

Abstract
Translesion DNA synthesis (TLS) performed by human DNA polymerase eta (hpol η) allows tolerance of damage from cis-diamminedichloroplatinum(II) (CDDP or cisplatin). We have developed hpol η inhibitors derived from N-aryl-substituted indole barbituric acid (IBA), indole thiobarbituric acid (ITBA), and indole quinuclidine scaffolds and identified 5-((5-chloro-1-(naphthalen-2-ylmethyl)-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6-(1H,5H)-dione (PNR-7-02), an ITBA derivative that inhibited hpol η activity with an IC50 value of 8 μM and exhibited 5–10-fold specificity for hpol η over replicative pols. We conclude rom kinetic analyses, chemical footprinting assays, and molecular docking that PNR-7-02 binds to a site on the little finger domain and interferes with the proper orientation of template DNA to inhibit hpol η. A synergistic increase in CDDP toxicity was observed in hpol η-proficient cells co-treated with PNR-7-02 (combination index values = 0.4–0.6). Increased γH2AX formation accompanied treatment of hpol η-proficient cells with CDDP and PNR-7-02. Importantly, PNR-7-02 did not impact the effect of CDDP on cell viability or γH2AX in hpol η-deficient cells. In summary, we observed hpol η-dependent effects on DNA damage/replication stress and sensitivity to CDDP in cells treated with PNR-7-02. The ability to employ a small-molecule inhibitor of hpol η to improve the cytotoxic effect of CDDP may aid in the development of more effective chemotherapeutic strategies.
Graphical Abstract
DNA lesions that remain unrepaired or that occur during S-phase when the genome is most vulnerable can be bypassed through a process known as translesion DNA synthesis (TLS).1 TLS provides the cell with a way to tolerate blocks to replication by relying upon specialized DNA polymerases (pols) to bypass DNA damage that arises from endogenous and exogenous sources.2 These enzymes can reduce the efficacy of anticancer drugs through bypass of what would otherwise be cytotoxic DNA damage. In addition to providing a mechanism for damage tolerance, TLS pols can increase the mutation frequency of tumors treated with DNA damaging agents, which has been shown to shorten the time to tumor relapse in cell culture and animal experiments.3 Several studies have reported increased TLS pol expression in bulk tumors, as well as in the stem cell niche, consistent with the idea that TLS pols contribute to resistance and relapse in human cancers.4–9 Discovering new ways to leverage our understanding of how TLS contributes to poor outcomes for cancer patients undergoing chemotherapy is an important area of research.
Y-family DNA pols are central mediators of TLS and DNA damage tolerance.10 As such, these enzymes can present a barrier to the effectiveness of anticancer drugs that act primarily through the induction of DNA damage. The notion of targeting TLS pols in cancer is one that has only developed in the past decade, as the diversity of form and function within the repertoire of the human DNA pol arsenal was not fully appreciated prior to the discovery of the Y-family pols.11 Since that time, structure–function studies have shown that Y-family members can bypass a variety of DNA lesions.11 However, the rationale for targeting human Y-family pols as a means of potentiating current cancer treatments awaited clinical studies showing that these enzymes were in fact impacting the prognosis and response of cancer patients.
The most abundant DNA adduct arising from the reaction between cis-diamminedichloroplatinum(II) (CDDP or cisplatin) and DNA is the intrastrand cross-link between two guanines (Pt–GG). Though not as cytotoxic as interstrand cross-links, intrastrand cross-links must still be repaired and/or bypassed. In this regard, human DNA polymerase eta (hpol η) seems to be an important player in the tolerance of unrepaired intrastrand cross-links generated by platinum drugs.4,6,8,12–14 In vitro kinetic analysis revealed that hpol η copies the 3′-dG and 5′-dG sites of a Pt–GG adduct with catalytic efficiencies of 82 and 35%, respectively, relative to unmodified DNA15 Cells derived from xeroderma pigmentosum variant (XPV) patients that do not express functional hpol η are considerably more sensitive to CDDP than cells proficient in hpol η activity.13 Another study showed that replication fork progression is slowed in hpol η-deficient cells treated with either CDDP or carboplatin and that this is accompanied by an increase in DNA strand breaks.14 The clinical relevance of hpol η activity in response to platinum agents has been illustrated in multiple cancers, including lung, stomach, and ovarian cancers.6,8,16 Most recently, hpol η overexpression and activity in the ovarian cancer stem cell niche was implicated in resistance to CDDP and tumor recurrence.8
We previously reported the identification of N-aroyl-substituted derivatives of indole thio-barbituric acid (ITBA) as DNA pol inhibitors, with N-naphthoyl-substituted ITBA analogues acting as the most effective inhibitors of hpol η (IC50 < 20 μM).17 In the current study, we have expanded the number of ITBA analogues and have examined the resulting structure–activity relationships for inhibition of hpol η in an effort to improve both the potency and specificity of our initial inhibitors. An important outcome of this study was the identification of PNR-7-02, an ITBA analogue with improved potency against hpol η. Additionally, experiments reported here provide new insights into the mechanism of hpol η inhibition by ITBA analogues and explore the potentiating effect of our most potent hpol η inhibitor, PNR-7-02, on CDDP cytotoxicity in human cells.
MATERIALS AND METHODS
Reagents.
The dNTPs used in both the fluorescence-based and gel-based pol assays were obtained from GE Healthcare Life Sciences (Piscataway, NJ). Oligonucleotides used in DNA binding and gel-based pol assays were synthesized by Integrated DNA Technologies (Coralville, IA) and purified using HPLC by the manufacturer, with analysis by matrix-assisted laser desorption time-of-flight MS. The oligonucleotides used in the fluorescence-based analysis of pol activity were synthesized by Biosearch Technologies (San Diego, CA). The synthetic procedures used to prepare the library of compounds have been reported previously.18,19
Protein Expression and Purification.
The core polymerase domain of the Y-family members hpol η (residues 1–437), hpol ι (residues 1–446), and hpol κ (residues 19–526) was expressed and purified as described previously.17 The Y-family member hRev1 (residues 330–833) was expressed and purified as described previously.20 The B-family pol Dpo1 and the Y-family pol Dpo4 from Sulfolobus solfataricus were expressed and purified as described previously21,22. The catalytic core of human DNA pol theta (hpol θ, residues 1792–2590) was expressed and purified as described previously.23 DNA pol I from Mycobacterium tuberculosis (MtbPol I) was expressed and purified as described previously.24 The purified core domain of human DNA pol epsilon (hpol ε, residues 1–1189/exo−) was a kind gift from Dr. Zachary Pursell (Tulane University). Purified human DNA pol beta (hpol β) was a kind gift from Dr. Samuel Wilson (NIEHS). The purified catalytic core of human DNA pol lambda (hpol λ, residues 1–325) was a kind gift from Dr. Miguel Garcia-Diaz (Stony Brook University).
DNA Binding Analysis.
Fluorescence anisotropy DNA binding assays were performed as described previously.25 Briefly, fluorescein (FAM)-labeled 13-mer primer (5′-(FAM-TTT)-GGGGGAAGGATTC-3′) and 18-mer template (5′-TCACGGAATCCTTCCCCC-3′) DNA were annealed (1:1.2 molar ratio, 95 °C for 5 min followed by slow cooling to room temperature). The FAM-labeled primer–template DNA substrate (2 nM) was incubated with increasing concentrations of hpol η, and fluorescence polarization was measured in a BioTek Synergy H4 plate reader using the appropriate filter sets (λex = 485 ± 20 nm and λem = 525 ± 20 nm). All titrations were performed at 25 °C in 50 mM HEPES buffer (pH 7.5) containing 10 mM KOAc, 10 mM MgCl2, 10 mM KCl, 2 mM β-mercaptoethanol (β-ME), 10% (v/v) dimethyl sulfoxide (DMSO), and 0.1 mg/mL BSA. The experiments were performed in the presence of 0, 1, 2, 4, 6, 8, 10, and 12 μM PNR-7-02. The resulting polarization data were fit to a quadratic equation to estimate the equilibrium dissociation constant (KD,DNA) for hpol η binding to primer–template DNA.
Fluorescence-Based Assay To Screen for Inhibition of DNA Polymerase Activity.
