

Abstract
We characterized mouse blood pressure and ion transport in the setting of commonly used rodent diets that drive K+ intake to the extremes of deficiency and excess. Male 129S2/Sv mice were fed either K+-deficient, control, high-K+ basic, or high-KCl diets for 10 days. Mice maintained on a K+-deficient diet exhibited no change in blood pressure, whereas K+-loaded mice developed an ~10-mmHg blood pressure increase. Following challenge with NaCl, K+-deficient mice developed a salt-sensitive 8 mmHg increase in blood pressure, whereas blood pressure was unchanged in mice fed high-K+ diets. Notably, 10 days of K+ depletion induced diabetes insipidus and upregulation of phosphorylated NaCl cotransporter, proximal Na+ transporters, and pendrin, likely contributing to the K+-deficient NaCl sensitivity. While the anionic content with high-K+ diets had distinct effects on transporter expression along the nephron, both K+ basic and KCl diets had a similar increase in blood pressure. The blood pressure elevation on high-K+ diets correlated with increased Na+-K+-2Cl− cotransporter and γ-epithelial Na+ channel expression and increased urinary response to furosemide and amiloride. We conclude that the dietary K+ maneuvers used here did not recapitulate the inverse effects of K+ on blood pressure observed in human epidemiological studies. This may be due to the extreme degree of K+ stress, the low-Na+-to-K+ ratio, the duration of treatment, and the development of other coinciding events, such as diabetes insipidus. These factors must be taken into consideration when studying the physiological effects of dietary K+ loading and depletion.
Keywords: blood pressure, kidney, potassium, sodium transport
INTRODUCTION
Potassium (K+) is an essential cation required for the maintenance of intracellular volume, while sodium (Na+) is essential for the maintenance of extracellular volume. Together, the balance between these two ions is critical for volume and blood pressure homeostasis. Dietary Na+ intake is well known to correlate positively with blood pressure; however, there is less appreciation regarding the inverse relationship between dietary K+ and blood pressure, despite ample evidence. For almost a century, K+ salts have effectively been used as diuretics to treat hypertension (31). Conversely, K+ depletion is associated with hypertension, nephropathy, and diabetes insipidus (48, 54). Yet, it remains unclear how K+ depletion leads to hypertension or how K+ excess opposes hypertension.
In 1958, Meneely and Ball (42) reported that rats fed toxic amounts of NaCl survived longer if coadministered KCl, proposing that it was not Na+ intake per se but rather the ratio of Na+ to K+ that was necessary for homeostasis. More recently, the INTERSALT study reported that the ratio of urinary K+ to Na+ had a stronger statistical relationship to blood pressure compared with either K+ or Na+ alone (28a). The importance of this ratio becomes evident when considering that our modern diets contain high Na+ and low K+ relative to ancient paleolithic diets, which contained up to fourfold more K+ and significantly less Na+ (43, 52). These primitive diets suggest that humans evolved to conserve Na+ and efficiently eliminate K+ but are inept at dealing with our modern diets that require the conservation of K+ and elimination of excess Na+.
There has been a resurgence of interest in understanding how dietary K+ intake contributes to blood pressure, endocrine signaling, acid/base balance, and renal ion transport (10). However, many animal studies have neglected to consider the K+ content of commercially available rodent diets and the integrated effects of those diets on volume status, acid/base status, hormonal changes, and electrolyte transport along the entire nephron. Here, we sought to address this knowledge gap by characterizing the effect of standard dietary K+ depletion and loading maneuvers on blood pressure, electrolyte balance, and renal Na+ transporter expression in mice. Notably, we found that under the conditions studied, these commonly used preformulated rodent diets do not capture the reported inverse relationship between blood pressure and dietary K+, likely due to a relative deficiency in NaCl content, the duration of the diet, and the severity of the K+ stress. By correlating the renal effects of these extreme diets with blood pressure, we identified multiple factors that warrant consideration when modeling the relationship between dietary K+ and hypertension.
MATERIALS AND METHODS
Animal Experiments
All animal protocols conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male 129/Sv-Elite mice [129S2/SvPasCrl (17), aged 10–20 wk, 25–30 g, Charles River Laboratories] were housed in a temperature-controlled room on a 12:12-h light-dark cycle. Elite status indicates a special health profile developed by Charles River Laboratories. Animals were fed with one of the following four matched synthetic commercially available diets (Teklad, Madison, WI): low K+ (TD.88239), control (TD.88238), high K+ “basic” (TD.07278), or high KCl (TD.09075). Dietary K+ content was as follows: low K+ (<0.003%), control (1% K+ with equal amounts of K+ citrate and KCl), high-K+ basic (5% K+ with a 1:1:1 ratio of K+ citrate, KCl, and K+ carbonate), and high KCl (5% K+). The Cl− content was as follows: low K+ (0.45%), control (0.9%), high K+ basic (2%), and high KCl (5.2%). The Na+ content of all diets was 0.3%.
During all telemetry experiments, mice were maintained on the pellet diets versus subsequent metabolic cage experiments, wherein gel diets were used to prevent pellet debris in the urine. Gels diets were derived from blended commercial diets combined with 1–2% agar. The electrolyte content of the agar in the gel diet was 0.006% Na+ and 0.008% K+. Gel diets provided a portion of the baseline water intake. Mice had free access to water unless otherwise noted. After 10 days on varying K+ diets, mice were anesthetized with isoflurane, and whole blood was collected with a heparinized syringe via cardiac puncture and analyzed immediately by iSTAT (Abbot). Kidneys were harvested and flash frozen for immunoblot analysis and paraformaldehyde treated for immunofluorescence. Urine was immediately collected from mouse bladders for urine pH measurements using AimStrip US-5 (Germaine, San Antonio, TX). Mice were euthanized by cervical dislocation. Blood plasma was isolated by centrifugation, and aldosterone levels were measured using ELISA kit (no. ADI-900-173, Enzo).
Telemetry
Mice were anesthetized with isoflurane, and DSI PA-C10 telemetry units (Data Sciences International, New Brighton, MN) were surgically implanted into the femoral artery as previously described (45). Mice recovered for 1 wk before dietary challenges commenced. Mice were fed varying K+ pellet diets for 10 days with >7 days of washout with control diet between each K+ variation, and blood pressures returned to baseline during washout. For the saline challenge, mice were maintained on varying K+ diets for 10 days and then challenged with 1% saline in their drinking water for 96 h. Telemetry data, including systolic blood pressure, diastolic blood pressure, heart rate, and pulse pressure, were collected over 22 h every other day for the duration of the diet challenge using Ponemah software (Data Sciences International). Hourly data points represent an average of 12 mean arterial pressure (MAP) measurements per hour obtained every 5 min. Daily data points represent an average of 264 MAP measurements per day.
Metabolic Cages
Mice were individually housed in metabolic cages (Tecniplast) on either days 4–5 or days 8–10. Before placement in the metabolic cage, mice consumed the pellet diet and were introduced to the gel diet + pellet diet 24 h before placement in the metabolic cages. Next, mice were individually placed into metabolic cages and switched to exclusive gel-based diets to prevent pellet debris from contaminating the urine. They acclimated for the first 24 h followed by 24-h measurement of food and water intake and urine collection. Urine Na+ and K+ concentrations were determined using flame photometry (Instrumentation Laboratory 943, Wefen, Bedford, MA). Urine osmolality was determined using a micro-osmometer (Precision Systems, Natick, MA).
Diuretic Challenge
Mice were individually housed in metabolic cages after 10 days of the respective K+ diets and acclimated for 24 h. After acclimation, mice were given intraperitoneal injections of either furosemide (30 mg/kg), hydrochlorothiazide (HCTZ; 25 mg/kg), or amiloride (5 mg/kg), and urine was collected from 0−6 h and 7–24 h. Values are shown as fold changes in urine output after diuretic compared with sham saline injection given to the same mouse on a different day.
Semiquantitative Immunoblot Analysis
For protein isolation, the whole kidney or isolated kidney cortex was flash frozen. The kidney was homogenized in ice-cold RIPA extraction buffer (Thermo Scientific) with freshly added protease and phosphatase inhibitor cocktail. Homogenized kidneys were agitated at 4°C for 30 min, and centrifugation was then performed to isolate the supernatant. Protein quantification was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). Equal amounts of protein (20–40 μg) were separated by SDS-PAGE using 4–15% Criterion TGX precast gels (Bio-Rad). Protein was transferred to a nitrocellulose membrane. Signals were measured using Bio-Rad ChemiDoc, and densitometry was quantified with ImageLab analysis software (Bio-Rad). Uniform protein concentrations were determined as previously described (41). Briefly, 12 μg protein from each sample was loaded onto the SDS-PAGE gel and then stained with Coomassie blue. Five random bands were quantified, and appropriate adjustments were made to the sample preparation until the loading was determined to be uniform.
