

Abstract
Cell-autonomous circadian clocks have emerged as temporal orchestrators of numerous biological processes. For example, the cardiomyocyte circadian clock modulates transcription, translation, posttranslational modifications, ion homeostasis, signaling cascades, metabolism, and contractility of the heart over the course of the day. Circadian clocks are composed of more than 10 interconnected transcriptional modulators, all of which have the potential to influence the cardiac transcriptome (and ultimately cardiac processes). These transcriptional modulators include BMAL1 and REV-ERBα/β; BMAL1 induces REV-ERBα/β, which in turn feeds back to inhibit BMAL1. Previous studies indicate that cardiomyocyte-specific BMAL1-knockout (CBK) mice exhibit a dysfunctional circadian clock (including decreased REV-ERBα/β expression) in the heart associated with abnormalities in cardiac mitochondrial function, metabolism, signaling, and contractile function. Here, we hypothesized that decreased REV-ERBα/β activity is responsible for distinct phenotypical alterations observed in CBK hearts. To test this hypothesis, CBK (and littermate control) mice were administered with the selective REV-ERBα/β agonist SR-9009 (100 mg·kg−1·day−1 for 8 days). SR-9009 administration was sufficient to normalize cardiac glycogen synthesis rates, cardiomyocyte size, interstitial fibrosis, and contractility in CBK hearts (without influencing mitochondrial complex activities, nor normalizing substrate oxidation and Akt/mTOR/GSK3β signaling). Collectively, these observations highlight a role for REV-ERBα/β as a mediator of a subset of circadian clock-controlled processes in the heart.
NEW & NOTEWORTHY Circadian clocks are composed of more than 10 interconnected transcriptional modulators, all of which have the potential to influence the cardiac transcriptome (and ultimately cardiac processes). Previous studies indicate that cardiomyocyte-specific BMAL1 knockout (CBK) mice exhibit a dysfunctional circadian clock (including decreased REV-ERBα/β expression) in the heart, associated with abnormalities in cardiac mitochondrial function, metabolism, signaling, and contractile function. Here we highlight decreased REV-ERBα/β as a mediator of glycogen synthesis, cardiomyocyte size, interstitial fibrosis, and contractile function abnormalities observed in CBK hearts.
Keywords: chronobiology, gene expression, heart, metabolism, mitochondria
INTRODUCTION
Virtually all biological processes are influenced by time of day. Day/night differences have been reported at whole body (e.g., behavior), organ (e.g., endocrine), and cellular (e.g., transcription) levels (45). The heart must contend with dramatic fluctuations in workload and neurohumoral stimuli over the course of the day, many of which are associated with sleep/wake and fasting/feeding cycles (11, 15). Therefore, it is not surprising that cardiac signaling [e.g., phosphorylation status of signal transduction kinases, such as Akt, AMP-activated protein kinase (AMPK), and mammalian target of rapamycin (mTOR)], electrophysiology (e.g., R-R and QTc intervals, as well as heart rate variability), metabolism (e.g., substrate reliance), and contractility (e.g., diastolic function) change over a 24-h period (53). Classically, daily fluctuations in many of these cardiac processes have been attributed to extra-cardiac stimuli/stresses, such as shear stress, autonomic/sympathetic tone, and various endocrine factors (33, 37, 48). More recently, circadian clocks have emerged as cell-autonomous molecular timekeeping mechanisms that temporally govern biological processes, many of which are critical for normal cardiac function (45). This is underscored by observations that genetic disruption of the murine circadian clock, either germline or cardiomyocyte specific, results in an age-onset cardiomyopathy and reduced lifespan (27, 52).
Circadian clocks are transcriptionally based molecular mechanisms composed of a series of positive and negative feedback loops (45). At the core of the mammalian mechanism are two transcription factors, BMAL1 and CLOCK (it is noteworthy that NPAS2 appears to have functional redundancy with CLOCK) (16, 21, 35). Upon binding to E-boxes, BMAL1:CLOCK heterodimers typically induce target genes, including a number of core clock components that form negative feedback loops, such as period (PER1/2/3), cryptochrome (CRY1/2), and REV-ERB (REV-ERBα/β) isoforms (26, 32, 40). More specifically, PER:CRY heterodimers bind directly to BMAL1:CLOCK, forming an inactive complex (26, 40). In contrast, REV-ERBα/β binds to the BMAL1 promoter, resulting in transcriptional repression (32). Importantly, oscillations in the activity/levels of clock components have a periodicity of ∼24 h, and positive components (BMAL1 and CLOCK) are essentially antiphase to negative components (PER, CRY, and REV-ERBα/β) (45). Circadian clock components not only target expression/activity of each other but also modulate expression of genes whose protein products do not directly feed back on the circadian clock mechanism. These are termed clock-controlled genes (CCGs). Between 3% and 10% of an organ’s transcriptome appears to be circadian regulated, and proteins encoded by CCGs impact a wide variety of critical cellular functions, ranging from transcription and translation to ion homeostasis, signal transduction, and metabolism (55).
Because of functional redundancy between many core circadian clock components, complete disruption of the mechanism consequent to single gene manipulations is possible only when specific components are targeted. For example, genetic manipulation of the BMAL1:CLOCK heterodimer typically results in disruption of the entire circadian clock mechanism (16, 21). Phenotypical characterization of cardiomyocyte-specific BMAL1-knockout (CBK) and cardiomyocyte-specific CLOCK mutant [CCM; involves overexpression of a dominant negative CLOCK mutant, thus overcoming neuronal PAS domain protein 2 (NPAS2) redundancy] mice has revealed that the circadian clock mechanism in the heart governs fundamentally important processes, including insulin signaling, substrate use, and Na+/K+ channel activity (5, 9, 29, 38, 39, 46). Moreover, unbiased transcriptomic analyses suggest that the cardiomyocyte circadian clock influences between 5 and 10% of the cardiac transcriptome (5, 52). What remains less clear are the molecular links between the clock mechanism and the processes that it governs. Genetic manipulation of BMAL1 and/or CLOCK alters expression of virtually all clock components, making it difficult to define which clock component serves as a mechanistic link to a distinct gene target and/or biological function (5, 52). Examples include REV-ERBα and REV-ERBβ, which are chronically repressed in the heart following disruption of the cardiomyocyte circadian clock (5, 52). Here, we hypothesized that decreased REV-ERBα/β activity observed in CBK hearts is responsible for distinct phenotypic alterations. We report that the selective REV-ERBα/β agonist SR-9009 attenuates abnormalities in glycogen synthesis, cardiomyocyte size, interstitial fibrosis, and contractile function that are observed in CBK hearts in the absence of effects on mitochondrial complex activities nor normalization of substrate oxidation and Akt/mTOR/GSK3β signaling. Collectively, these observations highlight REV-ERBα/β as an important mechanistic link between the cardiomyocyte circadian clock and distinct cardiac processes.
MATERIALS AND METHODS
Mice.
The present study used cardiomyocyte-specific Bmal1-knockout (CBK; Bmal1flox/flox/MHCαCre+/−) and littermate control (CON; Bmal1flox/flox/MHCαCre−/−) mice, as described previously (10). All experimental mice were male and were housed at the Center for Comparative Medicine at the University of Alabama at Birmingham (UAB), under temperature-, humidity-, and light-controlled conditions. A strict 12-h:12-h light-dark cycle regime was enforced [lights on at 6 am; zeitgeber time (ZT) 0]; the light-dark cycle was maintained throughout these studies, facilitating investigation of diurnal variations (as opposed to circadian rhythms). Mice were housed in standard microisolator cages and received food and water ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
SR-9009 administration.
Mice were administered with the REV-ERBα/β dual agonist SR-9009 (Cayman) at 100 mg·kg−1·day−1 ip. The agonist was dissolved in a DMSO/Kolliphor/water vehicle (10:15:75; % by volume). Mice received either SR-9009 or vehicle at a volume of 10 mg·kg−1·day−1 ip. For the acute (“disruption”) study, SR-9009 or vehicle was administered once at ZT0, followed by heart isolation 3 h later (i.e., ZT3). For the chronic (“normalization”) study, SR-9009 or vehicle was administered at ZT9 for a total of 8 days, followed by heart isolation 6 h after the last injection (i.e., ZT15 on day 8).
Quantitative RT-PCR.
RNA was extracted from hearts using standard procedures (8). Candidate gene expression analysis was performed by quantitative RT-PCR, using previously described methods (18, 20). For quantitative RT-PCR, specific Taqman assays were designed for each gene from mouse sequences available in GenBank or purchased from Applied Biosystems. All quantitative RT-PCR data are presented as fold change from an indicated control group.
Western blot analysis.
Qualitative analysis of protein expression and phosphorylation status was performed via standard Western blotting procedures as described previously (9). Briefly, 10–30 µg protein lysate was separated on polyacrylamide gels and transferred to PVDF membranes. Membranes were probed for the following targets: REV-ERBα (Cell Signaling, 13418) REV-ERBβ (Proteintech, 13906-1-AP), p-GSSer461 (Cell Signaling, 3891), GS (Cell Signaling, 3886), GP (Agrisera AS09455), p-GSK3βSer9 (Cell Signaling, 9336), GSK3α/β (Santa Crutz 7291), p-AKTSer473 (Cell Signaling, 9271), AKT (Cell Signaling, 9272), p-mTORSer2448 (Cell Signaling, 2971), mTOR (Cell Signaling 2983), LC3I/II (Cell Signaling, 12741), p62 (Novus Biologicals, H00008878-M01), STBD1 (Proteintech, 11842-1-AP), p-ERK1/2Thr202/Tyr204 (Cell Signaling, 9101), p-SMAD3(Ser423/Ser425) (Cell Signaling, 9520), and total OXPHOS Complexes (Abcam, ab110413). Rabbit and mouse HRP-conjugated secondary antibodies (Cell Signaling, 7074 and Santa Cruz sc-2005 respectively) were used for chemiluminescent detection with Luminata Forte Western Blotting substrate (Millipore, WBLUF0100). All densitometry data were normalized to amido black staining. Importantly, due to the nature of time course studies, to minimize the contribution that position on the gel might have on outcomes, samples were randomized on gels; samples were reordered postimaging, only for the sake of illustration of representative images (note, all bands for representative images for an individual experiment were from the same gel; original images are presented in Supplemental Fig. S1 (available online at 10.6084/m9.figshare.12030411).