The fluorescence-based assay was used to screen 85 IBA/ITBA derivatives for inhibition of hpol η activity (see Table S1 for a complete list of the chemical structures and inhibitory properties against hpol η). The DNA substrate was prepared as described previously. Briefly, a 5-carboxytetramethylrhodamine (TAMRA)-labeled reporter (or displaced) strand (5′-TTT TTT TTG C-TAMRA-3′) and unlabeled primer strand (5′-TCA CCC TCG TAC GAC TCT T-3′) were annealed to a Black Hole Quencher (BHQ)-labeled template strand (5′-BHQ2-GCA AAA AAA AAA GAG TCG TAC GAG GGT GA-3′) in a solution containing 10 mM Tris (pH 8.0), 50 mM NaCl, and 2 mM MgCl2. The template (T), primer (P), and displaced strand (D) oligonucleotides were mixed in a 1:1.5:1.5 (T:P:D) molar ratio for annealing. After an incubation period of 3 min at 95 °C, the DNA was allowed to slowly cool to room temperature overnight.
The assay conditions included 2.5 nM hpol η, 50 nM DNA, 20 μM compound, 100 μM dTTP, and 1 mM MgCl2. The reactions were performed in 50 mM Tris (pH 8.0) buffer containing 40 mM NaCl, 2 mM dithiothreitol (DTT), 10% (v/v) DMSO, and 0.01% (v/v) Tween-20. To perform the reactions, the enzyme, compound (or DMSO), and dTTP were combined with the reaction buffer in individual wells and incubated for 5–10 min. The reaction was initiated upon addition of DNA substrate, and the plate was read immediately using a BioTek Synergy H4 plate reader (λem = 525 nm, λem = 598 nm). The final reaction volume was typically 200 μL. Fluorescence was monitored for 60–90 min for most reactions. The initial portion of the velocity curve was analyzed by linear regression to calculate an observed rate of product formation. For each data set, we averaged eight DMSO control experiments to obtain our measure of 100% activity. Rates of product formation in the presence of each compound were then divided by the rate of the DMSO control to produce a relative measure of polymerase activity. The IC50 value was estimated by fitting the data to eq 1, which defines a four-parameter logistic model allowing a variable slope.
| (1) |
The mean (± standard deviation) of the IC50 values calculated for three independents experiments is reported.
The fluorescence-based assay, as described above, was used to determine the steady-state kinetic parameters for hpol η activity in the presence of multiple concentrations of dTTP (0, 0.5, 1, 2, 4, 8, 10, 20, 40, 60, 80, and 100 μM). The change in fluorescence was plotted as a function of time, and the linear portion of each curve was used to calculate the velocity for each dTTP concentration. The rate of product formation was then plotted as a function of dTTP concentration and fit to a hyperbola to estimate the Michaelis constant (KM,dTTP). The turnover number (kcat) was calculated after correcting for the enzyme concentration. These experiments were then repeated in the presence of PNR-7-02 (0–9 μM) to determine the effect of the small-molecule upon Michaelis–Menten kinetics and to help elucidate the mechanism of inhibition of hpol η1–437 by PNR-7-02.
Gel-Based Assay Measuring DNA Polymerase Activity.
As a second measure of enzyme inhibition, pol extension assays were performed. The gel-based assay was the only means of assaying for inhibition of hRev1, as we found that the cytidyl transferase activity of this enzyme was not readily amenable to the fluorescence-based assay. We determined the IC50 for PNR-7-02 against all four human Y-family pols using the gel-based assay. To prepare the DNA substrate (13/18-mer), the FAM-labeled primer was annealed to a complementary template oligonucleotide (1:2, primer:template molar ratio) by incubating at 95 °C for 5 min followed by slow cooling to room temperature. The polymerase extension assay was performed in 50 mM HEPES (pH 7.5) buffer containing 50 mM NaCl, 1 mM DTT, 0.1 mg/mL BSA, and 10% (v/v) glycerol. The enzyme (2 nM) was incubated with increasing concentrations of PNR-7-02 for 5 min, and the reaction was initiated by the addition of DNA·dNTP·Mg2+ (200 nM DNA, 10 μM of each dNTP and 5 mM MgCl2) at 37 °C. The reactions were terminated by adding quench solution (20 mM EDTA and 95% v/v formamide) to each reaction followed by heating at 95 °C. The product formation was analyzed as described previously. The experiments were performed in triplicate, and the mean (± standard deviation) of the IC50 values calculated for each data set is reported.
p-Hydroxyphenylglyoxal (HPG) Labeling of hpol η.
HPG labeling experiments were performed with hpol η (10 μM) in the presence and absence of PNR-7-02. Briefly, hpol η was preincubated with either 20 μM (1:2 molar ratio) or 100 μM (1:10 molar ratio) PNR-7-02 for 10 min at 37 °C in 50 mM HEPES buffer (pH 7.8) containing 200 μM NaCl, 10 mM MgCl2, and 10% v/v DMSO. The HPG labeling was carried out at 37 °C in the dark for 60 min by adding 500 μM HPG to the preincubated reaction mix, followed by addition of 200 mM free arginine to quench the reaction. The samples were run on an SDS-PAGE gel and submitted to the UAMS Proteomics Core Facility for analysis by mass spectrometry. In-gel trypsin digestion was used to produce peptides amenable for analysis by liquid chromatography mass spectrometry (LC-MS). A list of the peptides identified in the MS analysis is provided in Table S2.
Quantification of HPG Labeled Peptides.
Unmodified and HPG-modified peptides were both identified using Scaffold Viewer (Proteome Software Inc.). The modified peptides have a molecular weight of the peptide fragment plus the 132 Da HPG adduct. The peak intensity was then quantified manually using the corresponding m/z value, and the relative abundance for each modified peptide was calculated as a function of area under the peak in Xcalibur software (ThermoFisher Scientific). The percentage of modified arginine measured by mass spectrometry was calculated as a fraction of total arginine (i.e., unmodified + HPG modified) quantified for a specific residue. The data for each peptide was normalized to the hpol η-HPG alone sample to calculate the change in HPG modification after treatment with PNR-7-02.
Molecular Docking.
Docking was performed using SwissDOCK (www.swissdock.ch) as described previously. Only the coordinates for the protein were used for docking analysis.
Human Cell Culture.
HAP-1 cells, including hpol η null cells generated using CRISPR–Cas9 technology (product ID HZGHC000655c007), were purchased from Horizons Discovery Group LLC in March of 2016 (Cambridge, UK). The company using Sanger sequencing confirmed genome editing, and we confirmed elimination of hpol η protein expression by immunoblotting. HAP-1 cells were cultured in IMDM (Iscove’s modified Dulbecco’s medium) supplemented with 10% FBS (fetal bovine serum) and antibiotic/antimycotic solution (1%) (Sigma-Aldrich, St. Louis, USA). DAPI staining was performed periodically (last test performed March of 2017) to test for the presence of Mycoplasma. Both cell lines were at passage seven upon receipt from the company. Most experiments were performed with cells at passage 8–10. HAP-1 (hpol η+ and hpol η−) cells were co-treated with PNR-7-02 and CDDP to determine if the combination treatment affected cell growth. Briefly, cells were seeded at 5000 cells per well (96-well plate). The following day, CDDP (±PNR-7-02) was added to each well, and the cells were incubated for 48 h. In co-treatment experiments, cells were treated with 0–100 μM CDDP and 0.1 or 1 μM PNR-7-02. Cells were washed twice with Dulbecco’s phosphate-buffered saline (DPBS, pH 7.4) and incubated with 2 μM Calcein-AM (Thermo-Scientific, Waltham, MA, USA) for 30 min at 25 °C, and fluorescence was measured (λex = 490 nm and λem = 520 nm). The fluorescence intensity of control (untreated) cells was taken as 100% viability. The relative cell viability compared to control was calculated by using the following formula: cell viability (%) = fluorescence intensity of treated cells/fluorescence intensity of untreated cells × 100. The experiments were repeated for a total of 6–12 biological replicates, and the mean (± standard deviation) of the EC50 values is reported.
Immunoblotting.
