

Abstract
Background and Purpose
Acute intermittent porphyria (AIP) results from haplo‐insufficiency of the porphobilinogen deaminase (PBGD) gene encoding the third enzyme in the haem biosynthesis pathway. As liver is the main organ of pathology for AIP, emerging therapies that restore enzyme hepatic levels are appealing. The objective of this work was to develop a mechanistic‐based computational framework to describe the effects of novel PBGD mRNA therapy on the accumulation of neurotoxic haem precursors in small and large animal models.
Experimental Approach
Liver PBGD activity data and/or 24‐hr urinary haem precursors were obtained from genetic AIP mice and wild‐type mice, rats, rabbits, and macaques. To mimic acute attacks, porphyrogenic drugs were administered over one or multiple challenges, and animals were used as controls or treated with different PBGD mRNA products. Available experimental data were sequentially used to build and validate a semi‐mechanistic mathematical model using non‐linear mixed‐effects approach.
Key Results
The developed framework accounts for the different biological processes involved (i.e., mRNA sequence, release from lipid nanoparticle and degradation, mRNA translation, increased PBGD activity in liver, and haem precursor metabolism) in a simplified mechanistic fashion. The model, validated using external data, shows robustness in the extrapolation of PBGD activity data in rat, rabbit, and non‐human primate species.
Conclusion and Implications
This quantitative framework provides a valuable tool to compare PBGD mRNA drug products during early preclinical stages, optimize the amount of experimental data required, and project results to humans, thus supporting drug development and clinical dose and dosing regimen selection.
Abbreviations
- AIA
2‐allyl‐2‐isopropylacetamide
- AIP
acute intermittent porphyria
- ALA
aminolevulinic acid
- ALAS1
5‐aminolaevulinic acid synthase 1
- Ce,Pheno
concentrations of phenobarbital in effect site
- Circi
virtual circulating levels of haem precursors
- hPBGD
human PBGD
- KDEG
first‐order rate constant of PBGDL degradation
- KE
first‐order rate constant of mRNA elimination
- Kex,i
urinary excretion rate constant
- KLP
first‐order rate constant of distribution between liver and peripheral compartments
- KPL
first‐order rate constant of distribution between peripheral and liver compartments
- KREL
first‐order rate constant of mRNA release
- KSYN
first‐order rate constant of PBGDL synthesis
- LNPs
lipid nanoparticles
- mRNAENC
mRNA encapsulated
- NHPs
non‐human primates
- PBG
porphobilinogen
- PBGD
porphobilinogen deaminase
- PBGDL
liver PBGD activity
- PBGDL(0)
PBGDL at baseline
- SEQ
mRNA sequence
- tPOR
total porphyrins
- Ui
24‐hr amount of urinary precursors
- URO
uroporphyrin
- VPC
visual predictive check
- WT
wild‐type
- θPheno
phenobarbital effect parameter
- θmRNA
mRNA effect parameter
What is already known
PBGD mRNA‐based therapy can represent a potential therapy for unmet needs in acute intermittent porphyria.
Modelling and simulation are useful tools to support drug development and clinical use.
What this study adds
A modelling framework characterizing kinetic and pharmacological data across animal species for PBGD mRNA‐based therapies.
A tool to compare across novel therapies and optimize preclinical experiments supporting early drug development.
What is the clinical significance
A translational framework to project preclinical results to clinical situations for novel PBGD mRNA‐based therapies.
1. INTRODUCTION
Porphyrias constitute a group of rare metabolic disorders affecting the haem biosynthesis pathway. Among them, acute intermittent porphyria (AIP) represents the most frequent porphyria in Europe, with a prevalence of 5.9 per 1,000,000 inhabitants (Elder, Harper, Badminton, Sandberg, & Deybach, 2013). AIP is caused by mutations in the porphobilinogen deaminase (PBGD) gene resulting in a marked loss of activity of PGBD, the third enzyme of the haem biosynthesis pathway (Chen et al., 2016).
Haem production in the liver is controlled by the 5‐aminolaevulinic acid synthase 1 (ALAS1) enzyme, the first and rate‐limiting enzyme of the pathway (Karim et al., 2015; Puy, Gouya, & Deybach, 2010). Porphyria attacks result from external or internal factors that increase the demand for haem or strongly induce ALAS1 in the liver in individuals carrying an inherited deficiency of PBGD. The consequence is a marked overproduction and accumulation of aminolevulinic acid (ALA) and porphobilinogen (PBG), the intermediate products between both enzymes.
The pathogenesis of acute attacks is still not well understood, but it is believed that ALA and/or their metabolites overproduced by the liver are neurotoxic (Anderson et al., 2005; Stein, Badminton, & Rees, 2017). AIP appears in the form of acute attacks, typically consisting of severe abdominal pain, gastrointestinal manifestations (such as nausea, vomiting, or constipation), and in some cases accompanied by psychological disorders (anxiety depression, confusion, or seizure, among others), sensory loss, or motor neuropathy, which can be potentially life threatening (Fontanellas, Ávila, Anderson, & Deybach, 2019; Karim et al., 2015; Puy et al., 2010).
Treatment during acute attacks is mainly symptomatic, including in some cases the administration of human haemin, which replenishes the regulatory haem pool in hepatocytes and down‐regulates the over‐expression of ALAS1 in the liver by negative feedback mechanism. Preventive treatment largely relies on patient education to identify and avoid precipitating factors. The prophylactic use of haemin has also been reported in patients with recurrent attacks (Anderson et al., 2005; Balwani & Desnick, 2012). However, this entails frequent intravenous administration that can lead to iron overload and contribute to hepatic damage and fibrosis (Marsden et al., 2015).
Different emerging therapies targeting the liver include RNA interference, targeting ALAS1 mRNA (Sardh et al., 2019) and gene/mRNA therapy to express functional human PBGD (hPBGD) enzyme in the liver (Fontanellas, Ávila, & Berraondo, 2016) and are currently under development. The administration of hPBGD mRNA formulated into biodegradable lipid nanoparticles (LNPs), as a vehicle for drug delivery, was recently shown to be well tolerated and induce the expression of functional hPBGD protein in hepatocytes across small and large animal species, as well as normalize levels of urinary haem precursors in a transgenic AIP mouse model (Jiang et al., 2018). Therefore, hPBGD mRNA therapy may represent a therapeutic strategy for AIP, as an alternative to rAAV‐gene therapy (D'Avola et al., 2016).
Development of treatments for rare diseases such as AIP are challenging due to limited knowledge available, interindividual heterogeneity of symptoms, and the difficulty of conducting epidemiological studies involving large numbers of patients to understand and explore the disease (Dunoyer, 2011; Griggs et al., 2009). In this context, computational modelling and simulation can provide relevant support to integrate and maximize existing knowledge and optimize drug development.
Indeed, the Food and Drug Administration highlighted the opportunities offered by the application of mathematics, statistics, and computational analysis to biological data in order to construct robust and quantitative models as part of its critical path initiative (US Department of Health and Food Administration, 2007; Woodcock & Woosley, 2008). These models constitute useful tools to integrate existing information originated from multiple sources, into a quantitative framework allowing us to (a) improve the understanding of the interplay and temporal evolution of the components of a system, (b) explore the impact of different therapeutic strategies or dosing regimens, and (c) project preclinical response outcomes to clinical situations using in silico simulation techniques, thus supporting and guiding drug development.
Unfortunately, these modelling approaches are still scarce in the field of gene therapy (Mac Gabhann, Annex, & Popel, 2010; Parra‐Guillén, González‐Aseguinolaza, Berraondo, & Trocóniz, 2010) and rare diseases like AIP (Siegert & Holt, 2008; Vera‐Yunca et al., 2019), where most of the efforts focus on characterizing the non‐clinical or clinical pharmacokinetic profiles but are lacking in predictions of exposure–response.
The objectives of this work were to expand upon existing preclinical proof‐of‐concept studies for PBGD mRNA therapy as a treatment for AIP by (a) developing a comprehensive mechanistic computational model capable of integrating data on temporal liver PBGD activity (PBGDL) and urinary levels of haem precursors, collected across different preclinical species for the novel hPBGD mRNA compounds mentioned above and (b) extrapolating model predictions to the human condition in order to support and optimize clinical development. This mechanistic framework will allow the integration of new data gathered during the course of a development program to increase our quantitative understanding of the biological system, model robustness, and model applicability.
2. METHODS
2.1. Data and experimental designs
The data used to develop the mechanism‐based computational model were obtained from experiments conducted at the Center for Applied Medical Research, University of Navarra, as described in Jiang et al. (2018), in which either the (a) PBGD activity in the liver or the (b) amount of haem precursors (ALA, PBG, and total porphyrins [tPOR]) excreted in urine was measured in control and treated animals.
