

Abstract
The DARPin® drug platform was established with a vision to expand the medical use of biologics beyond what was possible with monoclonal antibodies. It is based on naturally occurring ankyrin repeat domains that are typically building blocks of multifunctional human proteins. The platform allows for the generation of diverse, well-behaved, multifunctional drug candidates. Recent clinical data illustrate the favorable safety profile of the first DARPin® molecules tested in patients. With the positive phase III results of the most advanced DARPin® drug candidate, abicipar, the DARPin® drug platform is potentially about to achieve its first marketing approval. This review highlights some of the key milestones and decisions encountered when transforming the DARPin® platform from an academic concept to a biotech drug pipeline engine.
Key Points
| Clinical data indicate that the DARPin® platform can yield drug candidates with a safety profile matching that of fully human monoclonal antibodies. |
| Abicipar clinical phase III data indicate that the drug candidate has the potential to reduce the need for frequent injections in ophthalmic diseases. |
| Several multifunctional DARPin® drug candidates are in clinical and preclinical development, all taking advantage of the DARPin® format flexibility to produce effective therapeutics for indications with high medical need. |
| Most recently, the DARPin® platform was used to generate extremely potent viral entry inhibitors against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the context of the worldwide coronavirus 2019 (COVID-19) pandemic in just a matter of weeks, illustrating the advantages of the DARPin® platform when it comes to very rapid generation of drug candidates. |
The Origin
Building on Antibody Know How
Monoclonal antibodies [1, 2] have contributed significantly to the progress of effective therapies thanks to their high affinity, high specificity, and long half-life. In the search to push the boundaries of antibody applications, novel protein engineering methods have been explored [3–5] and awarded with the 2018 Nobel Prize. These methods avoid immunizing animals and thus allow thinking beyond the immunoglobulin fold as a therapeutic modality and hence were key for the generation of nonimmunoglobulin-binding proteins, also referred to as alternative scaffolds [6]. Table 1 lists a few distinguishing properties of the immunoglobulin and the DARPin® architecture. Designed ankyrin repeat proteins are one example of such a scaffold. The initial work on designed ankyrin repeat molecules was performed by Prof. Andreas Plückthun and colleagues at the University of Zürich, Switzerland. Subsequently, the team elaborating the technology founded the company Molecular Partners to commercialize the DARPin® platform. The following sections describe (1) the DARPin® platform and (2) the process of transforming the platform from an academic concept into a commercial drug pipeline.
Table 1.
Comparison of monoclonal antibodies and a DARPin® candidate
| Classical IgG | MP0250 DARPin® drug candidate | |
|---|---|---|
| Molecular weight (kDa) | 150 | 62 |
| Number of polypeptide chains | 4 | 1 |
| Disulfide bonds | Yes | No |
| Effector function | Yes | No |
| Engineered target specificities | 1 | 3 |
| Binding mode | Flexible loops | Rigid secondary structure elements |
| Half-life extension | Fc | Serum albumin binding |
| Production in | Mammalian cells | Escherichia coli |
IgG immunoglobulin G
DARPin® Platform
Designed ankyrin repeat domains (see Fig. 1) are based on naturally occurring ankyrin repeat domains [7, 8]. These domains occur in various phylae at high abundance [9]. In humans, for example, 145 genes encode a total of 404 ankyrin repeat domains [10]. It is particularly interesting that, in nature, ankyrin repeat domains often occur in the context of multifunctional proteins, i.e., proteins having, for example, multiple ankyrin repeat domains or ankyrin repeat domains combined with other protein domains. This inspired us to test them as a basis for a therapeutic platform.
Fig. 1.

Representations of a single designed ankyrin repeat domain and a multidomain DARPin® drug candidate. a Ribbon representation of an ankyrin repeat domain (PDB ID: 1MJ0) consisting of five repeats, with randomized interaction colored in orange and a transparent surface superimposed. b Molecular model of MP0250, a DARPin® drug candidate consisting of four DARPin® domains (molecular weight approximately 62 kDa). The scaffold is dark blue, and the potential target interaction residues of the individual domains are orange (human serum albumin [HSA]), cyan (hepatocyte growth factor [HGF]), and red (vascular endothelial growth factor [VEGF])
Through a consensus design approach, designed ankyrin repeat domains were created based on naturally occurring ankyrin repeat domains [7, 8, 11]. The ankyrin repeat fold appears to be particularly stable, in terms of thermodynamic, thermal, and storage stability, which is an ideal basis for its use as a scaffold [7, 8, 12, 13]. This stability is also a good starting point to generate multispecific proteins, where often the weakest part determines the overall stability. Single designed ankyrin repeat domains as well as multidomain DARPin® drug candidates can be expressed at high levels, for example 1 g/L in simple shake-flask expression or 15 g/L in high-cell-density fermentation, in soluble form in Escherichia coli [13]. A high solubility is a further interesting feature of the fold [13], especially when it comes to high-concentration applications such as subcutaneous or intravitreal applications.
Very large and diverse ribosome display libraries of designed ankyrin repeat domains (theoretical diversity of 5.2 × 1015 for a four-repeat construct) [7] have been generated and successfully used for the generation of high-affinity DARPin® binding proteins [14, 15]. Affinities in the picomolar range are routinely achieved for single-domain monospecific binders [13, 16, 17], which typically exhibit excellent target specificity. The libraries are amenable to use with different selection systems such as ribosome display [14], phage display [18], or yeast-surface display [19]. The favorable biophysical properties observed for the scaffold typically are not compromised by the selections. Natural ankyrin repeat domains rely on a rigid scaffold with a clearly defined secondary structure on which specified surface amino acids interact with the target [7, 8]. To visualize the mode of interaction of selected DARPin® molecules, a number of co-crystal structures were determined, demonstrating that the high affinity and high specificity achieved is indeed a result of the very rigid scaffold of the ankyrin repeat domain fold, emphasizing that the scaffold is an excellent starting point for drug development. Another interesting feature of DARPin® molecules is that they can also be used to detect or even freeze very specific conformations of targets [20–22]. The strength of the DARPin® platform compared with antibodies and other similar approaches has been reviewed in detail [6, 15, 23, 24]. Additional applications of the DARPin® platform than those mentioned in the following have been summarized elsewhere [15]; this review focuses on how the DARPin® platform was used as a basis for building the drug pipeline of Molecular Partners.