Frozen HAP-1 cell pellets were lysed at 4 °C in a buffer containing 40 mM HEPES (pH 7.5), 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate, 10 mM sodium β-glycerophosphate, and 1% (v/v) Triton X-100 with 1× protease inhibitor cocktail (Sigma-Aldrich) added. The lysate was centrifuged for 15 min at 13 000g. The protein concentration of the supernatant was estimated using a Pierce bicinchoninic acid (BCA) protein assay kit (ThermoFisher Scientific). Samples containing 30–40 μg total protein were loaded on a 4–20% gradient SDS-PAGE gel, and electrophoresis was performed at 100 V for 80 min. The separated proteins were transferred to a 0.45 μm nitrocellulose membrane (Bio-Rad) at 4 °C at 200 mA for 90 min. After blocking with 5% (w/v) nonfat milk in 1× Tris-buffered saline (TBS), western blotting to detect the presence of specific proteins was performed by probing the membranes with the following antibodies: anti-hpol η antibody (Santa Cruz; catalog no. sc-17770) and anti-phosphorylated-histone H2AX (S139; Abcam catalog no. ab81299). Dilution of the primary antibody was 1:1000 for anti-hpol η and 1:1500 for γH2AX in TBS containing 1% (w/v) BSA. A horseradish peroxidase-coupled anti-rabbit secondary antibody (Thermo Scientific; catalog no. PI32460) was used at a 1:4000 dilution. The blots were also probed using a horseradish peroxidase-conjugated β-actin antibody (Abcam, catalog no. ab197277) at a 1:5000 dilution as a loading control. Protein bands were visualized using chemiluminescence (BioRad, Hercules, CA) detection on either an ImageQuant LAS 4000 (GE Healthcare Life Sciences, Piscataway, NJ) or X-ray film (Denville Scientific). Quantification of the bands was performed using ImageJ software.
Statistics.
Statistical analyses were performed using Prism software (GraphPad, San Diego, CA) where appropriate. To evaluate the statistical significance of differences between measured IC50 and EC50 values, 95% confidence intervals were used. In the case of single-dose pol activity assays, a two-tailed Student’s t test was used to calculate P values.
RESULTS AND DISCUSSION
Structure–Activity Studies Identify PNR-7-02 as a Potent Inhibitor of hpol η.
We utilized a fluorescence-based pol assay developed by researchers at NIH to evaluate a library of 85 indole barbituric acid (IBA) and indole thiobarbituric acid (ITBA) derivatives for inhibition of hpol η (Figure 1A,B).26 We originally identified 5-((1-(2-bromobenzoyl)-5-chloro-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (PNR-3-51) as an inhibitor of hpol η activity.17 The first compound identified (PNR-3-51, referred to as ITBA 12 in our previous publication) inhibited hpol η with an IC50 value of near 30 μM with less than 2-fold specificity for hpol η over other TLS and DNA repair pols. We went on to show that changing the N-substituent to a naphthoyl moiety (PNR-3-84, referred to as ITBA 16 in our previous publication) reduced the IC50 against hpol η to ~15 μM and that there was, again, about 2-fold specificity for hpol η over other pols (hpol κ IC50 = 33, μM).
Figure 1.
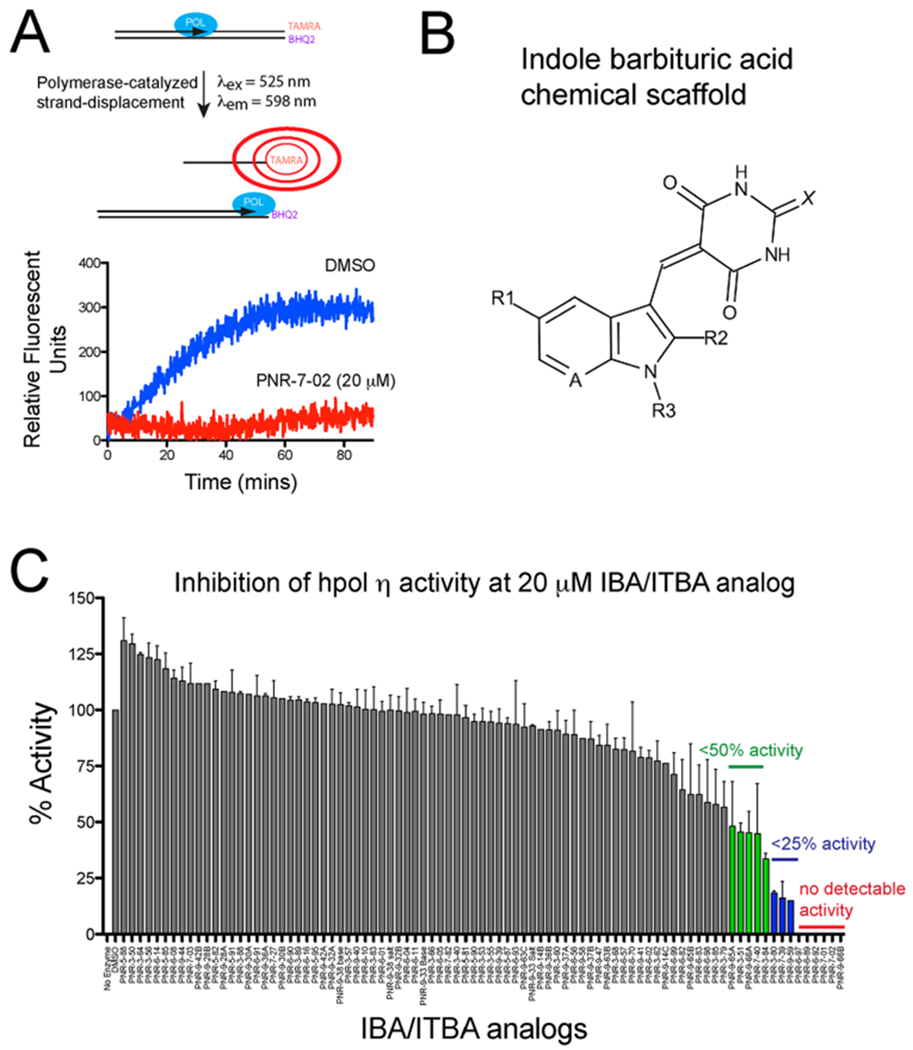
Structure–function studies identified PNR-7-02 as an inhibitor of hpol η with improved potency relative to other indole-derived compounds. (A) Schematic illustration of the pol activity assay. (B) General chemical structure of the parent indole barbituric acid (IBA) scaffold that incorporates all analogues tested for inhibition of TLS pol action. The availability of multiple sites of modification (R1–R3, A, and X positions are noted on the parent scaffold) allowed a useful structure–activity study to be performed. (C) Catalytic activity of hpol η (2.5 nM) was measured in the presence of 85 IBA and indole thiobarbituric acid (ITBA) derivatives (20 μM) to identify structure–activity relationships important for inhibition of this TLS pol. The results represent the mean (±SD) for experiments performed in triplicate. See Supporting Information Table S1 for the chemical structure and details regarding pol activity for each compound.
Since our first publication, we have examined an additional 77 IBA/IBTA derivatives and 8 indole quinuclidinones for activity against hpol η (Figure 1C and Supporting Information Table S1). From these experiments, several molecules were found to exhibit potency similar to or greater than that of PNR-3-84 (i.e., IC50 < 15 μM). Of the inhibitors identified in the pool of 85 compounds tested for activity against hpol η, 19 out of the top 20 were found to possess N-substituted naphthyl moieties on the indole ring (Table S1). Eleven out of the top 20 inhibitors possessed a 5-chloroindole substitution (Table S1). One of these compounds, PNR-7-02 was found to inhibit hpol η with an IC50 of 8 μM (Figure 2A,B). PNR-7-02 retains the 5-chloroindole moiety, but the substitution at the indolic N-position is a 2-naphthylmethylene moiety rather than the 2-naphthoyl substitution we reported previously with PNR-3-84. Thus, replacing a carbonyl group between the indole and naphthalene rings with a methylene moiety serves to reduce the IC50 against hpol η by 2-fold.
Figure 2.
