

Abstract
Macrophages play a pivotal role in tissue repair following myocardial infarction (MI). In response to injury, they exist along a spectrum of activation states tightly regulated by their microenvironment. Cardiosphere-derived cells (CDCs) have been shown to mediate cardioprotection via modulation of the macrophage response. Our study was designed to gain mechanistic insight into the role of CDC-derived extracellular vesicles (EVs) in modulating macrophage phenotypes and operant signaling pathways to better understand their potential contribution to immunomodulatory cardioprotection. We found that CDC-derived EVs alter the functional phenotype of macrophages, modifying levels of phagocytosis and efferocytosis without changing viability or proliferation. Interestingly, extracellular vesicles differentially regulate several M1/M2 genes dependent on macrophage activation before EV treatment but consistently upregulate arginase 1 regardless of macrophage origin or polarization state. CDC-derived EVs polarize M1 macrophages to a proangiogenic phenotype dependent on arginase 1 upregulation and independent of VEGF-A. In addition, EV-dependent arginase 1 upregulation downregulates nitric oxide (NO) secretion in activated macrophages. These data suggest a novel urea-cycle-dependent mechanism in macrophages that promotes angiogenesis and provides additional mechanistic insight into the potential contribution of CDC-derived extracellular vesicles in immunomodulatory cardioprotection.
NEW & NOTEWORTHY We hypothesized that in the window of therapeutic extracellular vesicle (EV) administration, inflammatory M1 macrophages are likely the primary target of cardiosphere-derived cell (CDC)-derived EVs. The effect of CDC-EVs on this population, however, is currently unknown. In this study, we demonstrate that CDC-derived EVs polarize M1 macrophages to a proangiogenic phenotype dependent on arginase 1 upregulation. These results provide insight into an immunomodulatory mechanism of CDC-EVs in a more physiologically relevant model of post-myocardial infarction (post-MI) macrophage polarization.
Keywords: angiogenesis, arginase 1, exosomes, extracellular vesicles, macrophages
INTRODUCTION
Macrophages play a crucial role in maintaining the myocardial milieu under normal conditions and modulating the immune response following cardiac injury. They represent a remarkably heterogenous cell population that exists along a spectrum of polarization states, from classically activated M1 inflammatory macrophages to alternatively activated M2 reparative macrophages (25, 37). Both their polarization and temporal phenotypic transitions are highly adaptable and closely regulated by cues from their environment (41). Following a myocardial infarction (MI), monocytes from the bone marrow and spleen are recruited to the ischemic and border zone where they differentiate into macrophages, replacing an obliterated resident cardiac macrophage population. Here, they contribute to the clearance of debris, tissue repair, resolution of inflammation, and angiogenesis (10, 15). The nonischemic remote zone also experiences an inflammatory response post-MI, mediated by both local macrophage renewal and monocyte recruitment from the circulation (33). While properly orchestrated macrophage-mediated wound healing results in restoration of tissue integrity post-MI, dysregulation of this process can result in chronic inflammation or pathological fibrosis (11). Recently, as our understanding of the mechanistic processes regulating macrophage plasticity has improved, attention has focused on macrophage phenotype modulation as a potential therapeutic strategy in the treatment of heart disease.
Extracellular vesicles (EVs), such as exosomes, are secreted lipid-bilayer nanovesicles containing cell-specific miRNA and protein cargo. They are important vectors for paracrine and autocrine cellular communication in both steady-state and disease and have been identified as the main paracrine mediators for numerous cellular therapeutics (23). Cardiosphere-derived cells (CDCs) are an example of a cell therapy that has been widely studied over the last decade for their applications in autologous and allogenic cell-based regeneration and remodeling. They have demonstrated safety and efficacy in numerous preclinical and clinical trials of heart disease. Previously, our group and others have established that the cardioprotective effects of CDCs, including stimulation of angiogenesis, induction of endogenous cardiomyocyte proliferation, and modulation of cardiomyocyte apoptosis and hypertrophy, are mediated by CDC-derived EVs (16, 20).
Recently, CDCs have been shown to facilitate cardiac repair by modulating M1/M2 polarization toward a more anti-inflammatory macrophage phenotype (8, 14). CDC-EVs have also been postulated to play a similar immunomodulatory role (7). When bone marrow-derived macrophages (BMDMs) were treated with CDC-EVs in vitro, they were polarized toward a unique anti-inflammatory phenotype, with a gene expression profile distinct from M1 and M2 macrophages. Our current results support these findings and provide additional functional insight into CDC-EV modulation of macrophage phagocytosis and efferocytosis.
While suggestive for a role of CDC-EVs in macrophage modulation post-MI, prior studies have focused on the response of naïve BMDMs to EV treatment. Early in an ischemic myocardial injury response, factors such as IFNγ, TNFα, and IL-1b are actively released into the tissue microenvironment, polarizing macrophages toward a proinflammatory M1 phenotype that predominates in the heart for the first 3 days following a MI. We hypothesized that in the window of therapeutic EV administration, inflammatory M1 macrophages are likely the primary target of CDC-derived EVs. The effect of CDC-EVs on this population, however, is currently unknown. In this study, we demonstrate that CDC-derived extracellular vesicles polarize M1 macrophages to a proangiogenic phenotype dependent on arginase 1 upregulation. These results provide insight into an immunomodulatory mechanism of CDC-EVs in a more physiologically relevant model of post-MI macrophage polarization.
MATERIALS AND METHODS
All animal experiments were performed according to protocols approved by the University at Buffalo Institutional Animal Care and Use Committee (IACUC). Human cardiosphere-derived cells were obtained from patients who provided written consent to tissue use under protocols approved by the University at Buffalo Research Subjects Institutional Review Board. Methods were carried out in accordance with the relevant guidelines and regulations.
Statistical Analysis
Quantitative end points are summarized with means ± SE. Qualitative data are summarized with percentages. Studies assessing differences in quantitative end points between treatment groups were assessed using a Student’s unpaired t test (two groups), one-way ANOVA and Tukey’s post hoc test (3 or more groups with 1 independent variable), and two-way ANOVA (2 independent variables). The cutoff for statistical significance was set at P < 0.05.
Extracellular Vesicle Isolation and Characterization
EVs were isolated from human cardiosphere-derived cells (CDCs) using either a precipitation-based method (ExoQuick-TC, System Biosciences) as previously described (20) or an ultrafiltration-based method. For the ultrafiltration isolation method, supernatants were first collected from human CDC culture flasks, passed through a 0.2-um filter, and subsequently concentrated with an Amicon Ultra-15 centrifugal filter with a 10-kDa molecular mass cutoff. EVs isolated from both methods were characterized by Western blot, nanoparticle tracking analysis, and cryo-transmission electron microscopy (cryo-TEM) as previously described (20, 22). Protein content was quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific) for downstream applications.
Cell Culture
Cardiosphere-derived cells.
Human cardiosphere-derived cells (CDCs) and nSMase2 siRNA knockdown CDCs were generated and characterized as previously described (20).
Peritoneal macrophages.
Two milliliters of 3% thioglycolate medium were injected into the peritoneal cavity of C57BL/6 mice. Two days later, mice were euthanized in accordance with IACUC guidelines and peritoneal macrophages isolated following intraperitoneal lavage with cold RPMI 1640 (Thermo Fisher). The sample was spun at 400 g for 10 min at 4°C. The supernatant was discarded, and the pellet was resuspended in 5 ml of cold RPMI 1640 medium. The cells were plated at a concentration of 1.5 × 106/mL, incubated for 2 h at 37°C, and subsequently washed with warm PBS. Attached peritoneal macrophages were incubated with RPMI 1640 for further culture and downstream analysis. Peritoneal macrophages were polarized in vitro with 1) 10 µg of CDC-EVs, 2) 100 µg of CDC-EVs, or 3) vehicle control for 6 h.
Bone marrow-derived macrophages.
Femurs were isolated from 3- to 4-mo-old C57BL/6 mice. The cellular fraction of the bone marrow was isolated by flushing PBS through the medullary cavity, filtering the isolate through a 70-µm mesh filter, and centrifuging at 300 g for 10 min. Cells were resuspended in RPMI 1640 media enriched with 30 ng/mL of M-CSF (Sigma-Aldrich) and plated at 4 × 105 cells/mL. After 2 days in culture, additional media were added to the wells with a complete media change completed at day 4 and repeated every other day thereafter. Starting at day 5, this population was referred to as naïve BMDM. On day 7 of BMDM culture, cells were activated for 24 h by 1) 100 ng/mL LPS (Sigma-Aldrich) and 50 ng/mL IFNγ (R&D Systems) toward an M1-like phenotype, 2) 10 ng/ml IL-4 and 10 ng/mL IL-13 (R&D Systems) toward an M2-like phenotype, 3) 10 µg CDC-EVs, or 4) a combination of M1 prepolarization followed by treatment with 10 µg CDC-EVs.
Human umbilical vein endothelial cells.
Human umbilical vein endothelial cells (HUVECs) were cultured on rat tail collagen-coated plates in MesoEndo Cell Growth Medium (Sigma-Aldrich). All cells were incubated at 37°C in 5% CO2.
