

Abstract
Background and Purpose
Alzheimer's disease (AD) is the most prevalent disease associated with cognitive dysfunction. Current AD therapeutic agents have several gastrointestinal or psychological adverse effects and therefore, novel therapeutic agents with fewer adverse effects must be developed. Previously, we demonstrated that oleanolic acid, which is similar in chemical structure to maslinic acid, ameliorates cognitive impairment through the activation of tropomyosin receptor kinase (TrkB)–ERK–cAMP response element‐binding protein (CREB) phosphorylation and increased levels of brain‐derived neurotrophic factor (BDNF). In the present study, we investigate the effect of maslinic acid on cholinergic blockade‐induced memory impairment in mice.
Methods and Key Results
Maslinic acid reversed scopolamine‐induced memory impairment, as determined by the Y‐maze, passive avoidance and Morris water maze tests. In addition, we also observed that ERK–CREB, PI3K and PKB (Akt) phosphorylation levels were increased by maslinic acid administration in the mouse hippocampus. Moreover, we determined that the effects of maslinic acid on scopolamine‐induced memory impairment in the passive avoidance test were abolished by a specific TrkB receptor antagonist (ANA‐12). Additionally, we observed similar temporal changes in the expression levels between BDNF and tissue plasminogen activator in the hippocampus.
Conclusion and Implications
These findings suggest that maslinic acid enhances cognitive function through the activation of BDNF and its downstream pathway signalling in the hippocampus and that it might be a potential therapeutic agent for cognitive decline, such as that observed in AD.
Abbreviations
- AD
Alzheimer's disease
- Akt
PKB
- BDNF
brain‐derived neurotrophic factor
- CREB
cAMP response element‐binding protein
- mBDNF
mature brain‐derived neurotrophic factor
- q‐PCR
quantitative PCR
- tPA
plasminogen activator, tissue type
- TrkB
tropomyosin receptor kinase
What is already known
Maslinic acid ameliorates the cognitive dysfunctions induced by NMDA blockade in mice.
Maslinic acid has neuroprotective effects via Akt–GSK3β signalling pathway.
What this study adds
Maslinic acid reverses the scopolamine‐induced memory impairments in mice
Maslinic acid increases the BDNF expression levels in the hippocampus.
What is the clinical significance
Maslinic acid can be a potential therapeutic agent for cognitive decline, as observed in AD.
1. INTRODUCTION
Alzheimer's disease (AD) is the most prevalent cause of dementia, which is related to ageing and cognitive dysfunction (Bishop, Lu, & Yankner, 2010; Samanez‐Larkin & Knutson, 2015). The accumulation of polymerized β‐amyloid (Aβ) monomers or the phosphorylation of tau protein, resulting in the formation of amyloid plaques or neurofibrillary tangles that induce neuronal cell death, is observed in AD (Ballatore, Lee, & Trojanowski, 2007; Mawuenyega et al., 2010). Although the aetiology of AD is unclear, AChE inhibitors or NMDA receptor antagonists are prescribed for treating AD (Hampel et al., 2018; Recanatini & Valenti, 2004). However, they have several gastrointestinal or psychological adverse effects, such as vomiting, anorexia, diarrhoea, nausea or insomnia (Gao, Yu, Chen, & Zhou, 2016; Talesa, 2001). Since the approval of galantamine, which is derived from Galanthus caucasicus, many researchers have attempted to identify natural products or natural product‐derived active compounds as new AD therapeutic agents because of their fewer adverse effects (Williams, Sorribas, & Howes, 2011).
Triterpenoids isolated from herbal materials have been shown to have many pharmacological actions (Gurovic et al., 2010; Jiang, Gao, & Turdu, 2017). Previously, we and others have reported that some triterpenes and their saponins from natural herbal medicines, such as lancemaside A, amyrin and ginsenoside, ameliorate cognitive dysfunction such as in scopolamine‐induced memory impairment in mice (Jung et al., 2012; Park et al., 2014; Yang, Han, Ryu, Jang, & Kim, 2009). Furthermore, we observed that oleanolic acid, a triterpenoid compound contained in Syzygium aromaticum, enhances cognitive functions by increasing levels of brain‐derived neurotrophic factor (BDNF) in the hippocampus (Jeon et al., 2017). As a triterpenoid, maslinic acid ((2α,3β)‐dihydroxyolean‐12‐en‐28‐oic acid), which is similar to oleanolic acid in structure, is found in olive fruit and in traditional herbal medicines, such as S. aromaticum (clove) or Crataegus pinnatida along with oleanolic acid. Maslinic acid has neuroprotective properties against apoptotic cell death and Aβ protein‐induced neurotoxicity (Lozano‐Mena, Sanchez‐Gonzalez, Juan, & Planas, 2014; Yang, Tsai, Mong, & Yin, 2015). In addition, maslinic acid also ameliorates object recognition memory deficits under NMDA receptor blockade in mice (Jeon et al., 2017). Based on these findings, if maslinic acid has similar cognition‐enhancing effects as oleanolic acid, its effects may be associated with BDNF expression and it may also be a good candidate for reversing memory impairment. However, no studies have been conducted on the memory‐enhancing effects of maslinic acid.
In the present study, we elucidated the ameliorating effect of maslinic acid on cognitive impairment induced by cholinergic blockade through several behavioural tests, including the Y‐maze test, the passive avoidance test and the Morris water maze test. Many researchers have suggested that scopolamine (hyoscine) was used to mirror the cognitive defects and molecular changes as observed in the brain of AD patient (Bajo et al., 2015; Tang, 2019). Herein, we used scopolamine to block cholinergic neurotransmitter system as a muscarinic receptor antagonist, which is a useful pharmacological tool to study the cellular and molecular changes associated with AD pathogenesis. In addition, we conducted an electrophysiology study, Western blotting, and quantitative PCR (q‐PCR) to identify the possible mechanism(s) by which maslinic acid is associated with ameliorating cognitive impairment.
2. METHODS
2.1. Animals
Male ICR mice (5 weeks old, 25–30 g) were purchased from the Orient Co., Ltd., a branch of Charles River Laboratories (Gyeonggi‐do, Korea) and habituated for 1 week before each experiment. The mice were housed five per cage, provided with water and food ad libitum, and maintained at a constant temperature (23 ± 1°C) and humidity (60 ± 10%) under a 12‐hr illumination cycle (lights on from 07:30 to 19:30). Animal treatments and maintenance were carried out in accordance with the Animal Care and Use Guidelines issued by Kyung Hee University and Dong‐A University, Republic of Korea. All experimental protocols using mice were approved by the IACUC of Kyung Hee University (Approval Number KHUASP (SE)‐18‐008) and Dong‐A University (Approval Number DIACUC‐approvr‐17‐20). We used a total of 392 mice in these experiments (Y‐maze task, n = 10 per group; step‐through passive avoidance task, n = 10 per group; Morris water maze task, n = 10 per group; field EPSP recording, n = 7 per group; Western blot analysis, n = 5 per group; and q‐PCR, n = 5 per group). The effect group size was estimated by power analysis (Charan & Kantharia, 2013). Each mouse was randomly allocated for one experiment. All efforts were made to minimize the number of animals and their suffering. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
2.2. Materials
Maslinic acid, scopolamine, donepezil, piracetam and ANA‐12 were purchased from Sigma Chemical Co. (St. Louis, MO). For q‐PCR, TRIzol reagent (Cat# 15596018) and reverse transcriptase (Cat# Ep0442) were purchased from Thermo Fisher Scientific (Waltham, MA) and SYBR Premix Ex Taq (Cat# RR420a) was purchased from TaKaRa Bio Inc. (Tokyo, Japan). Primary antibodies against PKB (Akt, rabbit polyclonal, Cat# sc‐8312, RRID:AB_671714), phosphorylated Akt at tyrosine 308 (p‐Akt, rabbit polyclonal, Cat# sc‐101629, RRID:AB_2224735), ERK (mouse monoclonal, Cat# sc‐514302, RRID:AB_2571739), cAMP response element‐binding protein (CREB, rabbit polyclonal, Cat# sc‐58, RRID:AB_631314) and GAPDH (mouse monoclonal, Cat# sc‐47724, RRID:AB_627678) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against PI3K (rabbit polyclonal, Cat# 4292, RRID:AB_329869), phosphorylated PI3K at tyrosine 199 (p‐PI3K, rabbit polyclonal, Cat# 4228, RRID:AB_659940), phosphorylated CREB at serine 133 (p‐CREB, rabbit monoclonal, Cat# 9198, RRID:AB_2561044) and phosphorylated ERK at threonine 202 and tyrosine 204 (p‐ERK, mouse monoclonal, Cat# 9106, RRID:AB_331768) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). An anti‐BDNF antibody (mouse monoclonal, Cat# ab203573, RRID:AB_2631315) was purchased from Abcam (Cambridge, UK). The HRP‐conjugated secondary antibodies for anti‐rabbit (Cat# GTX213110‐01, RRID:AB_10618573) and anti‐mouse (Cat# GTX213111‐01, RRID:AB_10618076) were purchased from GeneTex Inc. (Irvine, CA). Other materials used for the experiments were obtained from general commercial resources and were of the highest grade.