RNA sequencing.
Transcriptomic analysis was performed in biventricular samples through the use of RNA sequencing in the UAB Genomics Core facility. The quality of the RNA samples was tested using the Agilent BioAnalyzer, and RNA with RIN values >7.0 were used in downstream library preparation. The RNA was DNAse treated before library preparation. The RNA-sequencing libraries were generated using the NEBNext Ultra II RNA kit (NEB, Ipswich, MA), following the manufacturer’s protocol. The resulting libraries were sequenced on the Illumina NextSeq 500 (Illumina, Inc., San Diego, CA) using paired end 75-bp sequencing reads per standard methods.
Mitochondrial complex activities.
Mitochondrial complex activities were assessed as recently described (1). Briefly, snap-frozen heart tissues were pulverized in a liquid nitrogen and subsequently homogenized in MAS buffer (70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, and 2 mM HEPES, pH 7.4; 10 μL/mg tissue) using a glass-glass dounce homogenizer. Homogenates were centrifuged at 1,000 g for 10 min at 4°C, followed by supernatant collection. Protein concentration was determined by DC Protein Assay (Bio-Rad). Homogenates were diluted and loaded into Seahorse XF96 microplate (Agilent, Santa Clara, CA) in 20 µL of MAS (final concentration of 1 µg/well). The loaded plate was centrifuged at 2,000 g for 20 min at 4°C. After centrifugation, 160 µL of MAS prepared with cytochrome c (10 µM), and alamethicin (10 µg/ml) was added to each well. Substrate concentrations to measure complex activities were as follows: 1 mM NADH (C-I), 10 mM succinate with 2 μM rotenone (C-II), 0.5 mM duroquinol (C-III), or 2 mM ascorbate with 0.5 mM TMPD (C-IV). Complex inhibitors were used at the following concentrations: 2 µM rotenone (C-I), 10 µM antimycin A (AA) (C-II and C-III), or 20 mM azide (C-IV). Citrate synthase activity was assessed as described previously (1).
Working mouse heart perfusions.
Myocardial substrate use was measured ex vivo through isolated working mouse heart perfusions, as described previously (5, 9, 46, 47). All hearts were perfused in the working mode (nonrecirculating manner) for 40 min with a preload of 12.5 mmHg and an afterload of 50 mmHg. Standard Krebs-Henseleit buffer was supplemented with 8 mM glucose, 0.4 mM oleate conjugated to 3% BSA (fraction V, fatty acid-free; dialyzed), 10 μU/ml insulin (basal/fasting concentration), 2 mM β-hydroxybutyrate, 0.2 mM acetoacetate, 0.05 mM l-carnitine, and 0.13mM glycerol. Metabolic fluxes were assessed through the use of distinct radiolabeled tracers: 1) [U-14C]glucose (0.12mCi/L; glycolysis, glucose oxidation); and 2) [9,10-3H]oleate (0.067 mCi/L; β-oxidation). Measurements of cardiac metabolism (e.g., oxygen consumption) and function (e.g., cardiac power) were monitored as described previously (5, 9, 46, 47). At the end of the perfusion period, hearts were snap-frozen in liquid nitrogen and stored at −80°C before analysis. Data are presented as steady-state values (i.e., values during the last 10 min of the perfusion protocol). Heart perfusion conditions were chosen for consistency with a prior study describing the metabolic phenotype of CBK hearts (52).
Glycogen content.
Glycogen content was assessed using a spectrophotometric-based assay, as described previously (31).
Histologic assessment.
Cross-sections from the medial heart were taken immediately upon removal of heart and fixed in formalin for 24 h. Wheat germ agglutinin (WGA) staining was used for measurement of myocyte cross-sectional area; at least 45 myocytes were assessed per heart using ImageJ software (NIH), as described previously (23). Picrosirius Red staining of collagen fibers was used for semiquantitative measurement of left ventricular fibrosis, using ImagePro Plus software (Media Cybernetics, Inc., Rockville, MD), as described previously (10).
Statistical analysis.
Statistical analyses were performed using two-way ANOVA, as described previously (5, 6). Briefly, analyses were performed on Prism statistical software to investigate main effects of time, genotype, and/or treatment, followed by Bonferroni post hoc analyses for pairwise comparisons (indicated in the figures). In all analyses, the null hypothesis of no model effects was rejected at P < 0.05. RNA-seq data from each experimental group (CBK ± SR-9009; CON ± SR-9009) were curated into an Excel file list. This list was imported into GeneSpring Version 14.9-Build 11939 (Agilent Technologies, Inc.), generating expression data from 26,988 entities. First, a new experiment was launched using NGS analyses, on a gene expression experiment type, with input parameters set as mouse and linear scale. File format validation was set as tab separator, with no text qualifier, no missing value indicators, and no comment indicators. Data were annotated by Ensembl ID using the Genes and Transcript Model annotation source, Mouse mm10 (UCSC) build, and Ensembl Genes Annotation (2015.10.05). All original annotations were included with the import. Experiment parameters of genotype (CON or CBK) and treatment (SR-9009 or vehicle) were defined for each sample. Preprocess baseline options were set to the median of all samples. Next, an interpretation was created based on experimental parameters (genotype and treatment), and the profile plot display mode was set to categorical, with the conditions defined to include both comparator conditions, and such that we could view the chip averages or individual data points. We then performed a statistical analysis using all entities and genotype-treatment interpretation by two-way analysis of variance (ANOVA) with an asymptotic P value computation, no multiple testing corrections, and across four condition pairs. We selected entities at P < 0.01 for treatment, treatment/genotype, and genotype. From these lists, the entity IDs, normalized data values, fold change, and statistical P values from GeneSpring were exported into a Microsoft Excel file. The Ensembl gene IDs were imported into DAVID (Database for Annotation, Visualization, and Integrated Discovery) version 6.8 for Gene Ontology (GO) biological processes analyses (22). For cell type-specific analyses, we performed in silico digital cytometry using CIBERSORTx, a machine learning platform designed to infer single-cell abundance and gene expression profiles from bulk tissue samples (30). First, a reference data set was obtained from open access fluorescence-activated cell sorting (FACS) and RNA-seq data of single cell populations isolated from male C57Bl/6 mouse left ventricular tissue (GSE109774) (44). Raw gene expression data from this reference population was then imported into CIBERSORTx to generate a signature matrix consisting of barcode genes that discriminate individual cell types of interest (CM = cardiomyocyte, ET = endothelial cell, F = fibroblast). In silico cell enumeration and transcriptional analyses were performed from our bulk tissue samples from each of our four experimental groups (CON ± SR-9009; CBK ± SR-9009) using the impute cell fraction and impute group level gene expression modules in CIBERSORTx. Cells from the reference data set were visualized using two-dimensional t-distributed Stochastic Neighbor Embedding (RStudio version 1.2.5001). For this cell-specific gene expression from whole heart RNA-seq, data were imported into GeneSpring and differentially expressed genes plotted using heat maps and fold change scatter plots.
RESULTS
Aberrant temporal expression of circadian clock genes in CBK hearts.
We initially assessed gene expression of 10 core circadian clock components (bmal1, clock, npas2, per1, per2, per3, cry1, cry2, rev-erbα, and rev-erbβ) and two established direct clock-controlled genes (dbp and e4bp4) in hearts isolated from CBK and littermate control (CON) mice at 3-h intervals across a 24-h period. Cosinor analysis of the data revealed significant 24-h oscillations in all genes investigated in CON hearts (Fig. 1A and Table 1) in a temporal pattern that is consistent with operation of a functional circadian clock. Importantly, CBK hearts exhibit either a significant attenuation (i.e., decreased amplitude) or loss of oscillation in all clock components (Fig. 1A and Table 1). Moreover, daily average values for bmal1, per1, per3, rev-erbα, rev-erbβ, and dbp are significantly decreased in CBK hearts (CON-to-CBK ratio of 2.6, 1.4, 5.4, 2.3, 3.0, and 2.8, respectively), whereas clock, npas2, cry1, cry2, and e4bp4 are significantly increased (CBK-to-CON ratio of 1.5, 2.6, 1.9, 1.2, and 2.2, respectively; Fig. 1A and Table 1). When differential expression is averaged for core clock components within a redundant group (based on established transcriptional targets), the period (per1/2/3) and rev-erb (rev-erbα/β) isoforms are decreased by an average of 2.7- and 2.7-fold, respectively, whereas clock/npas2 and cryptochrome (cry1/2) isoforms are increased by an average of 2.0- and 1.5-fold respectively. As such, of the core circadian clock components investigated, period and rev-erb isoforms were differentially expressed to the greatest extent in CBK hearts (relative to CON hearts) and were at a similar magnitude of change compared with bmal1 (the gene specifically targeted in CBK hearts). It is noteworthy that per1 and per2 differential expression is relatively minor (1.4- and 1.1-fold, respectively) compared with per3 (5.4-fold). For these reasons, REV-ERBα and -β were considered the most consistently differentially expressed clock components in CBK hearts and, therefore, were the subjects of subsequent investigation.