Evolving an Academic Platform into a Commercial Drug Pipeline Engine
Strategic Aspects
Molecular Partners was incorporated in 2004 in Zürich, Switzerland, and received a worldwide exclusive license from the University of Zürich to the DARPin® technology with the vision to help patients by developing therapeutic DARPin® drugs. Since the DARPin® platform can, in principle, be used to generate proteins for various applications, including therapeutic molecules, the first decision needed was to set the focus of the company. By analyzing similar companies and talking to experts in various fields, it became clear that the requirements for becoming a successful reagent company versus a diagnostic company or a therapeutic company were too different to be pursued under one roof. The choice to develop DARPin® drugs was based on the ambition that the platform could yield drug candidates beyond what was known at the time.
The second big decision was where to focus within the medical therapy space. This was approached systematically by assessing opportunities and risks considering different aspects: platform validation, molecular complexity of the drug to be developed, pharmacology, status of the medical field, and potential market need/value. Since the DARPin® platform was untested in the clinic, the most important initial focus was on the platform itself during the first program while following a rather established path in terms of biological and medical challenges. Another important consideration was that the program should allow building of a sustainable company in parallel with all the necessary discovery, manufacturing, preclinical, and clinical development capabilities. At the time, a program fulfilling these requirements was regarded as a first-horizon program, and abicipar is the result (Fig. 2). It was postulated that once a first clinical validation of the first-horizon program was achieved, the risk profile could be expanded for second-horizon programs. With minimal biology resources available, such a second-horizon program would comprise an increased complexity of the molecule, while not taking too high risks in terms of pharmacology, i.e., work in phase II clinically validated pathways. In other words, a second-horizon program should be a multidomain multifunctional drug candidate, a format often seen in natural ankyrin repeat proteins and perceived to be a potentially key strength of the DARPin® technology. MP0250 and MP0274 are examples of such programs. The third-horizon programs would comprise multifunctional molecules active in new biology. Figure 2 illustrates the three-horizon strategy and its drug candidates per horizon.
Fig. 2.
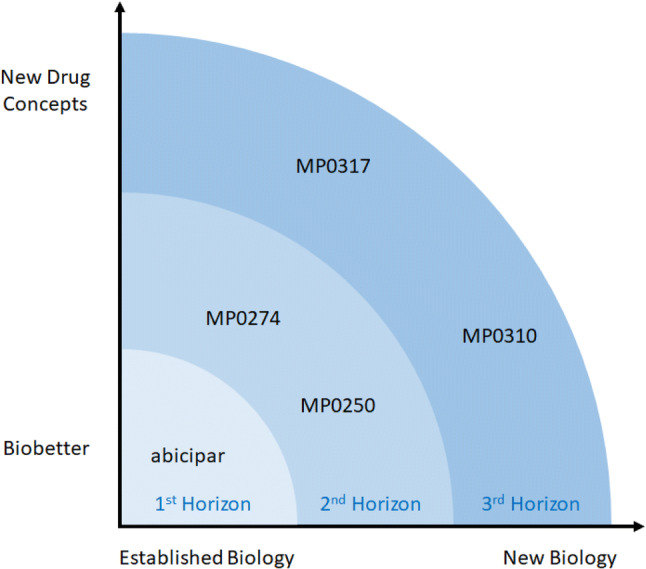
A schematic overview of all development-stage DARPin® drug candidates depicted on the two dimensions “degree of novel biology” vs. “degree of novel drug concept” on the drug development programs in the context of the three-horizon strategy of Molecular Partners. A first-horizon program such as abicipar is built on known biology and takes advantage of the favorable DARPin® drug properties for the generation of a “biobetter compound.” Second-horizon programs such as MP0250 and MP0274 are based on biology that has been validated in third-party phase II clinical trials; at the same time, they are multidomain drug candidates, a key strength of the DARPin® platform. In third-horizon programs, biology may be less validated and novel drug concepts may be applied, as illustrated by MP0310 (see Fig. 5) and MP0317
Abicipar
The success of both the monoclonal antibody bevacizumab in oncology and the Fab ranibizumab [25] in wet age-related macular degeneration (AMD) suggested vascular endothelial growth factor A (VEGF-A) to be a prime target candidate, in 2007, for generating the first therapeutic DARPin® drug candidate. The choice for ophthalmology was driven by two opportunities perceived at the time: first, a clear medical need expressed by physicians for a drug that allowed less frequent patient dosing and, second, an ideal match to the concept of a first-horizon program.
Three properties were identified as essential for the drug candidate to achieve the goal of less frequent dosing than ranibizumab (Fig. 3): (1) more potent inhibition of VEGF, enabling activity even at very low levels of drug in the eye (i.e., very high affinity); (2) high aqueous solubility and stability to allow delivery of a high dose into the vitreous, prolonging the time until clearance to nonpharmacologically active levels; and, perhaps most importantly, (3) a prolonged ocular half-life giving a longer residence time at relevant drug concentrations.
Fig. 3.