Antibodies Used
The following antibodies were used for immunoblot analysis: aquaporin 2 (Aqp2; 29 kDa unglycosylated and 35−45 kDa mature, sc-515770, Santa Cruz Biotechnology) (46), Na+/H+ exchanger 3 (NHE3; 84 kDa, SPC-400, Stressmarq) (28), Na+-bicarbonate cotransporter 1A (NBCe1A; 130 kDa denatured and 280 kDa dimer, AB3212-I, Millipore) (39, 49), Na+-glucose transporter 2 (SGLT2; 74 kDa, ab37296, Abcam) (44), Na+-K+-2Cl− cotransporter (NKCC2; 160 kDa, SPC-401, Stressmarq) (6), NaCl cotransporter (NCC; 130 kDa, kindly provided by David Ellison) (8), phosphorylated (p)NCC (Thr53; 130 kDa, P1311-53, Phospho-solutions) (40), γ-subunit of the epithelial Na+ channel (γ-ENaC; 83 kDa cleaved and 95 kDa uncleaved, SPC-405, Stressmarq) (6), renal outer medullary K+ channel (ROMK; 50−65 kDa complex glycosylated, R-80, kindly provided by James Wade) (61), electroneutral Cl− exchanger (pendrin; 85−110 kDa denatured and 220 kDa dimer, sc16894, Santa Cruz Biotechnology) (25, 62), and Na+-driven Cl−/bicarbonate exchanger (NDCBE; 123 kDa, no. 12531-1, ProteinTech) (53).
Immunofluorescence Confocal Microscopy
Paraformaldehyde-fixed kidney tissues were cryoprotected in 0.9 M sucrose overnight, embedded in blocks, and then stored at −20°C. Sections (6 μm) were sliced on a cryostat (Leica Microsystems) and collected on charged microscope slides (Fisher Scientific). Kidneys were rehydrated and treated with 1% SDS for 10 min for retrieval of antigenic sites. Slides were then washed with high-salt buffer + BSA before the addition of primary antibody. Primary antibodies were incubated overnight at 4°C, and subsequent incubation with secondary antibodies was used to visualize staining. Confocal imaging of the kidney tissue was performed using a Zeiss LSM 700 confocal imaging system. Three-dimensional z-stacks were acquired and maximum intensity projected to visualize apical versus diffuse ENaC localization. To quantify the distribution of ENaC within renal tubule epithelial cells from mice treated with varying diets, we performed a line profile analysis of ENaC immunofluorescence using FIJI ImageJ. Line segments five pixels wide and 6.5 μm long were drawn from the apical membrane to the basolateral membrane. A profile plot was used to measure the fluorescence intensity along the line, and data were exported to Graph Pad Prism for analysis. For each treatment group, line plots from 30 representative cells over different 3 experiments were averaged and plotted to compare the distribution between groups.
Data analysis
Data were analyzed using Graph Pad Prism software and are presented as means ± SE. Comparisons between two groups were determined by a Student’s t test. Comparisons between multiple groups were determined using one- or two-way ANOVA followed by the appropriate post hoc test. Significance was assumed to be P ≤ 0.05. Significant differences between KCl and K+ basic diets are indicated with in the figures.
RESULTS
Blood Pressure Was Significantly Elevated in Mice on High-K+ Diets
To determine the effect of dietary K+ on mean arterial pressure (MAP), male 129S2/Sv mice were fed one of the following four matched synthetic commercial diets with varied K+ content: low K+ (<0.003% K+), control (1% K+), high K+ basic (5% K+), or high KCl (5% K+). Blood pressure, heart rate, and activity were measured using radiotelemetry for 22 h every other day for 10 days per dietary condition (Table 1). Mice fed the low-K+ diet exhibited no significant difference in blood pressure compared with the control diet, whereas mice fed K+-loaded diets (either high-K+ basic or KCl) developed a significant increase in blood pressure within 8–10 days (Fig. 1A). As expected, all recordings demonstrated a circadian blood pressure rhythm over the 22-h period.
Table 1.
Telemetry values
| Low K+ | Ctrl | K+ Basic | KCl | |
|---|---|---|---|---|
| Day 4 (22 h) | ||||
| SBP, mmHg | 138 (2.2) | 133 (2.2) | 136 (1.6) | 137 (2.6) |
| DBP, mmHg | 96 (1.5) | 98 (1.5) | 97 (1.2) | 99 (2.6) |
| MAP, mmHg | 116 (1.7) | 115 (1.8) | 116 (1.3) | 117 (2.4) |
| HR, beats/min | 479 (12)† | 557 (8) | 545 (8) | 504 (5)*‡ |
| Pulse pressure, mmHg | 41 (1.0)† | 35 (0.8) | 39 (0.6) | 38 (1.4) |
| Activity, arbitrary units | 1.7 (0.2) | 2.0 (0.4) | 2.0 (0.2) | 2.5 (0.3) |
| Day 10 (22 h) | ||||
| SBP, mmHg | 136 (2.7) | 135 (1.4) | 149 (2.6)† | 145 (2.9)† |
| DBP, mmHg | 97 (2.1) | 97 (1.3) | 107 (2.0)† | 106 (3.7)* |
| MAP, mmHg | 115 (2.4) | 116 (1.3) | 128 (2.3)† | 125 (3.1)* |
| HR, beats/min | 430 (8)† | 540 (13) | 518 (4) | 510 (15) |
| Pulse pressure, mmHg | 39 (0.7) | 38 (0.4) | 42 (0.9) | 39 (2.5) |
| Activity, arbitrary units | 2.0 (0.3) | 2.0 (0.1) | 2.6 (0.2) | 3.5 (0.3)† |
Values are means (SE) and represent an average of 264 measurements per day; n = mice per diet. On day 4, varying dietary K+ had no significant effect on 22-h systolic blood pressure (SBP), diastolic blood pressure (DBP), or mean arterial pressure (MAP). Mice fed the low-K+ diet and KCl diet did have a significant decrease in heart rate (HR) compared with control. At day 10, both high-K+ diets elevated blood pressure compared with control [SBP (10–14 mmHg), DBP (9–10 mmHg), and MAP (9–12 mmHg)]. Additionally, at 10 days, the low-K+ diet caused a significant decrease in HR compared with control. Two-way ANOVA followed by Dunnett’s post hoc test was used.
P ≤ 0.05 and
P ≤ 0.01, significant difference from the control diet.
Significant difference between the K+ basic and KCl diets.
Fig. 1.
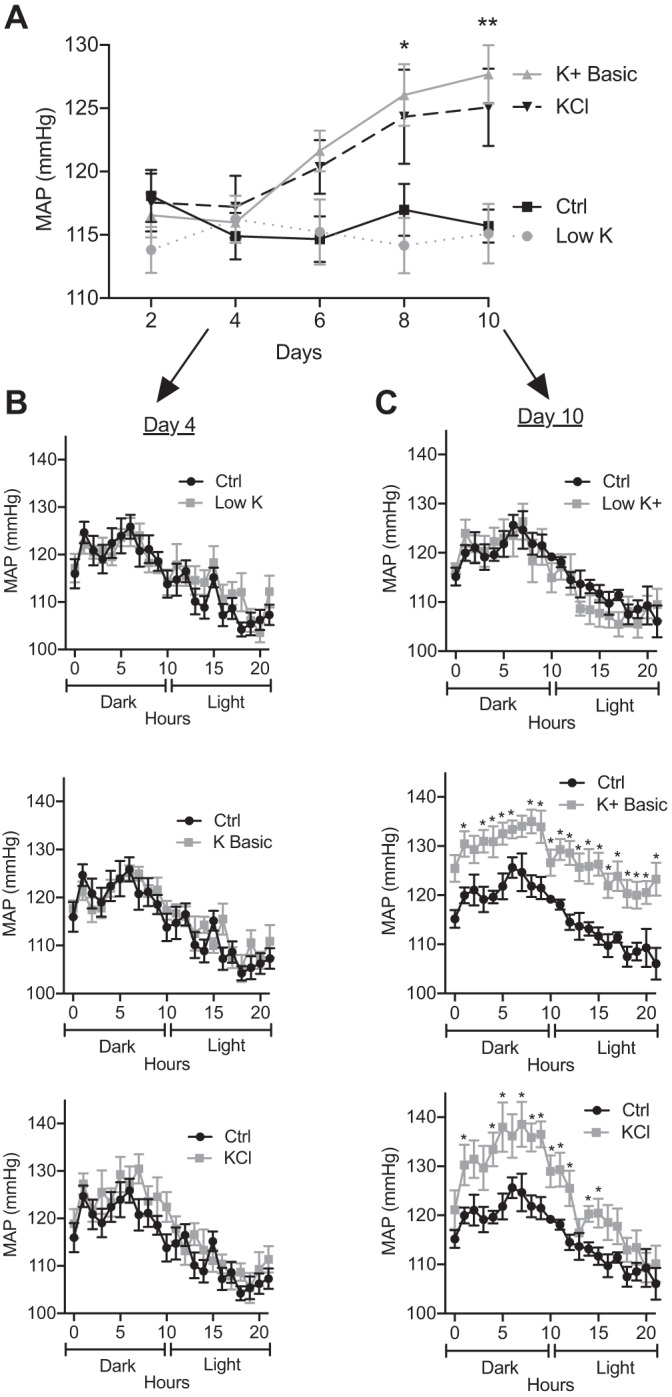
Mean arterial pressure (MAP) was significantly elevated in mice on high-K+ diets. Mice were treated with K+ control (Ctrl), low-K+, K+ basic, and KCl diets for 10 days. A: the time course of MAP (means ± SE) was significantly elevated within 8 days for mice on the K+ basic diet and within 10 days for the KCl diet compared with the control diet. The low-K+ diet had no effect on MAP (two-way ANOVA with Dunnett’s post hoc analysis, *P ≤ 0.05 and **P ≤ 0.01). n = 6 mice per diet. B: 4 days of varying dietary K+ had no effect on blood pressure. The graph shows the 22-h diurnal pattern of MAP (means ± SE) with varying K+ diets (multiple t tests). n = 6 mice per diet. C: 10 days of low-K+ diet had no effect on MAP, whereas 10 days of K+ basic diet significantly elevated blood pressure during both the dark/active and light/resting phases. The KCl diet significantly elevated MAP predominantly during the dark phase. The graph shows the 22-h diurnal pattern of MAP (means ± SE, multiple t tests, *P ≤ 0.05). n = 6 mice per diet.