Fig. 1.


Cardiomyocyte-specific deletion of BMAL1 alters diurnal variations of circadian clock components and output genes in the mouse heart. A: hearts were isolated from cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice at zeitgeber time (ZT)0, ZT3, ZT6, ZT12, ZT15, ZT18, and ZT21 (ZT0 and ZT24 are identical time points and data; data are duplicated only for illustration purposes, but not for statistical analysis), followed by assessment of gene expression by RT-PCR: bmal1 (i), clock (ii), npas2 (iii), per1 (iv), per2 (v), per3 (vi), cry1 (vii), cry2 (viii), rev-erbα (ix), rev-erbβ (x), dbp (xi), and e4bp4 (xii). B: hearts were isolated from CBK and CON mice at time points ZT0, ZT4, ZT8, ZT12, ZT16, and ZT20 (ZT0 and ZT24 are identical time points and data; data are duplicated only for illustration purposes for the line graphs, but not for statistical analysis), followed by assessment of protein expression by Western blotting (normalized to amido black staining): REV-ERBα (i and ii) and REV-ERBβ (i and iii). Data are reported for 5–8 mice/experimental group. Main effects for either time, genotype, and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs CBK (red squares) within a ZT.
Table 1.
Cosinor analysis of circadian clock genes and proteins in hearts isolated from CBK and littermate CON mice
| CON |
CBK |
|||||
|---|---|---|---|---|---|---|
| Mesor | Amplitude | Acrophase | Mesor | Amplitude | Acrophase | |
| Gene | ||||||
| bmal1 | 10.24 ± 0.40 | 11.29 ± 0.56 | 1.04 ± 0.19 | 3.84 ± 0.18* | 4.37 ± 0.26* | 1.03 ± 0.23 |
| clock | 1.41 ± 0.04 | 0.37 ± 0.06 | 0.98 ± 0.66 | Not rhythmic | Not rhythmic | Not rhythmic |
| npas2 | 3.32 ± 0.21 | 2.76 ± 0.29 | 1.28 ± 0.41 | 8.46 ± 0.41* | 2.12 ± 0.58 | 7.99 ± 1.04* |
| per1 | 3.09 ± 0.24 | 2.41 ± 0.33 | 11.36 ± 0.54 | 2.17 ± 0.14* | 1.46 ± 0.20* | 11.19 ± 0.53 |
| per2 | 3.78 ± 0.20 | 3.20 ± 0.28 | 13.63 ± 0.35 | 3.42 ± 0.18 | 2.33 ± 0.25* | 14.21 ± 0.41 |
| per3 | 10.40 ± 1.18 | 12.12 ± 1.64 | 13.39 ± 0.53 | 1.93 ± 0.18* | 2.47 ± 0.25* | 12.96 ± 0.39 |
| cry1 | 2.48 ± 0.17 | 1.41 ± 0.25 | 18.95 ± 0.65 | Not rhythmic | Not rhythmic | Not rhythmic |
| cry2 | 1.58 ± 0.06 | 0.46 ± 0.09 | 13.46 ± 0.71 | Not rhythmic | Not rhythmic | Not rhythmic |
| rev-erbα | 5.03 ± 0.35 | 4.90 ± 0.51 | 6.85 ± 0.38 | 2.03 ± 0.20* | 2.00 ± 0.29* | 8.53 ± 0.54* |
| rev-erbβ | 2.48 ± 0.16 | 1.18 ± 0.23 | 11.29 ± 0.74 | 0.80 ± 0.05* | 0.68 ± 0.08* | 10.62 ± 0.42 |
| dbp | 6.86 ± 0.46 | 7.91 ± 0.64 | 10.72 ± 0.31 | 2.34 ± 0.18* | 2.46 ± 0.25* | 9.91 ± 0.39 |
| e4bp4 | 2.08 ± 0.13 | 1.16 ± 0.18 | 21.99 ± 0.59 | Not rhythmic | Not rhythmic | Not rhythmic |
| Protein | ||||||
| REV-ERBα | 3.08 ± 0.27 | 2.77 ± 0.39 | 9.96 ± 0.53 | 1.86 ± 0.20* | 1.68 ± 0.28* | 10.16 ± 0.63 |
| REV-ERBβ | Not rhythmic | Not rhythmic | Not rhythmic | Not rhythmic | Not rhythmic | Not rhythmic |
Values are means ± SE. CBK, cardiomyocyte-specific BMAL1-knockout; CON, control. Not rhythmic, no significant rhythmicity observed following cosinor analysis.
P < 0.05 for CON vs. CBK.
Although BMAL1 protein levels have been investigated previously in CBK and CON hearts (52), REV-ERBα and/or REV-ERBβ protein levels have not. Accordingly, we assessed protein levels of REV-ERBα and REV-ERBβ in CBK and CON hearts isolated at 4-h intervals across a 24-h period. Time-of-day-dependent variations in REV-ERBα, but not REV-ERBβ, significantly fit a cosine curve (with a periodicity of 24 h) in CON hearts; this oscillation was significantly attenuated (i.e., 39% amplitude decrease) in CBK hearts (Fig. 1B and Table 1). Moreover, time-of-day-independent protein levels for REV-ERBα and REV-ERBβ were significantly decreased in CBK hearts (relative to CON hearts) by 36% and 18%, respectively (Fig. 1B). Collectively, these data are consistent with disruption of the circadian clock in CBK hearts, which is associated with significant repression of REV-ERBα/β protein levels.
Pharmacologic activation of REV-ERBα/β at ZT0 modulates clock genes in the heart.
Given that both REV-ERBα and REV-ERBβ are repressed in CBK hearts (Fig. 1B), we reasoned that lower activity of these transcription factors may contribute toward distinct phenotypical changes described previously in this model of cardiomyocyte circadian clock disruption. To address this possibility, we employed the use of the REV-ERBα/β dual agonist SR-9009 as a way to reactivate these nuclear receptors in CBK hearts. As an initial proof-of-principle study, CON and CBK mice were administered with SR-9009 at the beginning of the light phase (i.e., ZT0), when REV-ERBα/β activity is predicted to normally be low, based on 1) low REV-ERBα/β protein levels in CON hearts at ZT0 (Fig. 1B) and 2) high levels of e4bp4 mRNA at ZT0 [which is repressed by REV-ERBα/β (49); Fig. 1A]. Three hours after CBK and CON littermates were treated with SR-9009 or vehicle (i.e., ZT3), hearts were isolated for subsequent gene expression analysis. In both CON and CBK hearts, SR-9009 administration significantly increased expression of pdk4 [a predicted REV-ERBα/β target gene (54); Fig. 2A]. Interrogation of distinct core circadian clock components and clock-controlled genes revealed anticipated genotype main effects (Fig. 2B). Moreover, SR-9009 administration repressed clock and e4bp4 expression and concomitantly induced per1 (i.e., SR-9009 main effect; Fig. 2B). Collectively, these data indicate that SR-9009 treatment at ZT0 (when REV-ERBα/β activity is normally low) acutely alters circadian clock genes in the heart (i.e., perturbs the clock).
Fig. 2.

Effects of BMAL1 deletion and/or acute SR-9009 administration on cardiac gene expression from cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg/kg ip) or vehicle (Veh) at zeitgeber time (ZT)0, followed by heart isolation 3 h later (i.e., ZT3). A: pdk4 expression was assessed by RT-PCR. B: bmal1 (i), clock (ii), per1 (iii), cry2 (iv), rev-erbα (v), rev-erbβ (vi), dbp (vii), and e4bp4 (viii) were assessed by RT-PCR. Data are reported as means ± SE for 4–11 mice/experimental group. Main effects for either SR-9009, genotype, and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs. CBK (red squares) within a treatment group. $P < 0.05 for vehicle vs. SR-9009 administration within a genotype.
Minimal effects of pharmacological activation of REV-ERBα/β at ZT9 on clock gene expression in the heart.
Consistent with the primary goal of the study (to investigate the effects of REV-ERBα/β reactivation/normalization in CBK hearts), CON and CBK mice were treated with SR-9009 toward the end of the light phase (ZT9), when REV-ERBα/β activity is normally predicted to be high, given that 1) cosinor analysis indicates peak protein levels of REV-ERBα around ZT10 in CON hearts (Table 1) and 2) e4bp4 mRNA, which is repressed by REV-ERBα/β (49), exhibits the lowest expression levels in CON hearts around ZT10 (Table 1). Accordingly, mice were treated with SR-9009 or vehicle once daily at ZT9 (for 8 consecutive days); 6 h after the last treatment (i.e., ZT15), hearts were isolated for subsequent gene expression analysis. This intervention induced pdk4 in the heart, independent of genotype (i.e., SR-9009 main effect; Fig. 3A). In contrast, this treatment regime had minimal effects on expression of circadian clock components/output genes in the heart [with the exception of e4bp4, which was slightly increased; SR-9009 main effect (Fig. 3B)]. Collectively, these data indicate that SR-9009 treatment at ZT9 (when REV-ERBα/β activity is usually high) does not perturb the circadian clock in the heart.
Fig. 3.

Effects of BMAL1 deletion and/or chronic SR-9009 administration on cardiac gene expression. Cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15). A: pdk4 expression was assessed by RT-PCR. B: bmal1 (i), clock (ii), per1 (iii), cry2 (iv), rev-erbα (v), rev-erbβ (vi), dbp (vii), and e4bp4 (viii) were assessed by RT-PCR. Data are reported as means ± SE for 13–15 mice/experimental group. Main effects for either SR-9009, genotype, and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs. CBK (red squares) within a treatment group. $P < 0.05 for vehicle vs. SR-9009 administration within a genotype.
Transcriptome-wide effects of REV-ERBα/β agonist in the heart.