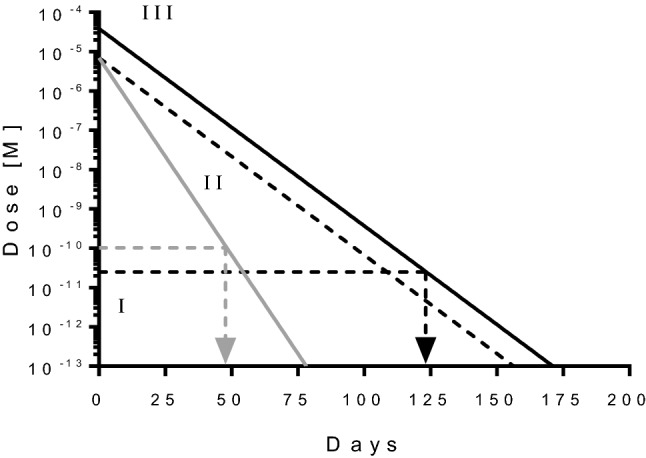
Schematic illustration of duration of action engineering of abicipar. Drug concentration of abicipar (black line) and ranibizumab (gray line) are shown over time. Assuming reference ranibizumab is administered 50 μl/0.5 mg intravitreally, and the drug has a half-life in the 1.5 ml rabbit eye of 3 days and an inhibition constant IC50 of about 100 pM, then the drug would stop being efficacious when reaching a concentration equivalent to the IC50, i.e. duration of action of about 48 days. Three aspects help increase the duration of action of abicipar: (I) improving the potency to lower values in combination with high drug stability ensure that the drug is also active at very low amounts; (II) engineering the half-life as such, requiring high drug stability; (III) applying higher doses. Assuming abicipar exhibits an IC50 of about 25 pM [26], has an ocular half-life of 6 days in the rabbit eye, and that it can be applied at about 5.5 times the molar amounts of ranibizumab, then it would take 124 days (4 months) for the vitreal concentration of abicipar reaches the IC50 concentration of 25 pM
The DARPin® platform allowed the rapid selection of high-affinity VEGF-A binders [16]: the final drug candidate has an affinity of approximately 394 fM to VEGF-A165, measured by kinetic exclusion assays (KinExA) and a potency of approximately 24.8 pM in a VEGF-A165–induced calcium mobilization assay [26]. The molecular weight of abicipar (34 kDa) is slightly smaller than that of ranibizumab (48 kDa) [27] and significantly smaller than that of bevacizumab (149 kDa) [27] or aflibercept (115 kDa) [27]. The biophysical properties of the VEGF-binding DARPin® molecule allowed the generation of a high-concentration formulation that enabled dosing at 1 or 2 mg [28], which is about 2.8 and 5.5 times higher in molar equivalents, respectively, than the 0.5 mg dose for ranibizumab. PEGylation of the VEGF-binding DARPin® molecule produced the drug candidate MP0112 with an ocular half-life in rabbit of 6.6 days, almost double that of ranibizumab (3.5 days) [29] and maintained high potency. Based on a simple mathematical model, these parameters supported the possibility that dosing humans once every 12 weeks (q12w) should provide continuous, complete VEGF inhibition [29, 30].
MP0112 was first tested in two open-label multicenter dose-escalation phase I/IIa clinical trials, one for the treatment of wet AMD (NCT01086761) [31] and one for the treatment of diabetic macular edema (DME; NCT01042678) [32]. Signs of efficacy were seen in terms of both best-corrected visual acuity and reduction of central retinal thickness. The ocular half-life of MP0112 in patients was determined to be about 2 weeks [32]. On the basis of these phase I/IIa data, the compound was partnered with Allergan and renamed abicipar (AGN 150,998).
Abicipar was tested in phase II trials in wet AMD (NCT01397409; REACH) [28] and in DME (NCT02186119; PALM), and the findings suggested that abicipar 2 mg every 8 weeks or q12w was likely to result in a treatment efficacy noninferior to ranibizumab administered every 4 weeks (q4w).
Based on the data of the REACH trial, two phase III studies in wet AMD were performed (CEDAR [NCT02462928] and SEQUOIA [NCT02462486]), and both have met their primary efficacy endpoints, with the 2-year results showing that > 90% of patients dosed at 12-week intervals experienced visual benefits similar to those receiving ranibizumab q4w, thus reducing the treatment burden by 60% [33]. In summary, the phase III data indicate that abicipar has the potential to be the first drug allowing convenient and fixed once-per-quarter dosing without reassessing patients in between. Marketing authorization applications are currently under review in both the USA and Europe.
MP0250
With the first clinical data of MP0112, and thus for the DARPin® platform, showing efficacy and safety in humans following local (intravitreal) dosing, it was decided that a second-horizon program involving systemic administration could be moved into clinical testing. The selection of the clinical indication and the target for a DARPin® drug candidate was based on preclinical studies and evaluation of the feasibility to meet an unmet medical need. DARPin® molecules targeting cytokines, growth factors, and receptors were investigated in preclinical studies in a range of potential clinical indications, with oncology eventually chosen as the strategic focus for building the Molecular Partners’ pipeline.
As a first systemic administration program and as extension to the expertise built with MP0112, the second drug candidate was created to bind and neutralize two soluble growth factors, VEGF-A and hepatocyte growth factor (HGF [34]) the ligand of cMET [35]. At the time, blocking the VEGF receptor (VEGFR)-2 pathway in mouse xenograft models had been shown to increase invasion and metastasis and upregulate the cMET pathway, and concurrent blocking of both the VEGFR-2 and cMET pathways reduced the invasion and metastasis [36]. Also, promising early clinical data were available for cMET pathway inhibitor antibodies [37] (however, these antibodies later failed to show efficiency in phase III clinical trials [38]). Thus, dual inhibition appeared to offer a means of countering the frequently encountered failure of single-agent VEGF inhibition to maintain control of tumor growth.