2.1.1. Experimental animals
All animal care and experimental protocols were approved by the Ethics Committee of the University of Navarra (CEEA050‐16 and CEEA142‐16) and the Institute of Public Health of Navarra (2016/292147 and 2017/50445) according to European Council Guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
Experiments were performed across different animal species which included AIP or wild‐type (WT) male C57BL mice (RRID:MGI:5657800), WT female Sprague Dawley rats (RRID:RGD_737903), WT female New Zealand rabbits, or WT female cynomolgus macaque (Macaca fascicularis). AIP mice exhibit 30% of normal PBGD activity levels and were generated by crossbreeding T1 (C57BL/6 Pbgdtm1(neo)Uam) and T2 (C57BL/6 Pbgdtm2(neo)Uam) mouse strains as previously described (Lindberg et al., 1996). Acute attacks are much more common in women than in men, potentially due to the frequent occurrence of precipitating factors such as menstrual cycle hormones. All the studies described in this work were carried out in females, except for mice. For unknown reasons, the induction of hepatic ALAS1 after phenobarbital challenge is greater in male mice than in females. Thus, the accumulation of precursors is higher in males than in females, which makes it possible to better discern the effect of small hepatic PBGD activity increases on the accumulation of porphyrin precursors.
Open‐label studies were designed to generate groups of equal size using randomization. To calculate the sample size, we used the computer application: http://www.biomath.info/power/ttest.htm. Assays could not be blind as administration, sample collection, and processing were carried out by the same researchers.
2.1.2. hPBGD mRNA drug product
hPBGD mRNA drug products consisted of different mRNA sequences encoding for the hPBGD enzyme encapsulated in different LNPs (Table S1). In addition to hPBGD mRNA, luciferase mRNA was also encapsulated in the different formulations to be used as negative controls. A detailed description of the PBGD mRNA production and formulation into LNP can be found in Jiang et al. (2018) and Sabnis et al. (2018).
All mRNA doses were administered intravenously. In the case of mice (weight range from 20 to 25 g), dose levels ranging from 0.05 to 2.5 mg·kg−1 were administered as a bolus (12 to 15 ml·kg−1) through the tail vein (see next section for detailed information). For the other species used, a dose of 0.5 mg·kg−1 was administered in a volume of 5 ml·kg−1. Rats (approximate weight of 250 g) were injected through the tail vein at a rate of 1 ml·min−1. Rabbits (3 kg of body weight) received the dose at a rate of 2 ml·min−1 through the ear vein. Finally, non‐human primates (NHPs, approximate weight of 3.5 kg) were injected, under anaesthesia, through the leg vein at a rate of 0.28 ml·min−1.
2.1.3. Studies evaluating hepatic PBGD activity
hPBGD mRNA drug products were administered as an intravenous single dose of 0.5 mg·kg−1. Animals in control groups received either no intervention or single intravenous injections of PBS or luciferase mRNA. No acute attack was induced in any of these animals.
Groups of three to five animals were killed at different times after administration (up to 10 days in the experiments performed in mice or 1 day after dosing in the other animal species; see Table S1 for additional information). Animals were perfused with 0.9% NaCl to eliminate circulating blood, and livers were removed. In the case of WT cyno, up to two liver biopsies (baseline and 24 hr post‐injection) were obtained. Mice and rats were anaesthetized with a combination of ketamine (50 mg·kg−1) and xylazine (5 mg·kg−1) prior to killing. Similarly, rabbits and cynos were anaesthetized with a combination of buprenorphine (0.01 mg·kg−1), ketamine (25 mg·kg−1), and diazepam (5 mg·kg−1) prior to intervention.
A total of 166 AIP mice, 7 rats, 8 rabbits, and 6 NHPs were used in these experiments. Liver samples of each animal were placed in cold 0.9% NaCl solution for immediate enzyme determination. PBGD activity in tissue homogenates was quantified measuring the conversion of PBG to uroporphyrin (URO) by spectrophotometry as previously described (Anderson, Reddy, Anderson, & Desnick, 1981). Results were expressed in terms of picomole of URO per milligram of protein per hour (Jiang et al., 2018).
2.1.4. Studies evaluating the accumulation of precursors in urine
Changes in urinary excretion of precursors, a disease marker elevated during acute attacks, were measured across one to three porphyrogenic challenges. Each challenge comprised three periods: baseline, induction of the acute attack and recovery.
To induce acute attacks in AIP mice, four to five increasing intraperitoneal (i.p.) doses of phenobarbital (75, 80, 85, and 90 mg·kg−1; i.p.) were administered in intervals of 24 hr (90 mg·kg−1 was the dose in case of the fifth administration of phenobarbital). In the case of WT animals, a daily subcutaneous morning administration of 300 or 350 mg·kg−1 of 2‐allyl‐2‐isopropylacetamide (AIA) alone or in combination with a daily i.p. evening administration of 200 mg·kg−1 of rifampicin were given during two to five consecutive days. During the induction period, animals receiving no intervention were used as controls.
In addition to the porphyrogenic drugs, induced animals received either the hPBGD mRNA drug product or 0.5 mg·kg−1 of luciferase mRNA with the same administration protocol (negative control). hPBGD mRNA drug products were administered according to different schedules across species. AIP mice were given a single dose of hPBGD mRNA (0.5 mg·kg−1, i.v.) together with the second (preventive protocol) or fourth dose of phenobarbital (treatment protocol) or multi‐dose of 0.05, 0.2, or 2.5 mg·kg−1 of hPBGD mRNA intravenously, one injection at each of the three challenges, together with the second dose of phenobarbital. WT mice received a single intravenous dose of 0.5 mg·kg−1 before the first or third AIA dose during the first challenge. Rats received the 0.5 mg·kg−1, i.v., dose together with the second or third dose of AIA/rifampicin also in the first challenge. Lastly, rabbits were treated either with a single intravenous dose of 0.5 mg·kg−1 administered before the first AIA dose (preventive protocol) or with the third dose of AIA/rifampicin (treatment protocol), or three intravenous doses, one per challenge administered together with the third dose of AIA/rifampicin (recurrent treatment protocol). A schematic illustration of the experimental treatment protocols across the different species can be found in Table S1.
A total of 201 animals (104 AIP mice, 16 WT mice, 23 WT rats, and 58 WT rabbits) were used in these experiments. Animals were housed in individual metabolic cages to collect the 24‐hr urine samples at baseline and after each day of induction. Subsequently, urine samples were frozen at −20°C until they were processed (no more than 1 week after collection). Care was taken to protect the urine samples from light.
Porphyrin precursors (ALA and PBG) were quantified using a quantitative ion exchange column method (BioSystems, Barcelona, Spain) and normalized to urine creatinine values. Briefly, the urine sample was passed through a column containing a buffered anionic exchange resin (which retains PBG) and subsequently through a column with a buffered cationic exchange resin (which retains ALA). Then ALA and PBG were eluted and spectrophotometrically quantified in an Ultrospec 3000 spectrophotometer (LS50B, PerkinElmer, Spain) by absorbance at 555 nm of the Ehrlich's reaction product. ALA and PBG measurements were converted to picomole per milligram of creatinine units using their molecular weights (131 and 226 g·mol−1, respectively). Urinary excretion of total porphyrins (tPOR) was analysed by the spectrofluorometric method (Westerlund, Pudek, & Schreiber, 1988) and normalized to urine creatinine values.
2.2. Data analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Data from multiple experiments were simultaneously analysed using non‐linear mixed‐effects approach, ensuring in all cases a group size of at least n = 5. Declared group size corresponds to the independent values (i.e., not replicates) used in the analysis. No available measurements were excluded from the analysis.
Figure 1 provides an overview of the structure of the computational model linking the time profiles of hepatic PBGD activity elicited by the mRNA treatments and their relationship with the 24‐hr urine excreted amounts of haem precursors established using the AIP mouse data. This figure also shows the translational approach followed, which is based on the established model and the data obtained in other species including humans.
FIGURE 1.
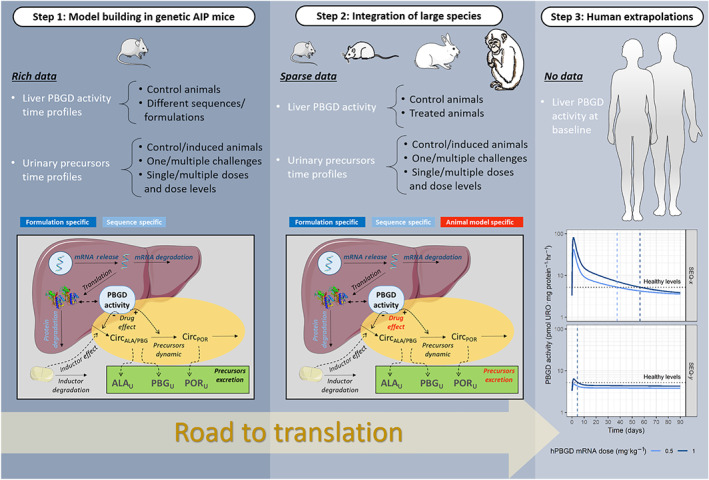
Translational framework illustrating data availability and modelling strategy
In the following, a model structure close to the one finally selected is described for clarity. In Section 3, the outcome of different model assumptions will also be presented justifying the selection of the final model.