Assessment of Macrophage Viability and Proliferation
Macrophages were seeded in a 24-well plate and incubated with or without CDC-EVs (10 µg) for 24 h. Calcein-AM (1 mg/mL, MilliporeSigma) was added to cells at a 1:300 dilution and incubated for 30 min. Cells were then fixed with 4% PFA, permeabilized, blocked with donkey serum, and incubated with rabbit polyclonal primary antibody against CD68 (Abcam, ab125212) at a 1:100 dilution overnight at 4°C. Cells were incubated with Alexa Fluor 555 donkey anti-rabbit secondary antibody (A-31572, Thermo Fisher) at a 1:400 dilution for 1 h at room temperature and subsequently counterstained with DAPI for quantification of cell viability (%calcein+/DAPI+ cells).
For assessment of proliferation, macrophages were pulsed for 4 h with ethynyl deoxyuridine (EdU; Click-iT EdU, Thermo Fisher). Following fixation and permeabilization, macrophages were stained with Alexa Fluor 555 donkey anti-rabbit secondary antibody (A-31572, Thermo Fisher) at a 1:400 dilution and counterstained with DAPI. The percentage of EdU+/DAPI+ cells was quantified for assessment of the proliferation index.
Macrophage Coculture Assay
Peritoneal macrophages were cocultured with either media alone, nSMase2 siRNA knockdown CDCs, or scrambled CDCs on transwell inserts (0.4-µm Pore Polyester Membrane Insert, Corning) for 24 h. After 24 h in culture, peritoneal macrophages were trypsinized for RNA extraction and qPCR analysis of Arg1, with relative expression normalized to GAPDH. To ensure CDC-EV uptake in macrophages, a subset was incubated with CDC-EVs labeled with DiR (Thermo Fisher) and visualized with fluorescent microscopy.
Isolation and Apoptosis Induction in Neutrophils
Human polymorphonuclear neutrophils (PMNs) were purified from whole blood through adaptation of a previously published protocol (27). Briefly, 5 mL of whole blood were carefully layered over 5 mL of neutrophil isolation media [Lymphocyte Poly(R), Cedarlane Laboratories] and centrifuged for 35 min at 500 g. The layer of neutrophils was isolated, diluted with HBSS (−/−), and centrifuged at 350 g for 10 min. Residual red blood cells in the neutrophil samples were lysed with RBC Lysis Buffer and the sample was washed with HBSS (−/−) and resuspended in HBSS/Human Serum Albumin Solution (2%) for counting. To induce apoptosis and stain neutrophils for flow cytometry detection, neutrophils were resuspended in RPMI 1640 medium containing 10 µM CellTracker Green CMFDA Dye (Thermo Fisher Scientific), incubated on ice for 15 min, washed, and subsequently incubated on ice for 2 h in RPMI 1640 supplemented with 10% human serum albumin (HSA). Apoptosis was quantified with annexin-V FITC and propidium iodide (Abcam) staining according to the manufacturer’s protocol, followed by flow cytometry analysis.
Efferocytosis Assay
Approximately 2 × 106 BMDMs were seeded in a 24-well plate, polarized toward each distinct phenotype (M1, M2, or MEV) or left as naïve BMDMs, and incubated with CMFDA-stained apoptotic neutrophils (1:1) for 1 h at 37°C. Fluorescence from membrane bound, but nonengulfed, apoptotic neutrophils were quenched by trypan blue (0.4% in PBS) at room temperature for 5 min. BMDMs were washed and analyzed by flow cytometry (BD Accuri CSampler). In our flow analysis, we first gated on macrophages based on size [forward scatter (FSC) vs. side scatter (SSC)] to exclude any free neutrophils. To separate positive signal from background fluorescence, we performed flow cytometry on BMDMs alone, setting the green fluorescent protein (GFP)+ gate to just past the tail of BMDM FITC autofluorescence. Efferocytosis was calculated as the percentage of BMDMs engulfing one or more CMFDA-labeled apoptotic neutrophils or the percentage of GFP+ BMDMs versus all BMDMs.
Phagocytosis Assay
BMDMs (naive, M1, M2, or MEVs) were incubated with FITC-conjugated latex beads (Cayman Chemical) according to the manufacturer’s protocol. Briefly, latex beads were added to BMDM cultures at a dilution of 1:100. After a 3.5-h incubation period, cells were transferred to 96-well U-bottom plates and treated with trypan blue quenching solution (1–2 min) to remove nonspecific surface staining. Following a wash with assay buffer, cells were analyzed via flow cytometry (BD Accuri CSampler) to assess phagocytosis in each group. To separate positive signal from background fluorescence, we ran BMDMs through the flow cytometer alone and analyzed their expression of FITC, setting the FITC+ gate to just past the tail of BMDM autofluorescence. Phagocytosis was calculated as the percentage of BMDMs engulfing one or more FITC-conjugated latex beads or the percentage of FITC+ BMDMs versus all BMDMs.
Immunocytochemistry
Cells were fixed with 4% PFA for 15 min, permeabilized for 20 min, blocked with donkey serum for 2 h, and incubated overnight at 4°C with primary antibodies at 1:100 dilutions. Secondary antibodies were incubated with cells at a 1:400 dilution for 1 h at room temperature in the dark and counterstained with DAPI. Images were obtained on an EVOS FL Auto Imaging System or a Leica DMi8 inverted fluorescence microscope for higher magnification images.
Flow Cytometry
Macrophages were characterized by flow cytometry for expression of CD68. Cells were incubated with rabbit CD68 (Abcam) at a 1:100 dilution for 1 h on ice, followed by a wash with Sort Wash Buffer (PBS + 2% FBS), and subsequent incubation with Alexa Fluor 555 donkey anti-rabbit secondary antibody (Thermo Fisher) at a 1:400 dilution for 30 min on ice.
Arginase 1 siRNA Knockdown
BMDMs were transfected with small interfering RNAs against Arg1 (MSS202169, MSS202170, MSS273267) or a negative scrambled control using Lipofectamine RNAiMAX (Thermo Fisher) following the manufacturer’s protocol, with BLOCK-iT Fluorescent Oligo (Thermo Fisher) used as a loading control. Following a 24-h incubation, transfected BMDMs were used for in vitro assays or isolated for RNA extraction and downstream RT-qPCR analysis.
Quantitative Real-Time PCR
RNA was extracted from cells using an E.Z.N.A. Total RNA Kit I (Omega Bio-tek). cDNA was synthesized using SuperScript III reverse transcriptase (Thermo Fisher). Quantitative real time PCR was performed using SsoFast EvaGreen Supermix (Bio-Rad) for genes associated with macrophage M1/M2 polarization (Arg1, IL-1b, IL-6, IL-10, Fpr2, Nos2, Pparγ, and TNFα). Primers used for SYBR Green-based RT-PCR are provided in Table 1. Samples were run in triplicate and gene expression calculated by ΔΔCt analysis using GAPDH as a reference. Statistical significance was tested on log2-transformed data using ANOVA.
Table 1.
Primers used for RT-PCR
| Primer | Sequence (5′→3′) |
|---|---|
| TNFα | |
| Forward | TCTTCTCATTCCTGCTTGTGG |
| Reverse | GGTCTGGGCCATAGAACTGA |
| Nos2 | |
| Forward | ACCTTGTTCAGCTACGCCTT |
| Reverse | CATTCCCAAATGTGCTTGTC |
| Il1b | |
| Forward | TGTAATGAAAGACGGCACACC |
| Reverse | TCTTCTTTGGGTATTGCTTGG |
| Il6 | |
| Forward | CCGGAGAGGAGACTTCACAG |
| Reverse | GGAAATTGGGGTAGGAAGGA |
| Fpr2 | |
| Forward | TCTACCATCTCCAGAGTTCTGTGG |
| Reverse | TTACATCTACCACAATGTGAACTA |
| Arg1 | |
| Forward | CTCCAAGCCAAAGTCCTTAGAG |
| Reverse | AGGAGCTGTCATTAGGGACATC |
| Il10 | |
| Forward | ATTTGAATTCCCTGGGTGAGAAG |
| Reverse | CACAGGGGAGAAATCGATGACA |
| Pparγ | |
| Forward | GGAAGACCACTCGCATTCCTT |
| Reverse | TCGCACTTTGGTATTCTTGGAG |
| NF-κB | |
| Forward | GCTGCCAAAGAAGGACACGACA |
| Reverse | GGCAGGCTATTGCTCATCACAG |
| TRAF6 | |
| Forward | TAAGGGATGCAGGGCACAAG |
| Reverse | GGCACTTTACCGTCAGGGAA |
| 18S | |
| Forward | CTTAGTTGGTGGTGGAGCATTTG |
| Reverse | GGCTGAACGCCACTTGTCC |
| Gapdh | |
| Forward | ACCACAGTCCATGCCATCAC |
| Reverse | CACCACCCTGTTGCTGTAGCC |
Generation of Drosha Knockdown EVs
Human CDCs were transfected with small interfering RNAs against Drosha (Thermo Fisher HSS178992) or a Medium GC Content Negative Control (Thermo Fisher) using Lipofectamine RNAiMAX (Thermo Fisher) or TransIT TKO (Mirus) following manufacturer's protocols, with BLOCK-iT Fluorescent Oligo (Thermo Fisher) used as a loading control. Following a 24-h incubation, media were changed to remove the transfection complex. Media were changed again on day 5 to serum free media, and exosomes were collected on day 12 posttransfection. RNA was extracted from exosomes using a miRNeasy Mini Kit (Qiagen). cDNA was prepared using the miRCURY LNA PCR Starter Kit (Qiagen) following manufacturer's protocols. PCR was run on a Bio-Rad CFX using a miR-146a miRCURY LNA PCR Primer Assay (Qiagen) and U6 snRNA miRCURY LNA PCR Primer Assay (Qiagen), following the manufacturer's protocol.
