch DJ, Gimeno RE, Punreddy S, Ge P, Watson N, Patel S, Kotler M, Raimondi A, Tartaglia LA, Lodish HF. Identification of the major intestinal fatty acid transport protein. Mol Cell 4: 299–308, 1999. doi: 10.1016/S1097-2765(00)80332-9. [DOI] [PubMed] [Google Scholar]
- 78.Takayama K, Qureshi N, Raetz CR, Ribi E, Peterson J, Cantrell JL, Pearson FC, Wiggins J, Johnson AG. Influence of fine structure of lipid A on Limulus amebocyte lysate clotting and toxic activities. Infect Immun 45: 350–355, 1984. doi: 10.1128/IAI.45.2.350-355.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tamai H, Kato S, Horie Y, Ohki E, Yokoyama H, Ishii H. Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol Clin Exp Res 24: 390–394, 2000. doi: 10.1111/j.1530-0277.2000.tb04629.x. [DOI] [PubMed] [Google Scholar]
- 80.Taveira da Silva AM, Kaulbach HC, Chuidian FS, Lambert DR, Suffredini AF, Danner RL. Brief report: shock and multiple-organ dysfunction after self-administration of Salmonella endotoxin. N Engl J Med 328: 1457–1460, 1993. doi: 10.1056/NEJM199305203282005. [DOI] [PubMed] [Google Scholar]
- 81.Thulesen J, Knudsen LB, Hartmann B, Hastrup S, Kissow H, Jeppesen PB, Ørskov C, Holst JJ, Poulsen SS. The truncated metabolite GLP-2 (3-33) interacts with the GLP-2 receptor as a partial agonist. Regul Pept 103: 9–15, 2002. doi: 10.1016/S0167-0115(01)00316-0. [DOI] [PubMed] [Google Scholar]
- 82.Tso P, Balint JA. Formation and transport of chylomicrons by enterocytes to the lymphatics. Am J Physiol Gastrointest Liver Physiol 250: G715–G726, 1986. doi: 10.1152/ajpgi.1986.250.6.G715. [DOI] [PubMed] [Google Scholar]
- 83.Tso P, Buch KL, Balint JA, Rodgers JB. Maximal lymphatic triglyceride transport rate from the rat small intestine. Am J Physiol Gastrointest Liver Physiol 242: G408–G415, 1982. doi: 10.1152/ajpgi.1982.242.4.G408. [DOI] [PubMed] [Google Scholar]
- 84.Vaishnava S, Hooper LV. Alkaline phosphatase: keeping the peace at the gut epithelial surface. Cell Host Microbe 2: 365–367, 2007. doi: 10.1016/j.chom.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 85.van der Hoven B, van Pelt H, Swart EL, Bonthuis F, Tilanus HW, Bakker J, Gommers D. Noninvasive functional liver blood flow measurement: comparison between bolus dose and steady-state clearance of sorbitol in a small-rodent model. Am J Physiol Gastrointest Liver Physiol 298: G177–G181, 2010. doi: 10.1152/ajpgi.90688.2008. [DOI] [PubMed] [Google Scholar]
- 86.van Deventer SJH, ten Cate JW, Tytgat GNJ. Intestinal endotoxemia. Clinical significance. Gastroenterology 94: 825–831, 1988. doi: 10.1016/0016-5085(88)90261-2. [DOI] [PubMed] [Google Scholar]
- 87.Wang JH, Inoue T, Higashiyama M, Guth PH, Engel E, Kaunitz JD, Akiba Y. Umami receptor activation increases duodenal bicarbonate secretion via glucagon-like peptide-2 release in rats. J Pharmacol Exp Ther 339: 464–473, 2011. doi: 10.1124/jpet.111.184788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology 129: 902–912, 2005. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 89.Wellmann W, Fink PC, Benner F, Schmidt FW. Endotoxaemia in active Crohn’s disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut 27: 814–820, 1986. doi: 10.1136/gut.27.7.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamada T, Inui A, Hayashi N, Fujimura M, Fujimiya M. Serotonin stimulates endotoxin translocation via 5-HT3 receptors in the rat ileum. Am J Physiol Gastrointest Liver Physiol 284: G782–G788, 2003. doi: 10.1152/ajpgi.00376.2002. [DOI] [PubMed] [Google Scholar]
- 91.Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, Narvaez A, Honrada R, Ruvalcaba D, McGrath MS. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol 206: 121–124, 2009. doi: 10.1016/j.jneuroim.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]