2.2.1. Mechanistic‐based computational model
The full computational structure consisted of (a) a model describing the time course of PBGDL in response to mRNA administration, (b) a disease/induction model accounting for the impact of induced AIP attacks on the ALA, PBG, and tPOR excretion, and finally, (c) a model for the effects of PBGDL to normalize urine excretion of haem precursors. The three mentioned models were developed sequentially.
-
a
Kinetic PBGDL model
A total of 159 PBGD activity observations from five different mRNA sequences (SEQ‐1 corresponds to mRNA construct 8 in Jiang et al., 2018) in three different formulations (LNP‐A, LNP‐B, and LNP‐C) were measured in the liver of AIP mice and sequentially incorporated into model development.
Three main mechanisms were considered to describe the PBGDL profiles as a response to the administration of the mRNA drug products.
The first one is related to the distribution and mRNA release from its LNP encapsulated form (mRNAENC) into the liver. LNPs are internalized into the hepatocytes through a process mediated by an interaction between the LDL receptor and apolipoprotein E and other opsonins. Once invaginated, LNPs are optimized to provide high delivery rates and better endosomal escape once invaginated (Sabnis et al., 2018). Data of LNP or mRNA levels in the liver were not available, and thus, liver distribution and mRNA release from its encapsulated form (mRNAENC) were model as a single process (Equation 1). Once released, free mRNA can be degraded as shown in Equation 2. d/dt accounts for the rate of change of the different entities in the model (mRNAENC and mRNA) and K REL and K E represent the first‐order rate constants of mRNA release and degradation, respectively.
| (1) |
| (2) |
Because mRNA measurements were not available, a common estimate was also assumed for both parameters. The initial conditions of the described system are the dose level (expressed as pmol kg‐1) and 0 for mRNAENC and mRNA, respectively.
The third mechanism describes the way mRNA is translated to the encoded PBGD protein and is represented by Equation 3, assuming proportionality between PBGD protein levels and PBGDL.
In absence of mRNA drug product, the endogenous levels of PBGDL [PBGDL(0)]—set to 3 pmol·mg protein−1·hr−1 (value derived from control non‐injected AIP mice)—are the result of the balance between synthesis and degradation processes characterized by the zero and first‐order rate constants K SYN and K DEG, respectively. K SYN parameter is thus derived from Equation 3 to ensure system homeostasis at baseline (absence of mRNA).
Released mRNA increases the levels of PBGDL through θ mRNA, a parameter accounting for the relative increase on PBGD activity synthesis triggered per unit of free mRNA in liver.
| (3) |
In the PBGDL model described by Equations 1–3, differences between LNP formulations were investigated in the parameter K REL, whereas differences and mRNA sequences were explored in the K DEG and θ mRNA parameters.
-
b
Disease/induction model
A total of 991 observations from 33 AIP mice were used to develop the model describing the dynamics of haem precursors (i.e., ALA, PBG, and tPOR) in the presence or absence of phenobarbital induction.
The rate of change of 24‐hr urine excretion (Ui) of the three haem precursors is mediated by the excretion constant (K ex,i) and the amounts of non‐measured circulating levels of the precursors (Circi), as represented in Equation 4, where the parameter γ i modulates the relationship between Circi levels and excretion rates (i = ALA, PBG, or tPOR).
| (4) |
Expression 4 implies that the dynamics of the urinary excretion of the biomarkers are independent from each other. The dependency between the three components of the haem pathway has been kept at the circulating levels by assuming that circulating ALA is the precursor of circulating PBG and the latter of circulating tPOR, as indicated in Equations 5 and 6 as follows:
| (5) |
| (6) |
Circulating levels were not measured, so their corresponding values at baseline were set arbitrarily to 1. K ENZ,1–3 represents first‐order enzymatic rate constant of reaction. Finally, it was observed that the time profiles of 24‐hr excreted amounts of ALA and PBG evolved in parallel; therefore, they were described with just a single expression.
The AIP attacks increase ALA and PBG as the result of an effect mediated by the predicted concentrations of phenobarbital in the target organ (C e,Pheno) on K ENZ,1 through the θ Pheno parameter. Supporting Information describes the pharmacokinetic and effect site models used to predict C e,Pheno.
-
c
PBGDL effects model
A total of 576 observations from 12 AIP mice receiving the mRNA sequence 1 (SEQ‐1) formulated as LNP‐A (n = 4) or as LNP‐B (n = 8) were used to characterize the effect of encoded PBGD enzyme on the accumulation of precursors after phenobarbital induction. Data from the rest of the treatment groups (1,732 observations and 59 AIP mice) were used to validate the model.
The models for the time profiles of PBGDL and phenobarbital‐induced AIP attacks were linked to characterize the capability of PBGDL to restore the functionality of the haem pathway. When PBGD activity levels were not available from any species, the mean K DEG estimate across formulations was used.
The increase in PBGDL with respect to the endogenous activity (ΔPBGDL) caused by mRNA administration resulted in a decrease in the induced effects of phenobarbital on K ENZ,1, and enhanced K ENZ,2, characterized by the parameters ΔPBGDL,50, γ PBGD, and θ PBGD as shown in Equations 7 and 8.
| (7) |
| (8) |
2.3. Translational framework
2.3.1. Integration of data from WT animal species
Kinetic PBGDL model
The time profiles of PBGDL in WT species were described using the model structure developed for AIP mice (Equations 1–3). Model parameters estimated in AIP mice were assumed to represent also the corresponding processes across species with the exception of (a) the endogenous PBGDL that was adjusted using the mean value obtained from the different species in the absence of treatment and (b) K DEG for the mRNA sequence 6, not available in AIP mice, and estimated using PBGDL data at Day 1 from rats, rabbits, and NHPs.
Disease/induction model
The model structure used for AIP mice and described by Equations 4–6 was maintained, and its model parameters were estimated for each of the different species. The effect of the inducer was modelled as in the case of AIP mice using the predicted concentrations of AIA and rifampicin in the target organ (C e,AIA/rifampicin) and estimating the drug effect parameter θ AIA/rifampicin. Supporting Information describes the kinetic–pharmacodynamic model used to predict C e,AIA/rifampicin.
A total of 775 observations from 61 WT animals—4 mice, 16 rats, and 41 rabbits—were used during this modelling process.
PBGDL effects model
As in the case of the AIP mice, the PBGDL and disease/induction models were linked to characterize the capability of PBGDL to restore the functionality of the haem pathway in the different WT animal species.
Similarly, the structure developed in AIP mice was carried forward to the WT species through Equations 7 and 8. Parameters ΔPBGD50, γ PBGD, and θ PBGD were allowed to be equal or different between AIP and WT animals. To normalize PBGD levels across species, relative increments of PBGD over baseline [(PBGDL − PBGDL_0)/PBGDL_0] were used as triggers of the PBGD effects.
A total of 504 observations (222, 222, and 60 ALA, PBG, and tPOR observations, respectively) from 12 WT C57BL mice, 7 WT Sprague Dawley rats, and 20 WT New Zealand rabbits were used to identify the PBGD activity effects in WT animals after the administration of SEQ‐6 or SEQ‐7 (LNP‐B and LNP‐C formulations).
2.3.2. Extrapolation to humans
PBGD activity levels in the liver were simulated after single or multiple administrations following every 2 weeks (Q2W), every 4 weeks (Q4W), or every 8 weeks (Q8W) intravenous dosing regimens for different mRNA drug products. To do so, the final developed AIP mouse model structure (Equations 1–3) together with their final parameter estimates (K REL, K DEG, and distribution rate constants [K PL, K LP] and θ SLP) was used, and the baseline PBGDL parameter was set to the measured levels of PBGDL in porphyric or healthy patients in the absence of treatment obtained from Jiang et al. (2018). Note that the synthesis rate constant of PBGD activity is derived using baseline PBGD activity levels and, thus, also varies across species and AIP patients.