Given that BMAL1, REV-ERBα, and REV-ERBβ are transcription factors, we reasoned that defining the transcriptome-wide effects of genetic deletion of BMAL1 (i.e., CBK) and pharmacological activation of REV-ERBα/β (i.e., SR-9009) may provide insight regarding the importance of these nuclear receptors in clock control of distinct cardiac processes. Accordingly, RNA-seq was performed for hearts isolated from CON and CBK mice that had been administered with SR-9009 or vehicle at ZT9 for 8 days (i.e., the “normalization” protocol). A two-way ANOVA analysis revealed main effects of genotype [of the 3,266 differentially expressed genes, 1,746 genes were induced in CBK hearts, and 1,520 genes were repressed in CBK hearts; Supplemental Table S1 (available online at 10.6084/m9.figshare.11306999)] and SR-9009 administration [of the 242 differentially expressed genes, 115 genes were induced by SR-9009, and 120 genes were repressed by SR-9009; Supplemental Table S2 (available online at 10.6084/m9.figshare.11306993)]. Consistent with prior studies (52), gene ontology analysis indicated that genetic deletion of BMAL1 in cardiomyocytes influenced biological processes such as cell signaling, growth/remodeling, transport, and metabolism, whereas SR-9009 administration influenced inflammation, cell signaling, metabolism, and transcription (Fig. 4A). The two-way ANOVA also revealed that 91 genes exhibited a significant genotype-treatment interaction [Supplemental Table S3 (available online at 10.6084/m9.figshare.11306996)]; these genes cluster in processes such as growth/remodeling, cell signaling, transcription, and transport (Fig. 4A). Comparison of differentially expressed genes based on genotype and SR-9009 administration main effects revealed that of the 3,487 total genes that were affected, 1,236 were either partially or fully normalized in CBK hearts in response to SR-9009 administration (i.e., if a gene was induced in CBK hearts, then SR-9009 decreased expression; or if a gene was repressed in CBK hearts, then SR-9009 increased expression). When stringent twofold cutoffs were applied, 26 “normalized” genes were identified (Table 2); examples of these genes include slc1a7, rgs1, cd27, and trim40 (Fig. 4B).
Fig. 4.


Global effects of BMAL1 deletion and/or SR-9009 administration on the cardiac transcriptome. Cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15). Gene expression was assessed by RNA-seq. A: gene ontology analysis for genotype main effects (i), SR-9009 administration main effects (ii), and genotype-SR-9009 interaction effects (iii). B: examples of genes partially normalized in CBK hearts following SR-9009 administration. Data are reported as means ± SE for 4 mice/experimental group. Main effects for either genotype and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs. CBK (red squares) within a treatment group. C: cell type-specific gene expression changes were interrogated by importing RNA-seq data into CIBERSORTx, an in silico digital cytometry platform (i), to reveal distinct clustering of genes in different heart cell types: CM, cardiomyocytes; ET, endothelial cells; F, fibroblasts (ii). Heat maps were generated (iii), and normalized genes in CBK-SR hearts are shown using scatter plots (green lines = 2-fold change; iv). Individual data values are in Supplemental Table S4.
Table 2.
Expression of genes that were partially normalized in CBK hearts following SR-9009 administration for 8 days at ZT9
| ENSEMBL ID | Gene Description | CBK Vehicle vs. CON Vehicle (Fold Change), | CBK SR-9009 vs. CBK Vehicle (Fold Change) |
|---|---|---|---|
| Up in CBK vehicle ≥ 2.0-fold | Down in CBK SR-9009 ≤ −2.0-fold | ||
| ENSMUSG00000101144 | Predicted gene 29054 | 5.6 | −3.7 |
| ENSMUSG00000098194 | Predicted gene 7286 | 4.4 | −2.4 |
| ENSMUSG00000024211 | Glutamate receptor, metabotropic 8 | 3.2 | −2.1 |
| ENSMUSG00000044322 | Desmocollin 1 | 3.1 | −2.2 |
| Down in CBK vehicle up to −2.0-Fold | Up in CBK SR-9009 ≥ 2.0-Fold | ||
| ENSMUSG00000041771 | Solute carrier family 24 (sodium/potassium/calcium exchanger), member 4 | −2.9 | 2.1 |
| ENSMUSG00000008932 | Solute carrier family 1 (glutamate transporter), member 7 | −3.5 | 2.6 |
| ENSMUSG00000085965 | RIKEN cDNA 2310016D23 gene | −3.6 | 2.4 |
| ENSMUSG00000092051 | Predicted gene 17229 | −3.7 | 2.2 |
| ENSMUSG00000016179 | Calcium/calmodulin-dependent protein kinase I gamma | −3.8 | 2.0 |
| ENSMUSG00000073399 | Tripartite motif-containing 40 | −4.0 | 2.6 |
| ENSMUSG00000086935 | Predicted gene 15956 | −4.5 | 2.0 |
| ENSMUSG00000044405 | Adipogenin | −4.6 | 2.0 |
| ENSMUSG00000105867 | Predicted gene 42517 | −4.6 | 2.2 |
| ENSMUSG00000025481 | Urate (5-hydroxyiso-) hydrolase | −4.8 | 2.3 |
| ENSMUSG00000026358 | Regulator of G protein signaling 1 | −5.1 | 2.5 |
| ENSMUSG00000050071 | Brain expressed X-linked 1 | −5.7 | 2.2 |
| ENSMUSG00000030336 | CD27 antigen | −6.0 | 3.4 |
| ENSMUSG00000030483 | Cytochrome P450, family 2, subfamily b, polypeptide 10 | −6.1 | 2.5 |
| ENSMUSG00000043999 | G protein-coupled receptor 75 | −6.1 | 2.5 |
| ENSMUSG00000061718 | Protein phosphatase 1, regulatory inhibitor subunit 1B | −7.0 | 2.1 |
| ENSMUSG00000051811 | Cytochrome c oxidase subunit 6B2 | −7.7 | 2.1 |
| ENSMUSG00000086645 | Predicted gene 15743 | −9.0 | 2.5 |
| ENSMUSG00000103476 | Predicted gene, 34302 | −9.1 | 2.1 |
| ENSMUSG00000079620 | Mucin 4 | −11.4 | 2.1 |
| ENSMUSG00000050423 | Protein phosphatase 1, regulatory subunit 3G | −18.6 | 2.0 |
| ENSMUSG00000028307 | Aldolase B, fructose-bisphosphate | −89.1 | 2.3 |
CBK, cardiomyocyte-specific BMAL1-knockout; CON, control; ZT9, zeitgeber time 9. Included are genes whose expression changed ≥2-fold in CBK vs. CON hearts (from vehicle-treated mice) and the same gene changed in the opposite direction ≥2-fold in CBK hearts following SR-9009 administration. RNA-seq data were used for this analysis.
To infer cell-specific changes from the whole heart RNA-Seq data, we used in silico digital cytometry analyses. The workflow is shown in Fig. 4Ci. Based on these analyses, we found distinct gene clustering attributable to cardiomyocytes (CM), endothelial cells (ET), and fibroblasts (F) (Fig. 4Cii). We next identified which cell type-enriched genes were differentially expressed in CON versus CBK hearts; 326 differentially expressed genes were identified in cardiomyocytes, 938 differentially expressed genes were identified in endothelial cells, and 1,500 differentially expressed genes were identified in fibroblasts (Fig. 4Ciii, Supplemental Table S4; available online at 10.6084/m9.figshare.12003210). Moreover, 158 genes (out of the 326 differentially expressed genes) were normalized in CBK cardiomyocytes following SR-9009 treatment, 421 genes (out of the 938 differentially expressed genes) were normalized in endothelial cells, and 922 (out of the 1,500 differentially expressed genes) were normalized in fibroblasts (Fig. 4Civ and Supplemental Table S4). Thus these RNAseq results suggest that gene expression changes in CBK hearts occur within different cell types.
REV-ERBα/β activation in CBK hearts selectively influences glycogen synthesis in CBK hearts.
Prior studies suggest that CBK hearts exhibit impairments in multiple metabolism-related parameters, including mitochondrial complex activities and substrate selection (19, 24, 29, 52). Moreover, SR-9009 increases mitochondrial biogenesis in skeletal muscle (50) and has previously been suggested to influence cardiac metabolism (based on transcriptional changes) (54). Indeed, our RNA-seq studies suggest that SR-9009 administration influences a number of metabolism-related genes in the heart, including partial restoration of cox6b2 mRNA (a complex IV subunit; Table 2). Collectively, these observations led us to hypothesize that SR-9009 may influence mitochondrial function in CBK hearts. Initial investigation of mitochondrial complex protein levels revealed decreased complex II levels in CBK hearts (i.e., genotype main effect; Fig. 5A). Somewhat surprisingly, complex IV levels were significantly higher in CBK hearts (i.e., genotype main effect), and complex III levels were significantly decreased by SR-9009 administration (i.e., SR-9009 main effect), whereas complex I levels were not influenced by either genotype or SR-9009 administration (Fig. 5A). Assessment of mitochondrial complex activities revealed increased complex IV activity in CBK hearts (i.e., genotype main effect) in the absence of significant differences in activity of complexes I, II, or III (Fig. 5B). Finally, citrate synthase activity was assessed, revealing no significant effects of either genotype or SR-9009 [although a trend for genotype main effect was observed (P = 0.052); Fig. 5C]. Importantly, no significant genotype-SR-9009 interactions were observed for complex activities/levels or citrate synthase activity (Fig. 5, A–C). Collectively, these data suggest that SR-9009 (and therefore REV-ERBα/β) has minimal impact on mitochondrial complexes in the heart.
Fig. 5.