Nitric Oxide Assay
BMDMs polarized to an M1 phenotype were transfected with Arg1 siRNA or scrambled control then further polarized with 100 µg of CDC-EVs or vehicle control for 24 h in a 24-well plate. The supernatant from each well was collected, centrifuged at 300 g to remove cellular debris, and stored at −80°C until needed. A Griess Reagent kit (Molecular Probes) was used to determine nitrite concentrations in each supernatant according to the manufacturer’s protocol. Briefly, Griess Reagent was prepared by mixing equal volumes of N-(1-naphthyl) ethylenediamine and sulfanilic acid. In a 96-well microplate, 150 µL of supernatant were combined with 20 µL of Greiss Reagent and 130 µL of deionized water and incubated for 30 min in the dark at room temperature and absorbance was read at 548 nm with a spectrophotometric microplate reader (Epoch, BioTek). Absorbance measurements were converted to nitrite concentrations by generating a standard curve of known nitrite standards. Data were analyzed using the Gen5 software package.
HUVEC Proliferation Assay
HUVECs were seeded on collagen-coated plates (Rat Tail Collagen Type 1, Thermo Fisher) at a density of 5,000 cells/96-well. Cells were incubated with polarized BMDM supernatant and proliferation assessed following a 4-h pulse with EdU (Click-iT EdU Cell Proliferation Kit, Thermo Fisher). HUVECs were counterstained with DAPI, imaged on an EVOS FL Auto (Thermo Fisher) at ×10, and analyzed using Fiji. The percentage of proliferating cells was calculated as Edu+DAPI+/DAPI+ cells.
HUVEC Angiogenesis Assay
Ice-cold Matrigel (50 μL) was loaded into a prechilled 96-well plate using chilled pipette tips and allowed to polymerize at room temperature for 1 h. HUVECs were seeded at 5,000 cells/well and concurrently incubated with media (control) or 100 µl of polarized macrophage supernatant for 24 h. Images were acquired with an EVOS FL Auto fluorescence microscope at 4 and 24 h at both ×4 and ×10. Tube length and branch points were quantified using ImageJ.
Data Availability
All data generated or analyzed during this study are included in this published article.
RESULTS
Bone Marrow-Derived Macrophage Viability and Proliferation Are Unchanged Following CDC-EV Treatment
In our previous studies, we characterized human CDCs and their secreted extracellular vesicles using several quantification methods (20, 22, 23). To ensure comparable downstream analysis, we performed similar characterizations on freshly isolated CDC-derived EVs used in our current studies. CDC-EVs demonstrated a typical size distribution profile (mean diameter: 87 nm) as assessed by nanoparticle tracking analysis (Fig. 1A) and an expected small, round morphology with a discernable lipid bilayer by cryo-TEM (Fig. 1B). CDC-EVs were positive for known exosome markers CD63, CD81, ALIX, FLOT1, ICAM1, EpCAM, ANXA5, and TSG101. EV preparations were negative for any cellular contamination as assessed by lack of GM130 (a Golgi matrix protein) expression (Fig. 1C).
Fig. 1.
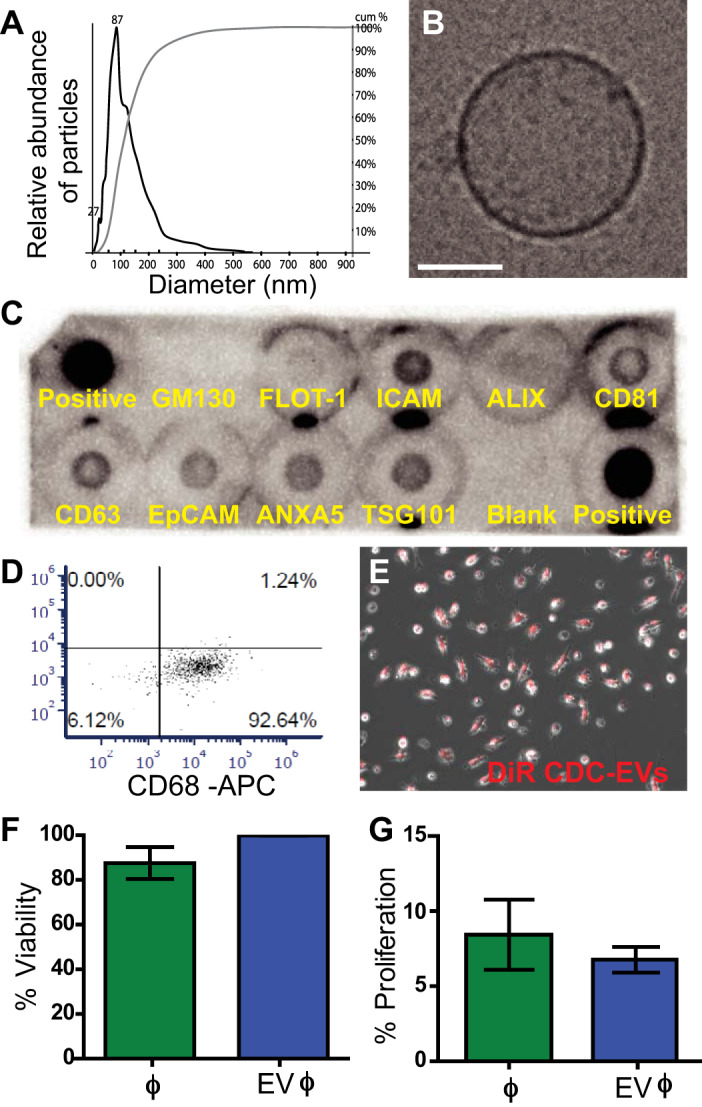
Characterization of cardiosphere-derived cell (CDC)-extracellular vesicles (EVs) and their effects on macrophage viability and proliferation. A: size distribution of CDC-EVs as determined by Nanoparticle Tracking Analysis. B: Exo-Check Exosome Antibody Array for exosome surface marker detection (FLOT-1, ICAM, ALIX, CD81, CD63, EpCAM, ANXA5, and TSG101) and assessment of potential cellular contamination (GM130). C: cryo-transmission electron microscopy (cryo-TEM) image of an isolated CDC-EV from serum free tissue culture. Scale bar = 50 µm. D: flow cytometric analysis of CD68 expression in bone marrow-derived macrophages (BMDMs). E: DiR-labeled CDC-EVs incubated with BMDMs display universal uptake in vitro. F and G: CDC-EV treatment did not significantly alter BMDM viability as assessed by calcein staining (F) or BMDM proliferation as assessed by 4-h ethynyl deoxyuridine (EdU) pulse (G) (n = 3 biological replicates per assay, means ± SE, P > 0.05, Student’s unpaired t test).
To determine if macrophage (ϕ) viability and proliferation were affected by CDC-EV therapy, we treated BMDMs with CDC-EVs for 24 h before labeling with calcein and EdU. Isolated and purified primary murine BMDMs were characterized by flow cytometry with the majority staining positive for CD68 (Fig. 1D). Macrophages demonstrated universal uptake of DiR labeled CDC-derived EVs following a 2-h incubation in vitro (Fig. 1E). There was no statistically significant difference in macrophage viability, as assessed by calcein staining (Fig. 1F), or proliferation, as quantified following a 4-h pulse with EdU (Fig. 1G), following 24-h EV exposure as compared with untreated cultures (n = 3 biological replicates for each assay, means ± SE, P > 0.05 using a Student’s unpaired t test).
CDC-Derived EVs Modulate Macrophage Efferocytosis and Phagocytosis
To investigate the effects of CDC-EVs on macrophage function, we polarized BMDMs with either 1) LPS/IFNγ toward an “M1” phenotype (M1 ϕ), 2) IL-4/IL-13 toward an “M2” phenotype (M2 ϕ), or 3) CDC-derived EVs (EV ϕ) for 24 h and assessed macrophage phagocytosis and efferocytosis (the clearance of apoptotic cells) by flow cytometry. As efferocytosis involves the recognition of apoptotic cells, we first generated a reproducible population of apoptotic neutrophils stained with CMFDA for downstream detection. Human PMNs were isolated from peripheral blood and apoptosis induced with ~50% of the neutrophil population staining annexin-V+PI−/weak at the time of the assay (early apoptotic cells) and 15% staining annexin-V+PI+ (late-apoptotic/necrotic cells) (Fig. 2A). Next, to verify engulfment of apoptotic neutrophils by BMDMs, we analyzed uptake over the course of 24 h with confocal microscopy. CD68+ BMDMs showed engulfment of CMFDA+ apoptotic neutrophils starting at 2-h (Fig. 2B). To quantify differences in efferocytosis, CMFDA-labeled apoptotic neutrophils were incubated with naïve ϕ, M1 ϕ, M2 ϕ, or EV ϕ for 2 h and analyzed with flow cytometry (Fig. 2C). CDC-EV-polarized BMDMs showed enhanced efferocytosis (42% ± 3%) as compared with either M1 (24% ± 2%)- or M2 (26% ± 2%)-polarized BMDMs. All groups engulfed apoptotic cells to a lesser degree than naïve BMDMs (67% ± 3%) (n = 3 biological replicates, means ± SE; P < 0.005 using one-way ANOVA followed by Tukey’s post hoc test) (Fig. 2, C and D).