2.4. Model selection and evaluation
The time profiles of PBGDL and 24‐hr urinary excretion profiles of the haem precursors were analysed using the naïve pool or the population approach depending on whether one or more time data points were available per subject, respectively (Mould & Upton, 2012). In the case of the naïve pool approach, all data are pooled together and considered to come from a single individual, and no distinction between inter‐subject and residual variabilities is made. In the population approach, inter‐subject variability in model parameters as well as residual error are estimated. The software NONMEM (RRID:SCR_016986) 7.3 and the first‐order conditional estimation method with interaction algorithm were used for the analysis. Data were logarithmically transformed during the analysis given the different scales of magnitude to improve the performance of the estimation algorithms. For plotting purposes, model predictions were back‐transformed to natural scale, and parameter estimates were obtained in the natural scale except for the error. Additive error model in the logarithmic scale was used to account for the discrepancies between model predictions and observations. When the population approach was applied, the inter‐subject variability was modelled exponentially (Kiang, Sherwin, Spigarelli, & Ensom, 2012; Mould & Upton, 2012).
Model selection was evaluated comparing the minimum value of the objective function provided by NONMEM, and approximately equal to −2×Log(likelihood) between two nested model, where a decrease in −2×Log(likelihood) of 3.84 is considered significant at 5% levels.
Model performance was evaluated using the simulated‐based diagnostics visual predictive checks (VPCs) and population prediction‐corrected VPCs (Bergstrand, Hooker, Wallin, & Karlsson, 2011). Briefly, 200 studies were simulated with the same design characteristics as the original experiment. For each stratification group (e.g., hPBGD mRNA drug product) and measurement time, the 10th, 50th, and 90th percentiles of the simulated biomarker values were calculated, and then the 80% prediction intervals of the above‐mentioned percentiles were obtained and presented graphically together with the 50th percentile of the raw data.
For the case of urinary biomarker data, different induction, as well as drug treatment, dosing schemes were followed in the different settings, and therefore, prediction‐corrected VPCs were used to evaluate simultaneously model performance from different scenarios. The procedure was the same as the one described for VPCs, but normalizing both the observed and simulated biomarkers by the corresponding population model predictions.
2.5. Nomenclature of targets and ligands
Key protein and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to Pharmacology 2019/2020 (Alexander et al., 2019).
3. RESULTS
3.1. Mechanistic‐based computational model
3.1.1. Kinetic PBGDL model
The delay in the increase of PBGD activity after hPBGD mRNA administration was well captured using compartments accounting for LNP‐encapsulated and free mRNA in the liver and assuming free mRNA to be responsible for the increase in PBGD activity. This is in agreement with the known physiological processes triggered upon mRNA administration (Equations 1–3). Incorporation of a reversible distribution from the PBGD activity liver compartment to an additional (peripheral) compartment controlled by first‐order rate constants (K LP and K PL, respectively) significantly improved the description of the data.
Best results in terms of parameter precision, model stability, and physiological plausibility were obtained assuming that the release parameter of encapsulated mRNA (K REL) was dependent on the formulation—and shared across sequences encapsulated in the same LNP—and the degradation rate constant (K DEG) was sequence specific (Table 1). Due to the limited amount of data for LNP‐B and LNP‐C formulations, a common K REL parameter was estimated for both. Similarly, SEQ‐1 and SEQ‐2 exhibited similar time profiles and were well characterized assuming the same set of model parameters.
TABLE 1.
Final parameter estimates of the kinetic PBGDL model
| Parameter | Estimate (RSE) | ||||
|---|---|---|---|---|---|
| θ SLP (kg·pmol−1) | 250 (35%) | ||||
| K LP (days−1) | 0.00892 (29%) | ||||
| K PL (days−1) | 0.00375 (43%) | ||||
| σ (residual error, %) | 28.8 (6%) | ||||
| Formulation specific | LNP‐A | LNP‐B and LNP‐C | |||
|---|---|---|---|---|---|
| K REL (days−1) | 0.323 (20%) | 0.129 (15%) | |||
| Animal species specific | AIP mouse | WT—rat | WT—rabbit | WT—cyno | |
|---|---|---|---|---|---|
| PBGDL(0) (pmol·mg protein−1·hr−1) | 3 FIX | 12.4 FIX | 9.37 FIX | 5.54 FIX | |
| mRNA sequence specific | SEQ‐1 and SEQ‐2 | SEQ‐3 | SEQ‐4 | SEQ‐5 | SEQ‐6 |
|---|---|---|---|---|---|
| K DEG (days−1) | 0.0056 (24%) | 0.0016 (22%) | 0.00889 (30%) | 0.00767 (37%) | 0.000256 (21%) a |
RSE, relative standard error.
Parameter estimated using data from WT animals.
LNP‐A formulation exhibited faster K REL than LNP‐B and LNP‐C formulations, indicating a faster transit (i.e., release and degradation). The slowest PBGD turnover was obtained for SEQ‐3. The final model was able to capture the median and dispersion of all experimental data (Figure 2a) using a common model structure. In addition, formulation‐ and sequence‐specific parameters were identified and estimated with adequate precision.
FIGURE 2.
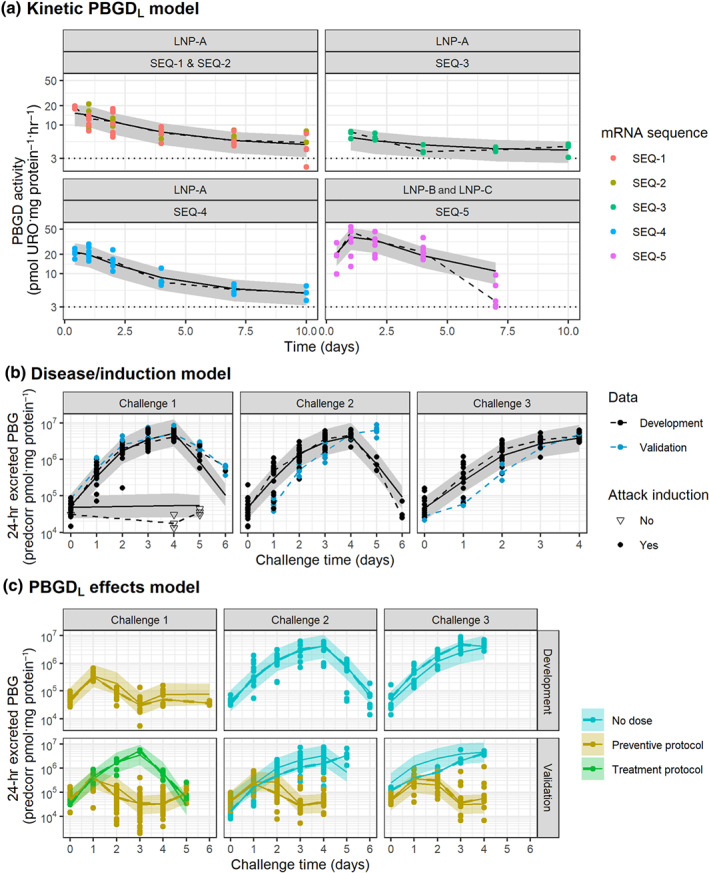
Evaluation of model performance in AIP mice. Grey areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median, and symbols represent raw observations. Dotted horizontal lines in top panels indicate baseline PBGD activity in the liver for AIP mice. Plots have been prediction corrected (predcorr) to enable representation of multiple experimental designs
3.1.2. Disease/induction model
The model that provided the most satisfactory overall performance was obtained when (a) excretion of precursors was controlled by the levels of unobserved circulating ALA/PBG and tPOR, (b) circulating ALA and PBG levels were assumed to be in equilibrium and combined into a unique circulating compartment controlling the excretion of ALA and PBG, and (c) phenobarbital effects increased the circulating levels of ALA/PBG through a delay compartment.
Estimating different enzymic rate constants for the different circulating compartments was not supported by the data available. However, inclusion of a feedback mechanism accelerating the catabolism of circulating ALA/PBG to tPOR in subsequent challenges resulted in a significant improvement of model performance. Final parameter estimates can be found in Table 2.
TABLE 2.