Effects of BMAL1 deletion and/or SR-9009 administration on mitochondrial parameters. Cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15). A: mitochondrial complex protein levels were assessed by Western blotting (and normalized to amido black). B: mitochondrial complex activities were assessed by following oxygen consumption rates in the presence of specific substrates. C: citrate synthase activity was assessed spectrophotometrically. Data are reported as means ± SE for 6–8 mice/experimental group. Main effects for either genotype or SR are reported at the top of each graph. ●, CON; red squares, CBK.
Consistent with our prior studies (29, 52), CBK hearts exhibit increased rates of fatty acid oxidation, concomitant with decreased rates of glucose oxidation, 14C-lactate release, and triglyceride synthesis (genotype main effects; Fig. 6, A and B). When substrate reliance is calculated, CBK hearts exhibit increased fatty acid oxidation reliance, whereas both glucose and other substrate [combination of unlabeled exogenous (i.e., β-hydroxybutyrate and acetoacetate) and endogenous (e.g., triglyceride and glycogen) substrates] oxidation reliance is decreased (genotype main effects; Fig. 6A). SR-9009 administration had no significant effect on rates of fatty acid oxidation, glucose oxidation, 14C-lactate release, or triglyceride synthesis in either CON or CBK hearts (Fig. 6, A and B). In contrast, a significant genotype-SR-9009 interaction was observed for glycogen synthesis; post hoc analysis revealed decreased glycogen synthesis in CBK vehicle hearts (compared with CON vehicle hearts) which, was reversed by SR-9009 administration (Fig. 6B,iii). Collectively, these observations suggest that REV-ERBα/β may influence cardiac glycogen metabolism.
Fig. 6.

Effects of BMAL1 deletion and/or SR-9009 administration on cardiac metabolism. Cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15). Metabolism was subsequently assessed in isolated working mouse hearts through use of radiolabeled tracers. A: oleate (i) and glucose (ii) oxidation rates as well as substrate reliance (iii). B: rates of [14C]lactate release (i), net triglyceride synthesis (ii), and net glycogen synthesis (iii). Data are reported as means ± SE for 6–8 mice/experimental group. Main effects for either genotype and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs. CBK (red squares) within a treatment group; $P < 0.05 for vehicle vs. SR administration within a genotype.
We next decided to investigate in greater depth the extent to which SR-9009 influences cardiac glycogen metabolism. First, we assessed glycogen content in freshly isolated hearts (i.e., hearts that were not subjected to ex vivo perfusions) from the four experimental groups. Contrary to expectation, we observed that SR-9009 significantly decreased glycogen content independent of genotype (i.e., SR-9009 main effect; Fig. 7A). Moreover, although not statistically significant, glycogen content tended to be higher in CBK hearts (relative to CON hearts; Fig. 7A). To interrogate the latter further, glycogen content was assessed in CON and CBK hearts collected at 3-h intervals over the course of the day. This analysis revealed main effects for both time and genotype; in the latter case, glycogen levels were statistically increased in CBK hearts (Fig. 7B). The net synthesis of glycogen is determined not only by the rate of synthesis (via glycogen synthase) but also by its degradation (via glycogen phosphorylase and glycophagy). Accordingly, we next investigated key components of these pathways at protein and posttranslational levels. Neither glycogen synthase nor phosphorylase total protein levels differed between the four experimental groups (Fig. 7, C and D). In contrast, the phosphorylation status of glycogen synthase at Ser461 (inhibitory site) tended to be decreased in CBK hearts (P = 0.084, genotype main effect when normalized to amido black; genotype main effect P value was 0.086 when normalized to total glycogen synthase levels; Fig. 7C). We have previously suggested that the cardiomyocyte circadian clock modulates cardiac glucose use via the Akt/mTOR/GSK3β signaling axis (29). Consistent with prior reports (29), Akt and mTOR phosphorylation (at Ser473 and Ser2448, respectively) was higher in CBK hearts, whereas GSK3β phosphorylation (at Ser9) was decreased (genotype main effect; Fig. 7, E–G). Interestingly, SR-9009 increased p-Akt levels (SR9009 main effect; Fig. 7E). In contrast, SR-9009 had no significant effect on either mTOR or GSK3β phosphorylation (Fig. 7, F and G). mTOR is an established repressor of autophagy, and we have previously reported attenuated autophagy in CBK hearts (29). Given that glycophagy impacts glycogen turnover, we next investigated autophagy/glycophagy components. Consistent with prior observations (29), p62 levels were increased in CBK hearts (genotype main effects) in the absence of alterations in LC3II (Fig. 7, H and I). In contrast, STBD1 levels were not altered in CBK hearts (Fig. 7J). SR-9009 did not significantly influence LC3II, p62, or STBD1 levels in the heart (Fig. 7, H–J). Collectively, these observations suggest that SR-9009 is unable to normalize perturbations in the Akt/mTOR/GSK3β signaling axis observed in CBK hearts.
Fig. 7.




Effects of BMAL1 deletion and/or SR-9009 administration on parameters involved in cardiac glycogen metabolism. A: cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15) and subsequent assessment of glycogen content. B: hearts were isolated from CBK and littermate control (CON) mice at ZT0, ZT3, ZT6, ZT12, ZT15, ZT18, and ZT21 (ZT0 and ZT24 are identical time points and data; data are duplicated only for illustration purposes, but not for statistical analysis), followed by assessment of glycogen content. C: CBK and littermate control (CON) mice challenged with either SR (100 mg·kg−1·day−1 ip) or vehicle (Veh) at ZT9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15) and subsequent assessment of protein expression by Western blotting (normalized to amido black staining): phosphorylated glycogen synthase (pGS; i and ii), total glycogen synthase (GS; i and iii), and the phospho-to-total ratio (pGS/GS; iv). D: samples from C used for assessment of glycogen phosphorylase (i and ii). E: samples from C used for assessment of phosphorylated Akt (p-Akt; i and ii), total Akt (Akt; i and iii), and the phospho-to-total ratio (p-Akt/Akt; iv). F: samples from C used for assessment of phosphorylated mTOR (p-mTOR; i and ii), total mTOR (mTOR; i and iii), and the phospho-to-total ratio (p-mTOR/mTOR. G: samples from C used for assessment of phosphorylated GSK3β (p-GSK3β; i and ii), total GSK3β (GSK3β; i and iii), and the phospho-to-total ratio (p-GSK3β/GSK3β; iv). H: samples from C used for assessment of LC3II (i and ii). I: samples from C used for assessment of p62 (i and ii). J: samples from C used for assessment of STBD1 (i and ii). Data are reported as means ± SE for 11–13 mice/experimental group. Main effects for SR-9009 and/or genotype are reported at the top of each graph.
SR-9009 influences adverse cardiac remodeling in CBK Mice.
CBK mice exhibit age-onset adverse cardiac remodeling, precipitating in development of a severe hypertrophic cardiomyopathy and reduced lifespan (52). We hypothesized that decreased REV-ERBα/β activity in CBK hearts potentially contributes toward this pathological phenotype. Although CBK mice do not exhibit contractile dysfunction in vivo (as assessed by echocardiography) at 16 wk of age (the age at which mice were investigated in the current study), adverse remodeling is observed at histological and molecular levels as well as during ex vivo assessment of contractility (52). Consistent with these previous reports, CBK hearts exhibit decreased rate pressure product (assessed ex vivo), increased cardiomyocyte size, and hypertrophic markers (anf, mhcβ), as well as increased interstitial fibrosis (Fig. 8, A–D). Interestingly, SR-9009 administration for 8 days (at ZT9) normalized rate pressure product in CBK mice and concomitantly decreased cardiomyocyte size and interstitial fibrosis (Fig. 8, A–D). SR-9009 did not affect these parameters in CON hearts (Fig. 8, A–D). Somewhat surprisingly, SR-9009 administration increased anf and mhcβ expression (genotype main effect; Fig. 8C). Neither genotype nor SR-9009 significantly influenced the phosphorylation status of ERK1/2 or SMAD3 (Figs. 8, E and F). Collectively, these observations are consistent with the concept that decreased REV-ERBα/β activity in CBK hearts contributes, at least partially, toward adverse cardiac remodeling.
Fig. 8.


Effects of BMAL1 deletion and/or SR-9009 administration on contractile function, markers of hypertrophy, and markers/mediators of fibrosis. Cardiomyocyte-specific BMAL1-knockout (CBK) and littermate control (CON) mice were challenged with either SR-9009 (SR; 100 mg·kg−1·day−1 ip) or vehicle (Veh) at zeitgeber time (ZT)9 for 8 consecutive days, followed by heart isolation 6 h after the last administration (i.e., ZT15). A: assessment of contractile function parameters in isolated working mouse hearts: cardiac power (i) and rate pressure product (ii). B: cardiomyocyte area assessment by wheat germ agglutinin (WGA) staining (i and ii). C: assessment of anf (i) and mhcβ (ii) expression by RT-PCR. D: interstitial fibrosis assessment by Picrosirius Red staining (i and ii). E: assessment of phosphorylated ERK1/2 (pERK1/2; i and ii), total ERK1/2 (ERK1/2; i and iii), and the phospho-to-total ratio (pERK1/2/ERK1/2; iv). F: assessment of phosphorylated SMAD3 (pSMAD; i and ii), total SMAD3 (SMAD3; i and iii), and the phospho-to-total ratio (pSMAD/SMAD3; iv). Data are reported as means ± SE for 7–14 mice/experimental group. Main effects for SR, genotype, and/or interaction are reported at the top of each graph. *P < 0.05 for CON (●) vs. CBK (red squares) within a treatment group; $P < 0.05 for vehicle vs. SR administration within a genotype; #P = 0.075 for vehicle vs. SR administration within a genotype.