On day 4, the various dietary K+ maneuvers did not affect blood pressure compared with control diets (Table 1 and Fig. 1B). By day 10, MAP of mice on the low-K+ diet remained unchanged compared with mice on the control diet, whereas mice fed K+-loaded diets developed a significant rise in blood pressure (Fig. 1C). Mice fed a high-K+ basic diet developed significantly elevated pressures (~12 mmHg) throughout the 22-h cycle. On day 10, mice fed a high-KCl diet exhibited a similar elevation in MAP (~9 mmHg) during the dark/active phase, although this effect waned during the resting period (Fig. 1C). This suggests that the anionic content of K+ loading differentially effects the circadian blood pressure pattern.
K+ Depletion Induces Saline Sensitivity
The results shown in Fig. 1 indicate that the inverse relationship between dietary K+ and blood pressure is not absolute, and we reasoned that the commercial rodent diets and experimental design might explain this problem. Although the commercial rodent diets used in these experiments have been used by several groups to model the physiological effects of K+ loading and restriction, the differences in K+ content relative to the control diet are extreme. Moreover, the diets lack enrichment in NaCl. Prior experiments in mice on high-NaCl diets (3–6%) challenged with dietary K+ restriction shown that the mice developed an increase in blood pressure (57, 60). Because our mice were maintained on diets containing lower amounts of Na+ (0.3%) with varied K+ concentrations, we challenged mice with 1% NaCl in the drinking water for 96 h after 10 days of K+ manipulation to evaluate the effects of NaCl on blood pressure. As shown in Fig. 2A, only K+-deficient mice developed a significant (7–8 mmHg) rise in blood pressure within 24 h of saline challenge. The NaCl-sensitive rise coincided with the dark/active phase (Fig. 2B), similar to prior reports (14, 60). In contrast, mice maintained on control or high-K+ diets did not develop a significant salt-sensitive increase in blood pressure throughout the 96-h saline challenge. These results indicate that the increase in blood pressure in response to a Na+ load is only manifested when mice are on a low-K+ diet; even control levels of K+ are sufficient to prevent the rise in blood pressure, and increasing dietary K+ beyond this has no further effect.
Fig. 2.

Low-K+ diet induces salt sensitivity. A: mice were treated with various K+ diets as indicated for 10 days and then challenged with 1% NaCl (supplemented to drinking water) for 96 h. The graph shows the change in mean arterial pressure (MAP; means ± SE). Mice on the low-K+ diet developed a significant change in MAP compared with the control (Ctrl) diet, whereas mice fed high-K+ basic or KCl diets had no significant change compared with the control diet (two-way ANOVA with Dunnett’s post hoc analysis, *P ≤ 0.05 and **P ≤ 0.01). n = 6 mice per diet. B: graph of the 22-h diurnal pattern showing MAP after 10 days on K+ diets and after 24 h of saline challenge. Mice maintained on the low-K+ diet had a significant elevation in MAP during the dark/active phase in response to saline challenge; however, there was no significant elevation during the light/resting phase (multiple t tests, *P ≤ 0.05). n = 6 mice per diet.
Effect of Dietary K+ on Blood and Urine Parameters
Next, we conducted metabolic cage experiments to verify that the mice were consuming the diets and had appropriate blood and urine changes. Mice were equilibrated in the metabolic cages for 24 h, and a 24-h collection of intake, output, and urine was then performed (Fig. 3A). These experiments were conducted on days 5 and 10 on the various K+ diets, and whole blood was obtained at the time of euthanization on day 10 (Table 2). As predicted, whole blood and urine K+ concentrations from day 10 appropriately reflected the changes in dietary K+ intake (Table 2 and Fig. 3C). Other significant changes to whole blood chemistries over 10 days included an increase in Cl− and decrease in total CO2 (representing a decrease in ) in mice that consumed the KCl diet compared with the control diet. There were also significant differences between mice maintained on K+ basic versus KCl diets; notably, mice on the KCl diet had higher Ca2+ and lower urine pH (Table 2). All of these changes are consistent with the KCl diets generating an acid load in these mice. Not surprisingly, control mice and KCl diet-fed mice had a urine pH of 6.5 and 6.0, respectively (Table 2). In contrast, both K+-deficient mice and high-K+ basic diet-fed mice exhibited a significant increase in urine pH. The increase in urine pH during K+ depletion has been attributed to an increase in ammonia production (32, 55).
Fig. 3.

Dietary K+ maneuvers induce polyuria. A: diagram indicating the type of cage and diet for each maneuver. Throughout the telemetry experiment, mice were maintained in telemetry cages and fed pellet diet (P) ad libitum. On days 11–14, their drinking water was switched from free water to 1% saline. For the 5- and 10-day urine and whole blood experiments, mice were maintained on the pellet diet. Then, 24 h before placement in metabolic cages, mice were introduced to the gel diet [access to both pellet and gel diets (P&G)] and next placed in metabolic cages exclusively on the gel diet (G) and acclimatized for 24 h, and urine was collected for 24 h. *Time point when mice were euthanized and whole blood was collected. For the diuretic experiments, mice were placed in the metabolic cages and on exclusive gel diets on day 10. B: mice were maintained on varying K+ diets, and 24-h intakes and outputs (I/O) were obtained on days 5 and 10. Mice maintained on the K+ basic diet had decreased consumption at day 5 compared with mice on the other diets, but by day 10 this difference was attenuated. C: 24-h urine trends were similar on both days 5 and 10. Urinary K+ (mmol/day) was appropriately decreased with the low-K+ diet. The KCl diet had more urinary K+ excretion than the K+ basic diet; both were appropriately increased compared with the control (Ctrl) diet. D: urinary Na+ (mmol/day) was significantly decreased on day 5 with the low-K+ diet compared with control diet-fed mice. On day 10 only the K+ basic diet significantly increased urine Na+ (mmol/day) compared with control. Mice maintained on K+ basic and KCl diets had significant differences in daily Na+ excretion. E: on day 5, mice fed the high-K+ diets had increased water intake compared with control; by day 10, mice fed the low-K+ diet also began to have increased water intake. F: on day 5, mice fed the low-K+ diet had a decrease in urine volume, whereas mice fed the KCl diet had an increase in urine volume. On day 10, urine volume was significantly increased by low-K+ and high-K+ diets compared with the control diet. G: on day 10, only the low-K+ diet significantly decreased urine osmolality (osms) compared with the control diet. Day 5: n = 5–6 mice per diet; day 10: n = 16–18 mice per diet. One-way ANOVA with Sidak’s post hoc analysis, *P ≤ 0.05 and **P ≤ 0.01, significant difference from the control diet and φsignificant difference between the K+ basic and KCl diets.
Table 2.
Effect of 10 days of dietary K+ on whole blood parameters and urine pH
| Low K+ | Control | K+ Basic | KCl | |
|---|---|---|---|---|
| Change in weight at 10 days, g | −0.55 (0.53) | +0.94 (0.34) | −0.15 (0.43) | −0.71 (0.51) |
| Day 10 whole blood | ||||
| Na+, mmol/L | 147.4 (0.61)* | 144.2 (0.41) | 144.3 (0.66) | 146.2 (0.92) |
| K+, mmol/L | 2.88 (0.08)† | 4.06 (0.09) | 4.93 (0.21)† | 5.34 (0.21)† |
| Cl−, mmol/L | 112 (0.48) | 112.5 (0.34) | 113.1 (0.83) | 119.5 (1.27)†‡ |
| Ionized Ca2+, mmol/L | 1.24 (0.02) | 1.29 (0.02) | 1.21 (0.03) | 1.31 (0.03)‡ |
| Total CO2, mmol/L | 24.3 (0.62) | 23.5 (0.29) | 24.4 (0.74) | 20.8 (0.48)†‡ |
| Glucose, mg/dL | 208.6 (4.66) | 217.8 (9.10) | 181.2 (7.06)† | 187.5 (9.60)* |
| Blood urea nitrogen, mg/dL | 25.83 (2.28) | 22.33 (0.89) | 23.08 (1.23) | 25.36 (1.70) |
| Hct, %packed cell volume | 41.67 (1.20) | 41.25 (0.76) | 38.00 (0.58)* | 37.27 (0.97)* |
| Aldosterone, pg/mL | 83 (17)* | 385 (66) | 929 (242)* | 2889 (390)†‡ |
| Day 10 (spot urine) pH | 7.2 (0.13)* | 6.5 (0.19) | 7.2 (0.26)* | 6.0 (0.04)‡ |
Values are means (SE). For the change in weight, n = 16–18 mice per diet; for whole blood and urine pH, n = 12 mice per diet. One-way ANOVA with Sidak’s post hoc analysis was used.
P ≤ 0.05 and
P ≤ 0.01, significant difference from the control diet.
Significant difference between K+ basic and KCl.