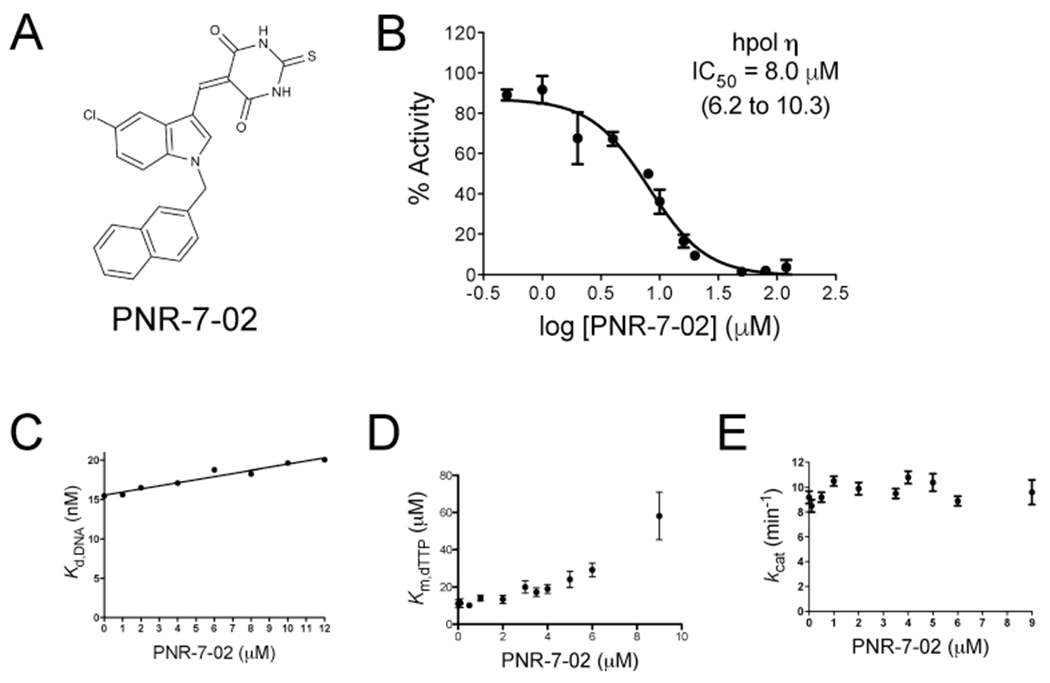
PNR-7-02 inhibits hpol η activity. (A) Chemical structure of PNR-7-02. (B) Dose–response curve for PNR-7-02 inhibition of hpol η. The IC50 value reported represents the best fit of the data (95% confidence interval). PNR-7-02 inhibits dNTP binding to hpol η through a partial competitive mechanism of action. (C) Kd,DNA for hpol η binding to primer–template DNA plotted as a function of PNR-7-02 concentration. (D) Michaelis constant (KM,dTTP) for hpol η activity exhibits a nonlinear increase as a function of PNR-7-02 concentration. (E) Turnover number (kcat) does not change at concentrations of PNR-7-02 at or below the IC50 for inhibition of hpol η activity. The results in panels B–E represent the mean (±SD) for three independent experiments.
We measured the IC50 value for the 10 most potent compounds identified in the single-dose assay (Table 1). In addition to having the N-substituted naphthyl ring system, 7 out of the 10 most potent compounds possessed the 5-chloroindole moiety. Replacing the thiobarbituric acid moiety of PNR-7-02 with a barbituric acid group (PNR-7-01) does not alter the IC50 value against hpol η. Changing the position of attachment for the naphthyl moiety from 2-naphthyl for PNR-7-02 to 1-naphthyl for PNR-6-89 does not significantly alter the IC50 value either. Replacing the indole moiety with a 7-azaindole moiety also does little to either increase or decrease the inhibitory properties against hpol η (Table 1, compare PNR-7-02 with PNR-9-66B), whereas introducing a methyl group at the 2-position of the indole ring (Table 1 compare PNR-7-02 with PNR-7-39) affords a 2-fold decrease in inhibitory potency.
Table 1.
IC50 Values for the Top 10 IBA/ITBA Inhibitors of hpol ηa
| compound ID | A | X | R1 | R2 | R3 | IC50 (μM) | 95% confidence interval |
|---|---|---|---|---|---|---|---|
| PNR-7-02 | CH | S | Cl | H | CH2-2-naphthyl | 8 | 7–11 |
| PNR-6-92 | CH | S | Br | H | CH2-1-naphthyl | 8 | 8–10 |
| PNR-7-01 | CH | O | Cl | H | CH2-2-naphthyl | 9 | 8–11 |
| PNR-6-89 | CH | S | Cl | H | CH2-1-naphthyl | 11 | 10–12 |
| PNR-6-97 | CH | O | H | H | CH2-2-naphthyl | 12 | 11–14 |
| PNR-9-66B | N | S | Cl | H | CH2-2-naphthyl | 12 | 11–13 |
| PNR-3-84b | CH | S | Cl | H | CO-1-naphthyl | 16 | 15–18 |
| PNR-3-80b | CH | S | Cl | H | CO-2-naphthyl | 17 | 14–19 |
| PNR-7-39 | CH | S | Cl | CH3 | CH2-1-naphthyl | 18 | 17–19 |
| PNR-3-79b | CH | S | H | H | CO-2-naphthyl | 25 | 22–27 |
See Figure 1B in the main text for notation of substitution positions. All IC50 values represent the mean (n = 3), and the 95% confidence interval was derived from the fit of the data to a four-parameter logistic model.
These compounds were identified in our previous publication.17 IC50 values were redetermined to ensure reproducibility.
DNA Binding by hpol η Is Not Impaired in the Presence of PNR-7-02.
The ability of PNR-7-02 to interfere with hpol η DNA binding was tested by measuring the equilibrium dissociation constant (Kd,DNA) using fluorescence anisotropy. The concentration of inhibitor was varied (1–12 μM). The Kd,DNA value for hpol η binding to DNA changes less than 20% even at values above the IC50 for PNR-7-02-mediated inhibition of pol activity (Figure 2C). From these results, we concluded that hpol η was able to effectively form binary complexes in the presence of the ITBA inhibitor. Our findings with PNR-7-02 are comparable to those obtained with the indole-based pol inhibitor MK-886 and the benzimidazole-containing TLS pol inhibitor candesartan cilexetil, both of which were found to have a negligible impact on pol binding to DNA.25,27 We next went on to investigate the effect of PNR-7-02 on ternary complex formation by hpol η.
PNR-7-02 Acts To Inhibit hpol η through a Partial Competitive Mechanism of Action.
Lack of an effect on DNA binding by hpol η led us to investigate whether PNR-7-02 interfered with dNTP binding and formation of a ternary complex. Steady-state kinetic analysis of hpol η activity was performed to examine the mechanism of inhibition by PNR-7-02. Previous results from kinetic analyses with PNR-3-51-mediated inhibition of hpol η were suggestive of a partial competitive mechanism of action, which was consistent with in silico docking results placing the ITBA inhibitors near sites that would not be predicted to compete directly with the incoming dNTP.17 We measured the turnover number (kcat) and the Michaelis constant (KM,dTXP) for hpol η catalysis in the presence of nine different concentrations of inhibitor (0–9 μM PNR-7-02). As with PNR-3-51, we observed a nonlinear increase in the KM,dTTP as a function of increasing concentration of PNR-7-02 (Figure 2D). There was no change in kcat at concentrations of PNR-7-02 near or below the IC50 value for inhibition of hpol η (Figure 2E). The nonlinear increase in KM,dTTP is consistent with a partial competitive mechanism of action for PNR-7-02 against hpol η.
PNR-7-02 Inhibits hpol η, hRev1, and hpol λ with Equal Potency.
We measured the IC50 value for PNR-7-02 against the other three Y-family members to elucidate specificity for hpol η. These IC50 values were determined using a gel-based pol assay, owing to the fact that hRev1 is a nonprocessive, cytidyl transferase and not readily amenable to the fluorescence-based pol assay. First, we found that the IC50 value for PNR-7-02 inhibition of hpol η determined using the gel-based assay (8 μM) was identical to that measured with the fluorescence assay (Figure 3A, compare results for hpol η with those shown in Figure 2B). The IC50 value for PNR-7-02-mediated inhibition of hRev1 was also 8 μM (Figure 3A). There was an approximate 3-fold increase in the IC50 value for PNR-7-02 against hpols η and κ, indicative of less potency against these two Y-family members (Figure 3A). It is worth noting here that hpol η and hRev1 have both been implicated in bypassing damage incurred by platinum-derived drugs.28
Figure 3.