DISCUSSION
The purpose of the present study was to investigate whether pharmacological activation of REV-ERBα/β normalizes distinct phenotypical alterations observed in the heart following cardiomyocyte-specific BMAL1 deletion (i.e., CBK mice). In doing so, we hypothesized that the contribution of REV-ERBα/β as a mediator of clock control of cardiac processes might be unmasked. Consistent with previously published observations, we report that CBK hearts exhibit profound transcriptomic alterations (including decreased rev-erbα/β expression) associated with perturbations in metabolism, cellular signaling, cardiomyocyte size, interstitial fibrosis, and contractility. Here, we report that administration of CBK mice with a REV-ERBα/β dual agonist (SR-9009) for only 8 days significantly attenuates abnormalities in glycogen synthesis, cardiomyocyte size, interstitial fibrosis, and contractile function in the absence of effects on mitochondrial function, substrate reliance, and the Akt/mTOR/GSK3β signaling axis. Collectively, these observations highlight REV-ERBα/β as a potential mechanistic link between the cardiomyocyte circadian clock and distinct cardiac processes.
Cell-autonomous circadian clocks have emerged as critical regulators of numerous biological functions (45). Being composed of more than 10 transcriptional modulators, the mammalian mechanism has the capability of modulating expression of thousands of genes in a temporally orchestrated manner (55). Although much progress has been made regarding the processes under circadian governance in the heart, unanswered questions remain regarding the mechanistic links involved. Here, we focus on REV-ERBα/β as putative mediators between the cardiomyocyte circadian clock and cardiac processes. Previous studies investigating the roles of these nuclear receptors have focused primarily on extracardiac tissues (12). The importance of REV-ERBα for temporal governance over the hepatic transcriptome was demonstrated through transgenic overexpression of REV-ERBα in the liver, which inactivated time-of-day-dependent oscillations of essentially all mRNA species (with the exception of only 31 genes, which still oscillated) (25). In addition to established circadian clock gene targets, genome-wide ChIPseq studies reveal that REV-ERBα binds to promoter/enhancer regions for various genes involved in lipid metabolism (13). Indeed, genetic deletion of REV-ERBα leads to hepatic steatosis, which is exacerbated when REV-ERBβ is also knocked down in these mice (consistent with some redundancy between α- and β-isoforms) (7). In other tissues, REV-ERBα has been suggested to play a role in oxidative metabolism. For example, REV-ERBα modulates brown adipose tissue-mediated thermogenesis (17), whereas it promotes mitochondrial biogenesis and function in skeletal muscle (50). Such observations, coupled with knowledge that heme serves as the natural ligand for REV-ERBα/β (51), are consistent with this nuclear receptor playing a prominent role in energy metabolism. In contrast, only a few studies have investigated the role of REV-ERBα/β in the heart. Zhang et al. (54) have recently reported that the REV-ERBα/β dual agonist SR-9009 influences expression of a number of fatty acid metabolism genes in cardiomyocytes, particularly in the presence of prohypertrophic stimuli. However, these studies did not observe any effects of the agonist on mitochondrial function; substrate oxidation was not investigated. Interestingly, SR-9009 attenuates pressure overload induced hypertrophy, interstitial fibrosis, and contractile dysfunction (54). Similarly, Alibhai et al. (2) have reported that SR-9009 attenuates age-onset cardiac hypertrophy. Collectively, such studies suggest that REV-ERBα/β plays important roles in cardiac physiology and pathophysiology.
The present study investigated to what extent REV-ERBα/β serves as a mechanistic link between the cardiomyocyte circadian clock and cardiac processes. Here, we employed a mouse model of cardiomyocyte-specific circadian clock disruption (namely CBK mice). Similar to previously published reports (52), circadian clock gene oscillations are severely attenuated or completely abolished in CBK hearts (Fig. 1A and Table 1). Importantly, protein levels of both REV-ERBα and REV-ERBβ are decreased in CBK hearts (consistent with decreased levels of their corresponding mRNAs; Fig. 1, A and B). These studies also revealed striking time-of-day-dependent oscillations in REV-ERBα levels in control hearts, which peak ∼10 h into the light phase (i.e., ZT10); oscillations in REV-ERBβ levels did not reach statistical significance, although maximal proteins levels tended to be observed around ZT12 (i.e., the light-to-dark phase transition). These observations are similar to those reported previously in the liver, wherein both REV-ERBα and REV-ERBβ protein levels peak around ZT10, with greater oscillations observed for REV-ERBα (compared with REV-ERBβ) (7). Indeed, liver ChIPseq studies indicate very low occupancy of REV-ERBα on target gene promoters at ZT22 (i.e., the end of the dark phase, when REV-ERBα levels are lowest) (14). We hypothesized that pharmacological activation of REV-ERBα/β around this time would have dramatic effects on target genes, given that baseline activity is usually low. Indeed, SR-9009 administration at ZT0 markedly altered expression of circadian clock components in the heart (Fig. 2). In contrast, SR-9009 administration at ZT9 (when REV-ERBα/β activity is already high) had essentially no effect on these target genes (Fig. 3). Such observations are consistent with prior studies indicating that SR-9009 treatment at the beginning of the light phase disrupts sleep cycles in mice (3). Thus, SR-9009 administration toward the end of the light phase (e.g., ZT9) is predicted to augment normal REV-ERBα/β activity rhythms.
BMAL1, REV-ERBα, and REV-ERBβ are all transcription factors. Therefore, we hypothesized that genetic deletion of BMAL1 from cardiomyocytes and/or pharmacological activation of REV-ERBα/β would impact cardiac processes primarily through transcriptional alterations. Accordingly, RNA-seq studies were performed. Consistent with previously published studies, CBK hearts exhibit >3,000 differentially expressed genes (compared with control hearts), which cluster in processes such as cellular signaling, growth/repair, and metabolism. SR-9009 also influenced the expression of 242 genes (compared with vehicle hearts) with known functions in processes such as inflammation, cellular signaling, and metabolism. Given that REV-ERBα/β levels are low in CBK hearts and that our 8-day SR-9009 administration strategy was designed to augment REV-ERBα/β activity at the correct time of the day (i.e., ZT9), we next looked for genes whose levels were (partially or fully) normalized in CBK hearts by SR-9009 administration. We also inferred from the RNA-seq data that distinct gene expression alterations following SR-9009 administration appear to have occurred in cell-specific populations within the heart, including many known to influence cellular signaling and metabolism. Also of note is that mitochondrial genes such as cox6b2 (a cytochrome c oxidase subunit; Table 2) were influenced by both BMAL1 deletion and SR-9009 administration. Interestingly, previously published studies suggest that genetic deletion of BMAL1 leads to mitochondrial dysfunction in the heart (24). Although SR-9009 did not appear to affect mitochondrial function in normal hearts (54), this agonist does promote mitochondrial biogenesis in skeletal muscle (50). Contrary to studies by Kohsaka et al. (24), the current study did not observe a signature of overt mitochondrial dysfunction in CBK hearts; the only genotype-specific alterations observed included decreased complex II levels and increased complex IV activity/levels in CBK hearts (Fig. 5, A and B). Similarly, 8 days of SR-9009 administration had only minimal effects these parameters (i.e., only a modest decrease in complex III levels; Fig. 5, A and B). Collectively, these studies are not consistent with a major effect of REV-ERBα/β on overall mitochondrial complex levels/activities in the heart.
Circadian clocks dramatically influence metabolism in multiple tissues, including the heart. For example, the cardiomyocyte circadian clock increases cardiac glucose use (oxidation, glycolysis, and glycogen synthesis) and triglyceride synthesis during the active period (5, 9, 46). CBK hearts exhibit chronically decreased ketone body and glucose oxidation rates and concomitant increased fatty acid oxidation (52). Here, we report that SR-9009 is unable to normalize perturbations in substrate reliance observed in CBK hearts (Fig. 6A). We have previously postulated that elevated fatty acid oxidation rates observed in CBK hearts are secondary to diminished rates of both ketone body (due to BMAL1-mediated regulation of BDH1; see Ref. 52) and glucose (due in part to BMAL1-mediated regulation of AS160 activation and glucose uptake; see Ref. 29) oxidation. Consistent with these concepts, SR-9009 administration did not normalize either decreased BDH1 or phospho-AS160 levels in CBK hearts (data not shown). Similarly, SR-9009 treatment failed to normalize either glycolytic flux (using 14C-labeled lactate release as an indirect marker) or triglyceride synthesis in CBK hearts (Fig. 6B). We have previously suggested that decreased triglyceride synthesis in cardiomyocyte circadian clock disruption is due in part to repression of dgat2 (46); we found that SR-9009 administration did not influence dgat2 expression (data not shown). However, SR-9009 administration did normalize glycogen synthesis rates in CBK hearts (Fig. 6B), an effect that appears to be independent of changes in proteins involved in glycogen turnover (glycogen synthase and phosphorylase) and glycophagy (LC3II, p62, and STBD1) (Fig. 7, C–J). Interestingly, we found that glycogen content is increased in freshly isolated CBK hearts (Fig. 7B) and that SR-9009 decreases cardiac glycogen content (Fig. 7A), leading to speculation that low glycogen reserves following SR-9009 administration may prime the heart for glycogen repletion during an ex vivo heart perfusion. It is also important to note that abnormalities in Akt/mTOR/GSK3β signaling reported previously in CBK hearts persist after SR-9009 administration (Fig. 7B), suggesting that changes occurring in this signaling cascade following BMAL1 loss are independent of REV-ERBα/β (29).