Parameter estimates of the disease/induction model
| Parameter | Estimate (RSE) | |||
|---|---|---|---|---|
| K ENZ (hr−1) | 0.328 (16.2%) | |||
| K FEED (hr−1) | 0.00039 (37.9%) | |||
| γ ALA (unitless) | 13.7 (13.2%) | |||
| γ PBG (unitless) | 20.6 (12.7%) | |||
| γ tPOR (unitless) | 11.3 (13.7%) | |||
| σ ALA (log [pmol·mg Crea−1]) | 0.391 (6%) | |||
| σ PBG (log [pmol·mg Crea−1]) | 0.567 (5%) | |||
| σ POR (log [pmol·mg Crea−1]) | 0.602 (4%) |
| Inductor specific | Phenobarbital | AIA/rifampicin | ||
|---|---|---|---|---|
| K E,inductor (hr−1) | 0.05 (6.7) | 0.0105 (13%) | ||
| θ Inductor (L·mg−1) | 0.0062 (16.1) | 0.12 (8%) | ||
| IAV_ θ Inductor (%) | 7.1 (13.7) | 32.7 (16%) |
| Animal species specific | AIP mice | WT—mice | WT—rat | WT—rabbit |
|---|---|---|---|---|
| K ex,ALA (pmol·mg Crea−1·hr−1) | 7,600 (4%) | 4,580 (7%) | 707 (11%) | 362 (6%) |
| K ex,PBG (pmol·mg Crea−1·hr−1) | 1,110 (6.9%) | 366 (24%) | 36.5 (25%) | 315 (7%) |
| K ex,POR (pmol·mg Crea−1·hr−1) | 0.0544 (6.3%) | — | — | 0.104 (19%) |
RSE, relative standard error.
The final model provided an adequate description of control data in the presence or absence of phenobarbital induction across the different induction cycles, suggesting no time‐dependent kinetics (Figures 2b and S1). In addition, an adequate description of data not included in the model development and used to validate the model was observed (Figure 2b), increasing model robustness.
3.1.3. PBGDL effects model
The effects of increased PBGD activity in the liver were evaluated on different processes of the disease model, such as the appearance of circulating ALA/PBG, transit to circulating tPOR, or the ability of phenobarbital to trigger the AIP attacks. The best description was obtained assuming a competitive interaction between PBGD and phenobarbital, decreasing the inducer effect. Interestingly, the obtained estimates indicated that an increment of PBGD activity of 8.9 pmol URO·mg protein−1·hr−1—which would generate PBGD levels close to those of WT mice—would be sufficient to prevent more than 90% of the phenobarbital‐induced effect in AIP mice.
Incorporating an additional effect on the transit between circulating compartments was significant, but its contribution over the normalization of ALA and PBG levels was minor, compared to the PBGD effect mentioned above. Final parameter estimates can be found in Table 3.
TABLE 3.
Parameter estimates of the PBGD effects model
| Parameter | Estimate (RSE) | |
|---|---|---|
| σ ALA (log [pmol·mg Crea−1]) | 0.454 (6%) | |
| σ PBG (log [pmol·mg Crea−1]) | 0.691 (7%) | |
| σ POR (log [pmol·mg Crea−1]) | 0.534 (4%) | |
| Animal model specific | AIP | WT |
|---|---|---|
| ΔPBGDL,50 (pmol URO·mg protein−1·hr−1) | 6.76 (3%) | — |
| γ PBGD (unitless) | 8 FIX | — |
| θ PBGD (mg Crea × hr·pmol URO−1) | 0.0068 (33%) | 0.407 (11%) |
RSE, relative standard error.
The final model successfully characterized the effects of hPBGD mRNA across the different experimental settings, assuming that the PBGD activity was preserved across treatments (i.e., same efficacy parameter; Figures 2c and S2). Consequently, differences on the accumulation of precursors are explained by the distinct hPBGD mRNA kinetics. The largest differences in the urinary PBG effects were noticed across formulations, observing a higher efficacy after the first challenge and still some protection during the second challenge when LNP‐B and LNP‐C formulations (higher PBGD activity levels achieved) were used (Figure 3).
FIGURE 3.
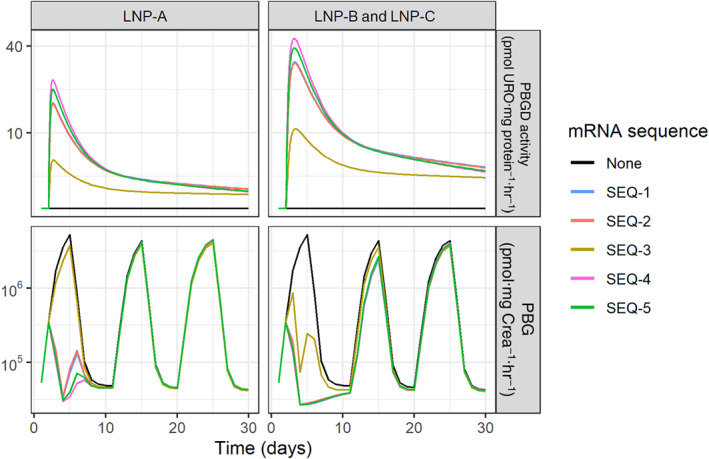
Predicted time course of liver PBGD activity and its effect on urinary levels of PBG in AIP mice during repeated acute attacks. Acute attacks were induced by i.p. injections of four daily increasing doses of phenobarbital (75, 80, 85, and 90 mg·kg−1) over three consecutive challenges (Days 1, 11, and 20). Solid lines represent typical profiles predicted by the final model in AIP mice after the administration of no treatment or a single dose of 0.5 mg·kg−1 of the different hPBGD mRNA sequences formulated as LNP‐A, LNP‐B, or LNP‐C administered 24 hr after the first dose of phenobarbital in Challenge 1
3.2. Translational framework: Integration of data from WT animals
3.2.1. Kinetic PBGDL model
PBGD liver activity from larger WT species (rats, rabbits, and NHPs) was obtained 1 day after administration of SEQ‐6, sequence for which the specific degradation rate constant (K DEG) was not available. Therefore, and to enable model predictions of large animal species, the sequence‐specific K DEG was estimated using the structure of the PBGD activity model developed for the AIP mice and only adjusting the baseline PBGD activity to reflect the specific value of the different animal species (Table 1).
Alternative models including allometric terms were also explored. However, taking into account that dose was already normalized to the animal weight (mg kg‐1), no further modifications were made, and model performance of the model mentioned above was considered sufficient (Figure 4a).
FIGURE 4.
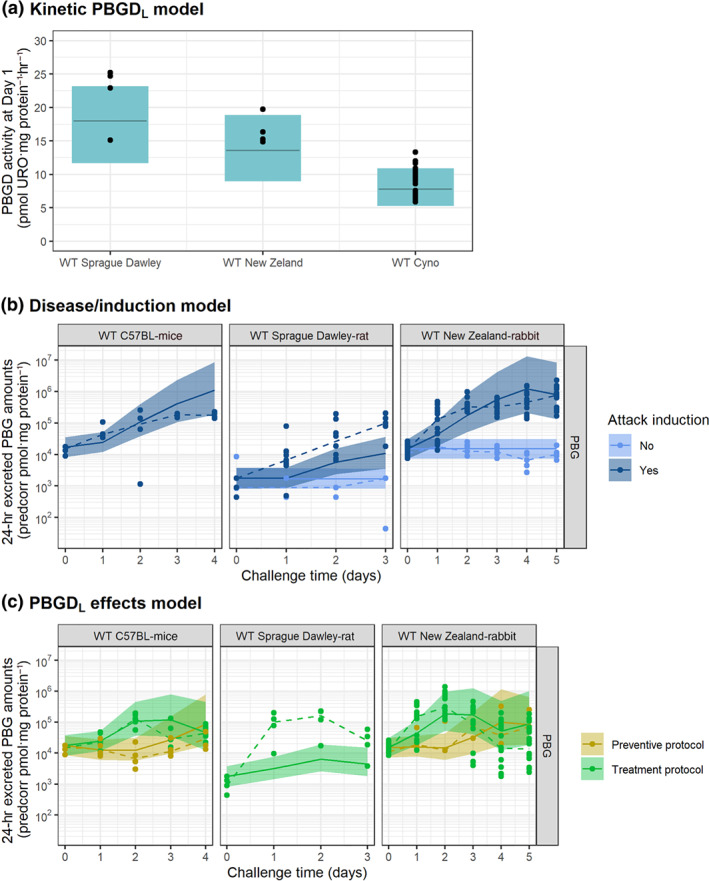
Evaluation of model performance in WT animals untreated or after administration of hPBGD mRNA drug product. Coloured areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median, and symbols represent raw observations. Plots have been prediction corrected (predcorr) to enable representation of multiple experimental designs
3.2.2. Disease/induction model
An adequate description of the urinary accumulation of haem precursors in WT species was obtained when (a) using a kinetic–pharmacodynamic model to describe the kinetics of AIA/rifampicin (see Supporting Information), (b) estimating a common effect of AIA/rifampicin levels over the synthesis of circulating ALA/PBG levels for all WT species, and (c) estimating animal‐specific excretion rate constants (physiological parameter reflecting half of the amount of each precursor excreted in urine every 24 hr) per animal species (Figure 4b).
Although the accumulation of PBG in WT rats after induction was slightly under‐predicted, the overall performance of the model was satisfactory (Figure S3). Table of specific model parameters for WT animals can be found in Table 2.