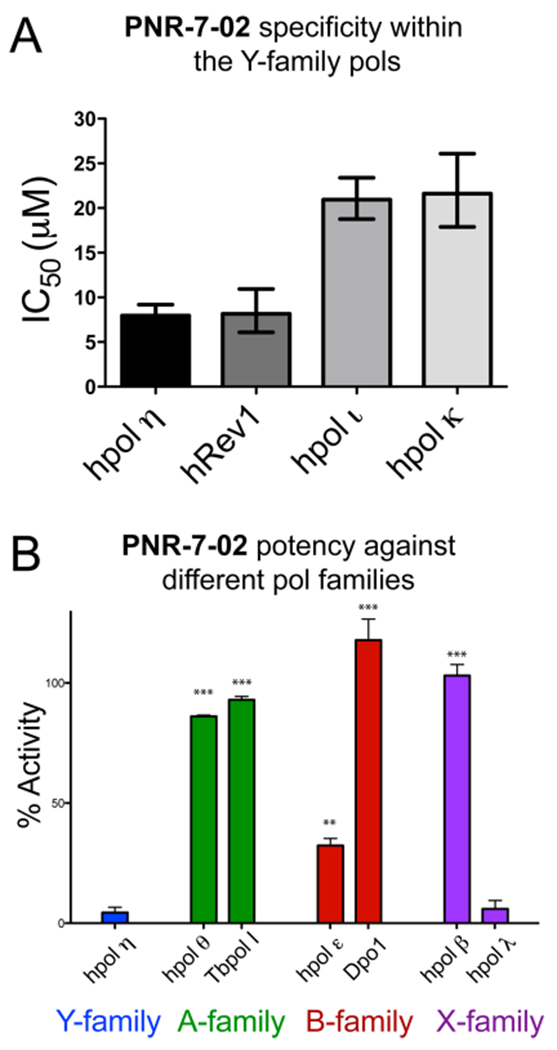
PNR-7-02 inhibits hpol η, hRev1, and hpol λ with similar potency. (A) IC50 value for PNR-7-02-mediated inhibition of the four human Y-family pols measured using gel-based analysis of pol activity. The IC50 values were 8 (7–9) μM for hpol η, 8 (6–11) μM for hRev1, 21 (19–23) μM for hpol β, and 22 (18–26) μM for hpol κ, where the values reported represent the best-fit value (95% confidence interval) derived from the results of three independent experiments. (B) Pol activity measured in the presence of PNR-7-02 (20 μM) using the fluorescence-based assay. Pol activity was 4.4 (±3.8)% for hpol η, 86 (±1)% for hpol θ, 93 (±2)% for TbPol I, 32 (±5)% for hpol ε, 118 (±15)% for Dpo1, 103 (±8)% for hpol β, and 5.9 (±2.1)% for hpol λ. The values reported represent the mean (±SD) activity relative to the DMSO control for three independent experiments. A two-tailed Student’s t test was used to compare hpol η activity in the presence of PNR-7-02 to that observed for each of the other pols (** = P < 0.001, *** = P < 0.0001).
We next tested the activity of pols from A-, B-, and X-families in the presence of 20 μM PNR-7-02 (Figure 3B). The A-family members hpol θ and Tb Pol I had 86 (±1)% and 93 (±2)% activities in the presence of 20 μM PNR-7-02, respectively (Figure 3B), where the reported values represent the mean (±SD) for three replicates. In contrast to similar effects on A-family members, we observed some differences in the effect of PNR-7-02 on two B-family pols from disparate organisms. Catalysis by the archaeal B-family pol Dpo1 from Sulfolobus solfataricus was not affected at all by PNR-7-02, whereas the activity of the hpol ε catalytic core was decreased to 32 (±5)% in the presence of 20 μM PNR-7-02 (Figure 3B). By way of comparison, hpol η activity is <5% at this concentration of inhibitor, which suggests that PNR-7-02 retains a 5–10-fold specificity for hpol η over the leading strand enzyme, hpol ε.29,30 Finally, PNR-7-02 exerted no effect on the X-family member hpol β, but it strongly inhibited catalysis by hpol λ (Figure 3B). We went on to measure an IC50 value of 8 (7–9) μM for PNR-7-02 against hpol λ (where the value represents the best-fit value and 95% confidence interval obtained from three replicates; see Figure S1 for results), which is similar to the IC50 values measured for PNR-7-02 against hpol η and hRev1.
PNR-7-02 Protects the L-Helix in the Little Finger Domain of hpol η from p-Hydroxyphenylglyoxal (HPG) Modification.
We previously identified the cleft between the little finger and finger domains of hpol η as a potential binding site for ITBA derivatives using molecular docking.17 Yet, experimental evidence to support this in silico prediction has hitherto been lacking. We performed a set of mass spectrometry (MS)-based protein footprinting experiments in an effort to identify the PNR-7-02 binding site on hpol η. The arginine-reactive probe HPG was incubated with hpol η alone or in the presence of two concentrations of PNR-7-02 (20 and 100 μM). Following quenching of the reaction with excess L-arginine, the modified protein was digested with trypsin and the resulting peptides were analyzed by LC-MS/MS (Table S2). In order to estimate the fraction of HPG-modified arginines at each site on the protein, the peak area of the parent ion was determined for both unmodified and HPG-modified peptides.
We were able to identify regions of the protein that were less reactive (i.e., “protected”) in the presence of PNR-7-02 by quantifying the extent of HPG modification at each arginine residue (Figure 4). While not every arginine residue was modified with HPG under the conditions used here, there was at least one modified arginine in each of the four domains of hpol η (Figure 4A). The addition of PNR-7-02 completely abrogated HPG modification of Arg351 at both concentrations tested (Figure 4B). Notably, Arg351 resides in the L-helix of the little finger domain and is in close proximity to the binding site previously predicted by molecular docking for PNR-3-84.1 In addition to the protective effects of PNR-7-02, we also observed an interesting “exposure” of several arginine residues in the palm domain when the concentration of PNR-7-02 was increased from 20 to 100 μM (Figure 4A,B).
Figure 4.
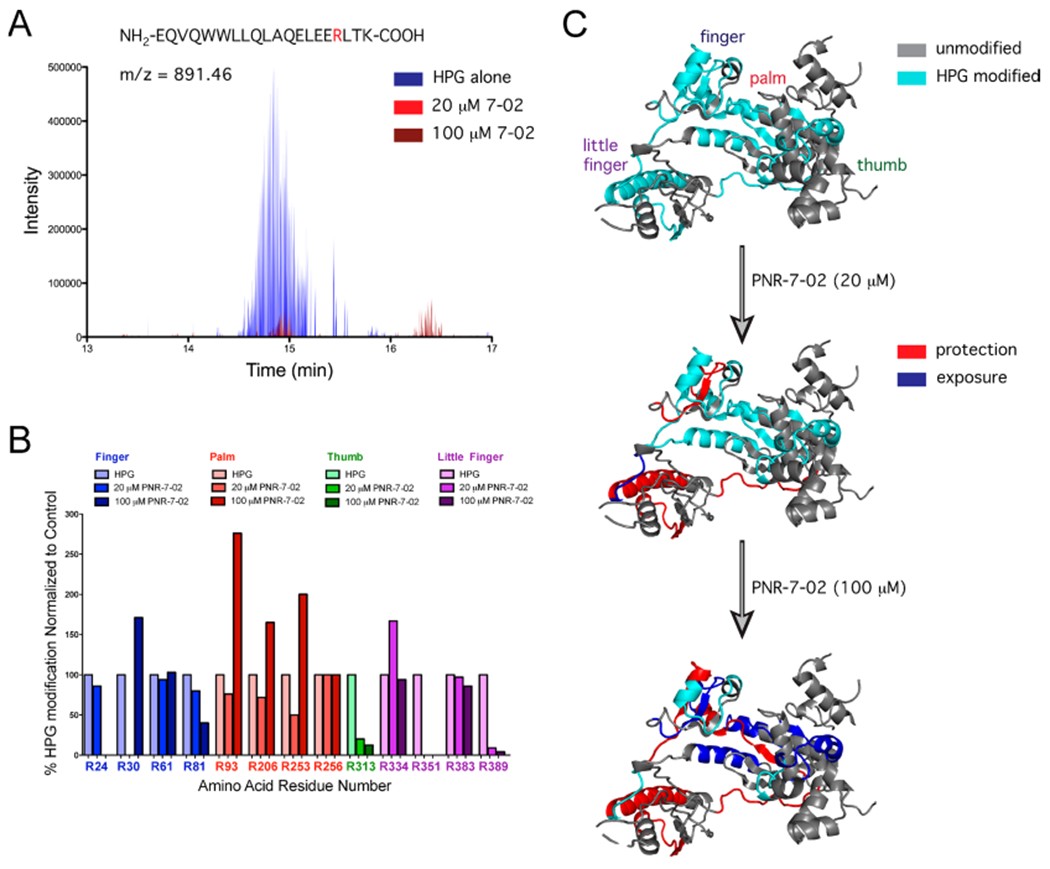
Protein footprinting identifies the PNR-7-02 binding site on the hpol η little finger. (A) Selected ion chromatogram for the HPG-modified peptide containing Arg351 in the presence of HPG alone (blue), HPG + 20 μM PNR-7-02 (red), and 100 μM PNR-7-02 (dark red). (B) Relative abundance of HPG-modified arginine was quantified from MS data. The fraction of HPG-modified arginine observed in the presence of PNR-7-02 was normalized to that observed in the absence of the compound. The modified arginine residues in the finger, palm, thumb, and little finger domains of hpol η are shown in blue, red, green, and purple, respectively. (C) Cartoon representation of hpol η (PDB ID 3MR2) shown with regions of the protein containing HPG-modified residues in cyan and regions that were not modified by HPG in gray. The addition of PNR-7-02 caused changes in HPG reactivity for multiple arginines. Peptides with arginine residues that displayed a greater than 2-fold reduction in HPG reactivity (i.e., protection) are shown in red, whereas regions that exhibited a greater than 2-fold increase in HPG reactivity (i.e., exposure) are shown in blue.