2.3. Drug administration
Scopolamine, donepezil and piracetam were dissolved in a 0.9% saline solution and maslinic acid was suspended in 10% Tween 80 solution as previously described (Jeon, Kim, et al., 2017). In all experiments, maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.), donepezil (5 mg·kg−1, p.o.), piracetam (200 mg·kg−1, i.p.) or the same volume of vehicle (10% Tween 80 solution for oral administration and 0.9% saline solution for intraperitoneal administration) was administered 1 hr before the behavioural tests or killed for sample collection (Jeon, Kim, et al., 2017; Jeon, Lee, et al., 2017). Scopolamine (1 mg·kg−1, i.p.) was administered 30 min before the behavioural test or killed for brain sample collection to generate a cognitive impairment mice model. For the temporal profiling of BDNF expression levels, maslinic acid (1 or 3 mg·kg−1, p.o.) was administered 3, 6, 9 or 12 hr before kill.
2.4. Y‐maze task
The Y‐maze task was used to evaluate the working memory performance in mice, as described elsewhere (Anisman, 1975) with slight modification in the platform and exploration time (Park et al., 2015). The Y‐maze has three non‐transparent horizontal arms (40 × 3 × 12 cm) arranged at 120° angles from one another. Each mouse was initially placed in the centre of the Y‐maze and the sequence of arm entries (i.e. ABACAB) over 8 min was recorded for each mouse. An actual alternation was defined as an entry into all three arms consecutively (i.e. ACB, BAC or ABC, but not BBA, AAC or CCA). Recording and analysis of actual alternation were manually conducted by a person who was blind to the treatment. After each trial, the Y‐maze was cleaned with 70% EtOH spray to remove residual odours and residues. The alternation score (%) for each mouse was defined as the ratio of the number of actual alternations to the possible number of alternations (the total number of arm entries minus 2) multiplied by 100, as shown in the following equation: % alternation = [(number of alternations)∕(total arm entry numbers − 2)] × 100. The number of arm entries was used as an indicator of locomotor behaviour (Sarter, Bodewitz, & Stephens, 1988).
2.5. Step‐through passive avoidance task
The step‐through passive avoidance task was employed to examine the long‐term memory performance of the mice, as previously described (Jarvik & Kopp, 1967). The task had two trials, an acquisition trial and a retrieval trial with slightly modification in the platform and foot shock (Kim et al., 2009). In the acquisition trial, male ICR mice were trained in a box that contained two identical chambers (20 × 20 × 20 cm) separated by a guillotine door (5 × 5 cm). One of the boxes was illuminated with a 50‐W bulb and the other was non‐illuminated. The boxes had stainless steel rods (diameter of 2 mm) under them. The mice were placed in the illuminated compartment during the acquisition trial. The door between the two compartments was opened 10 s later. After the mouse entered the non‐illuminated compartment, the door automatically closed and a 3‐s electric foot shock (0.5 mA) was given through the stainless steel rods. The mice that did not enter the non‐illuminated compartment for 60 s after the door opened were gently introduced into the non‐illuminated compartment and the latency was recorded as 60 s. The retention trial was performed 24 hr after the acquisition trial. The latency to enter the dark compartment was used to measure memory performance in the mice. In the retention trial, the latency was recorded up to 300 s. Each latency of the acquisition or retention trial was analysed by a person who was blind to the treatments. Other procedures were the same as those for the acquisition trial.
For the antagonism study against TrkB receptor, maslinic acid (3 mg·kg−1, p.o.) was administered 1 hr before the acquisition trial. The mice were treated with a sub‐effective dose of ANA‐12 (0.3 mg·kg−1, i.p.) 30 min after the administration of maslinic acid, as described elsewhere (Jeon, Lee, et al., 2017). Scopolamine (1 mg·kg−1, i.p.) was administered 3 min after ANA‐12 treatment. The following other behavioural procedures were similar with those described above.
For the memory enhancement study, the mice were given maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.) or piracetam (200 mg·kg−1, i.p.) as a positive control 1 hr before the acquisition trial, as previously described (Liao et al., 2019). When the mice entered the non‐illuminated chamber, the door was closed and a sub‐effective electroshock (3 s, 0.25 mA) was delivered to the mice through the stainless steel rods. In the retention trial, the latency up to 600 s was recorded. If the mice did not enter the non‐illuminated chamber within 600 s after the door was opened, the latency was recorded as 600 s. The other procedures were performed as described above.
2.6. Morris water maze task
The Morris water maze task was employed to investigate the long‐term spatial memory performance in mice, as described elsewhere (Morris, 1984) with slight modification in training trials and environmental controls (Kim et al., 2019). The maze consisted of a round tank (45 cm in height and 90 cm in diameter) with visual cues on the wall in a dimly lit room (50 lx). The tank was artificially divided into four quadrants. A round black platform (a width of 6 cm and a height of 29 cm) was placed in the centre of one quadrant of the tank. The water (24 ± 1°C) was dyed black with 30 cm deep, so that the platform was invisible. The test was conducted for 6 days and consisted of one habituation day, four training trial days and one probe test day. On the habituation day, the mice swam freely for 60 s without the platform in the tank. During the four consecutive training days, the animals were trained to search for the hidden platform in the tank within 60 s. When the platform was found within 60 s, the mouse would be allowed to stay on the platform for an additional 10 s. If the mouse stook more than 60 s to find the platform, it was led towards the platform and allowed to stay on the platform for 10 s. Then, the mouse was put back to its home cage and dried under the IR light. Each mouse underwent two training trials per day and the interval between each trial was 30 min. The amount of time spent searching for the platform (the latency) in every trial was manually recorded who was blind to the grouping. For each trial, each mouse was arbitrarily placed in the water facing a visual cue on the wall. On the probe test day, the animals were required to search for the platform for 60 s, however the platform had been removed from the tank. During the probe test, the swimming speed and the amount of time spent swimming in the target zone where the platform was originally located were recorded with an EthoVision video system (Noldus, Wageningen, The Netherlands, RRID:SCR_000441) and analysed by a person who was blind to the treatments.
2.7. Hippocampal slice preparation and field EPSP recording
The artificial CSF (ACSF) comprised 124‐mM NaCl, 3‐mM KCl, 26‐mM NaHCO3, 1.25‐mM NaH2PO4, 2‐mM CaCl2, 1‐mM MgSO4 and 10‐mM d‐glucose. We rapidly removed the brains and isolated the hippocampus. Mouse hippocampal tissues were sliced using a McIlwain tissue chopper. We collected four slices from an animal and used one slice among them. Hippocampal slices (400‐μm‐thick) were made and incubated in ACSF (20–25°C) for 1 hr before the experiment.
Field potential responses were recorded from the Schaffer–collateral–commissural pathway in the CA1 area. Stimuli (constant voltage) were delivered at 30‐s intervals. The slope of the evoked field potential responses (fEPSP) was averaged over consecutive recordings evoked at 30‐s intervals. To induce LTP, two trains of high‐frequency stimulation (HFS: 100 Hz, 100 pulses in 1 s, 30 s interval) were introduced 20 min after the initiation of a stable baseline. LTP was quantified by comparing the mean fEPSP slope at 80 min after the high‐frequency stimulation period with the mean fEPSP slope during the baseline period and calculating the percentage change from the baseline. For the experiments, slices were incubated in the ACSF containing vehicle or drugs for 120 min before recording.
2.8. Western blot analysis
Western blot analyses were conducted to identify the changes of signalling molecules in the hippocampus (Towbin, Staehelin, & Gordon, 1979). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. In the present study, the mice were killed at a specific time point through a cervical dislocation. Thereafter, hippocampal tissues were isolated from the brains and homogenized in ice‐cold Tris–HCl buffer (20 mM, pH 7.4) containing 0.32‐M sucrose, 1‐mM EDTA, 1‐mM EGTA, 1‐mM PMSF, 1‐mM sodium orthovanadate and one protease inhibitor cocktail tablet (Roche, Seoul, Korea) per 50 ml of buffer. The homogenates containing 15 μg of total protein were loaded on SDS‐PAGE gels for electrophoresis. Thereafter, the separated proteins were transferred to PVDF membranes at 300 mA for 2 hr at 4°C in transfer buffer (25‐mM Tris–HCl [pH 7.4] containing 192‐mM glycine and 20% v/v methanol). The procedures for probing the membranes were the same as those described previously (Liao et al., 2019). The membranes were blocked by 5% skimmed milk and the primary antibodies were incubated overnight at 4°C, respectively, as follows: ERK or p‐ERK (1:5,000), CREB or p‐CREB (1:3,000), PI3K or p‐PI3K (1:2,000), Akt or p‐Akt (1:3,000), BDNF (1:2,000) and GAPDH (1:5,000). After the incubation, the membranes were washed out with Tris‐buffered saline/Tween 20. Then, HRP‐conjugated secondary antibodies (1:2,000) were incubated with membranes 2 hr at room temperature. Finally, the bands were enhanced by using ECL‐detection system (Amersham Pharmacia Biotech, Sunnyvale, CA) and images were acquired by ChemiDoc XRS+ system application (Bio‐Rad Laboratories, Hercules, CA, RRID:SCR_014210). The densitometric analysis of immunoreactive bands was performed using ImageJ software (NIH, Bethesda, MD, RRID:SCR_003070).