3.2.3. PBGDL effects model
The inhibition exerted by SEQ‐6 and SEQ‐8 mRNA sequences (LNP‐B and LNP‐C formulations) on the accumulation of haem precursors in urine could be described, considering that PBGDL promoted the transit of ALA/PBG to circulating tPOR (Figures 4 and S4).
An estimate of 0.407 mg Crea · hr·pmol URO−1 (relative standard error of 11%) was obtained when assuming that the effect was driven by the relative increases in the levels of PBGD activity over baseline (relative gain of activity), rather than the absolute increases in PBGD activity, to account for the different PBGD activity levels across species.
3.3. Extrapolation to humans
PBGD activity profiles of two selected sequences, both formulated as LNP‐B, are presented in Figure 5 as an illustration. After a single dose of SEQ‐5, activity levels are predicted to remain above PBGD activity levels of normal subjects for up to 37 or 56 days after for a dose of 0.5 or 1 mg·kg−1, respectively. On the contrary, SEQ‐6, for which a lower turnover was estimated, PBGD activity levels barely reached the threshold of healthy subjects after a single dose but remained constant for a longer period of time.
FIGURE 5.
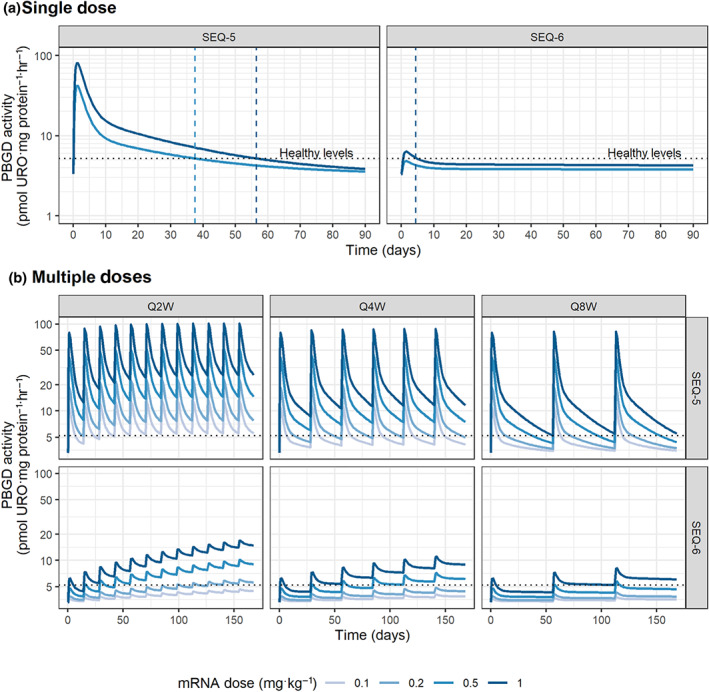
Extrapolated liver PBGD activity time profiles for chronic AIP patients. (a) Single or (b) multiple mRNA doses of two selected sequences in LNP‐B or LNP‐C formulations. QxW stands for every x weeks dosing scheme, for x = 2, 4, or 8. Dotted line indicates PBGD activity levels in healthy subjects
When exploring the effects of multiple dose administrations through model simulations (Figure 5b), it was clear that a steady state was rapidly reached for SEQ‐5, and sustained levels above the healthy threshold for at least 2 weeks are predicted already at low doses of 0.2 mg·kg−1, regardless of the dosing schema. On the other hand, lower maximum concentrations and larger accumulation—in agreement with its lower degradation rate constant—are achieved for SEQ‐6, suggesting that higher doses or longer times to reach sustained healthy levels would be needed in the absence of loading doses.
4. DISCUSSION
Despite the vast amount of preclinical and clinical information generated during drug development programs, high attrition rates are still observed across all therapeutic areas (Hay, Thomas, Craighead, Economides, & Rosenthal, 2014). Model‐informed drug development and discovery has grown over the past decades as a valuable tool to decrease these attrition rates and accelerate the translation of research from bench to bedside. Indeed, nowadays, PKPD modelling represents a key contributor to many drug discovery and development programs across modalities (Gibbs, Menon, & Kasichayanula, 2018; Milligan et al., 2013; Wang et al., 2019).
Mechanistic‐based modelling can have even a greater role in the development of new therapies in rare disease areas, where the low incidence and prevalence of the disease makes patient recruitment difficult for clinical trials. In these situations, it is ideal to maximize the information obtained during early development phases in order to project and predict the potential success or failure of the new drug.
Examples in which computational models have been developed and applied for rare diseases are scarce (Aguda, Marsh, Thacker, & Crouser, 2011; Pradhan et al., 2015; Yoneyama et al., 2018), particularly for those dealing with gene‐ and mRNA‐based therapeutics, where models are mainly constrained to in vitro or limited in vivo conditions (Berraondo, González‐Aseguinolaza, & Trocóniz, 2009; Moriguchi, Kogure, & Harashima, 2008; Varga, Hong, & Lauffenburger, 2001; Yamada, Kamiya, & Harashima, 2005). To the best of our knowledge, this work represents the first translational framework integrating temporal data in the porphyric preclinical arena across animal species and therapeutic mRNA‐based products to project results to humans.
Haem biosynthesis is a complex process that is not fully understood yet. This complexity is reflected in the fact that some patients with the underlying genetic defect remain latent while others are symptomatic (Lenglet et al., 2018). Furthermore, it is postulated that the acute attack is likely to be due to a combination of factors including the toxic effects of porphyrin precursors and enzyme malfunction due to exhaustion of the essential haem cofactor (e.g., tryptophan pyrrolase, NOS, and mitochondrial and microsomal cytochromes of the P450 complex). All these factors are intimately connected, and a strong up‐regulation of the rate‐limiting ALAS1 gene, caused mainly by hepatic haem deficiency, results in an over‐accumulation of haem precursors in circumstances of PBGD deficiency. However, the biochemical disorder resulting from the acute attack has not been fully described.
The development of a predictive framework involves the use of multi‐scale models with the ability to integrate data from multiple sources. In the current investigation, we have integrated mechanistic data not only from different animal species and different mRNA treatments but also from information reflecting intrahepatic processes (i.e., liver PBGD activity) and their effects on systemic dynamics (i.e., urinary excretion of haem precursors) within a single species. Importantly, PBGD activity data and the quantification of ALA, PBG, and tPOR, across the different animal species were obtained using the same analytical method from a single laboratory, providing a greater robustness to the developed model.
In data‐driven approaches, model granularity and complexity is constrained by the information available. Therefore, model assumptions as well as major simplifications of the underlying physiological mechanisms are often required. In this case, no information regarding mRNA levels or protein levels in the liver were available. Therefore, distribution of LNPs and mRNA release in the liver could not be identified as two processes and are simultaneously represented by the release process. In addition, incomplete liver bioavailability or differences in hepatocyte transfection are indirectly reflected in the translation efficacy parameter.
Similarly, haem precursors were only collected in urine; thus, virtual compartments connected between them and simultaneously representing liver and plasma levels were implemented. A similar model structure and parameter estimates were recently identified to characterize haem precursor accumulation in AIP mice (Vera‐Yunca et al., 2019), highlighting the robustness of the model to describe these types of data.
Interestingly, this “limited” information but gathered in a longitudinal manner across multiple experimental settings was sufficient to develop a model that accounted satisfactorily for the different biological processes of delivery and degradation of the mRNA material at the site of action, protein encoding and expression, and biomarker excretion in a simplified but mechanistic manner. The finally developed computational model was supported by the precise parameter estimates and adequate description of the data under the different experimental conditions, including those only used for model validation (not in model building), thus increasing model robustness and adequacy for data extrapolation.
The richness of experimental perturbations in small species (i.e., AIP mice) was especially relevant, as it permitted the development of complex structures that represent, in a mechanistic fashion, biological processes not directly observed (e.g., mRNA release or translation). Those structures provided then the infrastructure needed to integrate data from larger species, where the amount of information is commonly more limited due to experimental, ethical, and economic reasons, enabling the use of single hepatic PBGD activity measurements. In addition, data availability after the administration of different hPBGD mRNA sequences encapsulated in different formulations across species has enabled the identification of those processes that are dependent on the animal species (i.e., baseline PBGD activity), from those that are dependent on the drug (i.e., sequence and formulation specific).
This integrative approach provides highly interesting translational properties to the framework developed, which can be exploited at different levels of drug development. From the preclinical side, the model could be used to optimize new experimental settings reducing the number of animals and samples needed to evaluate new sequences and formulations. The model also enables the in silico projection of PBGD activity levels in AIP patients or healthy subjects for different mRNA sequences, LNP formulations, and dosing schemes, thus representing a potential tool to support candidate and dose selection in clinical settings. Certainly, these results represent a simulation exercise given the lack of clinical scenarios and need to be further validated and confronted by real clinical data.