The dual inhibitor of VEGF-A and HGF was named MP0250; the generation and characterization of MP0250 has been described in detail [13, 39]. In brief, antitumor activity was demonstrated in VEGF- and HGF-dependent xenograft and syngeneic models with activity superior to that of individual VEGF- and HGF-blocking DARPin® molecules. Combination therapy studies showed potentiation of the antitumor activity of chemotherapy and immunotherapy agents. Of note, in addition to the DARPin® domains binding VEGF-A and HGF, MP0250 contains two further DARPin® domains that bind to serum albumin to extend the plasma half-life of the molecule. A two-domain DARPin® molecule would have a size of approximately 30 kDa and thus would be rapidly cleared by kidney filtration. The addition of albumin-binding activity overcomes this and extends the half-life to be in the range of that of serum albumin [13]: a half-life of 2–3 weeks was predicted in humans [18], indicating the potential for dosing every 2–4 weeks. During the preclinical testing of MP0250, the addition of the second human serum albumin (HSA) binding DARPin® domain was explored and increased the observed half-life. Thus, MP0250 comprises a total of four designed ankyrin repeat domains that can bind VEGF-A, HGF, and albumin simultaneously [13]. Importantly, VEGF, HGF, and albumin are bound with low-picomolar half-maximal effect (EC50) or half-maximal inhibition (IC50) and with cellular potencies of mid-pM for both VEGF and HGF [13, 39]. MP0250 was tested in various mouse xenograft models and exhibited strong efficacy as monotherapy and in combination with standard-of-care drugs [39].
In addition to attempting to improve cancer treatment, a key objective for the MP0250 program was to further assess the safety and tolerability of the DARPin® platform in humans. MP0250 was the first DARPin® molecule dosed intravenously, the first with the serum albumin half-life extension, and the first multidomain DARPin® drug candidate. The phase I outcome of MP0250 in patients with cancer (NCT02194426) was thus key for the drug itself and, at the same time, pivotal for the company’s strategic direction, including further pipeline building. Initial results of the study are available [40–42]. A total of 45 patients with various advanced cancer indications were treated with doses ranging from 0.5 to 12 mg/kg either every 2 weeks (q2w) or every 3 weeks (q3w). MP0250 was well-tolerated, with most adverse events—for instance hypertension and proteinuria—consistent with inhibition of the VEGF pathway. The maximum tolerated dose/recommended dose was 8 mg/kg q2w or 12 mg/kg q3w. Exposure was dose proportional and sustained throughout the dosing period for all patients (up to 15 months). The half-life was about 2 weeks. Signs of single-agent antitumor activity were observed: one partial response and 25 patients with stable disease, with the longest duration of 65 weeks and a median duration of about 10 weeks, indicating that MP0250 has some potential as a monotherapy. Figure 4 shows the duration of treatment for all patients. These findings indicated that MP0250, and thus the multidomain DARPin® platform as such, can have a safety profile comparable to monoclonal antibodies such as, for example, bevacizumab [43].
Fig. 4.

Duration of treatment of patients with MP0250 in phase I clinical trial NCT02194426. The weeks of treatment are shown for every patient of the trial, and patients are grouped in the different cohorts. Graph
adapted from Azaro et al. [40]. q2w every 2 weeks, q3w every 3 weeks
Following the positive phase I data, a phase II study of MP0250 in combination with bortezomib is in progress: This trial was initiated based on preclinical studies with a multiple myeloma model showing that MP0250 enhanced the activity of the standard-of-care drug bortezomib [44] and on the finding that MP0250 had a profound effect on cell signaling in bone marrow isolates from patients with multiple myeloma [45]. In addition, published analyses of biopsies from patients with multiple myeloma indicated that both the VEGF and the HGF pathways are key in multiple myeloma [46–51]. The phase II trial is evaluating the combination of MP0250 with bortezomib and dexamethasone for the treatment of patients with refractory and relapsed multiple myeloma who experienced resistance to bortezomib/dexamethasone treatment as the most recent treatment (NCT03136653) [52, 53]. Interim results of this trial were recently presented [54] and confirmed the initial observations: 9 of the 20 patients treated and evaluated at the cut-off date reached an objective response. More importantly, several patients who were refractory (i.e., had never responded) to proteasome inhibitor treatment derived clinical benefit or responded upon addition of MP0250, confirming that dual VEGF/HGF inhibition can help these patients.
In summary, clinical data generated to date support further development of MP0250 as they have shown antitumor activity and an acceptable safety and pharmacokinetic profile. The current working hypothesis is that VEGF and HGF are key factors in the maintenance of the tumor microenvironment and thus the support for the proliferation and survival of tumor cells. In this case, inhibition of the growth factors breaks this support and results in inhibition of tumor growth, particularly if the tumor cells are concomitantly damaged by standard-of-care treatments. Such a mechanism may well apply to both untreated tumors and those that have developed drug resistance; in both cases, the microenvironment has developed to give appropriate support to the tumor cells.