2.9. Quantitative PCR
Hippocampal tissues were homogenized with TRIzol reagent to extract crude RNA and 3 μg of isolated RNA was converted into cDNA by oligo dT primer according to the manufacturer's protocol. The following primers for BDNF (Accession Number AY057907, Tm = 57.4°C), plasminogen activator, tissue type (tissue plasminogen activator, tPA, Accession Number J03520, Tm = 57.4°C) and GAPDH (Accession Number GU214026, Tm = 56.4°C) were used for PCR amplification:
-
1.
BDNF,
forward primer: 5′‐GACGACATCACTGGCTGACA‐3′.
reverse primer: 5′‐GCTGTGACCCACTCGCTAAT‐3′.
-
2.
tPA,
forward primer: 5′‐CTCAGTGCCTGTCCGAAGTT‐3′.
reverse primer: 5′‐CCAAGGTCTGGCATCACCAT‐3′.
-
3.
GAPDH,
forward primer: 5′‐CTACCCCCAATGTGTCCGTC‐3′.
reverse primer: 5′‐TGAAGTCGCAGGAGACAACC‐3′.
PCR amplification was conducted by using an ABI StepOnePlus™ Real‐Time PCR System (Applied Biosystems, Foster City, CA, RRID:SCR_018051) with SYBR Premix Ex Taq. The cycle condition of PCR amplification was conducted as the following conditions: initial denaturation for 5 min at 95°C, followed by 50 cycles of amplification at 95°C for 15 s, annealing at 57.4°C (BDNF and tPA) and 56.4°C (GAPDH) for 60 s and extension at 72°C for 60 s. The expression of the PCR amplicons was normalized to that of GAPDH and relative gene expression data were analysed using the (Livak & Schmittgen, 2001).
2.10. Data and statistical analysis
All values are presented as the means ± SEM and analysed by GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA). All the data were analysed by D'Agostino–Pearson normality test and followed a Gaussian distribution. Using one‐way ANOVA, we analysed the spontaneous alternations in the Y‐maze, latencies in the passive avoidance test, swimming time in the target zone in the Morris water maze test and the data from Western blot analysis. To analyse the LTP data, we used repeated measures ANOVA. The Student–Newman–Keuls test was used for multiple comparisons. The latencies in the training trial during the Morris water test, the expression levels of BDNF and the data from q‐PCR were analysed by using two‐way ANOVA, followed by Bonferroni's post hoc test. The day was the first variable for the Morris water maze test and the time was the first variable for the BDNF level and treatment was the second variable. The group size is the number of independent values and statistical analysis was done using these independent values. Post hoc tests were conducted only if F in ANOVA achieved P < .05 and there was no significant variance inhomogeneity. The data normalized by fold of the control mean were presented in the Western blot analysis and q‐PCR. There were no excluded data in data analysis. Statistical significance was set at P < .05. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology.
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Curtis et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Maslinic acid attenuated short‐term and long‐term memory in the scopolamine‐induced memory impairment model
In the cholinergic blockade‐induced memory impairment mouse model, we examined whether maslinic acid ameliorates short‐term and long‐term memory using the Y‐maze, the passive avoidance and the Morris water maze tests. First, we measured spontaneous alternation behaviours in the Y‐maze (Figure 1a). There was significant difference in the percentage of spontaneous alternations among all the groups (P < .05). The percentage of spontaneous alternations in the scopolamine treatment group was significantly lower than that in the vehicle‐treated group (P < .05) and this reduction was reversed by maslinic acid (1 and 3 mg·kg−1, p.o.) or donepezil (5 mg·kg−1, p.o.; P < 0.05). The total number of arm entries was similar among all experimental groups (Figure 1b), indicating that the ameliorating effect of maslinic acid in the Y‐maze task was not caused by the activation of general locomotor behaviour.
FIGURE 1.
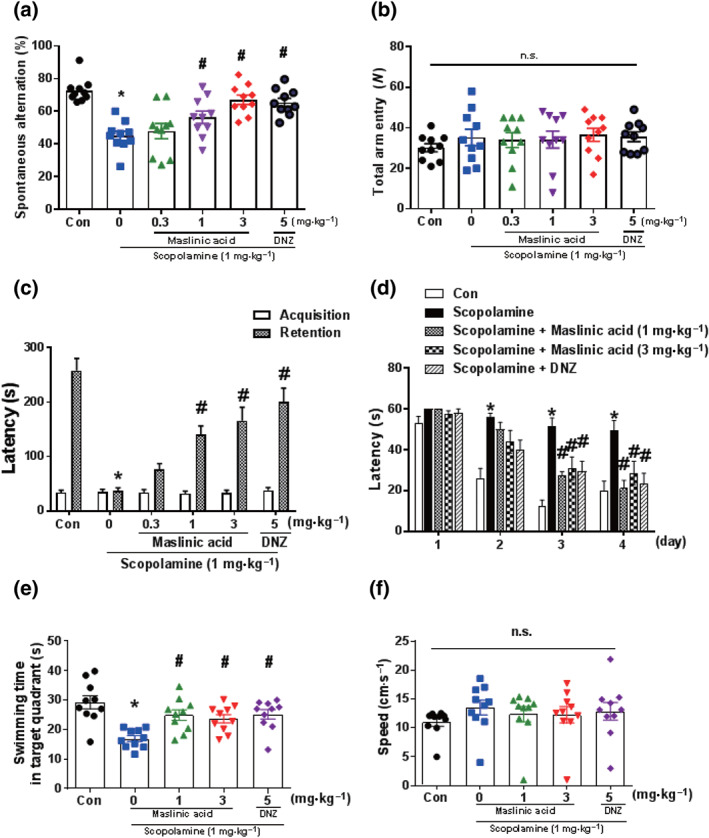
Maslinic acid ameliorates cognitive impairment induced by scopolamine treatment in mice. Mice were administered with maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.) or donepezil (DNZ; 5 mg·kg−1, p.o.) 1 hr before the Y‐maze test (a, b). Scopolamine (1 mg·kg−1, i.p.) was treated 30 min before the Y‐maze test to induce memory impairment. Spontaneous alternation behaviour (a) and the number of total arm entries (b) are presented. In addition, mice were treated with maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.) or DNZ (5 mg·kg−1, p.o.) 1 hr before the acquisition trial of the passive avoidance test, whereas scopolamine (1 mg·kg−1, i.p.) was treated 30 min before the acquisition trial to induce memory impairment. Twenty‐four hours after the acquisition trail, the retention trail was conducted for 300 s. The latency time is presented (c). Furthermore, mice were administered with maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.) or DNZ (5 mg·kg−1, p.o.) 1 hr before the first training trial per day for 4 days during the Morris water maze test. Scopolamine (1 mg·kg−1, i.p.) was treated 30 min before the first training trial per day to induce memory impairment (d–f). The escape latencies during the training trial (d), swimming time in the target zone (e) and swimming speed (f) during the probe test after the training trial are presented. Data were showed as means ± SEM (n = 8–10 per group; *P < .05, compared with the vehicle‐administered control [Con] group; # P < .05, compared with the scopolamine‐administered group; n.s., not significant)
Next, we conducted the passive avoidance task to determine the effects of maslinic acid on the scopolamine‐induced contextual long‐term memory impairment (Figure 1c). There was no significant difference in the latency at the acquisition trial between all treatment groups. However, in the retention trial, there were significant changes in the step‐through latency between treatment groups (P < .05). The cholinergic blockade‐reduced latency was significantly attwenuated by the administration of maslinic acid (1 and 3 mg·kg−1, p.o.) in a dose‐dependent manner (P < .05). Similar effects were observed at the donepezil (5 mg·kg−1, p.o.)‐treated group as a positive control. These results indicated that maslinic acid reduces scopolamine‐induced long‐term memory impairment.
To investigate whether maslinic acid can inhibit scopolamine‐induced long‐term spatial memory impairment, we conducted the Morris water maze task. There were significant group differences in day and treatment (P < .05). The scopolamine (1 mg·kg−1, i.p.)‐treated group took a longer time to escape than the control group after the second day of training (P < .05). Maslinic acid (1 or 3 mg·kg−1, p.o.) and donepezil (5 mg·kg−1, p.o.) treatment significantly decreased the escape latencies at the third and fourth days of training (P < .05; Figure 1d). The time spent swimming in the target quadrant during the probe trial (on Day 5) was obviously shorter in the scopolamine (1 mg·kg−1, i.p.) treatment group than that in the control group. Additionally, the maslinic acid (1 or 3 mg·kg−1, p.o.)‐treated and donepezil (5 mg·kg−1, p.o.)‐treated groups spent a longer time than the scopolamine‐treated group in the target quadrant (P < .05; Figure 1e). The speed of swimming was not affected by the administration of maslinic acid (Figure 1f). These results indicated that maslinic acid effectively reverses long‐term spatial memory impairment in cholinergic blockade mice.