Fig. 2.

Cardiosphere-derived cell (CDC)-extracellular vesicle (EV) treatment modulates macrophage efferocytosis and phagocytosis. A: representative flow cytometry characterization of neutrophil population used in efferocytosis assay. Neutrophils were stained with propidium iodide (PI) and annexin-V (A-V) to characterize the percentage of living (PI−AV−), apoptotic (PI−AV+), and necrotic cells (AV+) following the apoptosis induction protocol. B: confocal imaging highlighting engulfment of CMFDA-labeled apoptotic neutrophils by CD68-labeled bone marrow-derived macrophages (BMDMs). C and D: quantification of efferocytosis, expressed as the efferocytic index (percentage of BMDMs which engulfed an apoptotic neutrophil), as assessed by flow cytometry. The capacity of macrophages stimulated with CDC-EVs (“MEVs”) to efferocytose apoptotic neutrophils was enhanced as compared with both LPS/IFNγ (“M1”)- and IL4/IL13 (“M2”)-activated macrophages. E and F: quantification of macrophage phagocytosis, expressed as the phagocytic index, of IgG-FITC-conjugated latex beads by flow cytometry. M1-polarized BMDMs demonstrated the highest level of phagocytosis, followed by MEVs, unpolarized macrophages, and M2 macrophages (n = 3 biological replicates for each assay, means ± SE, *P < 0.005, one-way ANOVA followed by Tukey’s post hoc test).
In addition to modulating efferocytosis, CDC-EVs enhanced BMDM phagocytosis of IgG-FITC-conjugated latex beads (17% ± 1%) as compared with naïve BMDMs (11% ± 0.6%) and M2 (11% ± 0.4%)-polarized macrophages. There was no statistically significant difference between phagocytosis of M1 (19% ± 1%)- and CDC-EV-polarized BMDMs (17% ± 1%) (n = 3 biological replicates, means ± SE; P < 0.005 using one-way ANOVA followed by Tukey’s post hoc test) (Fig. 2, E and F).
Arginase 1 Is Upregulated by CDC-EVs across Macrophage Polarization States and Sites of Origin
To investigate if treatment with CDC-derived EVs results in unique patterns of differential gene expression depending on macrophage origin and polarization state at the time of EV exposure, we treated three different macrophage populations with CDC-EVs and looked at gene regulation of typical M1 and M2 markers: 1) naïve BMDMs (Fig. 3A), 2) BMDMs stimulated toward an inflammatory/M1 phenotype with LPS/INFγ (Fig. 3B), and 3) thioglycolate-activated peritoneal macrophages (PMs) (Fig. 3C) were treated with CDC-EVs for 24 h and assessed for differential regulation of TNFα, Nos2, Il1b, Il6, Fpr2, Arg1, Il10, and Pparγ. Stimulation of all three macrophage populations by CDC-EVs resulted in differential regulation of M1- and M2-associated genes by real-time RT-PCR. While EV-treated naïve BMDMs significantly upregulated TNFα (1.87 ± 0.27-fold, P = 0.0048), Nos2 (8.94 ± 3.75-fold increase, P = 0.047), and Il1b (2.17 ± 0.69-fold, P = 0.043) compared with their untreated control (Fig. 3A), LPS/INFγ -stimulated BMDMs showed significant downregulation of TNFα (0.17% ± 0.01%), Il10 (0.79 ± 0.004), and Pparγ (0.042 ± 0.003) with significant upregulation of Il6 (10.8% ± 1.5%) and Fpr2 (1.3% ± 0.07) compared with their untreated control (Fig. 3B). In contrast to both BMDM populations, CDC-EV-stimulated peritoneal macrophages did not demonstrate differential regulation of any M1 genes compared with untreated PMs (Fig. 3C). Interestingly, among the variable regulated M1 and M2 genes between macrophage classes, arginase 1 (Arg1) was unique in being universally upregulated in response to CDC-EV treatment across all three macrophage populations. (n = 3 biological replicates, means ± SE, P < 0.05 using a Student’s unpaired t test).
Fig. 3.
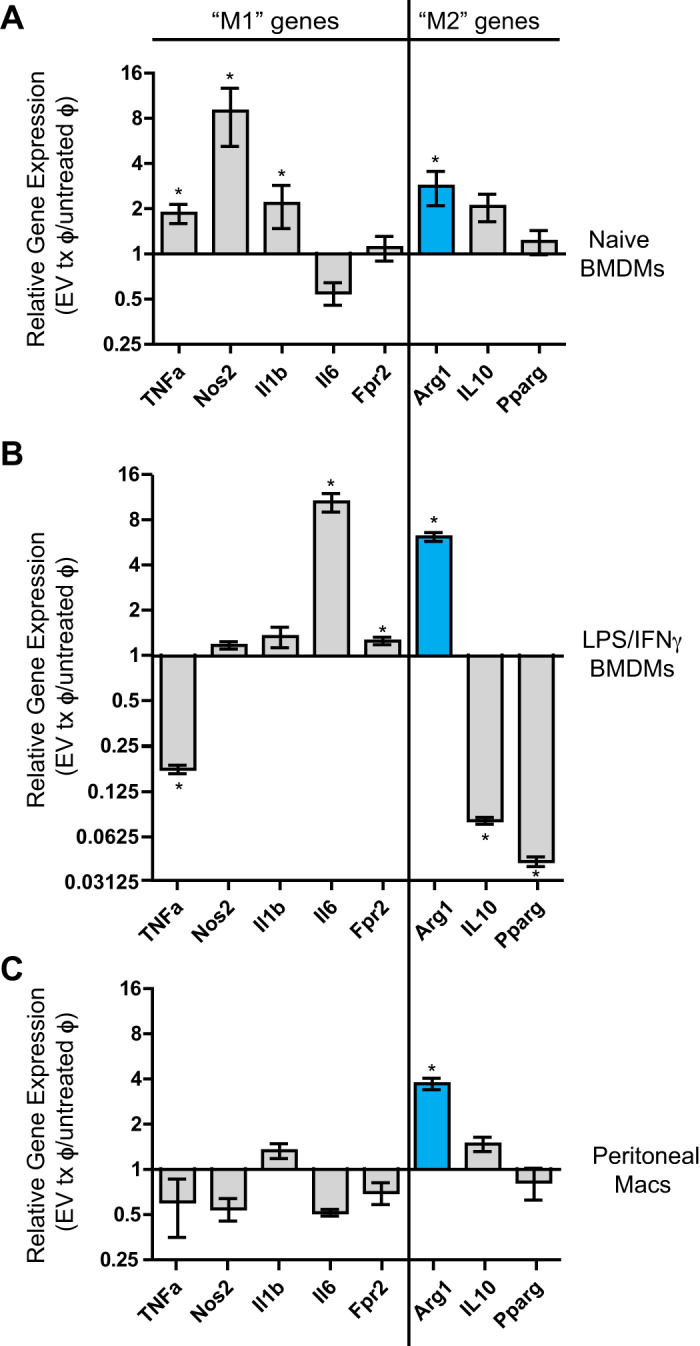
Macrophage gene expression is differentially regulated by cardiosphere-derived cell (CDC)-extracellular vesicle (EV) stimulation. M1- and M2-associated genes were differentially regulated by EV-stimulated naïve bone marrow-derived macrophages (BMDMs; A), LPS/IFNγ-activated BMDMs (B), and thioglycollate-activated peritoneal macrophages (C). Arginase 1 (Arg1) was uniquely universally upregulated by macrophages from different origins and activation states following treatment with CDC-derived EVs (n = 3 biological replicates for each assay, means ± SE; *P < 0.05 compared with untreated macrophages, Student’s unpaired t test).
CDC-EVs Are Necessary and Sufficient for the Upregulation of Macrophage Arginase 1
To determine if CDC EVs exert a dose-dependent effect on macrophage Arg1 upregulation, we exposed BMDMs to 0.1, 1, 10, and 100 µg of CDC-EVs per 2 × 105 cells for 24 h before RNA extraction and RT-qPCR. Macrophages demonstrated a dose-dependent increase in Arg1 expression in response to increasing CDC EV treatment, with an EC50 = 50.6 µg (Fig. 4A).
Fig. 4.

Cardiosphere-derived cell (CDC)-extracellular vesicles (EVs) are necessary and sufficient for the upregulation of arginase 1. A: CDC-EV incubation enhanced expression of arginase 1 (Arg1) in macrophages in a dose-dependent manner. B: macrophages cocultured with nSMase2 knockdown (KD) CDCs demonstrated a reduction of Arg1 expression when compared with macrophages cocultured with scrambled CDCs. C: IL4/IL13 (“M2”)-activated macrophages transfected with Arg1 shRNAi demonstrated a reduction in Arg1 expression verifying our in vitro KD model. D: LPS/IFNγ-activated bone marrow-derived macrophages (BMDMs) were transfected with either Arg1 siRNA or a scrambled control before +/− treatment with CDC-EVs. Arg1 expression was enhanced in BMDMs exposed to CDC-EVs but significantly reduced in EV-treated Arg1 KD macrophages (n = 3 biological replicates for each assay, means ± SE, *P < 0.05, Student’s unpaired t test for A–C, one-way ANOVA followed by Tukey’s post-hoc test for D).