MP0274
In parallel to developing MP0250, another second-horizon program was pursued with the aim of further expanding the range of DARPin® drug applications. While MP0250 is directed to soluble targets, MP0274 binds to a cell surface receptor, human epidermal growth factor receptor (HER)-2 (ErbB2, neu) [55], a target well-known to be overexpressed in breast and other cancer types [56]. With drugs such as trastuzumab, pertuzumab, and T-DM1, good antibody therapies are available to target HER2 [57]. However, MP0274 has a different mechanism of action: it contains two DARPin® domains that bind to different domains of HER2, i.e., the compound is bi-paratopic. This bi-paratopic binding induces rapid internalization of HER2 and subsequently induces apoptotic killing of a variety of cell lines expressing HER2 at IHC3+ to IHC1+ levels. In multiple in vivo models, MP0274 showed antitumor activity similar or superior to that of trastuzumab and efficacy equivalent to that with the combination of both approved drugs trastuzumab and pertuzumab. While several investigators have shown that the dominant antitumor mechanism of the monoclonal antibodies in mice is antibody-dependent cellular cytotoxicity/phagocytosis, MP0274 lacks effector functions and acts solely through profound inhibition of HER2 signaling. This different antitumor mechanism of MP0274 constitutes a novel and promising approach to directly kill HER2-addicted tumor cells and provide additional clinical benefit alone or in combination to patients with HER2-positive cancers [58]. MP0274 is currently in a phase I clinical trial (NCT03084926) to determine the safety profile and a recommended phase II dose [58]. In addition to the two HER2-binding DARPin® domains, MP0274 has two serum albumin-binding DARPin® domains that confer a long systemic terminal half-life, similar to that observed for MP0250.
MP0310 (AMG 506)
With the first phase I safety data for the systemic application of MP0250 available, we decided to use the multidomain DARPin® platform for systemic application in an even broader approach and start to tackle novel biology within oncology, the third horizon. The rationale for MP0310 was based on the assumption that the therapeutic window of a drug can be increased by building in an activation mechanism that allows the drug to move from a less active to a very active form. MP0310 was thus designed to overcome limitations of the 4-1BB agonistic immune-modulatory antibody-based drugs currently being developed [59]. Such antibodies activate T cells, not only as intended in the tumor environment but also systemically [60], hence associating promising efficacy with significant toxicity (Fig. 5). In contrast, MP0310 is a drug candidate with preclinical evidence that efficacy can be uncoupled from toxicity, ultimately leading to a larger therapeutic window. To achieve this, MP0310 combines tumor-restricted fibroblast-activating protein (FAP) binding and 4-1BB agonism in one molecule, similar to an approach that has been presented elsewhere [61]. The 4-1BB agonism of MP0310 relies on “clustering,” that is, T cells are activated only when 4-1BB molecules on T cells are brought together in a high local density, i.e., clustered [62] (Fig. 5). As long as there is no moiety leading to clustering in the rest of the body, clustering can be exclusively induced by clustering on FAP-overexpressing tissues. Many tumor-associated fibroblasts over-express FAP, whereas expression in healthy tissue is limited [63]. MP0310 is thus expected to preferentially activate T cells in the tumor microenvironment. A detailed mouse study evaluated dosing aspects of the compound [64] to initiate formal preclinical and clinical development. The first patient was dosed in October 2019. The development of MP0310 (AMG 506) is being conducted in a collaboration between Molecular Partners and Amgen.
Fig. 5.

Schematic representation of the differentiation of localized immune cell modulator MP0310 from classical immune cell modulators. Antibodies activating immune cells via 4-1BB typically activate immune cells in the tumor as well as in healthy tissue, leading to promising efficacy linked to a problematic toxicology profile. MP0310 binds to fibroblast-activating protein (blue dots) on tumor-associated fibroblast and clusters, providing an opportunity for immune cell activation. In healthy tissue, with a lack of opportunity to cluster, immune cells are not modulated by MP0310. mAb monoclonal antibody
The same principle of targeted activation or also targeted inhibition could be envisaged for other targeting/effector combinations. Indeed, a number of targeting moieties (fibronectin extracellular domain B, epidermal growth factor receptor, FAP, and HER2) and immune activator/inhibitor moieties (cluster of differentiation CD40, CD134, CD137) have been tested [65]. The combination of FAP targeting with CD40 agonism [66] is currently being pursued further toward the clinic with a DARPin® molecule called MP0317.
Broadening the Application of DARPin® Drugs
With the clinical validation by abicipar and MP0250 and the initiation of the MP0310 and MP0317 third-horizon programs, the DARPin® platform is now poised to unfold its full potential. Although the current focus of Molecular Partners is the development of multifunctional biologics in (immuno-) oncology, the technology allows for the generation of biologics in many more indications. In particular, it could be used in transforming applications beyond classical biologics, including viral (gene) therapy [67–70], chimeric antigen receptor T-cell (CAR-T) approaches [71, 72], or intracellular inhibition [73]. The approach Molecular Partners is taking to enable the broader use of the DARPin® platform is to establish multiple strategic research alliances. Such alliances are exemplified by a research alliance with Allergan in ophthalmology beyond abicipar and various other research collaborations. Nevertheless, Molecular Partners is prepared to be opportunistic if a suitable opportunity arises. This is exemplified by the decision in early 2020 to rapidly implement the expertise built up over the preceding years to initiate an effort to generate multispecific DARPin® molecules to inhibit the infectivity of the severe acute respiratory syndrome coronavirus 2 [74]. Key advantages of the DARPin® technology for rapid development of a potential drug in this indication are the speed at which high-affinity molecules can be generated, the geometric flexibility matching the target protein, and the relative ease of manufacturing to rapidly provide batches at sufficient scale for multinational treatment of the disease [75].
Conclusions
Molecular Partners set out to help patients by developing novel drugs using the DARPin® platform. Several factors contributed to the progress of the platform and the growth of the company. Technically, the favorable properties of the DARPin® drug platform enabled the generation of highly potent drug candidates in ophthalmology and oncology. The versatility of the platform further enabled the generation of abicipar as a differentiated treatment option for wet AMD and potentially other retinal diseases. It also enabled the generation of MP0250, the favorable safety profile of which is comparable to the profile of monoclonal antibodies, suggesting that the platform as such is safe. MP0250 has also shown initial encouraging response data in patients with multiple myeloma.