3.2. Maslinic acid facilitated LTP induction in the mouse hippocampus
Synaptic plasticity is associated with cognitive function (Kastellakis & Poirazi, 2019). T,hus we investigated the effect of maslinic acid on synaptic plasticity by measuring the LTP in the hippocampal slices treated with maslinic acid (1, 10 and 30 μM) for 2 hr (Figure 2). In the control slices, high‐frequency stimulation induced LTP (124 ± 6% of baseline) and this was maintained for more than 1 hr (Figure 2a). Interestingly, maslinic acid‐treated slices showed higher LTP levels than that observed in control slices and this change was concentration‐dependent manner (1 μM of maslinic acid and 123 ± 4% of baseline, Figure 2a; 10 μM of maslinic acid and 149 ± 4% of baseline, Figure 2b; and 30 μM of maslinic acid and 156 ± 3% of baseline, Figure 2c). Figure 2d represented the LTP ratio compared with the baseline after each treatment (P < .05). These results suggested that maslinic acid enhanced hippocampal LTP.
FIGURE 2.
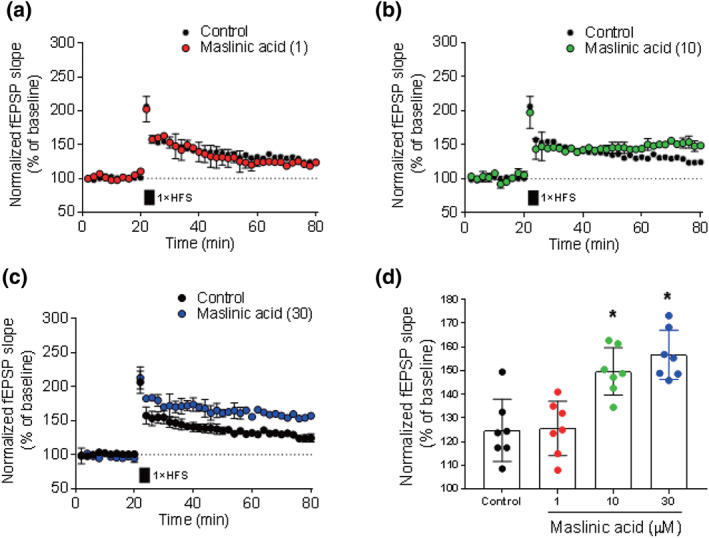
Maslinic acid facilitates LTP induction in the mouse hippocampus. Hippocampal slices were incubated with maslinic acid (1, 10 or 30 μM) for 2 hr. Extracellular field EPSPs (fEPSP) were recorded in Schaffer–collateral–commissural pathway in area CA1. LTP was induced by high‐frequency stimulation (HFS, each 100 Hz, 1 s). Normalized fEPSP slope for maslinic acid 1 μM (a), 10 μM (b) and 30 μM (c) and normalized LTP ratio at 80‐min time point (d) are presented. Data were showed as means ± SEM (n = 7 per group). *P < .05, compared with the vehicle‐administered control group. The number in the parentheses indicates the concentration of maslinic acid (μM)
3.3. Maslinic acid enhanced the ERK–CREB and PI3K–Akt signalling pathways in the hippocampus
We determined whether maslinic acid administration enhances the signalling pathway involved in cognitive function in the hippocampus of the mice (Figure 3). We found that the single administration of maslinic acid (1 or 3 mg·kg−1, p.o.) and donepezil (5 mg·kg−1, p.o.) enhanced the phosphorylation levels of ERK–CREB and PI3K–Akt in the hippocampal tissue (P < .05; Figure 3a). However, BDNF and glycogen synthase kinase‐3β, as the downstream signalling molecules of ERK–CREB and PI3K–Akt, were not significantly altered by the administration of maslinic acid (Figure S1). Consistent with our previous study (Jeon, Lee, et al., 2017; Park et al., 2012), scopolamine reduced the phosphorylation levels of ERK–CREB and PI3K–Akt signalling pathway molecules in hippocampal tissue (P < .05; Figure 3b). Those down‐regulated phosphorylation by scopolamine was significantly reversed by the administration of maslinic acid (1 or 3 mg·kg−1, p.o.) and donepezil (5 mg·kg−1, p.o.; P < .05). These findings suggested that maslinic acid enhances the ERK–CREB and PI3K–Akt signalling pathways under normal and the “hypocholinergic” state.
FIGURE 3.
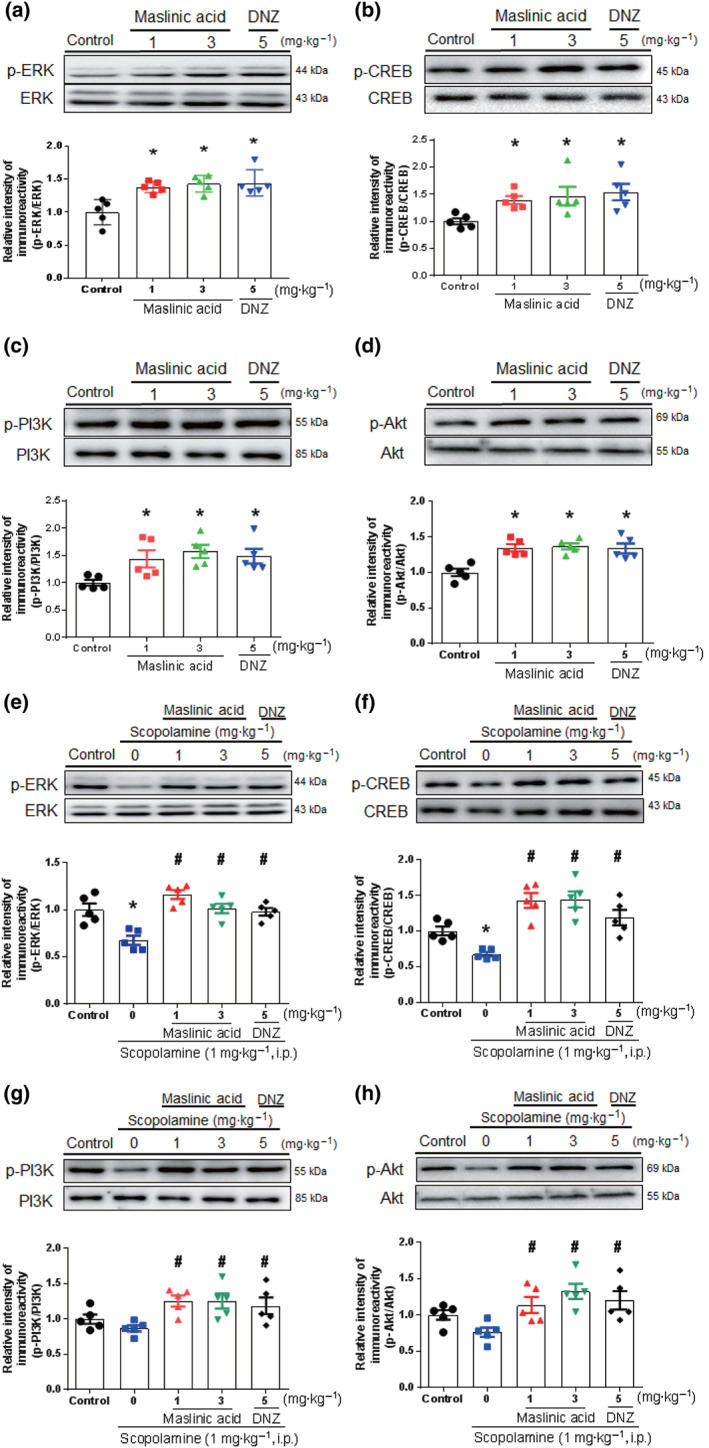
The effects of maslinic acid on the phosphorylation level of ERK, CREB, PI3K and Akt in hippocampal tissue. Mice were treated with maslinic acid (1 or 3 mg·kg−1, p.o.), donepezil (DNZ; 5 mg·kg−1, p.o.) or the same volume of vehicle solution 1 hr before the kill. To examine the effect of scopolamine on the signalling molecules, scopolamine was treated 30 min before kill. The levels of p‐ERK/ERK, p‐CREB/CREB, p‐PI3K/PI3K and p‐Akt/Akt were measured in the naïve (a–d) and scopolamine‐induced memory impairment model (e–h). The immunoreactivity of each band was normalized to the control (taken as 1.0). Data were showed as means ± SEM (n = 5 per group; *P < .05, compared with the vehicle‐administered control group; # P < .05, compared with the scopolamine‐administered group; n.s., not significant)
3.4. Maslinic acid enhanced BDNF and plasminogen activator, tissue type (tPA) at the specific time points in the hippocampus
Previously, we observed that the increased levels of mature brain‐derived neurotrophic factor (mBDNF) at specific time points, especially, 9 and 12 hr, after initial memory formation are required for memory consolidation or enhancement (Kim et al., 2012; Kim et al., 2014; Liao, Bae, Zhang, et al., 2019). Therefore, we conducted Western blot analysis to identify the expression levels of mBDNF in the mice hippocampi 3, 6, 9 or 12 hr after the administration of maslinic acid (1 or 3 mg·kg−1, p.o.; Figure 4a). There were significant group effects in time and treatment of mBDNF levels (P < .05). The expression levels of mBDNF at 9 and 12 hr after the administration of maslinic acid (1 and 3 mg·kg−1, p.o.) were significantly increased compared with those in the control (P < .05). These results indicated that the memory‐enhancing effect of maslinic acid correlates with the expression levels of mBDNF at 9 and 12 hr after the administration of maslinic acid.