Circadian disruption is associated with increased risk of cardiovascular diseases, including hypertension, atherosclerosis, myocardial infarction, and stroke (28). In animal models, genetic deletion of BMAL1 (in either a whole body or cardiomyocyte-specific manner) leads to severe adverse cardiac remodeling associated with age-onset dilated cardiomyopathy and decreased longevity (27, 52). Recent animal-based studies suggest that the REV-ERBα/β agonist SR-9009 exerts cardiovascular benefits. For example, SR-9009 increases tolerance of the heart to both ischemia-reperfusion and myocardial infarction, attenuates atherosclerosis progression, ameliorates pressure overload induced heart failure, and improves cardiac function during aging (2, 36, 41–43, 54). Therefore, we hypothesized that SR-9009 might reverse the adverse cardiac remodeling observed in CBK mice. Consistent with this concept, SR-9009 decreased both cardiomyocyte size and interstitial fibrosis, as well as increased contractility of CBK hearts (as assessed ex vivo; Fig. 8, A–D). However, somewhat surprisingly, SR-9009 increased molecular markers of cardiac hypertrophy (anf and mhcβ) in both control and CBK hearts (Fig. 8B); these observations are in contrast to those of Zhang et al. (54), which indicate that SR-9009 decreases anf expression in neonatal cardiomyocytes stimulated with phenylephrine. Collectively, these data reveal that SR-9009 reverses adverse cardiac remodeling in CBK hearts.
Although the current study has a several important strengths (e.g., highlighting REV-ERBα/β as an important contributing factor toward adverse remodeling in CBK hearts), a number of distinct weaknesses should be highlighted. First, a pharmacological approach was employed to activate REV-ERBα/β. Although both translational and transient in nature (the latter being important to activate REV-ERBα/β only at specific times of the day), such a strategy can limit mechanistic insight. More specifically, SR-9009 will activate REV-ERBα/β in a variety of cells/organs, leading to concern that extracardiac influences may contribute toward the outcomes observed. Indeed, evidence exists suggesting that CBK hearts are in a proinflammatory state (23), whereas REV-ERBα/β have anti-inflammatory properties (34). Interrogation of the RNA-seq data suggests that SR-9009 does influence inflammatory markers in the heart (Fig. 4A). A second concern regarding mechanism is that the current study is unable to establish causality between parameters measured. For example, which SR-9009-mediated gene expression changes contributed toward glycogen synthesis normalization and/or reversal of adverse cardiac remodeling in CBK hearts is uncertain. Moreover, recent studies suggest that REV-ERBα/β may exert some functions in a transcription-independent fashion (e.g., direct interaction with O-GlcNAc transferase and subsequent modulation of protein O-GlcNAcylation; see Ref. 4). The current study also primarily assessed end points at single time of the day (i.e., ZT15), leading to the possibility that genotype and/or SR-9009 effects may become evident at other distinct times. Moreover, mitochondrial activity measurements were performed in frozen tissues; use of fresh preparations may have revealed perturbations in efficiency. It is also noteworthy that Cre has been reported to exert phenotypical effects in the heart; although Cre-positive controls were not included in the present study, the impact of Cre-induced cardiotoxicity was minimized through investigation of mice at 16 wk of age (52). Finally, the current study did not assess contractile function in vivo (e.g., through the use of echocardiography); this is because contractile dysfunction is not observed in CBK mice in vivo at the current study target age (i.e., 16 wk). Future studies are required to determine whether SR-9009 attenuates diastolic dysfunction in CBK mice or whether prolonged SR-9009 administration prevents age-onset systolic dysfunction in CBK mice.
In summary, we report that acute (8 days) pharmacological activation of REV-ERBα/β is sufficient to normalize glycogen synthesis and ameliorate adverse cardiac remodeling in a genetic model of cardiomyocyte circadian clock disruption. In contrast, this pharmacological intervention did not normalize mitochondrial function, substrate oxidation, or the Akt/mTOR/GSK3β signaling axis. These observations suggest that REV-ERBα/β likely plays an important role in mediating clock control of a subset of cardiac processes (Fig. 9). These studies highlight further the importance of normal circadian clock function for the maintenance of cardiac function.
Fig. 9.
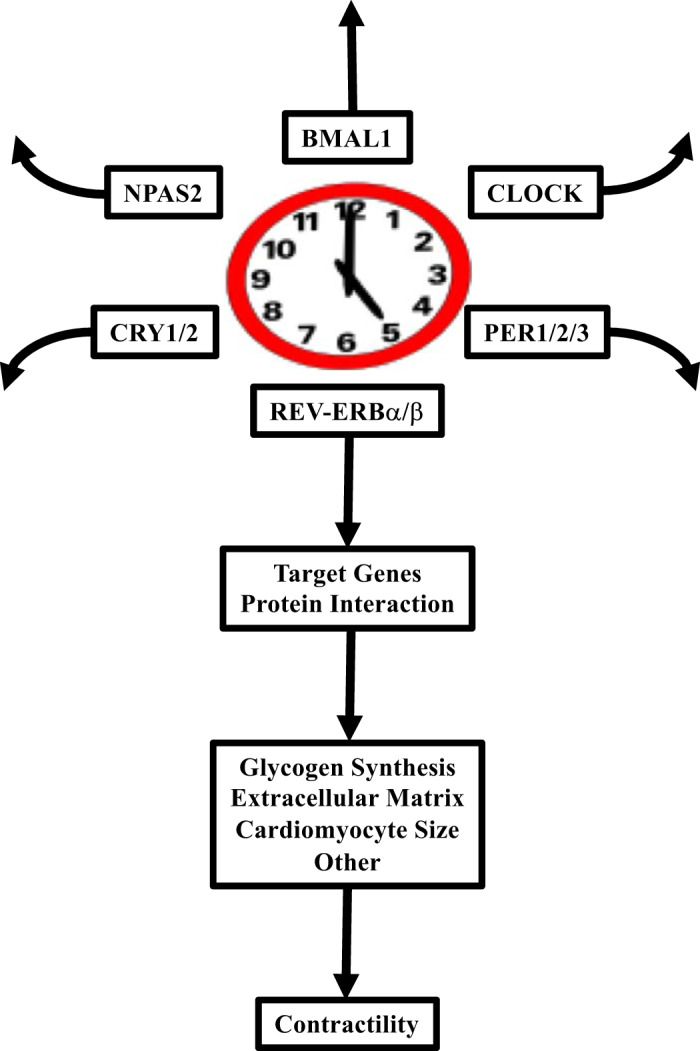
Summary of the study findings. Of the mammalian circadian clock components known, REV-ERBα/β appear to play important roles in the regulation of cardiac glycogen synthesis, cardiomyocyte size, interstitial fibrosis, and contractility. CRY1/2, cryptochrome 1/2; NPAS, neuronal PAS 2; PER1/2/3, period 1/2/3.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute Grants HL-123574 and HL-142216.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.E.Y. conceived and designed research; S.M., M.S.K., M.N.L., R.S., G.A.B., S.R.S., and M.E.Y. performed experiments; S.M., M.S.K., M.N.L., C.J.R., R.S., G.A.B., S.R.S., T.A.M., V.D.-U., and M.E.Y. analyzed data; S.M., M.S.K., M.N.L., C.J.R., S.R.S., S.J.F., T.A.M., J.Z., V.D.-U., and M.E.Y. interpreted results of experiments; C.J.R., T.A.M., and M.E.Y. prepared figures; M.E.Y. drafted manuscript; S.M., M.S.K., M.N.L., C.J.R., R.S., G.A.B., S.R.S., S.J.F., T.A.M., J.Z., V.D.-U., and M.E.Y. edited and revised manuscript; S.M., M.S.K., M.N.L., C.J.R., R.S., G.A.B., S.R.S., S.J.F., T.A.M., J.Z., V.D.-U., and M.E.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Maximiliano Grenett and Stephanie Reed for technical assistance.