There was no significant change in weight over the 10 days of varying K+ diets (Table 2). We did find that mice fed the K+ basic diet had a significant decrease in diet consumption on day 5 that began to normalize by day 10 (Fig. 3B), possibly due to the palatability of the K+ basic diet. Despite the decreased consumption, mice on the K+ basic diet had the anticipated increased urinary K+ excretion compared with mice on the control diet. The urinary excretion of K+ for all mice reflected dietary intake on both days 5 and 10 (Fig. 3C). Mice that consumed the K+ basic diet had significantly lower urinary K+ excretion compared with mice that consumed the KCl diet on both days 5 and 10. This may reflect changes in consumption or urinary handling (Fig. 3C). Next, we looked at urinary Na+ handling in mice fed varying K+ diets containing 0.3% Na+ content. On day 5, K+-deficient mice had a significant decrease in urinary Na+ excretion that reversed by day 10 (Fig. 3D). On day 10, only the K+ basic diet had a significant effect on urinary Na+ excretion, increasing urinary Na+ content.
Mice that consumed the high-KCl diet developed polydipsia and polyuria within 5 days of starting the diet and maintained this phenotype on day 10 (Fig. 3, E and F). Mice on the K+ basic diet developed polydipsia by day 5 and developed polyuria between days 5 and 10. On day 5, mice on the low-K+ diet had low urine volumes but over the course of 10 days developed polydipsia and polyuria (Fig. 3, E and F).
Mice Develop Diabetes Insipidus After 10 Days of Extreme Dietary K+ Restriction
By day 10, both K+ depletion and loading resulted in polyuria (Fig. 3F), associated with an increase in water intake (Fig. 3E). Interestingly, the type of diuresis, along with the apparent volume status, was dependent on the dietary K+ maneuver. Based on urine osmolality experiments, K+-deficient mice developed free water diuresis, whereas K+-loaded mice developed solute-rich diuresis (Fig. 3G). This solute-rich diuresis was driven by the increase in urinary K+ (Fig. 3C). Regardless of the anion, K+-loaded mice had a significant decrease in hematocrit, a rise in aldosterone, and elevated blood pressure (Table 2 and Fig. 1A), all consistent with a volume expanded state (24). In contrast, mice fed a K+-depleted diet became hypernatremic, with decreased urine osmolality consistent with the development of diabetes insipidus (38). One explanation is that K+ depletion for 10 days markedly decreased the expression of mature glycosylated AQP2 (Fig. 4). Given these findings, the commonly used method of K+ restriction used here qualifies as an extreme K+ deprivation maneuver.
Fig. 4.

Dietary K+ depletion decreases aquaporin 2 (Aqp2) abundance. The Western blot of the mouse kidney cortex demonstrates a significant reduction in the mature form (35 kDa) of Aqp2 during the low-K+ diet (one-way ANOVA with Sidak’s post hoc analysis, **P ≤ 0.01). n = 6 mice per diet.
Effect of Dietary K+ Maneuvers on Tubular Ion Transport Pathways
To gain further insight into the differential effects of these dietary K+ maneuvers on ion transport along the length of the cortical nephron, we evaluated the expression of physiologically relevant ion transport pathways extending from the proximal tubule to the cortical collecting duct (Fig. 5A).
Fig. 5.


Dietary K+ maneuvers affect the abundance of transporters and channels along the nephron. Kidney lysates from mice subjected to various dietary K+ maneuvers for 10 days were assessed by Western blot analysis. Equivalent protein loading was determined by BCA Protein Assay and Coomassie staining. Results are shown as means ± SE; n = 6 mice per diet. One-way ANOVA with Sidak’s post hoc analysis, *P ≤ 0.05 and **P ≤ 0.01 compared with control; φsignificant difference between K+ basic and KCl diets. A: we probed for a variety of channels and transporters expressed from the proximal tubule to the collecting duct, as indicated. B: Western blots of kidney homogenates from mice subjected to various K+ diets. All samples were of the kidney cortex unless noted [Na+-K+-2Cl− cotransporter (NKCC2) in the whole kidney]. #Multimerization of Na+-bicarbonate cotransporter 1A (NBCe1A) (39) and pendrin (62). C: graphical summary of the results. The dotted line depicts control values normalized to 1.0. DCT, distal convoluted tubule; γ-ENaC, γ-subunit of the epithelial Na+ channel; IC, intercalated cells; NCC, NaCl cotransporter; NHE3, Na+/H+ exchanger 3; ROMK, renal outer medullary K+ channel; NDCBE, Na+-driven Cl−/bicarbonate exchanger; PC, principal cells; PCT, proximal convoluted tubule; SGLT2, Na+-glucose transporter 2; TAL, thick ascending limb.
Proximal tubule.
First, we examined the effects of K+ on proximal tubule Na+-dependent net proton secretion. NHE3 accounts for a majority of the Na+ reabsorption and H+ secretion in the proximal tubule (7). K+ restriction was associated with a large (4-fold) increase in NHE3 expression, similar to previous reports (16, 21). NBCe1A, which facilitates the bicarbonate reclamation that accompanies luminal H+ secretion (39), exhibited a significant but less pronounced increase in response to K+ depletion (Fig. 5B) (35). KCl loading also had a trend toward increased NHE3 and NBCe1A expression, albeit to a lesser degree. In contrast, the alkaline load from the high-K+ basic diet resulted in an expected decrease in the expression of NHE3 and NBCe1A.
Next, we assessed the effect of dietary K+ on SGLT2. We hypothesized that with extreme K+ depletion, the expression of this cotransporter would increase to facilitate increased Na+ reabsorption in the upstream nephron. However, the results revealed a significant decrease in SGLT2 expression in K+-depleted mice (Fig. 4B). This is consistent with reports showing that pharmacological SGLT2 inhibition can induce a small increase in serum K+ levels, possibly through renal mechanisms (18, 66).
Thick ascending limb.
K+ loading did not affect cortical NKCC2 abundance (Fig. 5B); however, when total (cortical plus medullary) NKCC2 was quantified, K+ loading was associated with a significant increase in NKCC2 abundance (Fig. 5B). In contrast, K+ depletion decreased NKCC2 expression by 40–50% in both cortical and total lysates (Fig. 5B). This decrease is consistent with prior reports showing that hypokalemia downregulates NKCC2 mRNA, protein expression, and function in K+-depleted animals (2, 16, 27, 37). Due to antibody cross-reactivity with distal convoluted tubule thiazide-sensitive pNCC, we were unable to quantify the amount of phosphorylated NKCC2 (active NKCC2) despite using two different antibodies. However, we further evaluated the activity of NKCC2 by diuretic challenge (see Diuretic Challenges).
Distal convoluted tubule.
Several groups have reported a simple and highly reproducible inverse linear relationship between plasma K+ concentrations and NCC phosphorylation status (56, 59), providing an explanation for hypokalemia-induced NaCl sensitivity as a consequence of NCC overactivation (57). Consistent with these reports, we found that K+ restriction caused a threefold increase in NCC phosphorylation status, a surrogate for NCC activity (Fig. 5B). Moreover, we observed that KCl loading induced a more robust inhibition of NCC phosphorylation compared with the K+ basic diet (69% inhibition with KCl vs. 42% inhibition with K+ basic). This is consistent with prior reports indicating that the type of anion modulates the effect of dietary K+ loading on NCC activity (12, 56, 60).
Principal cells.
ENaC mediates electrogenic Na+ reabsorption via connecting tubule and collecting duct principal cells. Although ENaC is regulated by changes in extracellular fluid volume status and blood pressure, under certain circumstances it prioritizes K+ balance over Na+ and volume homeostasis (21). Dietary K+ loading is associated with increased cleavage (activation) of the γ-subunit of ENaC, whereas K+ depletion is associated with a shift to the inactive uncleaved form (21, 22). Our results reinforce prior studies, as K+-depleted mice exhibited a 3.5-fold increase in uncleaved γ-ENaC and a trend toward decreased cleavage (21, 22). Mice on high-K+ diets had a significant increase in cleaved γ-ENaC with the KCl diets having a more robust effect on γ-ENaC cleavage compared with the K+ basic diet (2.3 vs. 1.5-fold; Fig. 5B).
Immunofluorescence confocal microscopy confirmed that the high-KCl diet consistently induced ENaC localization to the apical membrane (Fig. 6, A and B). The high-K+ basic diet induced ENaC localization to the apical membrane in some segments; however, ENaC was also noted to be diffuse in other nephron segments (Fig. 6, top row).
Fig. 6.

Dietary K+ maneuvers affect epithelial Na+ channel (ENaC) localization. Kidney tissue from mice maintained on varying K+ diets for 10 days was fixed and immunostained for γ-ENaC (green). A: dietary K+ loading shifted γ-ENaC localization from diffuse to apical staining, which was more prominent for the KCl diet than for the K+ basic diet. Three-dimensional z-stacks were acquired and maximum intensity projected to visualize apical versus diffuse ENaC localization. B: quantification of γ-ENaC localization was performed using a fluorescence intensity line profile plot from 30 representative cells over different 3 experiments. Results were averaged and plotted to compare the fluorescence intensity distribution between groups. All statistics and analysis were performed using two-way ANOVA with Dunnett’s post hoc analysis to evaluate for significant changes between varying K+ diets and the control (Ctrl) diet. KCl- and K+ basic diet-fed mice displayed significantly different γ-ENaC distributions compared with control mice (P ≤ 0.0001), while low-K+ fed mice had a similar distribution to control. Specifically, KCl-fed mice exhibited γ-ENaC localization preference for the luminal membrane and decreased γ-ENaC within the cytoplasmic region. Compared with control mice, KCl diet-fed mice had γ-ENaC distribution that was significantly different from 2.0–6.4 µm from the lumen (*P ≤ 0.05, **P ≤ 0.01, and ****P ≤ 0.0001). K+ basic diet-fed mice displayed a slightly reduced cytoplasmic γ-ENaC distribution that was significantly different from control mice between 2.2–6.4 µm from the lumen. Low-K+ diet-fed mice had a similar γ-ENaC distribution compared with control mice, with a few significant points from 5.1–6.4 µm from the lumen.