The approach chosen by Molecular Partners to build the company around a few indications in a sustainable way enabled step-by-step building of the expertise needed for further growth. Alongside continuing to build low- and mid-risk approaches, Molecular Partners can now also start exploring compounds that tackle novel biology or known biology in novel ways. The versatility of the platform is attractive for use in indications beyond oncology, which is generally being explored via partnerships.
Acknowledgements
All current and former employees of Molecular Partners are acknowledged for their dedication for bringing innovative therapeutics to patients. DARPin® is a registered trademark owned by Molecular Partners AG.
Compliance with Ethical Standards
Funding
No sources of funding were used for the preparation of this manuscript.
Conflict of interest
MTS and KMD are employees and shareholders, and HKB is shareholder of, Molecular Partners AG, commercializing the DARPin® Technology.
References
- 1.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 3.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, Arnold FH. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci USA. 1993;90(12):5618–5622. doi: 10.1073/pnas.90.12.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321(6069):522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 6.Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23(10):1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 7.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332(2):489–503. doi: 10.1016/S0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 8.Kohl A, Binz HK, Forrer P, Stumpp MT, Plückthun A, Grütter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci USA. 2003;100(4):1700–1705. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bork P. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins. 1993;17(4):363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- 10.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 11.Forrer P, Binz HK, Stumpp MT, Plückthun A. Consensus design of repeat proteins. ChemBioChem. 2004;5(2):183–189. doi: 10.1002/cbic.200300762. [DOI] [PubMed] [Google Scholar]
- 12.Mosavi LK, Minor DL, Jr, Peng ZY. Consensus-derived structural determinants of the ankyrin repeat motif. Proc Natl Acad Sci USA. 2002;99(25):16029–16034. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binz HK, Bakker TR, Phillips DJ, Cornelius A, Zitt C, Göttler T, et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin(R) drug candidate. MAbs. 2017;9(8):1262–1269. doi: 10.1080/19420862.2017.1305529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22(5):575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 15.Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol. 2015;55:489–511. doi: 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- 16.Stahl A, Stumpp MT, Schlegel A, Ekawardhani S, Lehrling C, Martin G, et al. Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis. 2013;16(1):101–111. doi: 10.1007/s10456-012-9302-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahnd C, Wyler E, Schwenk JM, Steiner D, Lawrence MC, McKern NM, et al. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J Mol Biol. 2007;369(4):1015–1028. doi: 10.1016/j.jmb.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Steiner D, Merz FW, Sonderegger I, Gulotti-Georgieva M, Villemagne D, Phillips DJ, et al. Half-life extension using serum albumin-binding DARPin(R) domains. Protein Eng Des Sel. 2017;30(9):583–591. doi: 10.1093/protein/gzx022. [DOI] [PubMed] [Google Scholar]
- 19.Schütz M, Batyuk A, Klenk C, Kummer L, de Picciotto S, Gulbakan B, et al. Generation of fluorogen-activating designed ankyrin repeat proteins (FADAs) as versatile sensor tools. J Mol Biol. 2016;428(6):1272–1289. doi: 10.1016/j.jmb.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Kohl A, Amstutz P, Parizek P, Binz HK, Briand C, Capitani G, et al. Allosteric inhibition of aminoglycoside phosphotransferase by a designed ankyrin repeat protein. Structure. 2005;13(8):1131–1141. doi: 10.1016/j.str.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 21.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5(1):e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thieltges KM, Avramovic D, Piscitelli CL, Markovic-Mueller S, Binz HK, Ballmer-Hofer K. Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis. 2018;21(3):533–543. doi: 10.1007/s10456-018-9606-9. [DOI] [PubMed] [Google Scholar]
- 23.Hober S, Lindbo S, Nilvebrant J. Bispecific applications of non-immunoglobulin scaffold binders. Methods. 2019;154:143–152. doi: 10.1016/j.ymeth.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Gebauer M, Skerra A. Engineered protein scaffolds as next-generation therapeutics. Annu Rev Pharmacol Toxicol. 2020;60:391–415. doi: 10.1146/annurev-pharmtox-010818-021118. [DOI] [PubMed] [Google Scholar]
- 25.Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176(6):1248–1264. doi: 10.1016/j.cell.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigues GA, Mason M, Christie LA, Hansen C, Hernandez LM, Burke J, et al. Functional characterization of abicipar-pegol, an anti-VEGF DARP in therapeutic that potently inhibits angiogenesis and vascular permeability. Invest Ophthalmol Vis Sci. 2018;59(15):5836–5846. doi: 10.1167/iovs.18-25307. [DOI] [PubMed] [Google Scholar]
- 27.Avery RL, Castellarin AA, Steinle NC, Dhoot DS, Pieramici DJ, See R, et al. Systemic pharmacokinetics and pharmacodynamics of intravitreal aflibercept, bevacizumab, and ranibizumab. Retina. 2017;37(10):1847–1858. doi: 10.1097/IAE.0000000000001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Callanan D, Kunimoto D, Maturi RK, Patel SS, Staurenghi G, Wolf S, et al. Double-masked, randomized, phase 2 evaluation of abicipar pegol (an anti-VEGF DARPin therapeutic) in neovascular age-related macular degeneration. J Ocul Pharmacol Ther. 2018 doi: 10.1089/jop.2018.0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seal JR, Ekawardhani S, Schlegel A, Stumpp MT, Binz HK, Attar M, editors. Pegylation of abicipar increases vitreal half-life, supporting a potential for up to 3 month duration of action in the clinic. In: The Association for Research in Vision and Ophthalmology annual meeting; 2018; Honolulu, Hawaii.