Nonetheless, the simulation exercise suggests already the feasibility of the application of these PBGD mRNA drug products in humans given that lower doses than those tested in NHPs could be sufficient to achieved sustained PBGD activity levels for at least 2 weeks, a period longer than the standard duration of an acute attack in patients with sporadic presentation (Gouya et al., 2017). For those patients with recurrent attacks (5% of symptomatic individuals), modelling and simulation could guide the selection of optimal dosing protocols, which will differ depending on the kinetic properties of the selected sequence and LNP formulation.
In order to project the accumulation of precursors in humans, quantitative and longitudinal data of haem precursors in urine before and during acute attacks (in the absence of treatment) are needed. These data will enable the obtaining of human‐specific parameter estimates and further validate the model as a high‐quality translational framework. In this regard, collaborative efforts to collect these types of data and further support the development of novel mRNA therapies should be encouraged in the best interest of AIP patients.
Finally, given the mechanistic nature of the model and its ability to differentiate between drug and system parameters, additional data from new compounds, species, or even biomarkers could be easily integrated to further validate the framework and increase its predictive capability. Therefore, this computational model can serve as the basis of a future system model that integrates detailed knowledge of the kinetic parameters involved in the haem pathway, as well as biochemical changes associated to other porphyrias. This could include, but is not limited to, the inhibition of PBGD (deficient enzyme in AIP) triggered by the accumulation of coproporphyrinogen and protoporphyrinogen observed in the hereditary coproporphyria and variegate porphyria, respectively, or the oxidation of porphyrinogens to porphyrins responsible for the phototoxic manifestations in photocutaneous porphyrias. The resulting comprehensive framework of the haem biosynthetic pathway could then be used not only to support drug development and clinical use in AIP but also to develop new hypotheses for the pathophysiology of acute attack and, in general, for the biochemistry of the porphyrias.
In summary, a quantitative framework capable of describing the effects of novel mRNA compounds on the accumulation of haem precursors in urine during induced acute attacks, across different animal species has been proposed. This approach opens a wide range of new possibilities, including the optimization of new experimental settings, not only for the evaluated drug products but also for new investigational mRNA products (i.e., mRNA sequences and LNP formulations), and the projection of preclinical results to human clinical studies. These results should encourage experimentalists towards the value of sparse data collected longitudinally and strengthen collaborations between in silico and experimental disciplines to maximize the information to be extracted from preclinical and clinical data in order to support drug development and clinical use.
AUTHOR CONTRIBUTIONS
Z.P.P.‐G. and I.F.T. wrote the manuscript; A.F., L.J., P.M., M.H., and L.T.G. reviewed and edited the manuscript; Z.P.P.‐G., A.F., L.J., M.H., L.T.G., and I.F.T. designed the research and analysed the data; A.F. and D.J. performed the animal experiments; and Z.P.P.‐G., D.V.‐Y., and I.F.T. performed the analysis.
CONFLICT OF INTEREST
At the time the research was performed, Z.P.P.‐G., D.V.‐Y., and I.F.T. received research funding from Moderna, Inc. L.J., P.M., M.H., and L.T.G. are employees of, and receive salary and stock options from, Moderna, Inc.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Data S1. Supporting Information
Figure S1. Evaluation of the disease/induction model performance for ALA, PBG and tPOR in AIP mice. Grey areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Figure S2. Evaluation of final model performance for ALA, PBG and tPOR in AIP mice after administration of mRNA SEQ‐1. Coloured areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Dotted vertical lines indicate phenobarbital administration and black arrow mRNA administration.
Figure S3. Evaluation of disease/induction model performance for ALA, PBG and tPOR in WT large species. Colored areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Figure S4. Evaluation of final model performance for ALA, PBG and tPOR in WT animals after administration of mRNA SEQ‐6 in LNP‐C. Colored areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Table S1. Experimental design overview
ACKNOWLEDGEMENTS
This study was supported in part by grants from the Fundación Mutua Madrileña (Madrid, Spain), Fundación Eugenio Rodríguez Pascual (Madrid, Spain), and Spanish Institute of Health Carlos III (FIS) co‐financed by European FEDER funds (Grants PI15/01951 and PI18/00860) and the spanish Fundación Eugenio Rodríguez Pascual.
Parra‐Guillen ZP, Fontanellas A, Jiang L, et al. Disease pharmacokinetic–pharmacodynamic modelling in acute intermittent porphyria to support the development of mRNA‐based therapies. Br J Pharmacol. 2020;177:3168–3182. 10.1111/bph.15040
REFERENCES
- Aguda, B. D. , Marsh, C. B. , Thacker, M. , & Crouser, E. D. (2011). An in silico modeling approach to understanding the dynamics of sarcoidosis. PLoS ONE, 6, e19544 10.1371/journal.pone.0019544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Sharman, J. L. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, K. E. , Bloomer, J. R. , Bonkovsky, H. L. , Kushner, J. P. , Pierach, C. A. , Pimstone, N. R. , & Desnick, R. J. (2005). Recommendations for the diagnosis and treatment of the acute porphyrias. Annals of Internal Medicine, 142, 439 10.7326/0003-4819-142-6-200503150-00010 [DOI] [PubMed] [Google Scholar]
- Anderson, P. M. , Reddy, R. M. , Anderson, K. E. , & Desnick, R. J. (1981). Characterization of the porphobilinogen deaminase deficiency in acute intermittent porphyria. Immunologic evidence for heterogeneity of the genetic defect. The Journal of Clinical Investigation, 68, 1–12. 10.1172/jci110223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balwani, M. , & Desnick, R. J. (2012). The porphyrias: Advances in diagnosis and treatment. Blood, 120, 4496–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrand, M. , Hooker, A. C. , Wallin, J. E. , & Karlsson, M. O. (2011). Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. The AAPS Journal, 13, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berraondo, P. , González‐Aseguinolaza, G. , & Trocóniz, I. F. (2009). Semi‐mechanistic pharmacodynamic modelling of gene expression and silencing processes. European Journal of Pharmaceutical Sciences, 37, 418–426. 10.1016/j.ejps.2009.03.013 [DOI] [PubMed] [Google Scholar]
- Chen, B. , Solis‐Villa, C. , Hakenberg, J. , Qiao, W. , Srinivasan, R. R. , Yasuda, M. , … Desnick, R. J. (2016). Acute intermittent porphyria: Predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Human Mutation, 37, 1215–1222. 10.1002/humu.23067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Avola, D. , López‐Franco, E. , Sangro, B. , Pañeda, A. , Grossios, N. , Gil‐Farina, I. , … Schmidt, M. (2016). Phase I open label liver‐directed gene therapy clinical trial for acute intermittent porphyria. Journal of Hepatology, 65, 776–783. 10.1016/j.jhep.2016.05.012 [DOI] [PubMed] [Google Scholar]
- Dunoyer, M. (2011). Accelerating access to treatments for rare diseases. Nature Reviews. Drug Discovery, 10, 475–476. [DOI] [PubMed] [Google Scholar]
- Elder, G. , Harper, P. , Badminton, M. , Sandberg, S. , & Deybach, J.‐C. (2013). The incidence of inherited porphyrias in Europe. Journal of Inherited Metabolic Disease, 36, 849–857. [DOI] [PubMed] [Google Scholar]
- Fontanellas, A. , Ávila, M. A. , Anderson, K. E. , & Deybach, J.‐C. (2019). Current and innovative emerging therapies for porphyrias with hepatic involvement. Journal of Hepatology, 71, 422–433. [DOI] [PubMed] [Google Scholar]
- Fontanellas, A. , Ávila, M. A. , & Berraondo, P. (2016). Emerging therapies for acute intermittent porphyria. Expert Reviews in Molecular Medicine, 18, e17. [DOI] [PubMed] [Google Scholar]
- Gabhann, F. M. , Annex, B. H. , & Popel, A. S. (2010). Gene therapy from the perspective of systems biology. Current Opinion in Molecular Therapeutics, 12, 570–577. [PMC free article] [PubMed] [Google Scholar]