The close proximity of Arg351 to the template DNA provides a molecular rationale for the partial competitive mechanism of inhibition observed for PNR-7-02, as this mechanism of inhibition is associated with the formation of catalytically competent ternary complexes and the ability to tolerate a distorted template is a hallmark of Y-family pol activity. In agreement with this model, the top binding pose for PNR-7-01 and the second most favorable binding pose for PNR-7-02 identified by molecular docking with SwissDOCK were located between the finger and little finger domains of hpol η (Figure 5A,B). The most favorable docking pose for PNR-7-02 was in a cleft between the thumb and palm domains near Arg303 (Figure 5A). While we were able to identify peptides containing Arg303 in the MS data, a modified version of Arg303 was not detected, limiting our ability to measure changes in reactivity due to binding of PNR-7-02 at this site. Nevertheless, the top-binding poses for PNR-3-84 and PNR-7-1 placed these inhibitors near Arg351. The fact that molecular docking results for multiple compounds fit with the both HPG results and the kinetic mechanism of inhibition led us to propose a model where IBA/ITBA analogues inhibit hpol η by binding to the pocket between the finger and little finger domains, interfering with productive formation of ternary complexes (Figure 5C).
Figure 5.
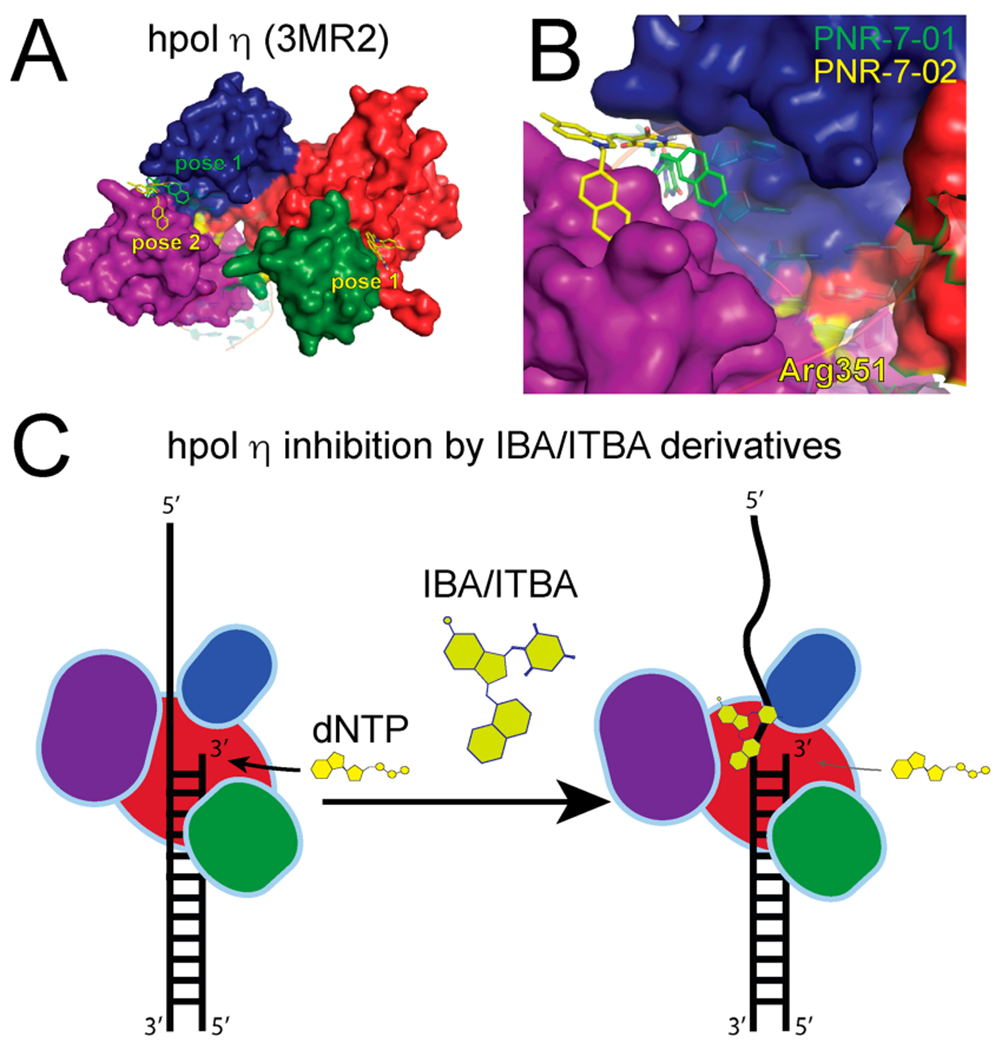
Molecular docking analysis of IBA/ITBA binding and model for inhibition of hpol η. (A) PNR-7-01 and PNR-7-02, the two most potent inhibitors of hpol η, were docked onto the target (PDB code 3MR2) using SwissDock. The finger, palm, thumb, and little finger domains of hpol η are shown in blue, red, green, and purple, respectively, as is the top binding pose for PNR-7-01 (green carbons) and the two top binding poses for PNR-7-02 (yellow carbons). (B) More detailed view of PNR-7-01 and PNR-7-02 (pose 2) binding to the cleft between the finger and little finger domains of hpol η. The location of Arg351 is noted as a yellow patch, and the primer–template DNA, which was removed for docking, is shown superimposed on the target in semitransparent cartoon form. (C) Model depicting the mechanism of hpol η inhibition by IBA/ITBA compounds based on the kinetic analysis, chemical footprinting, and molecular docking results.
PNR-7-02 Acts Synergistically with CDDP To Reduce Cell Viability and Induces γH2AX Formation in a Target-Dependent Manner.
We wanted to test the ability of PNR-7-2 to modulate the effects of CDDP in cell culture. To examine whether PNR-7-02 could exert a target-dependent effect on the antiproliferative effects of CDDP, we used two HAP-1 cell lines derived from the male chronic myeloid leukemia (CML) cell line KBM-7. One of these cell lines was hpol η-proficient (henceforth referred to as the “parental” cell line), whereas the other was edited using CRISPR–Cas9 technology such that the POLH gene was disrupted, knocking out hpol η expression (Figure S2). First, we established the EC50 values for PNR-7-02 in both parental and hpol-deficient cells. PNR-7-02 did not appear to exert hpol η-dependent effects on the viability of the two cell lines, as the EC50 value of PNR-7-02 was 4.6 (3.0–7.0) μM for the hpol η-proficient parental cell line and 5.6 (4.6–6.9) μM for the hpol η-deficient HAP-1 cell line (Figure S3).