REFERENCES
- 1.Acin-Perez R, Benador IY, Petcherski A, Veliova M, Benavides GA, Lagarrigue S, Caudal A, Vergnes L, Murphy A, Karamanlidis AG, Tian R, Reue K, Wanagat J, Sacks H, Amati F, Darley-Usmar VM, Liesa M, Divakaruni A, Stiles L, Shirihai OS. A novel approach to measure mitochondrial respiration in previously frozen biological samples. EMBO J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, Shulman A, Steinberg S, Burris TP, Oudit GY, Martino TA. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J Mol Cell Cardiol 105: 24–37, 2017. doi: 10.1016/j.yjmcc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Amador A, Huitron-Resendiz S, Roberts AJ, Kamenecka TM, Solt LA, Burris TP. Pharmacological Targeting the REV-ERBs in Sleep/Wake Regulation. PLoS One 11: e0162452, 2016. doi: 10.1371/journal.pone.0162452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berthier A, Vinod M, Porez G, Steenackers A, Alexandre J, Yamakawa N, Gheeraert C, Ploton M, Maréchal X, Dubois-Chevalier J, Hovasse A, Schaeffer-Reiss C, Cianférani S, Rolando C, Bray F, Duez H, Eeckhoute J, Lefebvre T, Staels B, Lefebvre P. Combinatorial regulation of hepatic cytoplasmic signaling and nuclear transcriptional events by the OGT/REV-ERBα complex. Proc Natl Acad Sci USA 115: E11033–E11042, 2018. doi: 10.1073/pnas.1805397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai JY, Bugger H, Zhang D, Rohrwasser A, Rennison JH, Dyck JR, Litwin SE, Hardin PE, Chow CW, Chandler MP, Abel ED, Young ME. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol 294: H1036–H1047, 2008. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 6.Bray MS, Ratcliffe WF, Grenett MH, Brewer RA, Gamble KL, Young ME. Quantitative analysis of light-phase restricted feeding reveals metabolic dyssynchrony in mice. Int J Obes 37: 843–852, 2013. doi: 10.1038/ijo.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, Lazar MA. Rev-erbα and Rev-erbβ coordinately protect the circadian clock and normal metabolic function. Genes Dev 26: 657–667, 2012. doi: 10.1101/gad.186858.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159, 1987. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 9.Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, Ratcliffe WF, Brewer RA, Nagendran J, Villegas-Montoya C, Zou C, Zou L, Johnson RL Jr, Dyck JR, Bray MS, Gamble KL, Chatham JC, Young ME. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem 286: 44606–44619, 2011. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durgan DJ, Tsai JY, Grenett MH, Pat BM, Ratcliffe WF, Villegas-Montoya C, Garvey ME, Nagendran J, Dyck JR, Bray MS, Gamble KL, Gimble JM, Young ME. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int 28: 187–203, 2011. doi: 10.3109/07420528.2010.550406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circ Res 106: 647–658, 2010. doi: 10.1161/CIRCRESAHA.109.209957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett LJ, Lazar MA. Nuclear receptor Rev-erbα: up, down, and all around. Trends Endocrinol Metab 25: 586–592, 2014. doi: 10.1016/j.tem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang B, Everett LJ, Jager J, Briggs E, Armour SM, Feng D, Roy A, Gerhart-Hines Z, Sun Z, Lazar MA. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell 159: 1140–1152, 2014. doi: 10.1016/j.cell.2014.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331: 1315–1319, 2011. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nat Rev Endocrinol 10: 466–475, 2014. doi: 10.1038/nrendo.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science 280: 1564–1569, 1998. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 17.Gerhart-Hines Z, Feng D, Emmett MJ, Everett LJ, Loro E, Briggs ER, Bugge A, Hou C, Ferrara C, Seale P, Pryma DA, Khurana TS, Lazar MA. The nuclear receptor Rev-erbα controls circadian thermogenic plasticity. Nature 503: 410–413, 2013. doi: 10.1038/nature12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res 6: 995–1001, 1996. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 19.He L, Hamm JA, Reddy A, Sams D, Peliciari-Garcia RA, McGinnis GR, Bailey SM, Chow CW, Rowe GC, Chatham JC, Young ME. Biotinylation: a novel posttranslational modification linking cell autonomous circadian clocks with metabolism. Am J Physiol Heart Circ Physiol 310: H1520–H1532, 2016. doi: 10.1152/ajpheart.00959.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res 6: 986–994, 1996. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 21.Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA 95: 5474–5479, 1998. doi: 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Ingle KA, Kain V, Goel M, Prabhu SD, Young ME, Halade GV. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am J Physiol Heart Circ Physiol 309: H1827–H1836, 2015. doi: 10.1152/ajpheart.00608.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohsaka A, Das P, Hashimoto I, Nakao T, Deguchi Y, Gouraud SS, Waki H, Muragaki Y, Maeda M. The circadian clock maintains cardiac function by regulating mitochondrial metabolism in mice. PLoS One 9: e112811, 2014. doi: 10.1371/journal.pone.0112811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol 5: e34, 2007. doi: 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98: 193–205, 1999. doi: 10.1016/S0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 27.Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol 303: H475–H485, 2012. doi: 10.1152/ajpheart.00238.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martino TA, Young ME. Influence of the cardiomyocyte circadian clock on cardiac physiology and pathophysiology. J Biol Rhythms 30: 183–205, 2015. doi: 10.1177/0748730415575246. [DOI] [PubMed] [Google Scholar]
- 29.McGinnis GR, Tang Y, Brewer RA, Brahma MK, Stanley HL, Shanmugam G, Rajasekaran NS, Rowe GC, Frank SJ, Wende AR, Abel ED, Taegtmeyer H, Litovsky S, Darley-Usmar V, Zhang J, Chatham JC, Young ME. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. J Mol Cell Cardiol 110: 80–95, 2017. doi: 10.1016/j.yjmcc.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37: 773–782, 2019. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peliciari-Garcia RA, Goel M, Aristorenas JA, Shah K, He L, Yang Q, Shalev A, Bailey SM, Prabhu SD, Chatham JC, Gamble KL, Young ME. Altered myocardial metabolic adaptation to increased fatty acid availability in cardiomyocyte-specific CLOCK mutant mice. Biochim Biophys Acta 1861: 1579–1595, 2016. doi: 10.1016/j.bbalip.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110: 251–260, 2002. doi: 10.1016/S0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 33.Prinz PN, Halter J, Benedetti C, Raskind M. Circadian variation of plasma catecholamines in young and old men: relation to rapid eye movement and slow wave sleep. J Clin Endocrinol Metab 49: 300–304, 1979. doi: 10.1210/jcem-49-2-300. [DOI] [PubMed] [Google Scholar]
- 34.Ramakrishnan SN, Muscat GE. The orphan Rev-erb nuclear receptors: a link between metabolism, circadian rhythm and inflammation? Nucl Recept Signal 4: e009, 2006. doi: 10.1621/nrs.04009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science 293: 506–509, 2001. doi: 10.1126/science.1060699. [DOI] [PubMed] [Google Scholar]
- 36.Reitz CJ, Alibhai FJ, Khatua TN, Rasouli M, Bridle BW, Burris TP, Martino TA. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol 2: 353, 2019. doi: 10.1038/s42003-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richards AM, Nicholls MG, Espiner EA, Ikram H, Cullens M, Hinton D. Diurnal patterns of blood pressure, heart rate and vasoactive hormones in normal man. Clin Exp Hypertens A 8: 153–166, 1986. doi: 10.3109/10641968609074769. [DOI] [PubMed] [Google Scholar]
- 38.Schroder EA, Burgess DE, Zhang X, Lefta M, Smith JL, Patwardhan A, Bartos DC, Elayi CS, Esser KA, Delisle BP. The cardiomyocyte molecular clock regulates the circadian expression of Kcnh2 and contributes to ventricular repolarization. Heart Rhythm 12: 1306–1314, 2015. doi: 10.1016/j.hrthm.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schroder EA, Lefta M, Zhang X, Bartos DC, Feng HZ, Zhao Y, Patwardhan A, Jin JP, Esser KA, Delisle BP. The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am J Physiol Cell Physiol 304: C954–C965, 2013. doi: 10.1152/ajpcell.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, Reppert SM. Interacting molecular loops in the mammalian circadian clock. Science 288: 1013–1019, 2000. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 41.Sitaula S, Billon C, Kamenecka TM, Solt LA, Burris TP. Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem Biophys Res Commun 460: 566–571, 2015. doi: 10.1016/j.bbrc.2015.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, Yoo SH, Takahashi JS, Butler AA, Kamenecka TM, Burris TP. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485: 62–68, 2012. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stujanna EN, Murakoshi N, Tajiri K, Xu D, Kimura T, Qin R, Feng D, Yonebayashi S, Ogura Y, Yamagami F, Sato A, Nogami A, Aonuma K. Rev-erb agonist improves adverse cardiac remodeling and survival in myocardial infarction through an anti-inflammatory mechanism. PLoS One 12: e0189330, 2017. doi: 10.1371/journal.pone.0189330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators . Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372, 2018. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 9: 764–775, 2008. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai JY, Kienesberger PC, Pulinilkunnil T, Sailors MH, Durgan DJ, Villegas-Montoya C, Jahoor A, Gonzalez R, Garvey ME, Boland B, Blasier Z, McElfresh TA, Nannegari V, Chow CW, Heird WC, Chandler MP, Dyck JR, Bray MS, Young ME. Direct regulation of myocardial triglyceride metabolism by the cardiomyocyte circadian clock. J Biol Chem 285: 2918–2929, 2010. doi: 10.1074/jbc.M109.077800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai JY, Villegas-Montoya C, Boland BB, Blasier Z, Egbejimi O, Gonzalez R, Kueht M, McElfresh TA, Brewer RA, Chandler MP, Bray MS, Young ME. Influence of dark phase restricted high fat feeding on myocardial adaptation in mice. J Mol Cell Cardiol 55: 147–155, 2013. doi: 10.1016/j.yjmcc.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turton MB, Deegan T. Circadian variations of plasma catecholamine, cortisol and immunoreactive insulin concentrations in supine subjects. Clin Chim Acta 55: 389–397, 1974. doi: 10.1016/0009-8981(74)90014-X. [DOI] [PubMed] [Google Scholar]
- 49.Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, Iino M, Shigeyoshi Y, Hashimoto S. A transcription factor response element for gene expression during circadian night. Nature 418: 534–539, 2002. doi: 10.1038/nature00906. [DOI] [PubMed] [Google Scholar]
- 50.Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, Hesselink MK, Paquet C, Delhaye S, Shin Y, Kamenecka TM, Schaart G, Lefebvre P, Nevière R, Burris TP, Schrauwen P, Staels B, Duez H. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med 19: 1039–1046, 2013. doi: 10.1038/nm.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science 318: 1786–1789, 2007. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 52.Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons BJ, Bray MS, Chatham JC, Wende AR, Yang Q, Chow CW, Martino TA, Gamble KL. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms 29: 257–276, 2014. doi: 10.1177/0748730414543141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Chatham J, Young ME. Circadian regulation of cardiac physiology: rhythms that keep the heart beating. Annu Rev Physiol 82: 79–101, 2020. doi: 10.1146/annurev-physiol-020518-114349Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Zhang R, Tien CL, Chan RE, Sugi K, Fu C, Griffin AC, Shen Y, Burris TP, Liao X, Jain MK. REV-ERBα ameliorates heart failure through transcription repression. JCI Insight 2: e95177, 2017. doi: 10.1172/jci.insight.95177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci USA 111: 16219–16224, 2014. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]