In addition to Na+ transporters, we also measured the expression of ROMK (Kir1.1). ROMK is a major K+ secretory channel in the downstream nephron. On a Western blot, ROMK migrates as an immature species that runs at ~37kDa and a post-Golgi complex-glycosylated mature band that runs at 50–65 kDa (61). The low-K+ diet significantly decreased expression of the complex band by 48% (Fig. 5B). Since ROMK is expressed in both the distal nephron and cortical thick ascending limb, these findings represent the combined expression of the channel across multiple nephron segments. However, the data are consistent with a prior report of K+-diet-mediated regulation of the ROMK-dependent K+ secretory machinery in the distal nephron (61).
Intercalated cells.
Intercalated cells in the distal segments of the kidney are associated with acid-base, Cl−, and K+ regulation along with ammonia secretion (50). Pendrin is an apical transporter essential for the secretion of bicarbonate and reabsorption of Cl− in type B intercalated cells. It is known to be upregulated with metabolic alkalosis and downregulated with Cl− loading and metabolic acidosis (13, 62). However, the effects of K+ are conflicting (34, 47, 62, 67). We found that extreme K+ restriction dramatically increased pendrin expression by 3.6-fold compared with control and promoted the appearance of multimers (Fig. 5B) (62). In the high-K+ diet conditions, pendrin protein expression was not significantly different from control (Fig. 5B). We could not make further conclusions regarding the significance of the effects of the K+ basic or KCl diets on pendrin protein expression due to low immunodetectable signal. Next, we measured the expression of NDCBE located in the apical membrane of intercalated cells (53). Both K+ restriction and K+ basic loading significantly decreased this transporter.
Diuretic Challenges
Finally, to assess the functional effects of dietary K+ on Na+ transport, we inhibited key apical Na+ transport pathways along the nephron and measured the change in urine output, natriuresis, and kaliuresis at 6 and 24 h. The diuretics exerted their effects within 6 h and then dissipated within 24 h.
Furosemide-sensitive NKCC2.
In our diuretic experiments, extreme K+ depletion diminished furosemide sensitivity (30 mg/kg ip) (Fig. 7A). Mice maintained on a low-K+ diet exhibited a smaller change in urine output and no significant change in urinary Na+ or urinary K+ excretion when injected with furosemide compared with injection with vehicle. This low K+-induced furosemide resistance corresponds with our observation that K+ restriction decreased expression of total NKCC2 (Fig. 5B). In contrast, K+-loaded mice (either K+ basic or KCl) treated with furosemide exhibited an increase in 6-h urine output (3- to 4-fold) and a significant change in urinary Na+ excretion (2- to 3-fold) compared with vehicle-injected mice, consistent with cotransporter activation during K+ loading (Fig. 7A).
Fig. 7.

Dietary K+ influences diuretic sensitivity. Mice were maintained on either low-K+, K+ basic, or KCl diets for 10 days. Mice were placed in metabolic cages 24 h before diuretic challenge. Mice were intraperitoneally injected with the diuretic furosemide (30 mg/kg), hydrochlorothiazide (HCTZ; 25 mg/kg), or amiloride (5 mg/kg), and urine was collected from 0–6 h and 7–24 h. Values are shown as fold changes in urine output after diuretic compared with sham saline injection given to the same mouse on a different day (two-way ANOVA with Sidak’s post hoc analysis, *P ≤ 0.05 and **P ≤ 0.01 compared with sham). A: mice fed a high-K+ diet exhibited increased sensitivity to furosemide compared with mice on the low-K+ diet with increased urine output and increased 6 h urinary Na+ excretion (UNa*V). Dietary K+ had no significant effect on urine K+ excretion (UK*V) in response to furosemide. n = 9–12 mice per diet. B: K+-restricted mice were more sensitive to HCTZ, with significantly increased urine output and 6-h urinary Na+. n = 4–6 mice per diet. C: mice fed a high-K+ diet had increased sensitivity to the K+-sparing effect of amiloride with a decrease in kaliuresis when treated with amiloride. n = 10–12 mice per diet.
HCTZ-sensitive NCC.
In the HCTZ diuretic challenge, K+-depleted mice subjected to intraperitoneal injection (25 mg/kg) developed a twofold increase in 6-h urine output compared with vehicle injection and a 4-fold increase in urine Na+ (Fig. 7B). There was a trend for the urinary K+ to increase with HCTZ on the low-K+ diet. However, this did not reach significance, presumably because the absolute values for urinary K+ on the low-K+ diet are near the lower limit of detection (Table 3). Mice maintained on high-K+ diets did not have a significant response to HCTZ injection, whereas mice subjected to K+ depletion exhibited increased HCTZ sensitivity, consistent with the increased expression of pNCC observed on immunoblots in K+-depleted mice (Fig. 5B).
Table 3.
Raw values for changes in urine Na+ and K+ after 6-h diuretic treatment
| Vehicle | Furosemide | Vehicle | Hydrochlorothiazide | Vehicle | Amiloride | |
|---|---|---|---|---|---|---|
| Low K+ | ||||||
| UNa, mmol/L | 106 (5) n = 11 |
82 (6) n = 12 |
101 (8) n = 6 |
189 (10)† n = 6 |
106 (5) n = 11 |
142 (13) n = 12 |
| UNa × V, mmol/6 h | 0.113 (0.015) n = 11 |
0.136 (0.013) n = 12 |
0.043 (0.007) n = 6 |
0.177 (0.028)† n = 6 |
0.116 (0.013) n = 11 |
0.142 (0.023) n = 12 |
| UK, mmol/L | 1.636 (0.157) n = 11 |
2.167 (0.428) n = 12 |
1.333 (0.198) n = 6 |
1.067 (0.169) n = 6 |
1.38 (0.190) n = 11 |
1.23 (0.187) n = 12 |
| UK × V, mmol/6 h | 0.002 (0.000) n = 11 |
0.004 (0.000) n = 12 |
0.0005 (0.0001) n = 6 |
0.0011 (0.0003) n = 6 |
0.002 (0.000) n = 11 |
0.002 (0.001) n = 12 |
| K+ basic | ||||||
| UNa, mmol/L | 162 (12) n = 9 |
131 (8) n = 11 |
158 (14) n = 5 |
172 (9) n = 5 |
127 (6) n = 10 |
308 (39)† n = 10 |
| UNa × V mmol/6 h | 0.081 (0.014) n = 9 |
0.162 (0.020)† n = 11 |
0.063 (0.012) n = 5 |
0.071 (0.017) n = 5 |
0.072 (0.013) n = 10 |
0.108 (0.022) n = 10 |
| UK, mmol/L | 447 (42) n = 9 |
198 (28)† n = 11 |
373 (41) n = 5 |
387 (68) n = 5 |
418 (42) n = 10 |
277 (41)† n = 10 |
| UK × V, mmol/6 h | 0.229 (0.042) n = 9 |
0.274 (0.055) n = 11 |
0.146 (0.028) n = 5 |
0.153 (0.033) n = 5 |
0.246 (0.053) n = 10 |
0.095 (0.021)† n = 10 |
| KCl | ||||||
| UNa, mmol/L | 90 (4) n = 12 |
84 (6) n = 12 |
106 (10) n = 4 |
124 (28) n = 6 |
87 (4) n = 12 |
258 (15)† n = 12 |
| UNa × V, mmol/6 h | 0.058 (0.008) n = 12 |
0.140 (0.014)† n = 12 |
0.069 (0.014) n = 4 |
0.098 (0.025) n = 6 |
0.073 (0.009) n = 12 |
0.126 (0.018) n = 12 |
| UK, mmol/L | 482 (13) n = 12 |
153 (14)† n = 12 |
560 (73) n = 4 |
479 (96) n = 6 |
493 (16) n = 12 |
268 (47)† n = 12 |
| UK × V, mmol/6 h | 0.308 (0.040) n = 12 |
0.278 (0.041) n = 12 |
0.364 (0.080) n = 4 |
0.371 (0.089) n = 6 |
0.408 (0.048) n = 12 |
0.135 (0.025)† n = 12 |
Values are means (SE); n, number of mice. Mice were treated with varying K+ diets for 10 days and then injected with a diuretic [either furosemide (30 mg/kg), hydrochlorothiazide (25 mg/kg), or amiloride (5 mg/kg)], and urine was collected after 6 h. UNa × V and UK × V, urinary Na+ and K+ excretion. Results were compared with 6-h urine collected from a sham vehicle-injected mouse on an alternate day. Two-way ANOVA with Sidak’s post hoc analysis was used.
P ≤ 0.01, significant difference between vehicle and diuretic, respectively.
Amiloride-sensitive ENaC.