- 30.Luu KT, Seal JR, Attar M. A mechanistic and translational pharmacokinetic-pharmacodynamic model of abicipar pegol and vascular endothelial growth factor inhibition. J Pharmacol Exp Ther. 2020;373(2):184–192. doi: 10.1124/jpet.119.263178. [DOI] [PubMed] [Google Scholar]
- 31.Souied EH, Devin F, Mauget-Faysse M, Kolar P, Wolf-Schnurrbusch U, Framme C, et al. Treatment of exudative age-related macular degeneration with a designed ankyrin repeat protein that binds vascular endothelial growth factor: a phase I/II study. Am J Ophthalmol. 2014;158(4):724–32e2. doi: 10.1016/j.ajo.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 32.Campochiaro PA, Channa R, Berger BB, Heier JS, Brown DM, Fiedler U, et al. Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: a phase I/II study. Am J Ophthalmol. 2013;155(4):697–704, e1–2. doi: 10.1016/j.ajo.2012.09.032. [DOI] [PubMed] [Google Scholar]
- 33.Kunimoto D, Yoon YH, Wykoff CC, Chang A, Khurana RN, Maturi RK. Efficacy and safety of abicipar in neovascular age-related macular degeneration: 52-week results of phase 3 randomized controlled study. Ophthalmology. 2020 doi: 10.1016/j.ophtha.2020.03.035. [DOI] [PubMed] [Google Scholar]
- 34.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10(10):2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 36.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2(3):270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH, Jr, Blumenschein GR, Jr, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spigel DR, Edelman MJ, O'Byrne K, Paz-Ares L, Mocci S, Phan S, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J Clin Oncol. 2017;35(4):412–420. doi: 10.1200/JCO.2016.69.2160. [DOI] [PubMed] [Google Scholar]
- 39.Fiedler U, Ekawardhani S, Cornelius A, Gilboy P, Bakker TR, Dolado I, et al. MP0250, a VEGF and HGF neutralizing DARPin((R)) molecule shows high anti-tumor efficacy in mouse xenograft and patient-derived tumor models. Oncotarget. 2017;8(58):98371–98383. doi: 10.18632/oncotarget.21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azaro A, Rodon J, Middleton MR, Baird RD, Herrmann R, Fiedler U et al., editors. First-in-class phase I study evaluating MP0250, a VEGF and HGF neutralizing DARPin® molecule, in patients with advanced solid tumors. In: American Society of Clinical Oncology annual meeting; 2018.
- 41.Rodon J, Omlin A, Herbschleb KH, Garcia-Corbacho J, Steiner J, Dolado I et al., editors. First-in-human phase I study to evaluate MP0250, a DARPin® blocking HGF and VEGF, in patients with advanced solid tumors. In: AACR-NCI-EORTC international conference on molecular targets and cancer therapeutics; Boston, MA; 2015.
- 42.Middleton MR, Azaro A, Kumar S, Niedermann P, Rodon J, Herbschleb KH et al., editors. Interim results from the completed FIH Phase I dose escalation study evaluating MP0250, a multi-DARPin® drug candidate blocking HGF and VEGF, in patients with advanced solid tumors. In: European Society for Medical Oncology annual meeting; 2016.
- 43.Fernando NH, Hurwitz HI. Targeted therapy of colorectal cancer: clinical experience with bevacizumab. Oncologist. 2004;9(Suppl 1):11–18. doi: 10.1634/theoncologist.9-suppl_1-11. [DOI] [PubMed] [Google Scholar]
- 44.Fiedler U, Dawson KM, Gilboy P, Stumpp MT, Tadjalli-Mehr K, Harstrick A, editors. MP0250, a bispecific VEGF- and HGF-blocking multi-DARPin*, in combination with bortezomib in multiple myeloma: preclinical rationale and phase 2 study outline. In: 21st Congress of the European Association of Hematology; Copenhagen, Denmark; 2016.
- 45.Rao L, De Veirman K, Giannico D, Saltarella I, Desantis V, Frassanito MA, et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin((R)) protein MP0250: a preclinical study. Oncotarget. 2018;9(17):13366–13381. doi: 10.18632/oncotarget.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moschetta M, Basile A, Ferrucci A, Frassanito MA, Rao L, Ria R, et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin Cancer Res. 2013;19(16):4371–4382. doi: 10.1158/1078-0432.CCR-13-0039. [DOI] [PubMed] [Google Scholar]
- 47.Giuliani N, Storti P, Bolzoni M, Palma BD, Bonomini S. Angiogenesis and multiple myeloma. Cancer Microenviron. 2011;4(3):325–337. doi: 10.1007/s12307-011-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrucci A, Moschetta M, Frassanito MA, Berardi S, Catacchio I, Ria R, et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin Cancer Res. 2014;20(22):5796–5807. doi: 10.1158/1078-0432.CCR-14-0847. [DOI] [PubMed] [Google Scholar]
- 49.White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer. 2013;119(2):339–347. doi: 10.1002/cncr.27745. [DOI] [PubMed] [Google Scholar]
- 50.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wader KF, Fagerli UM, Holt RU, Stordal B, Borset M, Sundan A, et al. Elevated serum concentrations of activated hepatocyte growth factor activator in patients with multiple myeloma. Eur J Haematol. 2008;81(5):380–383. doi: 10.1111/j.1600-0609.2008.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szarejko M, Knop S, Schreder M, Goldschmidt H, Raab MS, Jurczyszyn A et al., editors. MP0250 in combination with bortezomib and dexamethasone in patients with relapsed-and-refractory multiple myeloma: First ssafety and early efficacy analysis of MP0250-CP201. In: European Hematological Association annual meeting; 2018.
- 53.Raab MS, Ria R, Schlenzka J, Krahnke T, Haunschild J, Hermann F et al., editors. MP0250—a dual inhibitor of VEGF and HGF—plus bortezomib + dexamethasone in a phase 2 open-label, single-arm, multicenter trial in patients with refractory and relapsed multiple myeloma (RRMM). In: European Society for Medical Oncology annual meeting; 2017.