FIGURE 4.
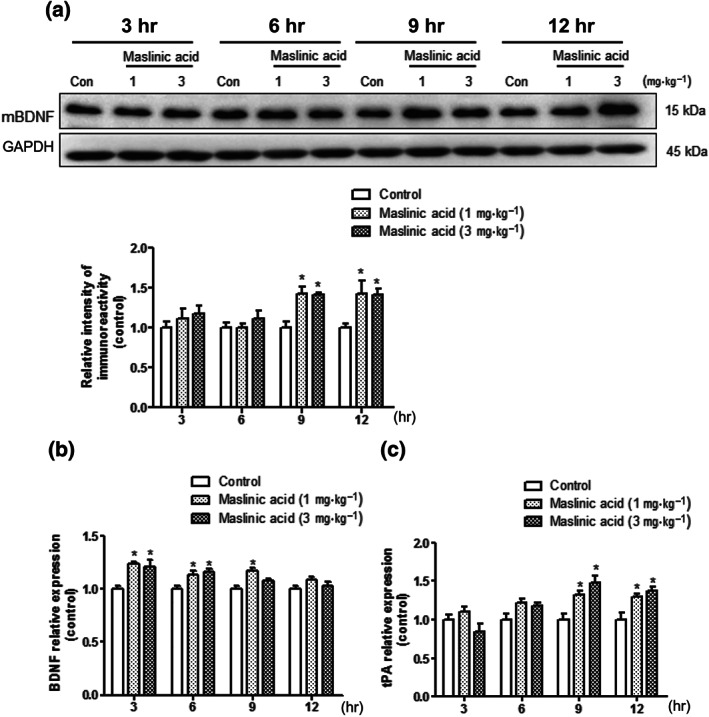
The effects of maslinic acid on the BDNF and plasminogen activator, tissue type (tPA) expression in the hippocampus. Mice were killed 3, 6, 9 and 12 hr after the administration of maslinic acid (1 or 3 mg·kg−1, p.o.) or the same volume of vehicle solution. The levels of immunoreactivity of BDNF and GAPDH were measured. The graph shows the ration of BDNF/GAPDH normalized to each time point control (a). By using q‐PCR, the mRNA expression levels of BDNF, tPA and GAPDH were measured in the hippocampus. The mRNA expression levels of BDNF (b) and tPA (c) are presented. The mRNA expression levels were normalized by GAPDH. Data were showed as means ± SEM (n = 5 per group; *P < .05, compared with the vehicle‐administered control [Con] group)
Next, we investigated whether the increased mBDNF expression levels might be derived from the increased BDNF mRNA levels. We conducted q‐PCR to determine the mRNA expression levels of BDNF in the hippocampus (Figure 4b). There were significant group effects on time and treatment of mRNA levels of BDNF (P < .05). The mRNA expression level of BDNF was slightly increased at 3 and 6 hr after the administration of maslinic acid (1 and 3 mg·kg−1, p.o.) and decreased in a time‐dependent manner. These results suggested that the conversion rate of mBDNF to proBDNF would be increased on 9 or 12 hr after the administration of maslinic acid. Therefore, we measured the mRNA expression level of tPA in mice hippocampus. tPA plays an important role in post‐translating process of BDNF in the brain (Melchor & Strickland, 2005). There were significant group effects in treatment and time on tPA mRNA levels (P < .05). The mRNA expression levels of tPA were significantly increased at 9 and 12 hr after the administration of maslinic acid (1 and 3 mg·kg−1, p.o.; P < .05; Figure 4c). These results indicated that the significant increase in mBDNF levels at 9 and 12 hr after the administration of maslinic acid may be caused by the increased expression levels of tPA.
3.5. Maslinic acid‐induced memory‐enhancing effects were mediated by TrkB activation
The increased mBDNF levels at 9 or 12 hr after the acquisition trial could enhance consolidation of memory, resulting in memory enhancement (Kim et al., 2012). Therefore, to confirm whether maslinic acid enhances cognitive function, we conducted the passive avoidance task in the naïve mice after a single administration of maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.). No significant change in latency was observed in the acquisition trial, but, in the retention trial, there were significant group effects on latency in all treated groups (P < .05; Figure 5a). Obviously, the single administration of maslinic acid (1 and 3 mg·kg−1, p.o.) and piracetam (200 mg·kg−1, p.o.) induced a longer latency compared with that in the vehicle‐treated control group (P < .05). These results suggested that the enhancement of mBDNF expression levels at specific time points (9 and 12 hr) after the administration of maslinic acid would induce memory enhancement in the passive avoidance test.
FIGURE 5.
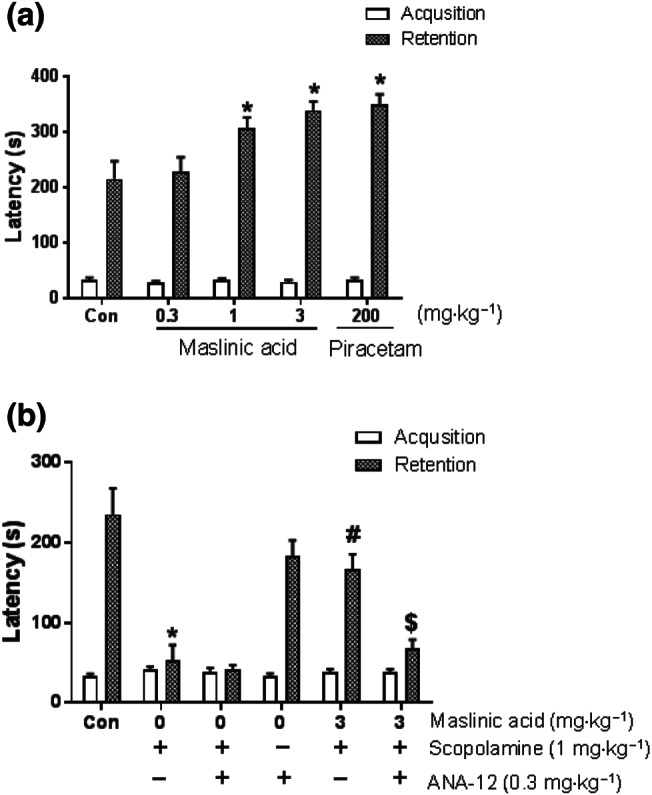
Memory‐enhancing activity of maslinic acid and TrkB activation involved in cognitive function. For the memory enhancement study, mice were treated with maslinic acid (0.3, 1 or 3 mg·kg−1, p.o.) or piracetam (200 mg·kg−1, p.o.) 1 hr before an acquisition trial. Twenty‐four hours after the acquisition trial, the retention trial was performed for 600 s. As piracetam‐treated group, maslinic acid (1 and 3 mg·kg−1, p.o.) enhanced the latency in a dose‐dependent manner (a). In the case of antagonism study, mice were treated with maslinic acid (3 mg·kg−1, p.o.) or the same amount of vehicle solution 1 hr before the acquisition trial. The specific TrkB receptor blocker, ANA‐12 (0.3 mg·kg−1, i.p.), was treated 30 min before the acquisition trial. Consecutively, scopolamine (1 mg·kg−1, i.p.) was treated 3 min after the ANA‐12 treatment. The graph represents the latency of the acquisition and retrieval trials (b). Data were showed as means ± SEM (n = 10 per group; *P < .05, compared with the vehicle‐administered control [Con] group; # P < .05, compared with the scopolamine‐administered group; $ P < .05, compared with the maslinic acid and scopolamine co‐treatment group)
If mBDNF signalling after binding to TrkB receptor is crucial for memory enhancement, such effect would be inhibited by the administration of a TrkB receptor antagonist. We performed the passive avoidance test to confirm the ameliorating effect of maslinic acid on TrkB blocking condition. After maslinic acid (3 mg·kg−1, p.o.) administration, ANA‐12 (0.3 mg·kg−1, i.p.), a sub‐effective dose of TrkB inhibitor, with or without scopolamine (1 mg·kg−1, i.p.), was concomitantly administered. The latency showed that scopolamine‐induced cognitive dysfunction was ameliorated by maslinic acid (3 mg·kg−1, p.o.) treatment (P < .05; Figure 5b). This was reversed by the co‐administration of ANA‐12. Neither ANA‐12 nor co‐treatment with ANA‐12 and scopolamine altered the cognitive behaviour in the passive avoidance test. These results indicated that the BDNF–TrkB system would be involved in the ameliorating effect of maslinic acid on cognitive dysfunction.