Next, we measured the effect of CDDP on cell viability in the presence and absence of PNR-7-02. The EC50 value of CDDP for the parental cell line was 0.31 (0.27–0.37) μM (Figure 6A). Co-treating the hpol η-proficient parental HAP-1 cells with PNR-7-02 decreased the EC50 value >2-fold for both concentrations of inhibitor tested (Figure 6A). We calculated the combination index value for the two treatments using the Chou–Talalay method31 in order to obtain a measure of synergy between CDDP and PNR-7-02. The combination index (CI) value was calculated to be 0.47 and 0.59 for 0.1 and 1 μM PNR-7-02, respectively, indicative of synergy between CDDP and PNR-7-02. The synergy between CDDP and PNR-7-02 was not only limited to HAP-1 cells, as similar results were obtained for the ovarian cancer-derived OVCAR3 cell line. Combined treatment of OVCAR3 cells with CDDP and PNR-7-02 resulted in a CI value of 0.4 (Figure S4).
Figure 6.
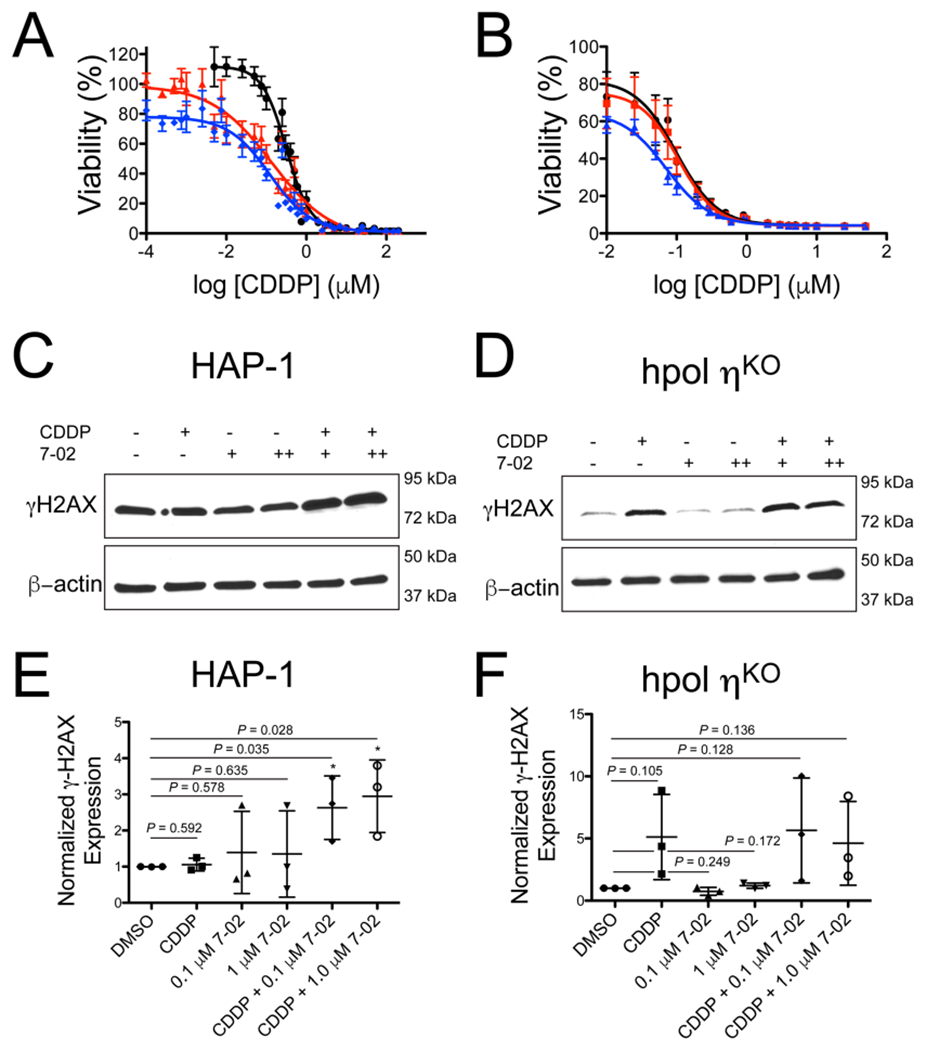
PNR-7-02 potentiates the cytotoxic effects of CDDP in a manner largely dependent on hpol η expression. Cell viability was measured for parental HAP-1 cells expressing hpol η (panel A) and hpol η-deficient cells (panel B) treated with CDDP alone (black circles), CDDP + 0.1 μM PNR-7-02 (red triangles), or CDDP + 1 μM PNR-7-02 (blue diamonds). The values reported represent the mean (±SEM) of n =12 for panel A and 6 for panel B. The EC50 values derived from the dose–response curves for HAP-1 cells expressing hpol η (panel A) were 0.31 (0.27–0.37) for CDDP alone, 0.14 (0.08–0.23) for CDDP + 0.1 μM PNR-7-02, and 0.12 (0.08–0.17) for CDDP + 1 μM PNR-7-02. The EC50 values derived from the dose–response curves for hpol η-deficient cells (panel B) were 0.10 (0.07–0.14) for CDDP alone, 0.10 (0.08–0.15) for CDDP + 0.1 μM PNR-7-02, and 0.07 (0.06–0.09) for CDDP + 1 μM PNR-7-02. The reported EC50 values represent the mean (95% confidence interval). hpol η-proficient HAP-1 (C) and hpol η-deficient cells (D) were treated with either DMSO or CDDP (0.5 μM) in combination with PNR-7-02 (0.1 and 1 μM) at the indicated concentrations. Immunoblotting was performed to assess changes in γH2AX levels. Quantification of γH2AX expression (normalized against β-actin and relative to DMSO-treated cells) is shown for hpol η-proficient HAP-1 (E) and hpol η-deficient knockout cells (F). The mean (±SD) is shown for three biological replicates. A Student’s t test was used to calculate the P values.
We then performed dose-response experiments with CDDP (±PNR-7-02) in hpol η-deficient cells. As expected, hpol η-deficient cells are more sensitive to CDDP than the parental cell line, as evidenced by the fact that loss of hpol η reduces the EC50 value of CDDP ~3-fold (Figure 6B). Adding PNR-7-02 does not have a notable effect on the toxicity of CDDP in hpol η-deficient cells (Figure 6B). The combination index values calculated for hpol η-deficient cells treated with 0.1 and 1 μM PNR-7-02 were 1.02 and 0.90, respectively. The small decrease in the combination index value observed when hpol η-deficient cells were treated with CDDP and 1 μM PNR-7-02 could indicate that another TLS pol involved in bypass of Pt-damage is being targeted. This may be related to the fact that hpol η is an important but not the only means of bypassing unrepaired intrastrand cross-links induced by CDDP. The most likely secondary target of PNR-7-02 is hRev1, as previous studies have shown that the activity of hRev1 (in coordination with pol Ξ) promotes tolerance of damage induced by platinum-based drugs.28,32 These results are consistent with the idea that hpol η is necessary for the synergy between CDDP and PNR-7-02 observed with the parental cell line. Thus, PNR-7-02 is the first small-molecule inhibitor of hpol η to have a target-dependent effect on the cytotoxicity of CDDP.
Next, we examined the effect of combining CDDP with the hpol η inhibitor, PNR-7-02, on genome stability by probing for activation of the DNA damage/replication stress marker μH2AX. The amount of μH2AX did not change substantially in hpol η-proficient HAP-1 cells treated with CDDP alone (Figure 6C). Treating either cell line with PNR-7-02 alone did not change μH2AX formation (Figure 6C,D). Combining CDDP with PNR-7-02 (either 0.1 or 1 μM) in hpol η-proficient HAP-1 cells increased μH2AX levels (Figure 6C). Quantification of μH2AX expression revealed that the combination treatment increased the DNA damage marker about 3-fold in cells expressing hpol η (Figure 6E). Treatment of hpol η-deficient cells with CDDP increased the amount of μH2AX (~5-fold) following exposure to CDDP (Figure 6D,F), consistent with the increased sensitivity of these cells to the DNA damaging effects of CDDP. Similar to the parental HAP-1 cells, treatment of hpol η-deficient cells with PNR-7-02 did not increase μH2AX levels above that of DMSO-treated cells (Figure 6D,F). The combination treatment did not increase the amount of μH2AX formed above that observed for hpol η-deficient cells treated with CDDP alone (Figure 6D,F). From these results, we concluded that the effects of PNR-7-02 on μH2AX formation (and by extension, DNA damage/replication stress) were largely hpol η-dependent in nature.