- Gibbs, J. P. , Menon, R. , & Kasichayanula, S. (2018). Bedside to bench: Integrating quantitative clinical pharmacology and reverse translation to optimize drug development. Clinical Pharmacology and Therapeutics, 103, 196–198. 10.1002/cpt.897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouya, L. , Bloomer, J. , Balwani, M. , Bissell, D. , Rees, D. , Stölzel, U. , … Deybach, J. C. (2017). EXPLORE: A prospective, multinational, natural history study of patients with acute hepatic porphyrias (AHP) with recurrent attacks. In 26 June 2017 I ICPP I Bordeaux, France, p. [DOI] [PMC free article] [PubMed]
- Griggs, R. C. , Batshaw, M. , Dunkle, M. , Gopal‐Srivastava, R. , Kaye, E. , Krischer, J. , … Rare Diseases Clinical Research Network . (2009). Clinical research for rare disease: Opportunities, challenges, and solutions. Molecular Genetics and Metabolism, 96, 20–26. 10.1016/j.ymgme.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S.D. , Sharman, J.L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 46, D1091‐1106. 10.1093/nar/gkx1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, M. , Thomas, D. W. , Craighead, J. L. , Economides, C. , & Rosenthal, J. (2014). Clinical development success rates for investigational drugs. Nature Biotechnology, 32, 40–51. [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Berraondo, P. , Jericó, D. , Guey, L. T. , Sampedro, A. , Frassetto, A. , … Fontanellas, A. (2018). Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nature Medicine, 24, 1899–1909. 10.1038/s41591-018-0199-z [DOI] [PubMed] [Google Scholar]
- Karim, Z. , Lyoumi, S. , Nicolas, G. , Deybach, J.‐C. , Gouya, L. , & Puy, H. (2015). Porphyrias: A 2015 update. Clinics and Research in Hepatology and Gastroenterology, 39, 412–425. 10.1016/j.clinre.2015.05.009 [DOI] [PubMed] [Google Scholar]
- Kiang, T. K. L. , Sherwin, C. M. , Spigarelli, M. G. , & Ensom, M. H. H. (2012). Fundamentals of population pharmacokinetic modelling. Clinical Pharmacokinetics, 51, 515–525. 10.1007/BF03261928 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenglet, H. , Schmitt, C. , Grange, T. , Manceau, H. , Karboul, N. , Bouchet‐Crivat, F. , … Gouya, L. (2018). From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Human Molecular Genetics, 27, 1164–1173. 10.1093/hmg/ddy030 [DOI] [PubMed] [Google Scholar]
- Lindberg, R. L. P. , Porcher, C. , Grandchamp, B. , Ledermann, B. , Bürki, K. , Brandner, S. , … Meyer, U. A. (1996). Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nature Genetics, 12, 195–199. 10.1038/ng0296-195 [DOI] [PubMed] [Google Scholar]
- Marsden, J. T. , Guppy, S. , Stein, P. , Cox, T. M. , Badminton, M. , Gardiner, T. , … Rees, D. C. (2015). Audit of the use of regular haem arginate infusions in patients with acute porphyria to prevent recurrent symptoms (pp. 57–65). Berlin, Heidelberg: Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan, P. A. , Brown, M. J. , Marchant, B. , Martin, S. W. , van der Graaf, P. H. , Benson, N. , … Lalonde, R. L. (2013). Model‐based drug development: A rational approach to efficiently accelerate drug development. Clinical Pharmacology and Therapeutics, 93, 502–514. 10.1038/clpt.2013.54 [DOI] [PubMed] [Google Scholar]
- Moriguchi, R. , Kogure, K. , & Harashima, H. (2008). Non‐linear pharmacodynamics in the transfection efficiency of a non‐viral gene delivery system. International Journal of Pharmaceutics, 363, 192–198. 10.1016/j.ijpharm.2008.07.020 [DOI] [PubMed] [Google Scholar]
- Mould, D. R. , & Upton, R. N. (2012). Basic concepts in population modeling, simulation, and model‐based drug development. CPT: Pharmacometrics & Systems Pharmacology, 1, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra‐Guillén, Z. P. , González‐Aseguinolaza, G. , Berraondo, P. , & Trocóniz, I. F. (2010). Gene therapy: A pharmacokinetic/pharmacodynamic modelling overview. Pharmaceutical Research, 27, 1487–1497. 10.1007/s11095-010-0136-4 [DOI] [PubMed] [Google Scholar]
- Pradhan, R. , Gastonguay, M. , Gao, X. , Monteleone, J. , Fisher, J. , Godfrey, C. , et al. (2015). Exposure–response modeling and simulation to support evaluation of efficacious and safe exposure and dose range for asfotase alfa in patients with hypophosphatasia. Journal of Bone and Mineral Research, 30(Suppl 1). [Google Scholar]
- Puy, H. , Gouya, L. , & Deybach, J.‐C. (2010). Porphyrias. Lancet, 375, 924–937. 10.1016/S0140-6736(09)61925-5 [DOI] [PubMed] [Google Scholar]
- Sabnis, S. , Kumarasinghe, E. S. , Salerno, T. , Mihai, C. , Ketova, T. , Senn, J. J. , … Benenato, K. E. (2018). A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non‐human primates. Molecular Therapy, 26, 1509–1519. 10.1016/j.ymthe.2018.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardh, E. , Harper, P. , Balwani, M. , Stein, P. , Rees, D. , Bissell, D. M. , … Anderson, K. E. (2019). Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. The New England Journal of Medicine, 380, 549–558. 10.1056/NEJMoa1807838 [DOI] [PubMed] [Google Scholar]
- Siegert, S. W. K. , & Holt, R. J. (2008). Physicochemical properties, pharmacokinetics, and pharmacodynamics of intravenous hematin: A literature review. Advances in Therapy, 25, 842–857. 10.1007/s12325-008-0094-y [DOI] [PubMed] [Google Scholar]
- Southan, C. , Sharman, J. L. , Benson, H. E. , Faccenda, E. , Pawson, A. J. , Alexander, S. P. H. , … NC‐IUPHAR . (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: Towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Research, 44, D1054–D1068. 10.1093/nar/gkv1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, P. E. , Badminton, M. N. , & Rees, D. C. (2017). Update review of the acute porphyrias. British Journal of Haematology, 176, 527–538. 10.1111/bjh.14459 [DOI] [PubMed] [Google Scholar]
- US Department of Health and Food Administration . (2007). Critical path opportunities report.
- Varga, C. M. , Hong, K. , & Lauffenburger, D. A. (2001). Quantitative analysis of synthetic gene delivery vector design properties. Molecular Therapy, 4, 438–446. 10.1006/mthe.2001.0475 [DOI] [PubMed] [Google Scholar]
- Vera‐Yunca, D. , Serrano‐Mendioroz, I. , Sampedro, A. , Jericó, D. , Trocóniz, I. F. , Fontanellas, A. , & Parra‐Guillén, Z. P. (2019). Computational disease model of phenobarbital‐induced acute attacks in an acute intermittent porphyria mouse model. Molecular Genetics and Metabolism, 128, 367–375. 10.1016/j.ymgme.2018.12.009 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Zhu, H. , Madabushi, R. , Liu, Q. , Huang, S. , & Zineh, I. (2019). Model‐informed drug development: Current US regulatory practice and future considerations. Clinical Pharmacology and Therapeutics, 105, 899–911. 10.1002/cpt.1363 [DOI] [PubMed] [Google Scholar]
- Westerlund, J. , Pudek, M. , & Schreiber, W. E. (1988). A rapid and accurate spectrofluorometric method for quantification and screening of urinary porphyrins. Clinical Chemistry, 34, 345–351. [PubMed] [Google Scholar]
- Woodcock, J. , & Woosley, R. (2008). The FDA critical path initiative and its influence on new drug development. Annual Review of Medicine, 59, 1–12. [DOI] [PubMed] [Google Scholar]
- Yamada, Y. , Kamiya, H. , & Harashima, H. (2005). Kinetic analysis of protein production after DNA transfection. International Journal of Pharmaceutics, 299, 34–40. 10.1016/j.ijpharm.2005.04.003 [DOI] [PubMed] [Google Scholar]
- Yoneyama, K. , Schmitt, C. , Kotani, N. , Levy, G. G. , Kasai, R. , Iida, S. , … Kawanishi, T. (2018). A pharmacometric approach to substitute for a conventional dose‐finding study in rare diseases: Example of phase III dose selection for emicizumab in hemophilia A. Clinical Pharmacokinetics, 57, 1123–1134. 10.1007/s40262-017-0616-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Figure S1. Evaluation of the disease/induction model performance for ALA, PBG and tPOR in AIP mice. Grey areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Figure S2. Evaluation of final model performance for ALA, PBG and tPOR in AIP mice after administration of mRNA SEQ‐1. Coloured areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Dotted vertical lines indicate phenobarbital administration and black arrow mRNA administration.
Figure S3. Evaluation of disease/induction model performance for ALA, PBG and tPOR in WT large species. Colored areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Figure S4. Evaluation of final model performance for ALA, PBG and tPOR in WT animals after administration of mRNA SEQ‐6 in LNP‐C. Colored areas represent the 80% model prediction interval, solid lines are the median model prediction, dashed lines are the observed median and symbols represent raw observations. Plots have been prediction‐corrected (predcorr) to enable representation of multiple experimental designs.
Table S1. Experimental design overview