4. DISCUSSION
In the present study, we examined the effect of maslinic acid on a mouse model of scopolamine‐induced cognitive impairment and observed that maslinic acid reversed cognitive dysfunction, as measured by the Y‐maze test, passive avoidance test and Morris water maze test. In addition, maslinic acid also activated BDNF signalling and enhanced LTP formation in the hippocampal tissues. We also observed that the effects of maslinic acid on cognitive function were blocked by specific TrkB receptor inhibition.
Maslinic acid is a pentacyclic triterpene that is found not only in traditional herbal medicines but also in vegetables and fruits. Recently, several studies elucidated that maslinic acid has many biological activities, such as anti‐oxidative (Montilla et al., 2003), cardio‐protective (Liu et al., 2018), anti‐diabetic (Li, Li, Shi, & Guo, 2017) or neuroprotective effect (Wang, Mong, Yang, & Yin, 2018; Yang et al., 2015). If maslinic acid attenuates cognitive dysfunction in an AD model, it would be a novel agent for treating cognitive dysfunction in AD. In the present study, we found that scopolamine‐induced deficits in working memory, contextual short‐term or long‐term memory deficits, were reversed by maslinic acid administration in a dose‐dependent manner in mice. In addition, we conducted an ex vivo study to determine whether maslinic acid inhibits AChE activity. Maslinic acid did not show any inhibitory effect on AChE activity (Figure S2), suggesting that the ameliorating effect of maslinic acid on scopolamine‐induced memory impairment might not be derived from the inhibition of AChE activity in the hippocampus, which is consistent with a previous report (Schwarz et al., 2015).
It is well acknowledged that the ERK–CREB signalling pathway plays a crucial role in memory acquisition and consolidation by activating several memory‐related genes (Lakhina et al., 2015; Silva, Kogan, Frankland, & Kida, 1998). In addition, PI3K–Akt signalling pathway is also involved in synaptic plasticity and formatting memory in the hippocampus (Yi et al., 2018; Yi et al., 2018). Moreover, the inhibition of the PI3K–Akt signal pathway induces memory impairment (Chen et al., 2005; Mizuno et al., 2003). Consistent with previous findings that the acute or chronic administration of maslinic acid altered the phosphorylation levels of ERK–CREB or PI3K–Akt signalling pathway (Jeon, Kim, et al., 2017; Qian et al., 2015), we found that maslinic acid affected the phosphorylation levels of ERK and CREB or PI3K and Akt in the hippocampus of the naïve or cholinergic blockade mice. However, the potency of maslinic acid to alter ERK, CREB and Akt phosphorylation levels was slightly different between our previous and present results (30 vs. 3 mg·kg−1). This inconsistent finding can be attributed to the purity difference of maslinic acid among studies (present, >98%; previous, >95%). We do not have any clue to explain these different results. Further study should be conducted to clarify this issue. Meanwhile, several studies have indicated that the ERK–CREB and PI3K–Akt signalling pathways mediate increasing mBDNF levels in the hippocampus and, vice versa, through TrkB receptor activation (Nakai et al., 2014; Revest et al., 2014). BDNF plays an important role in memory consolidation (Kim, Kim, Park, Cai, et al., 2012; Radiske et al., 2017) and, especially, activity‐dependent BDNF secretion is essential for learning and memory in the hippocampus via maintaining LTP formation (Karpova, 2014). Therefore, we hypothesized that the activation of BDNF and its downstream signalling pathways after the administration of maslinic acid would explain its cognitive‐improving effects. We then measured the mBDNF levels in the hippocampus 1 hr after the administration of maslinic acid. However, maslinic acid did not affect the levels of mBDNF (Figure S1). It has been reported that the level of BDNF at 9 to 12 hr after the memory acquisition is essential for memory consolidation and enhancement (Kim et al., 2014; Kim, Kim, Park, Lee, et al., 2012). Thus, we investigated the temporal profiles of BDNF at 3, 6, 9 or 12 hr after the administration of maslinic acid in the hippocampus. Excitingly, maslinic acid significantly increased the levels of mBDNF at 9 and 12 hr after the administration, suggesting that the increasing mBDNF level in the hippocampus at 9 and 12 hr after the administration of maslinic acid plays a crucial role in memory consolidation and enhancement.
BDNF is a post‐translational processing molecule, that it is initially translated into its precursor form, proBDNF. After this translation, proBDNF is converted into the mature form, mBDNF, by some proteolytic process (Lessmann & Brigadski, 2009). Based on this information, we investigated whether the increased mBDNF protein level induced by maslinic acid is caused by an increase in mRNA expression or the post‐translation process. Therefore, we conducted q‐PCR to determine the temporal profiles of mRNA expression level of BDNF in the hippocampus. We found that the mRNA expression level of BDNF was elevated at 3 and 6 hr after the administration of maslinic acid and decreased in a time‐dependent manner. These results suggested that maslinic acid increases the transcription level of BDNF in early stage of memory acquisition. However, it is unclear whether maslinic acid activates the conversion process of mBDNF. In the CNS, the converting process of proBDNF to mBDNF is dominantly regulated by plasmin or plasminogen activator, tissue type (tPA; Melchor & Strickland, 2005). Extracellular tPA elicits its proteolytic function by converting proBDNF into mBDNF, which is essential for memory consolidation (Pang et al., 2004). It also inhibits Aβ protein accumulation through its serine protease activity (Melchor, Pawlak, Chen, & Strickland, 2003). In the present study, we observed that the mRNA expression levels of tPA were increased at 9 and 12 hr after the administration of maslinic acid, which is consistent with the findings of mBDNF levels at 9 and 12 hr. These findings on the correlation between the levels of tPA and mBDNF had not been reported elsewhere yet, within our knowledge. In addition, as shown in Figure 2, maslinic acid facilitated the LTP induction in hippocampal tissue. Taken together, the results suggested that maslinic acid would regulate the mRNA expression levels of BDNF and tPA at specific time points to enhance cognitive function.
Previously, we showed that oleanolic acid activates the TrkB receptor, resulting in the stimulation of ERK–CREB–BDNF signalling pathway (Jeon, Lee, et al., 2017). Although oleanolic acid does not have a direct binding affinity for the TrkB receptor, activation of TrkB receptor by oleanolic acid may be important for memory enhancement. Thus, we hypothesized that maslinic acid reverses cognitive deficits induced by cholinergic blockade through TrkB receptor activation, as observed in oleanolic acid. As expected, the specific inhibitor for TrkB receptor, ANA‐12, blocked the reversing effect of maslinic acid on scopolamine‐induced cognitive dysfunction. Thus, TrkB activation after the binding of mBDNF and, thereafter, the activation of ERK–CREB or PI3K–Akt would be a major pathway through which maslinic acid improves cognitive deficits. Additional detailed studies are warranted to determine the exact target of maslinic acid.
In conclusion, the present study demonstrates that maslinic acid has ameliorating effects of cognitive impairment induced by cholinergic blockade and these effects are likely facilitated by the increased level of mBDNF at specific time points through the enhanced (tPA) expression. We propose that maslinic acid would be a potential therapeutic agent for treating cognitive impairments induced by a cholinergic dysfunction, such as dementia or the mild state of AD. However, the present findings may not in general provide clinically useful outcomes in patients; therefore, further studies using other animal models of age‐related or AD‐related cognitive impairments are needed to support the present findings.
AUTHOR CONTRIBUTIONS
H.J.B. conducted the experiments and wrote the manuscript; Jihyun. K., Jaehoon. K.,N.G.,K.C. and S.Y.J. analyzed the behavioral data; M.C. performed the western blotting; H.K. and D.H.K. conducted electrophysiologic study; D.S.J. provided expert guidance; J.H.R. designed the experiments and wrote the manuscript; All authors, B.H.J., Jiyun. K., Jaehoon. K.,N.G.,M.C.,K.C.,S.Y.J.,H.K.,D.H.K.,D.S.J. and J.H.R. reviewed and approved the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1 Statistical analysis of Figures.
Figure S1 The expression levels of phospho‐GSK3β and mBDNF in the hippocampus after a single administration of maslinic acid (0.3, 1 or 3 mg/kg, p.o). The mice were sacrificed 1 h after the maslinic acid (0.3, 1 or 3 mg/kg, p.o) treatment. The immunoreactivity of P‐GSK3 β/GSK3β (A) and mBDNF/Gapdh (B) was normalized with control (Con) group. Data showed as means ± S.E.M (n = 5/group) (* P < 0.05, compared to the vehicle‐administered control groups; n.s, not significant)
Figure S2. The inhibitory effect of AChE activity in the brain after the administration of maslinic acid. Maslinic acid (0.3, 1 or 3 mg/kg, p.o) or donepezil (5 mg/kg, p.o) were treated 1 h before sacrifice. The AChE inhibition activity was represented. Data showed as means ± S.E.M (n = 6/group) (* P < 0.05, compared to the vehicle‐administered control groups).