To assess if CDC-EVs, and more specifically CDC-derived exosomes, were both necessary and sufficient to cause Arg1 upregulation in macrophages, we used our previously characterized (20) nSMase2 knockdown (KD) CDC cell line (with significantly reduced exosome secretion) in a coculture assay. Macrophages were cocultured with either control CDCs or nSMase2-KD CDCs for 24 h and subsequently analyzed by qPCR for Arg1 expression. Macrophages cocultured with nSMase2 KD CDCs demonstrated a reduction in Arg1 compared with those cocultured with control CDCs (Fig. 4B).
To define the functional role of macrophage Arg1 expression on target cells, we transfected BMDMs with Arg1 siRNA. Using M2-polarized BMDMs, which are known to express high levels of Arg1 at baseline, we verified a siRNA mediated Arg1 knockdown efficiency of ~75% (Fig. 4C). Consistent with a functional knockdown, when Arg1 KD M1 ϕ were treated with CDC-EVs, they failed to recapitulate the level of Arg1 induction seen with the scrambled control (Fig. 4D).
EV miRNA Cargo Is Responsible for Arg1 Upregulation
Specific miRNA cargo has been shown to be integral in driving certain functional benefits of CDC-EV therapy (7, 16). As such, we hypothesized that miRNA cargo within CDC-EVs was responsible for the upregulation of macrophage arginase 1. To investigate if CDC-EV miRNA mediates arginase 1 activation, we transfected CDCs with Drosha siRNA and collected their miR-depleted EVs. Cells that lack Drosha, which is involved in the processing of primary miRNA, are unable to generate mature miRNA through canonical pathways (19). PCR analysis using a candidate miRNA (miR-146a) highly expressed in CDC-EVs demonstrated a significant reduction of >90% in EV miRNA content in CDC-EVs isolated from CDCs transfected with Drosha siRNA (Drosha-KD) as compared with a scrambled control (Fig. 5A) (relative gene expression of 1 ± 0.053 vs. 0.081 ± 0.011) (n = 3 biological replicates, means ± P < 0.05, Student’s unpaired t test). While control CDC-EVs activated arginase 1 in M1 BMDMs when compared with our vehicle control (relative gene expression of 2.02 ± 0.21 vs. 1.02 ± 0.03) (Fig. 5B), reduced miRNA content in Drosha-KD EVs translated to reduced upregulation of ϕ arginase 1 when compared with control CDC-EVs (Fig. 5B) (relative gene expression of 1.33 ± 0.16 vs. 2.02 ± 0.21) (n = 3 biological replicates, means ± SE; P < 0.05, one-way ANOVA).
Fig. 5.
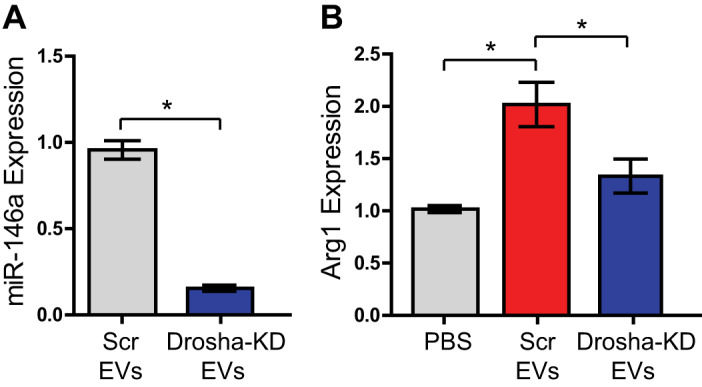
Cardiosphere-derived cell (CDC)-extracellular vesicle (EV) upregulation of arginase 1 (Arg1) is regulated by miRNA cargo. A: PCR analysis with candidate miRNA (miR-146a) revealed significantly lower miRNA expression in CDC-EVs isolated from CDCs with a Drosha knockdown (KD) when compared with scrambled (Scr) control (n = 3 biological replicates, means ± SE, *P < 0.05, Student’s unpaired t test). B: CDC-EVs with a Drosha knockdown are unable to upregulate arginase-1 expression to the same level as control CDC-EVs (n = 3 biological replicates, means ± SE, *P < 0.05, one-way ANOVA).
EV-Dependent Arg1 Upregulation Results in Decreased Macrophage Nitric Oxide Production
Given its universal upregulation following CDC-EV treatment, we hypothesized that Arg1 plays a key role in CDC-EV mediated cardioprotective immunomodulation. As l-arginine is the sole substrate for all forms of nitric oxie (NO) synthases, we predicted that CDC-EV induction of ϕ arginase 1 would decrease NO production in activated macrophages. To investigate the role of Arg1 expression in modulating macrophage NO levels, BMDMs were polarized to an inflammatory/M1 phenotype with LPS/INFγ and subsequently treated with CDC-EVs or vehicle control. Following treatment with CDC-EVs, macrophages exhibited a significant decrease in NO secretion [n = 3, means ± SE, main effect EV treatment F(2,3) = 24.9, P < 0.05, two-way ANOVA] (Fig. 6A). To further define the functional role of macrophage Arg1 expression on modulating NO levels following EV treatment, we transfected M1 BMDMs with Arg1 siRNA. We hypothesized that Arg1 KD would block CDC-EV-mediated depletion of macrophage NO. Following treatment with CDC-EVs, Arg1 KD M1 macrophages had no difference in NO production between the groups (13 µg ± 2 µg vs. 14 µg ± 2 µg) (n = 3 biological replicates, means ± SE; P > 0.05, two-way ANOVA) (Fig. 6B).
Fig. 6.
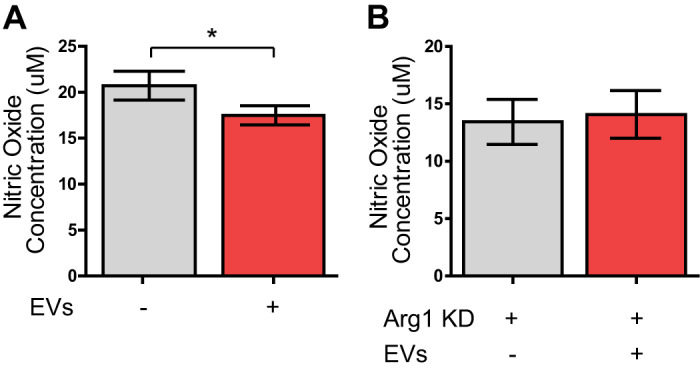
Extracellular vesicle (EV) upregulation of arginase 1 (Arg1) decreases nitric oxide (NO) production. A: a Griess assay was performed on supernatant collected from LPS/IFNγ-activated bone marrow-derived macrophages (BMDMs) treated with CDC-EVs or vehicle control. Following treatment with CDC-EVs, macrophages exhibited significantly decreased secretion of NO [n = 3, means ± SE, main effect EV treatment F(2,3) = 24.9, *P < 0.05, two-way ANOVA]. B: M1 BMDMs transfected with Arg1 siRNA and treated with CDC-EVs showed no difference in NO production compared with no EV vehicle control (n = 3, means ± SE, P > 0.05, two-way ANOVA). KD, knockdown.
Macrophages Exposed to CDC-EVs Stimulate Angiogenesis in an Arg1-Dependent Manner
After demonstrating a role for EV-mediated Arg1 upregulation in ϕ NO production, we focused on CDC-EV modulation of the alternate arm of l-arginine metabolism. Arginase 1 is a crucial enzyme in the urea cycle, responsible for converting l-arginine to urea and l-ornithine, which is subsequently metabolized into proline and polyamides. Prior studies have demonstrated a link between increased polyamide production and endothelial cell proliferation (39). As such, we hypothesized that CDC-EV mediated upregulation of ϕ Arg1 would result in increased polyamide production, which would enhance angiogenesis. We performed an HUVEC proliferation assay to investigate the role of CDC-EVs in modulating the macrophage secretome to enhance endothelial proliferation. Supernatant from macrophages grown under a variety of treatment conditions was collected and added to HUVECs in culture. Endothelial cells were pulsed for 4 h with EdU and proliferation assessed as the percentage of EdU+/DAPI+ cells. HUVECs grown in supernatant from M1 BMDMs exposed to CDC-EVs (Fig. 7, B and C) demonstrated enhanced proliferation when compared with their untreated BMDM control (19 ± 0.6% vs. 14 ± 0.4%) (n = 3 biological replicates, means ± SE, main effect: EVs P < 0.01, two-way ANOVA) (Fig. 7, A and C). This effect was dependent on Arg1 upregulation by EVs (interaction P < 0.01), as HUVECs grown in the presence of supernatant collected from Arg1 KD BMDMs treated with EVs failed to elicit the same level of endothelial proliferation as seen with HUVECS cultured in supernatant from EV-treated M1 BMDMs without Arg1 knockdown (16 ± 0.7% vs. 19 ± 0.5%). There was no effect of Arg1 KD alone on endothelial proliferation in the absence of EV treatment (P = 0.1) (Fig. 7C).
Fig. 7.