Our protein expression experiments indicate that dietary K+ intake directly correlates with ENaC cleavage and thus its activation status (Fig. 5B). Therefore, we expected to see significant changes in amiloride sensitivity between the varying K+ diets. In our experiments, amiloride (5 mg/kg ip) had no significant effect on the 6 h fold change of urinary volume or urine Na+ in mice on high-K+ diets (Fig. 7C), despite the high-K+ diets having robust effects on γ-ENaC protein expression and localization (Figs. 5B and 6). Analysis of raw values (Table 3) revealed that amiloride-treated mice on high-K+ diets did have a significant increase in the concentration of Na+ in the urine (mmol/L), although their actual 6-h urine output tended to be lower, negating any significant change in urinary Na+ output over 6 h (mmol/6 h). As expected, amiloride significantly affected urinary K+ excretion. Amiloride-treated mice maintained on high-K+ diets had a significant fold decrease in 6-h urinary K+ excretion compared with vehicle-injected mice. Mice maintained on low-K+ diets were resistant to amiloride, consistent with diffuse ENaC localization and decreased ENaC expression on low-K+ diets (Figs. 5B and 6).
DISCUSSION
Numerous human epidemiological studies have indicated an inverse correlation between K+ intake and blood pressure (5, 15, 19, 28a, 29, 51). Despite this strong association, studies in animal models using standardized diets have had conflicting results (1, 3, 15, 20, 24, 33, 36, 42, 60, 63, 68). To address this dilemma, researchers have turned to animal models and standardized diets to study the effect of dietary K+ on blood pressure. Most studies have focused on a limited number of renal transport pathways or have made inferences about blood pressure via measurements of renal Na+ transporters along the nephron, without measuring blood pressure itself. Moreover, studies have generally used readily available preformulated commercial rodent diets whose electrolyte compositions reside at the extreme ends of K+ deficiency and excess. While these dietary maneuvers have robust effects on renal ion transport, we postulated that they may not accurately model “Western” and high-K+ diets in humans.
With these issues in mind, we carried out an extensive analysis of commonly used dietary K+ maneuvers on blood pressure, electrolyte handling, and major renal tubular ion transport pathways extending from the proximal tubule to the collecting duct. Ten days was chosen based on our prior work and to allow for comparison with historical studies over the same time period (9, 60). Notably, the extreme dietary K+ maneuvers used here did not elicit an inverse relationship between K+ intake and blood pressure. Rather, K+ deprivation using a popular “K+-deficient” diet led to diabetes insipidus. Reports of the association between severe K+ deficiency and impaired urinary concentration first appeared in the 1950s (54). Further studies revealed this defect is due in part to vasopressin-resistance, decreased AQP2 expression, impaired NaCl reabsorption in the thick ascending limb, and downregulation of urea transporters (16, 27, 30, 37, 38). Our results confirm that K+ depletion decreased expression of NKCC2 and Aqp2, inducing polyuria and free water diuresis. This polyuria was associated with a volume-depleted state primed for a salt-sensitive pressor response, with increased expression of NHE3/NBCe1 in the proximal tubule, pNCC in the distal convoluted tubule, and pendrin in the cortical collecting duct. Of these proteins, we only confirmed an increase in NCC activity in diuretic experiments, but it is conceivable that the increased expression of NHE3/NBCe1 and pendrin also contributes to the NaCl-sensitive rise in blood pressure. Although severe K+ depletion induced salt sensitivity, it did not mimic the hypertension observed in humans on high-Na+, low-K+ (Western) diets. To replicate the Western diet in rodents, we propose that a more modest degree of K+ restriction of shorter duration with adequate NaCl loading is required. To ensure the validity of such models, we contend that any low-K+ dietary maneuver in rodents must be accompanied by a careful assessment of urine osmolality and output and/or renal AQP2 protein expression to rule out unintended diabetes insipidus.
Our present study builds on earlier work by Vitzthum et al. (60) investigating the role of dietary K+ on mouse blood pressure. Key differences between our study and theirs include the ratio of Na+ in the diet and the mouse strain. While Vitzthum et al. used a high-NaCl (3%) diet with varying amounts of K+, we performed our experiments in the setting of limited Na+ (0.3%) intake and supplemented NaCl via saline loading experiments. The results from our 1% saline-loaded mice most closely mimic their experimental 3% NaCl blood pressure model. In the setting of excess NaCl, both studies found that K+-depleted mice develop elevated nighttime blood pressure (14, 60) and that adding excess NaCl to mice on control diets has no effect on blood pressure.
Additionally, we report that a high-K+ (5%) basic diet modestly increases blood pressure. Although Vitzthrum et al.’s study and ours both reported a marked increase in aldosterone levels with K+ loading, in their study, amiloride and mineralocorticoid receptor antagonists did not lower blood pressure. On the other hand, we observed ENaC cleavage and increased total NKCC2 abundance along with an increase in urinary Na+ parameters in response to both amiloride and furosemide. These data would suggest that ENaC and NKCC2 are active during extreme K+ loading, but their contributions to the rise in blood pressure associated with this maneuver are unclear. Given the extreme nature of the K+ loads that were used, it is debatable as to whether exploring the mechanism underlying the modest relative hypertension in these mice warrants further investigation, although the observation is relevant for future study design. Our study also expands on the effects of high-K+ diets by demonstrating that K+-loaded mice do not have a further increase in blood pressure in the setting of excess NaCl, regardless of the K+ anion (K+ basic or KCl). Taken together, these findings indicate that the increase in blood pressure in response to the high-salt diet is only manifested when mice are on a low-K+ diet; even control levels of K+ are sufficient to prevent the rise in blood pressure. Therefore, K+ depletion induces NaCl sensitivity, moderate K+ supplementation opposes NaCl-sensitive hypertension (15, 26, 33, 57, 63) and extreme K+ loading causes a modest rise in blood pressure that is NaCl insensitive.
Our study was performed using 129S2/Sv mice, whereas Vitzthrum et al. (60) used C57bl6 mice. This is an important difference since 129S2/Sv mice have two renin genes and C57 mice have only one renin gene (65). Mice with two renin genes have previously been reported to have higher plasma renin concentration and activity as well as an elevated blood pressure and renal response to salt compared with mice with one renin gene (65). On average, our 129S2/Sv mice tended to have a 10-mmHg higher MAP compared with the published data on C57bl6 mice on control diets (diets with ~1% K+ and ~0.3% Na+) (60). However, it is difficult to speculate how much of an effect the two renin genes have without a direct comparison between the two strains of mice on varying K+ diets. Overall, our outcomes complement the results from Vitzthum et al. Taken together, the combined results highlight the importance of considering the Na+-to-K+ ratio of rodent diets and mouse strain when designing experiments to test effects of K+ on blood pressure.
Much of the recent attention toward dietary K+ and blood pressure has focused on the inverse relationship between K+ and NCC activity (57). Our results replicated the effects of K+ on NCC phosphorylation and thiazide sensitivity. Yet, the activation of NCC in severely K+-deficient mice was not sufficient to induce hypertension on the 0.3% Na+ diet; rather, an increase in blood pressure was only seen when K+-deficient mice were supplemented with 1% saline. Moreover, inhibition of NCC in mice subjected to extreme K+ loading with 0.3% Na+ did not attenuate blood pressure; instead, these mice developed an increase in blood pressure. However, the inhibition of NCC may have contributed to the observed resistance to further increases in blood pressure when K+-loaded mice were challenged with 1% saline. Thus, at the extremes of dietary K+ with moderate (0.3%) Na+ intake, the relationship between plasma K+, NCC activity, and blood pressure becomes uncoupled.
Previous literature indicates altering dietary K+ modulates electrogenic ENaC activity to restore K+ balance (21). Indeed, during dietary K+ restriction, Na+ is predominantly reabsorbed in the upstream nephron, limiting downstream delivery (and flow rate) to the ENaC-expressing K+-secreting segments (64). On the other hand, during K+ loading, NKCC2 and ENaC may be the main Na+-reabsorptive pathways. Na+ reabsorption is shifted downstream to sites where ENaC activity is high and voltage-dependent K+ secretion is enhanced (11, 23). Both our Western blot and immunofluorescence data suggest the KCl is a more robust inducer of ENaC activation compared with the K+ basic diet. We propose that KCl is a more potent due to the substantially increased aldosterone levels (2,889 pg/mL) compared with mice fed either control (385 pg/mL) or K+ basic (929 pg/mL) diets. Perhaps KCl induces a larger increase in serum aldosterone levels due to the concurrent metabolic acidosis. A possible alternative is that the mice fed KCl simply ate more of the high-K+ diet compared with mice fed a K+ basic diet and therefore had higher serum K+ and higher aldosterone levels.
Our results indicate that K+ is a primary determinant of Na+ transport along the length of the nephron. K+ intake has a direct correlation with NKCC2 and ENaC activity and an inverse relationship with NCC activity. However, it appears that the K+ anion is also important, as evidenced by the significant differences in transporter expression between KCl versus K+ basic diets. In the proximal tubule, high-KCl had similar trends with K+ depletion, presumably due to similar effects of acidosis and hypokalemia on NHE3 activity. In intercalated cells, high-K+ basic diets had similar trends to K+ restriction, likely due to the alkalotic stress present under both conditions (Fig. 8).
Fig. 8.