- 54.Grzasko N, Knop S, Goldschmidt H, Raab MS, Jurczyszyn A, Duerig J et al., editors. The MP0250-CP201 MiRRoR study: a phase 2 study update of MP0250 plus bortezomib and dexamethasone in relapsed/refractory multiple myeloma (RRMM) patients previously exposed to proteasome inhibitors and immunomodulatory drugs. In: American Society of Hematology; Orlando, FL; 2019.
- 55.Padhy LC, Shih C, Cowing D, Finkelstein R, Weinberg RA. Identification of a phosphoprotein specifically induced by the transforming DNA of rat neuroblastomas. Cell. 1982;28(4):865–871. doi: 10.1016/0092-8674(82)90065-4. [DOI] [PubMed] [Google Scholar]
- 56.Yan M, Schwaederle M, Arguello D, Millis SZ, Gatalica Z, Kurzrock R. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34(1):157–164. doi: 10.1007/s10555-015-9552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Escriva-de-Romani S, Arumi M, Bellet M, Saura C. HER2-positive breast cancer: current and new therapeutic strategies. Breast. 2018;39:80–88. doi: 10.1016/j.breast.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Baird RD, Omlin A, Kiemle-Kallee J, Fiedler U, Zitt C, Feuerstein D et al., editors. MP0274-CP101: a phase 1, first-in-human, single-arm, multi-center, open-label, dose escalation study to assess safety, tolerability, and pharmacokinetics of MP0274 in patients with advanced HER2-positive solid tumors. In: San Antonio Breast Cancer Symposium; San Antonio, TX; 2017.
- 59.Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131(1):49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 60.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res. 2017;23(8):1929–1936. doi: 10.1158/1078-0432.CCR-16-1272. [DOI] [PubMed] [Google Scholar]
- 61.Muller D, Frey K, Kontermann RE. A novel antibody-4-1BBL fusion protein for targeted costimulation in cancer immunotherapy. J Immunother. 2008;31(8):714–722. doi: 10.1097/CJI.0b013e31818353e9. [DOI] [PubMed] [Google Scholar]
- 62.Link A, Hepp J, Reichen C, Schildknecht P, Tosevski I, Taylor J et al., editors. Preclinical pharmacology of MP0310: a 4-1BB/FAP bispecific DARPin® drug candidate promoting tumor-restricted T cell co-stimulation. In: American Association for Cancer Research annual meeting; Chicago, Illinois; 2018.
- 63.Reichen C, Bessey R, DePasquale C, Imobersteg S, Béhé M, Blanc A et al., editors. FAP-mediated tumor accumulation of a T cell agonistic 4-1BB/FAP DARPin® drug candidate analyzed by SPECT/CT and quantitative biodistribution. In: American Association for Cancer Research annual meeting; Chicago, Illinois; 2018.
- 64.Tosevski I, Juglair L, Link A, Lemaillet G, Poulet H, Veitonmäki N et al., editors. Preclinical identification of the pharmacologically active dose range of the tumor targeted 4-1BB agonist MP0310. In: Society for ImmunoTherapy of Cancer annual meeting; Washington D.C.; 2018.
- 65.Fiedler U, Reichen C, Taylor J, Schildknecht P, Barsin S, Metz C et al., editors. Tumor-restricted immune-modulation by multispecific molecules from the DARPin® toolbox. In: American Association for Cancer Research annual meeting; Chicago, Illinois; 2018.
- 66.Rigamonti N, Schlegel A, Barsin S, Schwestermann J, Mangold S, Kaufmann Y et al., editors. Fibroblast activation protein (FAP)-selective delivery of CD40 agonistic DARPin® molecule for tumor-localized immune activation. In: Society for ImmunoTherapy of Cancer annual meeting; Washington D.C.; 2018.
- 67.Münch RC, Muth A, Muik A, Friedel T, Schmatz J, Dreier B, et al. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat Commun. 2015;6:6246. doi: 10.1038/ncomms7246. [DOI] [PubMed] [Google Scholar]
- 68.Pecqueur L, Duellberg C, Dreier B, Jiang Q, Wang C, Plückthun A, et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc Natl Acad Sci USA. 2012;109(30):12011–12016. doi: 10.1073/pnas.1204129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Friedrich K, Hanauer JR, Prufer S, Münch RC, Volker I, Filippis C, et al. DARPin-targeting of measles virus: unique bispecificity, effective oncolysis, and enhanced safety. Mol Ther. 2013;21(4):849–859. doi: 10.1038/mt.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Münch RC, Mühlebach MD, Schaser T, Kneissl S, Jost C, Plückthun A, et al. DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther. 2011;19(4):686–693. doi: 10.1038/mt.2010.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hammill JA, VanSeggelen H, Helsen CW, Denisova GF, Evelegh C, Tantalo DG, et al. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J Immunother Cancer. 2015;3:55. doi: 10.1186/s40425-015-0099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Siegler E, Li S, Kim YJ, Wang P. Designed ankyrin repeat proteins as Her2 targeting domains in chimeric antigen receptor-engineered T cells. Hum Gene Ther. 2017;28(9):726–736. doi: 10.1089/hum.2017.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verdurmen WP, Luginbuhl M, Honegger A, Plückthun A. Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J Control Release. 2015;200:13–22. doi: 10.1016/j.jconrel.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 74.Molecular Partners A. Molecular partners initiates anti-COVID-19 therapeutic program leveraging multi-target binding of DARPin® proteins to neutralize SARS-CoV-2 virus. www.molecularpartners.com. 2020.
- 75.Molecular Partners A. Molecular partners confirms ultra-potent inhibition of SARS-CoV-2 live virus by anti-COVID-19 DARPin® candidates. www.molecularpartners.com. 2020.