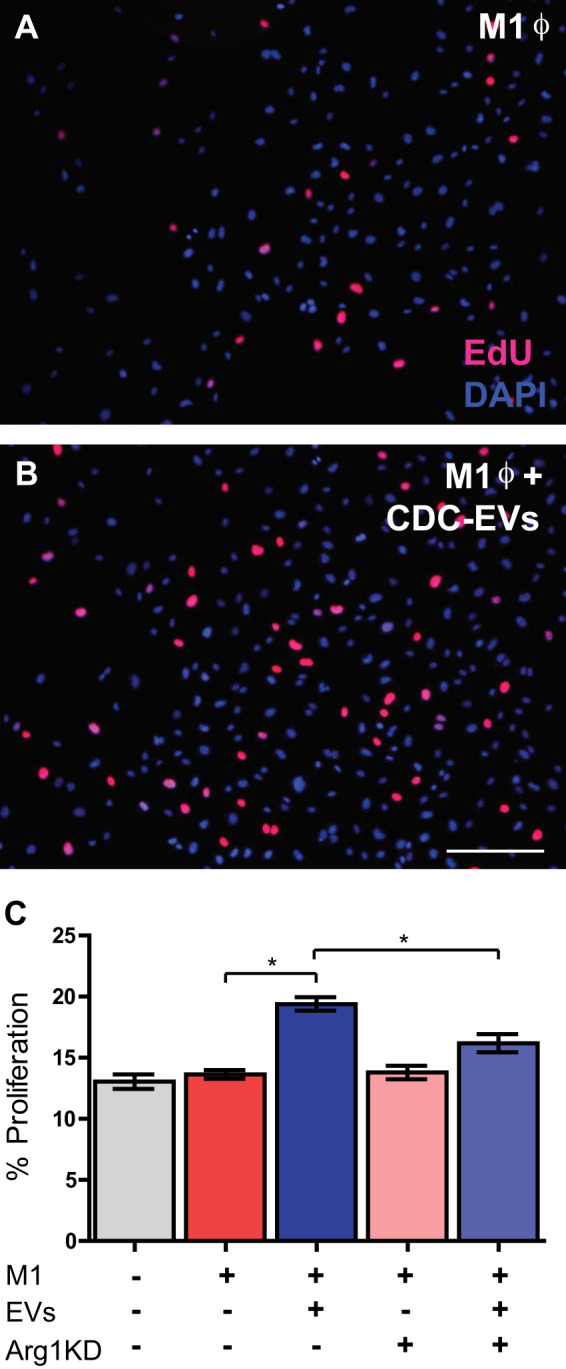
Cardiosphere-derived cell (CDC)-extracellular vesicles (EVs) reprogram bone marrow derived macrophages (BMDMs) to enhance endothelial proliferation. Representative images of 4-h EdU pulse endothelial proliferation assay following exposure to supernatant from LPS/IFNγ-stimulated (“M1”) (A) macrophages and M1 macrophages treated with CDC-EVs (B) and human umbilical vein endothelial cells (HUVECs) cultured with supernatant from M1 CDC-EV-treated macrophages (C) demonstrate an increase in proliferation as compared with HUVECs cultured with supernatant from control BMDMs and arginase 1 (Arg1) knockdown (KD) EV-treated M1 macrophages. Scale bar = 200 µm (n = 3 biological replicates, means ± SE, *P < 0.01, two-way AVOVA).
To further investigate the role of CDC-EVs in polarizing macrophages to a proangiogenic phenotype, we performed an in vitro angiogenesis assay. Supernatant was collected from control and CDC-EV-treated BMDMs (following a media change to remove any free CDC-EVs in the supernatant following BMDM polarization) and added to HUVECs. After a 4-h incubation, tube length (Fig. 8, C and D) and the number of branch points per node (Fig. 8, C and E) were calculated. Interestingly, supernatant collected from M1 BMDMs exposed to CDC-EVs (Fig. 8, B, D, and E) demonstrated a significant increase in tube length (134 ± 4% vs. 102 ± 6%) (n = 3 biological replicates, means ± SE, main treatment: EVs P < 0.05, two-way ANOVA) and branch points (149 ± 5% vs. 100 ± 6%) (n = 3 biological replicates, means ± SE, main treatment: EVs P < 0.05, two-way ANOVA) when compared with untreated M1 BMDMs (Fig. 8, A, D, and E). This effect was Arg1 dependent (interaction P < 0.05 for tube length and branch points), as Arg1 KD BMDMs treated with EVs failed to elicit enhanced the angiogenic properties of increased tube length and branch points that were seen with M1 BMDMs exposed to CDC-EVs. Importantly, Arg1 KD had no effect on tube length or branch points alone without EV addition (P > 0.05) (Fig. 8, D and E).
Fig. 8.

Macrophages stimulate angiogenesis in a human umbilical vein endothelial cell (HUVEC) angiogenesis assay following treatment with cardiosphere-derived cell (CDC)-extracellular vesicle (EVs). A and B: representative ×4 images of a 4-h HUVEC Matrigel tube assay following exposure to supernatant from LPS/IFNγ-stimulated (“M1”) macrophages (A) and M1 macrophages treated with CDC-EVs (B). C: overlay demonstrating the methods used for measurement of HUVEC tube length and quantification of branch points. Tube length was measured from the base of attachment on one cell to the next cell (red lines) using ImageJ software. The number of branch points was calculated as the number of tubes emanating from each node (white arrows). D and E: HUVECs cultured with supernatant from LPS/IFNγ-stimulated, 24-h CDC-EV-treated macrophages demonstrate an increase in tube length (D) and number of branch points (E) as compared with HUVECs cultured with supernatant from arginase 1 (Arg1) knockdown (KD) EV-treated M1 macrophages in a 4-h Matrigel in vitro tube assay. Scale bar = 250 µm (n = 3 biological replicates for each assay, means ± SE, *P < 0.05, two-way AVOVA).
To determine if this effect was mediated by enhanced expression of VEGF as has recently been implied in proangiogenic macrophages (10), we performed qPCR on M1 macrophages treated with CDC-EVs and compared their expression of VEGF-A with untreated M1 macrophages. We found no significant difference in VEGF-A gene expression between groups (relative expression of 0.25 ± 0.17 in M1 macrophages vs 0.20 ± 0.15 in M1 macrophages plus CDC-EVs, P = 0.8, n = 3 biological replicates, means ± SE, Student’s unpaired t test).
ϕ Arg1-Mediated Polyamide Secretion Is Responsible for Enhanced Endothelial Proliferation and Angiogenesis
Arg1 facilitates l-arginine metabolism to proline and polyamine production, the latter of which we hypothesized to play a role in modulating angiogenesis. We tested our hypothesis using DFMO (α-difluoromethylornithine) to block ornithine decarboxylase (ODC), an enzyme which catalyzes the decarboxylation of ornithine to putrescine (Fig. 9A). As before, we observed a significant effect of EV treatment on endothelial proliferation. HUVECs cultured with supernatant from M1 CDC-EV-treated macrophages demonstrated an increase in proliferation as compared with HUVECs cultured with supernatant from control BMDMs (25% ± 1% vs. 15% ± 1%) (n = 3 biological replicates, main effect: EVs P < 0.05, two-way ANOVA). Importantly, DFMO had no effect on proliferation alone without EV addition (P > 0.05) and we observed a highly significant interaction between DFMO and EV treatment (interaction P < 0.05), indicating that the proliferation effect of EVs was dependent on macrophage polyamide production (Fig. 9B).
Fig. 9.

Arginase 1 (Arg1)-mediated polyamide secretion is responsible for enhanced endothelial proliferation and angiogenesis. A: schematic of l-arginine metabolism focusing on ARG1 metabolism leading polyamine production [which can be blocked by DFMO targeting ornithine decarboxylase (ODC)]. B–D: human umbilical vein endothelial cells (HUVECs) cultured with supernatant from M1 cardiosphere-derived cell (CDC)-extracellular vesicle (EV)-treated macrophages demonstrate an increase in proliferation (B), tube length (C), and number of branch points (D) as compared with HUVECs cultured with supernatant from control bone marrow-derived macrophages (BMDMs) and DFMO EV-treated M1 macrophages. (n = 3 biological replicates for each assay, means ± SE, *P < 0.05, two-way AVOVA). NOHA, Nω-hydroxy-nor-l-arginine; OAT, ornithine aminotransferase; NOS2, nitric oxide synthase 2.
In addition to the dependence of endothelial cell proliferation on polyamide synthesis in response to CDC-EV exposure, we found that the angiogenic properties of HUVECs exposed to supernatant from CDC-EV-treated macrophages was also dependent on ϕ polyamide production (n = 3 biological replicates, means ± SE, main effect: EVs P < 0.05, interaction P < 0.05, two-way ANOVA). HUVECs cultured with supernatant from LPS/IFNγ-stimulated, 24-h CDC-EV-treated macrophages demonstrate an increase in tube length (116% ± 3% vs 106% ± 1%) (Fig. 9C) and number of branch points (132% ± 6% vs 113% ± 4%) (Fig. 9D) as compared with HUVECs cultured with supernatant from DFMO EV-treated M1 macrophages in a 4-h Matrigel in vitro tube assay. DFMO had no effect on angiogenic parameters alone without EV treatment (P = 0.3).
DISCUSSION
Macrophages are critical orchestrators of the inflammatory response following cardiac injury. Previous depletion studies have demonstrated their importance in cardiac repair post-MI, where treatment with clodronate liposomes has led to impaired wound healing, left ventricular dilation and wall thinning, and increased mortality (1, 12, 40). Systemic depletion of macrophages has also been shown to mitigate the cardioprotective effects of CDCs, demonstrating an essential role of macrophages in CDC-mediated cardioprotection (8).