Summary of the effects of extreme dietary K+ maneuvers on Na+ transport. The effect of dietary K+ depletion and loading are summarized based on transporter abundance and activity obtained from immunoblot and diuretic experiments. Extreme K+ depletion had a significant effect on all Na+ transporters and channels studied due to the combined effects of volume loss and K+ deficiency. K+ loading had similar stimulatory effects on Na+-K+-2Cl− cotransporter (NKCC2) and epithelial Na+ channel (ENaC) and inhibitory effects on NaCl cotransporter (NCC). The K+ anion (basic vs. Cl−) had opposing effects in the proximal tubule and intercalated cells. CCD, cortical collecting duct; CNT, connecting tubule; cTAL, cortical thick ascending limb; DCT, distal convoluted tubule; IC, intercalated cells; mTAL, medullary thick ascending limb; PC, principal cells; PT, proximal tubule.
Limitations
Notable issues in our study design include the administration of saline in the drinking water and the use of gel diets. We did not measure how much water the mice consumed while on telemetry; however, based on our metabolic cage experiments, mice on the control diets consumed less water compared with the mice on the low-K+ or high-K+ diets after 10 days. Thus, we assume that polydipsic mice received higher doses of saline. Despite the increased ingestion of saline by K+-depleted mice compared with controls, fluid intake was similar between K+-deficient and K+-loaded mice. Thus, our results suggest that the blood pressure increase that we observed in saline-treated K+-depleted mice was not due to NaCl intake per se but instead was due to an increase in NaCl sensitivity. These data complement previously published data where the NaCl load was contained in the diet and equal amounts were consumed (60). Furthermore, prior reports in mice and humans have similarly shown that low dietary K+ induces salt-sensitive hypertension (15, 43, 57, 58).
For our metabolic cage experiments, we switched mice to gel (agar)-based diets to prevent pellet particles from contaminating the urine. Agar contains trace amounts of K+ (0.008%), Na+ (0.006%), and protein. Thus, switching to gel diets could conceivably affect the results. Additionally, mice fed the K+ basic gel diet had significantly less intake at day 5, which began to recover by day 10. We propose that the decrease in K+ basic diet ingestion was due to decreased palatability of the basic diet, but we cannot rule out the effect of the agar. Although we propose that significant differences between mice fed KCl and K+ basic diets are due to the differences in anionic content, we cannot rule out the possibility that these discrepancies are simply due to the fact that the mice on the K+ basic diet consumed less food and thus consumed less K+.
Conclusions
In summary, we have shown that commercially available commonly used preformulated rodent diets that are designed to cause marked K+ deficiency and excess can have extreme effects that do not recapitulate human epidemiological studies. While K+ deprivation on these diets for 10 days can induce diabetes insipidus, K+ loading can drive an increase in blood pressure. Our data indicate that more moderate short-term maneuvers should be used to preserve water handling and that consideration should be given to the accompanying Na+ and anionic content when modeling the effect of K+ intake on renal physiology.
GRANTS
This work was supported, in whole or in part, by National Institutes of Health Grants K08-DK-118211-01 (to C. R. Boyd-Shiwarski), R01-DK-119252 (to A. R. Subramanya), R01-DK-098145 (to A. R. Subramanya), F32-HL-142229 (to D. J. Shiwarski), and P30-DK-079307 (to Pittsburgh Center for Kidney Research).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.R.B.-S and ARS conceived and designed research; C.R.B.-S, C.J.W., R.T.B., D.J.S., K.A.C., L.J.N., S.M.M., S.E.G., S.A.K., R.S.S., E.C.R., and A.L.M. performed experiments; C.R.B.-S, D.J.S., and A.R.S. analyzed data; C.R.B.-S, D.J.S., and A.R.S. interpreted results of experiments; C.R.B.-S, D.J.S., and A.R.S. prepared figures; C.R.B.-S drafted manuscript; C.R.B.-S and A.R.S. edited and revised manuscript; C.R.B.-S, C.J.W., R.T.B., D.J.S., K.A.C., L.J.N., S.M.M., S.E.G., S.A.K., R.S.S., E.C.R., A.L.M., and A.R.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank David Ellison and James Wade for antibodies and Sean Stocker and Thomas Kleyman for technical assistance and helpful discussions.
REFERENCES
- 1.Abbrecht PH. Cardiovascular effects of chronic potassium deficiency in the dog. Am J Physiol 223: 555–560, 1972. doi: 10.1152/ajplegacy.1972.223.3.555. [DOI] [PubMed] [Google Scholar]
- 2.Amlal H, Wang Z, Soleimani M. Potassium depletion downregulates chloride-absorbing transporters in rat kidney. J Clin Invest 101: 1045–1054, 1998. doi: 10.1172/JCI686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson DE, Kearns WD, Worden TJ. Potassium infusion attenuates avoidance-saline hypertension in dogs. Hypertension 5: 415–420, 1983. doi: 10.1161/01.HYP.5.4.415. [DOI] [PubMed] [Google Scholar]
- 5.Bernabe-Ortiz A, Sal Y Rosas VG, Ponce-Lucero V, Cárdenas MK, Carrillo-Larco RM, Diez-Canseco F, Pesantes MA, Sacksteder KA, Gilman RH, Miranda JJ. Effect of salt substitution on community-wide blood pressure and hypertension incidence. Nat Med 26: 374–378, 2020. doi: 10.1038/s41591-020-0754-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blass G, Klemens CA, Brands MW, Palygin O, Staruschenko A. Postprandial effects on ENaC-mediated sodium absorption. Sci Rep 9: 4296, 2019. doi: 10.1038/s41598-019-40639-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bobulescu IA, Moe OW. Luminal Na(+)/H (+) exchange in the proximal tubule. Pflugers Arch 458: 5–21, 2009. doi: 10.1007/s00424-008-0595-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499–509, 2018. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd-Shiwarski CR, Subramanya AR. The renal response to potassium stress: integrating past with present. Curr Opin Nephrol Hypertens 26: 411–418, 2017. doi: 10.1097/MNH.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandis M, Keyes J, Windhager EE. Potassium-induced inhibition of proximal tubular fluid reabsorption in rats. Am J Physiol 222: 421–427, 1972. doi: 10.1152/ajplegacy.1972.222.2.421. [DOI] [PubMed] [Google Scholar]
- 12.Castañeda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vázquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang JH, Kim S. Role of pendrin in acid-base balance. Electrolyte Blood Press 7: 20–24, 2009. doi: 10.5049/EBP.2009.7.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Combe R, Mudgett J, El Fertak L, Champy MF, Ayme-Dietrich E, Petit-Demoulière B, Sorg T, Herault Y, Madwed JB, Monassier L. How does circadian rhythm impact salt sensitivity of blood pressure in mice? a study in two close C57Bl/6 substrains. PLoS One 11: e0153472, 2016. doi: 10.1371/journal.pone.0153472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahl LK, Leitl G, Heine M. Influence of dietary potassium and sodium/potassium molar ratios on the development of salt hypertension. J Exp Med 136: 318–330, 1972. doi: 10.1084/jem.136.2.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkjær ML, Kwon TH, Wang W, Nielsen J, Knepper MA, Frøkiaer J, Nielsen S. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol 283: F1376–F1388, 2002. doi: 10.1152/ajprenal.00186.2002. [DOI] [PubMed] [Google Scholar]
- 17.Festing MF, Simpson EM, Davisson MT, Mobraaten LE. Revised nomenclature for strain 129 mice. Mamm Genome 10: 836, 1999. doi: 10.1007/s003359901099. [DOI] [PubMed] [Google Scholar]
- 18.Filippatos TD, Tsimihodimos V, Liamis G, Elisaf MS. SGLT2 inhibitors-induced electrolyte abnormalities: An analysis of the associated mechanisms. Diabetes Metab Syndr 12: 59–63, 2018. doi: 10.1016/j.dsx.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Filippini T, Violi F, D’Amico R, Vinceti M. The effect of potassium supplementation on blood pressure in hypertensive subjects: a systematic review and meta-analysis. Int J Cardiol 230: 127–135, 2017. doi: 10.1016/j.ijcard.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 20.Freed SC, Friedman M. Hypotension in the rat following limitation of potassium intake. Science 112: 788–789, 1950. doi: 10.1126/science.112.2922.788-a. [DOI] [PubMed] [Google Scholar]
- 21.Frindt G, Houde V, Palmer LG. Conservation of Na+ vs. K+ by the rat cortical collecting duct. Am J Physiol Renal Physiol 301: F14–F20, 2011. doi: 10.1152/ajprenal.00705.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frindt G, Shah A, Edvinsson J, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am J Physiol Renal Physiol 296: F347–F354, 2009. doi: 10.1152/ajprenal.90527.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. Hypertension of Kcnmb1-/- is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci USA 106: 11800–11805, 2009. doi: 10.1073/pnas.0904635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB, Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest 125: 2136–2150, 2015. doi: 10.1172/JCI78558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gritter M, Rotmans JI, Hoorn EJ. Role of dietary K+ in natriuresis, blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 73: 15–23, 2018. [DOI] [PubMed] [Google Scholar]