ACKNOWLEDGEMENTS
This research was supported by the Mid‐Career Researcher Program and the Medical Research Program through an NRF grant funded by the Ministry of Education, Science and Technology (2018R1A2A2A05023165), the Medical Research Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT, South Korea (NRF‐2017R1A5A2014768),and the BB21 Plus Project in 2019. We thank Dr Chrislean Jun Botanas (College of Pharmacy, Sahmyook University, Republic of Korea) for constructive discussions on the manuscript and editorial input.
Bae HJ, Kim J, Kim J, et al. The effect of maslinic acid on cognitive dysfunction induced by cholinergic blockade in mice. Br J Pharmacol. 2020;177:3197–3209. 10.1111/bph.15042
REFERENCES
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Southan, C. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisman, H. (1975). Dissociation of disinhibitory effects of scopolamine: Strain and task factors. Pharmacology, Biochemistry, and Behavior, 3, 613–618. [DOI] [PubMed] [Google Scholar]
- Bajo, R. , Pusil, S. , Lopez, M. E. , Canuet, L. , Pereda, E. , Osipova, D. , … Pekkonen, E. (2015). Scopolamine effects on functional brain connectivity: A pharmacological model of Alzheimer's disease. Scientific Reports, 5, 9748 10.1038/srep09748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore, C. , Lee, V. M. , & Trojanowski, J. Q. (2007). Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nature Reviews. Neuroscience, 8, 663–672. [DOI] [PubMed] [Google Scholar]
- Bishop, N. A. , Lu, T. , & Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature, 464, 529–535. 10.1038/nature08983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charan, J. , & Kantharia, N. D. (2013). How to calculate sample size in animal studies? Journal of Pharmacology and Pharmacotherapeutics, 4, 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Garelick, M. G. , Wang, H. , Lil, V. , Athos, J. , & Storm, D. R. (2005). PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nature Neuroscience, 8, 925–931. 10.1038/nn1482 [DOI] [PubMed] [Google Scholar]
- Gao, L. B. , Yu, X. F. , Chen, Q. , & Zhou, D. (2016). Alzheimer's disease therapeutics: Current and future therapies. Minerva Medica, 107, 108–113. [PubMed] [Google Scholar]
- Gurovic, M. S. , Castro, M. J. , Richmond, V. , Faraoni, M. B. , Maier, M. S. , & Murray, A. P. (2010). Triterpenoids with acetylcholinesterase inhibition from Chuquiraga erinacea D. Don. subsp. erinacea (Asteraceae). Planta Medica, 76, 607–610. [DOI] [PubMed] [Google Scholar]
- Hampel, H. , Mesulam, M. M. , Cuello, A. C. , Farlow, M. R. , Giacobini, E. , Grossberg, G. T. , … Khachaturian, Z. S. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain, 141, 1917–1933. 10.1093/brain/awy132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvik, M. E. , & Kopp, R. (1967). An improved one‐trial passive avoidance learning situation. Psychological Reports, 21, 221–224. 10.2466/pr0.1967.21.1.221 [DOI] [PubMed] [Google Scholar]
- Jeon, S. J. , Kim, E. , Lee, J. S. , Oh, H. K. , Zhang, J. , Kwon, Y. , … Ryu, J. H. (2017). Maslinic acid ameliorates NMDA receptor blockade‐induced schizophrenia‐like behaviors in mice. Neuropharmacology, 126, 168–178. 10.1016/j.neuropharm.2017.09.014 [DOI] [PubMed] [Google Scholar]
- Jeon, S. J. , Lee, H. J. , Lee, H. E. , Park, S. J. , Gwon, Y. , Kim, H. , … Ryu, J. H. (2017). Oleanolic acid ameliorates cognitive dysfunction caused by cholinergic blockade via TrkB‐dependent BDNF signaling. Neuropharmacology, 113, 100–109. 10.1016/j.neuropharm.2016.07.029 [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Gao, H. , & Turdu, G. (2017). Traditional Chinese medicinal herbs as potential AChE inhibitors for anti‐Alzheimer's disease: A review. Bioorganic Chemistry, 75, 50–61. 10.1016/j.bioorg.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Jung, I. H. , Jang, S. E. , Joh, E. H. , Chung, J. , Han, M. J. , & Kim, D. H. (2012). Lancemaside A isolated from Codonopsis lanceolata and its metabolite echinocystic acid ameliorate scopolamine‐induced memory and learning deficits in mice. Phytomedicine, 20, 84–88. 10.1016/j.phymed.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Karpova, N. N. (2014). Role of BDNF epigenetics in activity‐dependent neuronal plasticity. Neuropharmacology, 76(Pt C), 709–718. [DOI] [PubMed] [Google Scholar]
- Kastellakis, G. , & Poirazi, P. (2019). Synaptic clustering and memory formation. Frontiers in Molecular Neuroscience, 12, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. H. , Kim, J. M. , Park, S. J. , Cai, M. , Liu, X. , Lee, S. , … Ryu, J. H. (2012). GABAA receptor blockade enhances memory consolidation by increasing hippocampal BDNF levels. Neuropsychopharmacology, 37, 422–433. 10.1038/npp.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. H. , Kim, J. M. , Park, S. J. , Lee, S. , Shin, C. Y. , Cheong, J. H. , & Ryu, J. H. (2012). Hippocampal extracellular signal‐regulated kinase signaling has a role in passive avoidance memory retrieval induced by GABAA receptor modulation in mice. Neuropsychopharmacology, 37, 1234–1244. 10.1038/npp.2011.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. H. , Kim, S. , Jeon, S. J. , Son, K. H. , Lee, S. , Yoon, B. H. , … Ryu, J. H. (2009). Tanshinone I enhances learning and memory, and ameliorates memory impairment in mice via the extracellular signal‐regulated kinase signalling pathway. British Journal of Pharmacology, 158, 1131–1142. 10.1111/j.1476-5381.2009.00378.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. H. , Lee, Y. , Lee, H. E. , Park, S. J. , Jeon, S. J. , Jeon, S. J. , … Ryu, J. H. (2014). Oroxylin A enhances memory consolidation through the brain‐derived neurotrophic factor in mice. Brain Research Bulletin, 108, 67–73. 10.1016/j.brainresbull.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Kim, J. , Kim, J. , Huang, Z. , Goo, N. , Bae, H. J. , Jeong, Y. , … Ryu, J. H. (2019). Theracurmin ameliorates cognitive dysfunctions in 5XFAD mice by improving synaptic function and mitigating oxidative stress. Biomol Ther (Seoul), 27, 327–335. 10.4062/biomolther.2019.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhina, V. , Arey, R. N. , Kaletsky, R. , Kauffman, A. , Stein, G. , Keyes, W. , … Murphy, C. T. (2015). Genome‐wide functional analysis of CREB/long‐term memory‐dependent transcription reveals distinct basal and memory gene expression programs. Neuron, 85, 330–345. 10.1016/j.neuron.2014.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann, V. , & Brigadski, T. (2009). Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neuroscience Research, 65, 11–22. 10.1016/j.neures.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Li, F. , Li, Q. , Shi, X. , & Guo, Y. (2017). Maslinic acid inhibits impairment of endothelial functions induced by high glucose in HAEC cells through improving insulin signaling and oxidative stress. Biomedicine & Pharmacotherapy, 95, 904–913. [DOI] [PubMed] [Google Scholar]
- Liao, Y. , Bae, H. J. , Park, J. H. , Zhang, J. , Koo, B. , Lim, M. K. , … Ryu, J. H. (2019). Aster glehni extract ameliorates scopolamine‐induced cognitive impairment in mice. Journal of Medicinal Food, 22, 685–695. 10.1089/jmf.2018.4302 [DOI] [PubMed] [Google Scholar]
- Liao, Y. , Bae, H. J. , Zhang, J. , Kwon, Y. , Koo, B. , Jung, I. H. , … Ryu, J. H. (2019). The ameliorating effects of bee pollen on scopolamine‐induced cognitive impairment in mice. Biological & Pharmaceutical Bulletin, 42, 379–388. 10.1248/bpb.b18-00552 [DOI] [PubMed] [Google Scholar]