Reflective of their wide spectrum of activation states, macrophages play numerous roles in homeostasis and disease. In addition to serving as phagocytes during an innate immune response, they also mediate efferocytosis, the process of engulfing apoptotic cells. This prevents secondary necrosis and the release of inflammatory molecules from apoptotic bodies and is crucial in postinjury states such as myocardial infarction (42). As extracellular vesicles have been shown to mediate the cardioprotective effects of CDCs, there has been a focused interest on the interaction between CDC-derived EVs and macrophages (7). In our current study, we found that naïve bone marrow-derived macrophages treated with CDC-EVs (MEVs) exhibited enhanced phagocytosis when compared with M2 and naïve macrophages and resulted in higher levels of efferocytosis than M1- or M2-polarized macrophages. While altering levels of phagocytosis may not have a direct effect in a noninfectious cardiac injury state, modulation of macrophage efferocytosis has significant implications in the regulation of post-MI inflammation and restoration of tissue homeostasis. Statins, for example, have been shown to enhance efferocytosis, possibly in part by inhibition of membrane association and activation of RhoA (24) and PPARγ (46). The enhancement of efferocytosis following statin therapy may be a contributing factor to their additional survival benefit independent of lipid lowering activity. Prior studies have implicated a role for both exosomal miR-181b and miR-26a in modulating macrophage function, the latter of which has been shown to enhance efferocytosis via suppression of Adam17 and upregulation of MerTK expression in BMDMs (6, 7). Additionally, EVs may enhance efferocytosis via delivery of early acting mediators that increase Rac 1 activity or proresolution lipids such as resolvins, protectins or maresins (35, 36). They may also mediate efferocytosis through programming changes, such as activation of macrophage PPARδ signaling or interaction with nuclear receptors LXR and RXRα (31).
We hypothesized that CDC-derived EVs differentially regulate the phenotype and function of macrophages dependent on their origin and polarization state. Prior studies have solely focused on the interaction of EVs and naïve BMDMs. The ability to extrapolate and generalize these findings to an injury state is limited, however, as naïve BMDMs do not recapitulate the cardiac macrophage milieu post-MI. In the therapeutic window following a myocardial infarction, inflammatory macrophages are the most likely target of delivered EVs. We therefore designed our study to establish the effect of CDC-EVs on both naïve and inflammatory BMDMs to gain mechanistic insight into EV modulation of macrophage phenotype, function and immunomodulatory cardioprotection. The main findings of our work are as follows: 1) CDC-derived EVs modulate BMDM efferocytosis and phagocytosis to a unique functional phenotype distinct from either M1 or M2 macrophages, 2) the polarization state of BMDMs at the time of EV treatment influences their differential expression of inflammatory and anti-inflammatory genes, and 3) arginase 1 is universally upregulated in response to CDC-EVs and plays a role in downregulating ϕ NO secretion and modulating inflammatory macrophages to an angiogenic phenotype dependent on enhanced polyamide production.
Angiogenesis is a critical step in the healing process after MI. Following cardiac injury, circulating monocytes migrate into the infarcted myocardium and differentiate into macrophages. Early in the injury response (days 1–3), factors such as IFNγ, TNFα, and IL-1b are actively released into the tissue microenvironment, polarizing macrophages toward a proinflammatory (M1) phenotype (44). Activated macrophages secrete growth factors and cytokines such as TNFα and IFNγ, which regulate the expression of VCAM-1 and ICAM-1 on endothelial cells, promoting leukocyte adhesion and extravasation into damaged myocardium. This phase is followed by a reparative phase characterized by resolution of inflammation and angiogenesis. Reparative M2 macrophages play a defined role in angiogenesis via secretion of anti-inflammatory cytokines and angiogenic growth factors. For example, in human invasive breast carcinoma, macrophage number correlates with vascularization (21). Recently, proangiogenic macrophages have been shown to promote myocardial repair. The reparative macrophage phenotype was induced by myeloid cell-delivered annexin-A1 (AnxA1), which enhanced production and secretion of proangiogenic VEGF-A by macrophages (10). Given the universal upregulation of arginase 1 following CDC-EV treatment, we hypothesized that Arg1 plays a role in EV-mediated immune regulation that may contribute to cardioprotection following injury to the heart. In our study, treatment with CDC-EVs resulted in macrophage-mediated endothelial cell proliferation and angiogenesis that was blocked with Arg1 KD, suggesting a novel functional role of arginase 1 induction in regulating angiogenesis. Unlike prior studies that outlined a role for enhanced VEFG-A production in angiogenic macrophages, we saw no significant difference in VEGF-A expression following EV treatment suggesting an alternate mechanism for an Arg1-mediated proangiogenic macrophage phenotype.
Arginase 1 is an essential component of the hepatic urea cycle and catalyzes the conversion of l-arginine to urea and l-ornithine (4, 26). l-ornithine is further processed into l-proline and polyamines, which contribute to cell proliferation (18), collagen production (43), and angiogenesis (38). Under Th1 cytokine stimulation, inducible nitric oxide synthase (iNOS) outcompetes Arg1 for their shared substrate, l-arginine, and enhances the inflammatory response through production of nitric oxide and l-citrulline (30). Nitric oxide (NO) has been shown to inhibit the proliferation of vascular endothelial cells in vitro (45) and downregulate angiogenesis in the in vivo system of the chick embryo chorioallantoic membrane (28, 29). Arginase 1 activity has been shown to limit the supply of l-arginine needed for the formation of cytotoxic levels of NO by iNOS (32, 34). We hypothesized that CDC-EV-induced Arg1 upregulation would result in increased use of the common substrate l-arginine, resulting in decreased NO production. Consistent with prior studies that have shown arginase expression to modulate NO production in activated macrophages (3), we saw a significant decrease in macrophage NO levels following treatment with CDC-EVs. This effect was Arg1 dependent, as there was no difference in NO levels between Arg1 KD BMDMs treated with CDC-EVs versus vehicle control.
Prior studies have also demonstrated a link between polyamines and angiogenesis. Takigawa et al. (39) studied the effect of α-difluoromethylornithine (DFMO), an irreversible inhibitor of ornithine decarboxylase, on tumor-induced angiogenesis. Their findings suggest the antitumor activity seen with DFMO was secondary to inhibition of endothelial cell proliferation induced by polyamine depletion, rather than any direct effect on tumor cell proliferation (39). We hypothesized that upregulation of Arg1 by CDC-EVs would result in stimulation of polyamide synthesis and a subsequent polarization of macrophages to a proangiogenic phenotype. Treatment with CDC-EVs resulted in macrophage-mediated endothelial proliferation as well as increased tube length and branch points in an in vitro angiogenesis assay that was blocked with Arg1 KD. These data define a novel role for CDC-derived EV upregulation of ϕ Arg1 in modulating an angiogenic macrophage phenotype (Fig. 10). This appears to be mediated by EV miRNA, as opposed to the protein, mRNA, or long noncoding RNA (lncRNA) EV cargo, as miR-depleted CDC-EVs failed to illicit upregulation of Arg1 in inflammatory macrophages.
Fig. 10.

Schematic diagram illustrating the effects of cardiosphere-derived cell (CDC)-extracellular vesicles (EVs) on inflammatory macrophages and their regulation of a novel urea-cycle-dependent mechanism promoting angiogenesis. Arg1, arginase 1; NOHA, Nω-hydroxy-nor-l-arginine; OAT, ornithine aminotransferase; NOS2, nitric oxide synthase 2; ODC, ornithine decarboxylase.
A limitation of our study is that the results are specifically related to studying CDC-EVs in vitro. Additional long-term studies using CDC-EVs in transgenic models of myocardial infarction will be required to establish the impact of this mechanism in vivo. In addition, further work will be necessary to identify the functional contribution of specific candidate miRNAs in CDC-EVs and validate miR targets in vivo.
Aside from the proangiogenic mechanism we describe, CDC EV-mediated arginase 1 upregulation may play additional roles in macrophage immunomodulation of inflammation. Arg1 has been implicated in macrophage-mediated protection in preclinical animal models of spinal cord injury and stroke (2, 9). This has recently been attributed to STAT6/Arg1 regulation of efferocytosis-mediated inflammation resolution by brain microglia and macrophages (2). Interestingly, we saw that CDC-EV-polarized BMDMs had a higher level of efferocytosis when compared with M2-polarized BMDMs, despite expressing a lower relative level of Arg1. This suggests that additional mechanisms other than Arg1 upregulation are also involved in immunomodulation. Further studies are needed to elucidate the role of EV-mediated macrophage Arg1 upregulation in modulating efferocytosis and the inflammatory response post-MI.
While the initial process of inflammatory cell infiltration is crucial in debridement following ischemia and integral for restoring myocardial integrity, shifting the polarization state toward an anti-inflammatory, angiogenic phenotype earlier in the healing process has demonstrated therapeutic potential (5, 13, 47). Our study was designed to gain mechanistic insight into the role of CDC-EVs in modulating macrophage phenotypes and operant signaling pathways to better understand their potential contribution to immunomodulatory cardioprotection. We defined a novel role of Arg1 in modulating macrophages to a proangiogenic phenotype dependent on increased polyamide production. Understanding the underlying molecular mechanisms and associated mediators of EV immunomodulation and angiogenesis may provide for improved therapeutic options for the treatment of heart disease.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant K08-HL-130594 (to J.K.L.), Veterans Affairs Grant IK2BX004097 (to J.K.L.), Eugene R. Mindell and Harold Brody Clinical Translational Research Award (to K.I.M.), and Frawley Research Fellowship (to A.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.I.M., A.M., and J.K.L. conceived and designed research; K.I.M., A.M., J.D.S., L.M.E., and J.K.L. performed experiments; K.I.M., A.M., J.D.S., L.M.E., and J.K.L. analyzed data; K.I.M., A.M., J.D.S., L.M.E., and J.K.L. interpreted results of experiments; K.I.M. and J.K.L. prepared figures; K.I.M. and J.K.L. drafted manuscript; K.I.M. and J.K.L. edited and revised manuscript; K.I.M., A.M., J.D.S., L.M.E., and J.K.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Dr. Min Guo from Kent State provided technical assistance with cryo-transmission electron microscopy (cryo-TEM).