- 27.Gutsche HU, Peterson LN, Levine DZ. In vivo evidence of impaired solute transport by the thick ascending limb in potassium-depleted rats. J Clin Invest 73: 908–916, 1984. doi: 10.1172/JCI111314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris AN, Grimm PR, Lee HW, Delpire E, Fang L, Verlander JW, Welling PA, Weiner ID. Mechanism of hyperkalemia-induced metabolic acidosis. J Am Soc Nephrol 29: 1411–1425, 2018. doi: 10.1681/ASN.2017111163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Intersalt Cooperative Research Group Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ 297: 319–328, 1988. doi: 10.1136/bmj.297.6644.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson SL, Cogswell ME, Zhao L, Terry AL, Wang CY, Wright J, Coleman King SM, Bowman B, Chen TC, Merritt R, Loria CM. Association between urinary sodium and potassium excretion and blood pressure among adults in the United States: National health and nutrition examination survey, 2014. Circulation 137: 237–246, 2018. doi: 10.1161/CIRCULATIONAHA.117.029193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung JY, Madsen KM, Han KH, Yang CW, Knepper MA, Sands JM, Kim J. Expression of urea transporters in potassium-depleted mouse kidney. Am J Physiol Renal Physiol 285: F1210–F1224, 2003. doi: 10.1152/ajprenal.00111.2003. [DOI] [PubMed] [Google Scholar]
- 31.Keith NM, Binger MW. Diuretic action of potassium salts. JAMA 105: 1584–1591, 1935. doi: 10.1001/jama.1935.02760460020005. [DOI] [Google Scholar]
- 32.Kornandakieti C, Tannen RL. Hydrogen ion secretion by the distal nephron in the rat: effect of potassium. J Lab Clin Med 104: 293–303, 1984. [PubMed] [Google Scholar]
- 33.Krishna GG, Miller E, Kapoor S. Increased blood pressure during potassium depletion in normotensive men. N Engl J Med 320: 1177–1182, 1989. doi: 10.1056/NEJM198905043201804. [DOI] [PubMed] [Google Scholar]
- 34.Lazo-Fernandez Y, Aguilera G, Pham TD, Park AY, Beierwaltes WH, Sutliff RL, Verlander JW, Pacak K, Osunkoya AO, Ellis CL, Kim YH, Shipley GL, Wynne BM, Hoover RS, Sen SK, Plotsky PM, Wall SM. Pendrin localizes to the adrenal medulla and modulates catecholamine release. Am J Physiol Endocrinol Metab 309: E534–E545, 2015. doi: 10.1152/ajpendo.00035.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HW, Harris AN, Romero MF, Welling PA, Wingo CS, Verlander JW, Weiner ID. NBCe1-A is required for the renal ammonia and K+ response to hypokalemia. Am J Physiol Renal Physiol 318: F402–F421, 2020. doi: 10.1152/ajprenal.00481.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linas SL, Marzec-Calvert R. Potassium depletion ameliorates hypertension in spontaneously hypertensive rats. Hypertension 8: 990–996, 1986. [DOI] [PubMed] [Google Scholar]
- 37.Luke RG, Booker BB, Galla JH. Effect of potassium depletion on chloride transport in the loop of Henle in the rat. Am J Physiol 248: F682–F687, 1985. doi: 10.1152/ajprenal.1985.248.5.F682. [DOI] [PubMed] [Google Scholar]
- 38.Marples D, Frøkiaer J, Dørup J, Knepper MA, Nielsen S. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest 97: 1960–1968, 1996. doi: 10.1172/JCI118628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maunsbach AB, Vorum H, Kwon TH, Nielsen S, Simonsen B, Choi I, Schmitt BM, Boron WF, Aalkjaer C. Immunoelectron microscopic localization of the electrogenic Na/HCO(3) cotransporter in rat and ambystoma kidney. J Am Soc Nephrol 11: 2179–2189, 2000. [DOI] [PubMed] [Google Scholar]
- 40.McCormick JA, Nelson JH, Yang CL, Curry JN, Ellison DH. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension 58: 888–894, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonough AA, Veiras LC, Minas JN, Ralph DL. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol 308: C426–C433, 2015. doi: 10.1152/ajpcell.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meneely GR, Ball CO. Experimental epidemiology of chronic sodium chloride toxicity and the protective effect of potassium chloride. Am J Med 25: 713–725, 1958. doi: 10.1016/0002-9343(58)90009-3. [DOI] [PubMed] [Google Scholar]
- 43.Meneely GR, Battarbee HD. High sodium-low potassium environment and hypertension. Am J Cardiol 38: 768–785, 1976. doi: 10.1016/0002-9149(76)90356-8. [DOI] [PubMed] [Google Scholar]
- 44.Ng KM, Lau YM, Dhandhania V, Cai ZJ, Lee YK, Lai WH, Tse HF, Siu CW. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci Rep 8: 14872, 2018. doi: 10.1038/s41598-018-33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ong J, Kinsman BJ, Sved AF, Rush BM, Tan RJ, Carattino MD, Stocker SD. Renal sensory nerves increase sympathetic nerve activity and blood pressure in 2-kidney 1-clip hypertensive mice. J Neurophysiol 122: 358–367, 2019. doi: 10.1152/jn.00173.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pizzoni A, López González M, Di Giusto G, Rivarola V, Capurro C, Ford P. AQP2 can modulate the pattern of Ca2+ transients induced by store-operated Ca2+ entry under TRPV4 activation. J Cell Biochem 119: 4120–4133, 2018. doi: 10.1002/jcb.26612. [DOI] [PubMed] [Google Scholar]
- 47.Procino G, Milano S, Tamma G, Dossena S, Barbieri C, Nicoletti MC, Ranieri M, Di Mise A, Nofziger C, Svelto M, Paulmichl M, Valenti G. Co-regulated pendrin and aquaporin 5 expression and trafficking in Type-B intercalated cells under potassium depletion. Cell Physiol Biochem 32: 184–199, 2013. doi: 10.1159/000356638. [DOI] [PubMed] [Google Scholar]
- 48.Relman AS, Schwartz WB. The nephropathy of potassium depletion; a clinical and pathological entity. N Engl J Med 255: 195–203, 1956. doi: 10.1056/NEJM195608022550501. [DOI] [PubMed] [Google Scholar]
- 49.Romero MF, Hediger MA, Boulpaep EL, Boron WF. Expression cloning and characterization of a renal electrogenic Na+/ cotransporter. Nature 387: 409–413, 1997. doi: 10.1038/387409a0. [DOI] [PubMed] [Google Scholar]
- 50.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, Karanja N, Lin PH, Aickin M, Most-Windhauser MM, Moore TJ, Proschan MA, Cutler JA; DASH-Sodium Collaborative Research Group . Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med 344: 3–10, 2001. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 52.Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 26: 447–453, 2006. doi: 10.1016/j.semnephrol.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith SG, Lasater TE. A diabetes insipidus-like condition produced in dogs by a potassium deficient diet. Proc Soc Exp Biol Med 74: 427–431, 1950. doi: 10.3181/00379727-74-17928. [DOI] [PubMed] [Google Scholar]
- 55.Tannen RL. Relationship of renal ammonia production and potassium homeostasis. Kidney Int 11: 453–465, 1977. doi: 10.1038/ki.1977.63. [DOI] [PubMed] [Google Scholar]
- 56.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Treasure J, Ploth D. Role of dietary potassium in the treatment of hypertension. Hypertension 5: 864–872, 1983. [DOI] [PubMed] [Google Scholar]
- 59.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vitzthum H, Seniuk A, Schulte LH, Müller ML, Hetz H, Ehmke H. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol 592: 1139–1157, 2014. doi: 10.1113/jphysiol.2013.266924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011. doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP. Regulation of the expression of the Cl−/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int 62: 2109–2117, 2002. doi: 10.1046/j.1523-1755.2002.00671.x. [DOI] [PubMed] [Google Scholar]
- 63.Walter C, Tanfous MB, Igoudjil K, Salhi A, Escher G, Crambert G. H,K-ATPase type 2 contributes to salt-sensitive hypertension induced by K+ restriction. Pflugers Arch 468: 1673–1683, 2016. doi: 10.1007/s00424-016-1872-z. [DOI] [PubMed] [Google Scholar]
- 64.Walter SJ, Shore AC, Shirley DG. Effect of potassium depletion on renal tubular function in the rat. Clin Sci (Lond) 75: 621–628, 1988. doi: 10.1042/cs0750621. [DOI] [PubMed] [Google Scholar]
- 65.Wang Q, Hummler E, Nussberger J, Clément S, Gabbiani G, Brunner HR, Burnier M. Blood pressure, cardiac, and renal responses to salt and deoxycorticosterone acetate in mice: role of Renin genes. J Am Soc Nephrol 13: 1509–1516, 2002. doi: 10.1097/01.ASN.0000017902.77985.84. [DOI] [PubMed] [Google Scholar]
- 66.Weir MR, Kline I, Xie J, Edwards R, Usiskin K. Effect of canagliflozin on serum electrolytes in patients with type 2 diabetes in relation to estimated glomerular filtration rate (eGFR). Curr Med Res Opin 30: 1759–1768, 2014. doi: 10.1185/03007995.2014.919907. [DOI] [PubMed] [Google Scholar]
- 67.Xu N, Hirohama D, Ishizawa K, Chang WX, Shimosawa T, Fujita T, Uchida S, Shibata S. Hypokalemia and Pendrin Induction by Aldosterone. Hypertension 69: 855–862, 2017. [DOI] [PubMed] [Google Scholar]
- 68.Young DB, McCaa RE, Pan YJ, Guyton AC. The natriuretic and hypotensive effects of potassium. Circ Res 38, Suppl 2: 84–89, 1976. doi: 10.1161/01.RES.38.6.84. [DOI] [PubMed] [Google Scholar]