- Liu, Y. L. , Kong, C. Y. , Song, P. , Zhou, H. , Zhao, X. S. , & Tang, Q. Z. (2018). Maslinic acid protects against pressure overload‐induced cardiac hypertrophy in mice. Journal of Pharmacological Sciences, 138, 116–122. 10.1016/j.jphs.2018.08.014 [DOI] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the method. Methods, 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lozano‐Mena, G. , Sanchez‐Gonzalez, M. , Juan, M. E. , & Planas, J. M. (2014). Maslinic acid, a natural phytoalexin‐type triterpene from olives—A promising nutraceutical? Molecules, 19, 11538–11559. 10.3390/molecules190811538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega, K. G. , Sigurdson, W. , Ovod, V. , Munsell, L. , Kasten, T. , Morris, J. C. , … Bateman, R. J. (2010). Decreased clearance of CNS β‐amyloid in Alzheimer's disease. Science, 330, 1774 10.1126/science.1197623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor, J. P. , Pawlak, R. , Chen, Z. , & Strickland, S. (2003). The possible role of tissue‐type plasminogen activator (tPA) and tPA blockers in the pathogenesis and treatment of Alzheimer's disease. Journal of Molecular Neuroscience, 20, 287–289. 10.1385/JMN:20:3:287 [DOI] [PubMed] [Google Scholar]
- Melchor, J. P. , & Strickland, S. (2005). Tissue plasminogen activator in central nervous system physiology and pathology. Thrombosis and Haemostasis, 93, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno, M. , Yamada, K. , Takei, N. , Tran, M. H. , He, J. , Nakajima, A. , … Nabeshima, T. (2003). Phosphatidylinositol 3‐kinase: A molecule mediating BDNF‐dependent spatial memory formation. Molecular Psychiatry, 8, 217–224. 10.1038/sj.mp.4001215 [DOI] [PubMed] [Google Scholar]
- Montilla, M. P. , Agil, A. , Navarro, M. C. , Jimenez, M. I. , Garcia‐Granados, A. , Parra, A. , & Cabo, M. M. (2003). Antioxidant activity of maslinic acid, a triterpene derivative obtained from Olea europaea . Planta Medica, 69, 472–474. 10.1055/s-2003-39698 [DOI] [PubMed] [Google Scholar]
- Morris, R. (1984). Developments of a water‐maze procedure for studying spatial learning in the rat. Journal of Neuroscience Methods, 11, 47–60. 10.1016/0165-0270(84)90007-4 [DOI] [PubMed] [Google Scholar]
- Nakai, T. , Nagai, T. , Tanaka, M. , Itoh, N. , Asai, N. , Enomoto, A. , … Yamada, K. (2014). Girdin phosphorylation is crucial for synaptic plasticity and memory: A potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. The Journal of Neuroscience, 34, 14995–15008. 10.1523/JNEUROSCI.2228-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, P. T. , Teng, H. K. , Zaitsev, E. , Woo, N. T. , Sakata, K. , Zhen, S. , … Lu, B. (2004). Cleavage of proBDNF by tPA/plasmin is essential for long‐term hippocampal plasticity. Science, 306, 487–491. 10.1126/science.1100135 [DOI] [PubMed] [Google Scholar]
- Park, S. J. , Ahn, Y. J. , Oh, S. R. , Lee, Y. , Kwon, G. , Woo, H. , … Ryu, J. H. (2014). Amyrin attenuates scopolamine‐induced cognitive impairment in mice. Biological & Pharmaceutical Bulletin, 37, 1207–1213. 10.1248/bpb.b14-00113 [DOI] [PubMed] [Google Scholar]
- Park, S. J. , Kim, D. H. , Jung, J. M. , Kim, J. M. , Cai, M. , Liu, X. , … Ryu, J. H. (2012). The ameliorating effects of stigmasterol on scopolamine‐induced memory impairments in mice. European Journal of Pharmacology, 676, 64–70. 10.1016/j.ejphar.2011.11.050 [DOI] [PubMed] [Google Scholar]
- Park, S. J. , Lee, J. Y. , Kim, S. J. , Choi, S. Y. , Yune, T. Y. , & Ryu, J. H. (2015). Toll‐like receptor‐2 deficiency induces schizophrenia‐like behaviors in mice. Scientific Reports, 5, 8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, Y. , Huang, M. , Guan, T. , Chen, L. , Cao, L. , Han, X. J. , … Sun, H. (2015). Maslinic acid promotes synaptogenesis and axon growth via Akt/GSK‐3β activation in cerebral ischemia model. European Journal of Pharmacology, 764, 298–305. 10.1016/j.ejphar.2015.07.028 [DOI] [PubMed] [Google Scholar]
- Radiske, A. , Rossato, J. I. , Gonzalez, M. C. , Kohler, C. A. , Bevilaqua, L. R. , & Cammarota, M. (2017). BDNF controls object recognition memory reconsolidation. Neurobiology of Learning and Memory, 142, 79–84. [DOI] [PubMed] [Google Scholar]
- Recanatini, M. , & Valenti, P. (2004). Acetylcholinesterase inhibitors as a starting point towards improved Alzheimer's disease therapeutics. Current Pharmaceutical Design, 10, 3157–3166. [DOI] [PubMed] [Google Scholar]
- Revest, J. M. , Le Roux, A. , Roullot‐Lacarriere, V. , Kaouane, N. , Vallee, M. , Kasanetz, F. , … Piazza, P. V. (2014). BDNF–TrkB signaling through Erk1/2 MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Molecular Psychiatry, 19, 1001–1009. 10.1038/mp.2013.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanez‐Larkin, G. R. , & Knutson, B. (2015). Decision making in the ageing brain: Changes in affective and motivational circuits. Nature Reviews. Neuroscience, 16, 278–289. 10.1038/nrn3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter, M. , Bodewitz, G. , & Stephens, D. N. (1988). Attenuation of scopolamine‐induced impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and agonist β‐carbolines. Psychopharmacology, 94, 491–495. 10.1007/bf00212843 [DOI] [PubMed] [Google Scholar]
- Schwarz, S. , Loesche, A. , Lucas, S. D. , Sommerwerk, S. , Serbian, I. , Siewert, B. , … Csuk, R. (2015). Converting maslinic acid into an effective inhibitor of acylcholinesterases. European Journal of Medicinal Chemistry, 103, 438–445. 10.1016/j.ejmech.2015.09.007 [DOI] [PubMed] [Google Scholar]
- Silva, A. J. , Kogan, J. H. , Frankland, P. W. , & Kida, S. (1998). CREB and memory. Annual Review of Neuroscience, 21, 127–148. [DOI] [PubMed] [Google Scholar]
- Talesa, V. N. (2001). Acetylcholinesterase in Alzheimer's disease. Mechanisms of Ageing and Development, 122, 1961–1969. 10.1016/s0047-6374(01)00309-8 [DOI] [PubMed] [Google Scholar]
- Tang, K. S. (2019). The cellular and molecular processes associated with scopolamine‐induced memory deficit: A model of Alzheimer's biomarkers. Life Sciences, 233, 116695. [DOI] [PubMed] [Google Scholar]
- Towbin, H. , Staehelin, T. , & Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America, 76, 4350–4354. 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. H. , Mong, M. C. , Yang, Y. C. , & Yin, M. C. (2018). Asiatic acid and maslinic acid attenuated kainic acid‐induced seizure through decreasing hippocampal inflammatory and oxidative stress. Epilepsy Research, 139, 28–34. 10.1016/j.eplepsyres.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Williams, P. , Sorribas, A. , & Howes, M. J. (2011). Natural products as a source of Alzheimer's drug leads. Natural Product Reports, 28, 48–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. H. , Han, S. J. , Ryu, J. H. , Jang, I. S. , & Kim, D. H. (2009). Ginsenoside Rh2 ameliorates scopolamine‐induced learning deficit in mice. Biological & Pharmaceutical Bulletin, 32, 1710–1715. 10.1248/bpb.32.1710 [DOI] [PubMed] [Google Scholar]
- Yang, Y. W. , Tsai, C. W. , Mong, M. C. , & Yin, M. C. (2015). Maslinic acid protected PC12 cells differentiated by nerve growth factor against β‐amyloid‐induced apoptosis. Journal of Agricultural and Food Chemistry, 63, 10243–10249. 10.1021/acs.jafc.5b04156 [DOI] [PubMed] [Google Scholar]
- Yi, J. H. , Baek, S. J. , Heo, S. , Park, H. J. , Kwon, H. , Lee, S. , … Kim, D. H. (2018). Direct pharmacological Akt activation rescues Alzheimer's disease like memory impairments and aberrant synaptic plasticity. Neuropharmacology, 128, 282–292. 10.1016/j.neuropharm.2017.10.028 [DOI] [PubMed] [Google Scholar]
- Yi, J. H. , Zhang, J. , Ko, S. Y. , Kwon, H. , Jeon, S. J. , Park, S. J. , … Ryu, J. H. (2018). Fluoxetine inhibits natural decay of long‐term memory via Akt/GSK‐3β signaling. Molecular Neurobiology, 55, 7453–7462. 10.1007/s12035-018-0919-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Statistical analysis of Figures.
Figure S1 The expression levels of phospho‐GSK3β and mBDNF in the hippocampus after a single administration of maslinic acid (0.3, 1 or 3 mg/kg, p.o). The mice were sacrificed 1 h after the maslinic acid (0.3, 1 or 3 mg/kg, p.o) treatment. The immunoreactivity of P‐GSK3 β/GSK3β (A) and mBDNF/Gapdh (B) was normalized with control (Con) group. Data showed as means ± S.E.M (n = 5/group) (* P < 0.05, compared to the vehicle‐administered control groups; n.s, not significant)
Figure S2. The inhibitory effect of AChE activity in the brain after the administration of maslinic acid. Maslinic acid (0.3, 1 or 3 mg/kg, p.o) or donepezil (5 mg/kg, p.o) were treated 1 h before sacrifice. The AChE inhibition activity was represented. Data showed as means ± S.E.M (n = 6/group) (* P < 0.05, compared to the vehicle‐administered control groups).