REFERENCES
- 1.Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, Blum G, Epstein FH, Silman Z, Cohen S, Leor J. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol 62: 1890–1901, 2013. doi: 10.1016/j.jacc.2013.07.057. [DOI] [PubMed] [Google Scholar]
- 2.Cai W, Dai X, Chen J, Zhao J, Xu M, Zhang L, Yang B, Zhang W, Rocha M, Nakao T, Kofler J, Shi Y, Stetler RA, Hu X, Chen J. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 4: e131355, 2019. doi: 10.1172/jci.insight.131355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang CI, Liao JC, Kuo L. Arginase modulates nitric oxide production in activated macrophages. Am J Physiol Heart Physiol 274: H342–H348, 1998. doi: 10.1152/ajpheart.1998.274.1.H342. [DOI] [PubMed] [Google Scholar]
- 4.Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun 206: 667–673, 1995. doi: 10.1006/bbrc.1995.1094. [DOI] [PubMed] [Google Scholar]
- 5.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, Wojtkiewicz G, Dutta P, Sager HB, Borodovsky A, Novobrantseva T, Klebanov B, Fitzgerald K, Anderson DG, Libby P, Swirski FK, Weissleder R, Nahrendorf M. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol 63: 1556–1566, 2014. doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Couto G, Jaghatspanyan E, DeBerge M, Liu W, Luther K, Wang Y, Tang J, Thorp EB, Marbán E. Mechanism of enhanced MerTK-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler Thromb Vasc Biol 39: 2082–2096, 2019. doi: 10.1161/ATVBAHA.119.313115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP, Marbán E. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation 136: 200–214, 2017. doi: 10.1161/CIRCULATIONAHA.116.024590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marbán E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest 125: 3147–3162, 2015. doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J Neurosci 34: 8904–8917, 2014. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferraro B, Leoni G, Hinkel R, Ormanns S, Paulin N, Ortega-Gomez A, Viola JR, de Jong R, Bongiovanni D, Bozoglu T, Maas SL, D’Amico M, Kessler T, Zeller T, Hristov M, Reutelingsperger C, Sager HB, Döring Y, Nahrendorf M, Kupatt C, Soehnlein O. Pro-angiogenic macrophage phenotype to promote myocardial repair. J Am Coll Cardiol 73: 2990–3002, 2019. doi: 10.1016/j.jacc.2019.03.503. [DOI] [PubMed] [Google Scholar]
- 11.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11: 255–265, 2014. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frantz S, Hofmann U, Fraccarollo D, Schäfer A, Kranepuhl S, Hagedorn I, Nieswandt B, Nahrendorf M, Wagner H, Bayer B, Pachel C, Schön MP, Kneitz S, Bobinger T, Weidemann F, Ertl G, Bauersachs J. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 27: 871–881, 2013. doi: 10.1096/fj.12-214049. [DOI] [PubMed] [Google Scholar]
- 13.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci USA 108: 1827–1832, 2011. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasan AS, Luo L, Yan C, Zhang TX, Urata Y, Goto S, Mangoura SA, Abdel-Raheem MH, Zhang S, Li TS. Cardiosphere-derived cells facilitate heart repair by modulating m1/m2 macrophage polarization and neutrophil recruitment. PLoS One 11: e0165255, 2016. [Erratum in PLoS One 12: e0171892, 2017.] doi: 10.1371/journal.pone.0165255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 115: 284–295, 2014. [Erratum in Circ Res 115: e95, 2014.] doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim AG, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2: 606–619, 2014. doi: 10.1016/j.stemcr.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igarashi K, Kashiwagi K. Modulation of cellular function by polyamines. Int J Biochem Cell Biol 42: 39–51, 2010. doi: 10.1016/j.biocel.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Kim YK, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci USA 113: E1881–E1889, 2016. doi: 10.1073/pnas.1602532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang JK, Young RF, Ashraf H, Canty JM Jr. Inhibiting extracellular vesicle release from human cardiosphere derived cells with lentiviral knockdown of nSMase2 differentially effects proliferation and apoptosis in cardiomyocytes, fibroblasts and endothelial cells in vitro. PLoS One 11: e0165926, 2016. doi: 10.1371/journal.pone.0165926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res 56: 4625–4629, 1996. [PubMed] [Google Scholar]
- 22.Mentkowski KI, Lang JK. Exosomes engineered to express a cardiomyocyte binding peptide demonstrate improved cardiac retention in vivo. Sci Rep 9: 10041, 2019. doi: 10.1038/s41598-019-46407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mentkowski KI, Snitzer JD, Rusnak S, Lang JK. Therapeutic potential of engineered extracellular vesicles. AAPS J 20: 50, 2018. doi: 10.1208/s12248-018-0211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morimoto K, Janssen WJ, Fessler MB, Xiao YQ, McPhillips KA, Borges VM, Kench JA, Henson PM, Vandivier RW. Statins enhance clearance of apoptotic cells through modulation of Rho-GTPases. Proc Am Thorac Soc 3: 516–517, 2006. doi: 10.1513/pats.200603-073MS. [DOI] [PubMed] [Google Scholar]
- 25.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969, 2008. [Erratum in Nat Rev Immunol 10: 460, 2010.] doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158: 638–651, 2009. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh H, Siano B, Diamond S. Neutrophil isolation protocol. J Vis Exp 17: 745, 2008. doi: 10.3791/745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pipili-Synetos E, Sakkoula E, Haralabopoulos G, Andriopoulou P, Peristeris P, Maragoudakis ME. Evidence that nitric oxide is an endogenous antiangiogenic mediator. Br J Pharmacol 111: 894–902, 1994. doi: 10.1111/j.1476-5381.1994.tb14822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide is involved in the regulation of angiogenesis. Br J Pharmacol 108: 855–857, 1993. doi: 10.1111/j.1476-5381.1993.tb13476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol 5: 532, 2014. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rébé C, Raveneau M, Chevriaux A, Lakomy D, Sberna AL, Costa A, Bessède G, Athias A, Steinmetz E, Lobaccaro JM, Alves G, Menicacci A, Vachenc S, Solary E, Gambert P, Masson D. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ Res 105: 393–401, 2009. doi: 10.1161/CIRCRESAHA.109.201855. [DOI] [PubMed] [Google Scholar]
- 32.Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol 166: 2173–2177, 2001. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- 33.Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 119: 853–864, 2016. doi: 10.1161/CIRCRESAHA.116.309001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schleicher U, Paduch K, Debus A, Obermeyer S, König T, Kling JC, Ribechini E, Dudziak D, Mougiakakos D, Murray PJ, Ostuni R, Körner H, Bogdan C. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Reports 15: 1062–1075, 2016. doi: 10.1016/j.celrep.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447: 869–874, 2007. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8: 349–361, 2008. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787–795, 2012. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soda K. The mechanisms by which polyamines accelerate tumor spread. J Exp Clin Cancer Res 30: 95, 2011. doi: 10.1186/1756-9966-30-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takigawa M, Enomoto M, Nishida Y, Pan HO, Kinoshita A, Suzuki F. Tumor angiogenesis and polyamines: alpha-difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase, inhibits B16 melanoma-induced angiogenesis in ovo and the proliferation of vascular endothelial cells in vitro. Cancer Res 50: 4131–4138, 1990. [PubMed] [Google Scholar]
- 40.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 170: 818–829, 2007. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van den Bossche J, Saraber DL. Metabolic regulation of macrophages in tissues. Cell Immunol 330: 54–59, 2018. doi: 10.1016/j.cellimm.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 113: 1004–1012, 2013. doi: 10.1161/CIRCRESAHA.113.301198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu G, Bazer FW, Burghardt RC, Johnson GA, Kim SW, Knabe DA, Li P, Li X, McKnight JR, Satterfield MC, Spencer TE. Proline and hydroxyproline metabolism: implications for animal and human nutrition. Amino Acids 40: 1053–1063, 2011. doi: 10.1007/s00726-010-0715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62: 24–35, 2013. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 45.Yang W, Ando J, Korenaga R, Toyo-oka T, Kamiya A. Exogenous nitric oxide inhibits proliferation of cultured vascular endothelial cells. Biochem Biophys Res Commun 203: 1160–1167, 1994. doi: 10.1006/bbrc.1994.2304. [DOI] [PubMed] [Google Scholar]
- 46.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, Motoshima H, Taguchi T, Sonoda K, Kukidome D, Takuwa Y, Kawada T, Brownlee M, Nishikawa T, Araki E. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res 100: 1442–1451, 2007. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X, Gao L, Xie J, Xu B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc Res 115: 1205–1216, 2019. doi: 10.1093/cvr/cvz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.