CONCLUSIONS
Constitutive activation of the replication stress response is a contributing factor in multiple aspects of cancer etiology and resistance to anti-cancer drugs.33–36 During periods of replication stress, specialized enzymes perform TLS past impediments to the fork.37 TLS pols promote the direct bypass of blocks to the replication fork because they harbor active sites that are able to accommodate DNA adducts, distorted template, and non-Watson–Crick base pairing modes.10,11 Not surprisingly, TLS pols can render genotoxic anti-cancer drugs less effective through bypass of lesions incurred following treatment, and they are involved in mutagenesis following exposure to DNA damaging agents, which can also facilitate resistance. All these features make TLS pols attractive targets for the development of new anti-cancer therapies.
Inhibition of DNA repair and replication is an established approach to treating cancer,38 as the elevated expression of DNA repair and damage tolerance proteins can negatively affect patient response to genotoxic drugs. As an example, the Y-family member hpol η is overexpressed in the stem cell niche of ovarian cancer patients, and this overexpression decreases the effectiveness of CDDP, shortening the time to tumor recurrence.8 The ability to slow the development of resistance and increase efficacy could benefit many patients undergoing chemotherapy with platinum-based drugs. Toward this end, we developed a novel chemical scaffold for inhibition of translesion DNA polymerases. In doing so, PNR-7-02 was identified as a reasonably potent inhibitor of hpol η (IC50 = 8 μM). The first step in the pol catalytic cycle, DNA binding, was not strongly influenced by the presence of the inhibitor. Instead, PNR-7-02 increased the Michaelis constant for the incoming dNTP in a nonlinear fashion. From these results, we concluded that PNR-7-02 inhibits hpol η by competing with formation of the ternary complex (i.e., through a partial competitive mechanism of action), consistent with previous findings from our group using a different ITBA analogue. Importantly, PNR-7-02 displayed synergistic effects on cell viability when combined with CDDP, but this effect was limited to hpol η-proficient HAP-1 cells, as PNR-7-02 did not alter the sensitivity of knockout cells to CDDP (Figure 6). The ability of PNR-7-02 to increase the genotoxic effects of CDDP was further validated by immunoblotting for μH2AX. The hpol η-dependent nature of the effect was evidenced by the fact that combining CDDP with PNR-7-02 only increased μH2AX in HAP-1 cells (Figure 6D–F). All these results are consistent with the idea that PNR-7-02 is able to inhibit hpol η activity to a degree that is sufficient to increase the genotoxic effects of CDDP and induce replication stress response/DNA damage response (RSR/DDR) activation.
There are only a handful of instances where small molecules have been used to target enzymes involved in TLS.17,26,27 Inhibitors of the repair enzyme hpol λ have been identified, refined, and shown to modulate the cytotoxic effects of temozolomide and H2O2 in colorectal cancer cells.39,40 A recent study illustrated nicely that inhibiting monoubiquitination of PCNA, an important means of recruiting TLS pols to the replication fork, with a small molecule (T2AA) resulted in greater sensitivity to CDDP for HeLa and U2OS cells.41 Another study found that nucleotides with altered hydrogen bonding capacity are able to reduce DNA synthesis by hpol η at sites of intrastrand cisplatin adducts, but to the best of our knowledge, there are no small-molecule inhibitors of hpol η that have been shown to exert a target-dependent effect on an anti-cancer drug in human cells. Our results with PNR-7-02 represent an important step forward in developing a small-molecule inhibitor of a clinically relevant TLS pol that is involved in promoting resistance to drugs, such as CDDP. Moreover, hpol η expression in the ovarian cancer stem cell niche has been shown to increase resistance of this subpopulation of tumor cells to CDDP. In this capacity, successful inhibition of hpol η could render platinum-based treatments more effective by (i) eliminating the tumor cell population largely responsible for tumor relapse and (ii) suppressing the emergence of chemo-resistant tumor cells.
In summary, we have developed a novel small-molecule inhibitor of hpol η, an enzyme that impacts patient responses to platinum-based anti-cancer drugs. This compound, PNR-7-02, appears to disrupt the active site of hpol η by binding to the little finger domain near the template DNA, and it is a much stronger block to TLS pol activity than normal replicative pols. Moreover, PNR-7-02 is capable of potentiating the cytotoxic effects of CDDP in a target-dependent manner by inducing RSR/DDR components and diminishing the viability of tumor cells. The targeted inhibition of hpol η as an adjuvant to chemotherapeutic regimens could enhance patient response and limit time to relapse through depletion of the tumor stem cell population.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by U.S.P.H. Service grants CA183895 (R.L. Eoff) and GM109005 and PO1/NIA/NIH-P01AG012411 (P.A Crooks), as well as an Arkansas Research Alliance (ARA) Scholar award (P.A. Crooks).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b01176.
Summary of IBA/ITBA chemical structures and the inhibition of DNA polymerase η; hpol η-derived peptides identified by mass spectrometry; determination of the IC50 value for PNR-7-02 against hpol λ; validation of hpol η knockout in HAP-1 cells; determination of EC50 values for PNR-7-02 in hpol η-proficient and hpol η-deficient HAP-1 cells; PNR-7-02-mediated sensitization of ovarian cancer-derived cells to CDDP (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, and Wood RD (2006) DNA repair: from molecular mechanism to human disease. DNA Repair 5, 986–996. [DOI] [PubMed] [Google Scholar]
- (2).Chang DJ, and Cimprich KA (2009) DNA damage tolerance: when it’s OK to make mistakes. Nat. Chem. Biol 5, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Xie K, Doles J, Hemann MT, and Walker GC (2010) Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci U. S. A 107, 20792–20797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Albertella MR, Green CM, Lehmann AR, and O’Connor MJ (2005) A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 65, 9799–9806. [DOI] [PubMed] [Google Scholar]
- (5).Albertella MR, Lau A, and O’Connor MJ (2005) The overexpression of specialized DNA polymerases in cancer. DNA Repair 4, 583–593. [DOI] [PubMed] [Google Scholar]
- (6).Ceppi P, Novello S, Cambieri A, Longo M, Monica V, Lo Iacono M, Giaj-Levra M, Saviozzi S, Volante M, Papotti M, and Scagliotti G (2009) Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res 15, 1039–1045. [DOI] [PubMed] [Google Scholar]
- (7).Lemee F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire M-J, Bieth A, Gentil C, Baker L, Martin A-L, Leduc C, Lam E, Magdeleine E, Filleron T, Oumouhou N, Kaina B, Seki M, Grimal F, Lacroix-Triki M, Thompson A, Roche H, Bourdon J-C, Wood RD, Hoffmann J-S, and Cazaux C (2010) DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. U. S. A 107, 13390–13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, Zhao W, Zhang X, Yu J, and Wang Q-E (2015) Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. U. S. A 112, 4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang H, Wu W, Wang H-W, Wang S, Chen Y, Zhang X, Yang J, Zhao S, Ding H-F, and Lu D (2010) Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro-Oncol 12, 679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sale JE, Lehmann AR, and Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol 13, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang W, and Woodgate R (2007) What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S. A 104, 15591–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, and Cordeiro-Stone M (2004) The role of DNA polymerase eta in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 64, 6469–6475. [DOI] [PubMed] [Google Scholar]
- (13).Chen Y, Cleaver JE, Hanaoka F, Chang C, and Chou K (2006) A novel role of DNA polymerase eta in modulating cellular sensitivity to chemotherapeutic agents. Mol. Cancer Res 4, 257–265. [DOI] [PubMed] [Google Scholar]
- (14).Sokol AM, Cruet-Hennequart S, Pasero P, and Carty MP (2013) DNA polymerase η modulates replication fork progression and DNA damage responses in platinum-treated human cells. Sci. Rep 3, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhao Y, Biertümpfel C, Gregory MT, Hua Y-J, Hanaoka F, and Yang W (2012) Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. U. S. A 109, 7269–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Teng K, Qiu M, Li Z, Luo H, Zeng Z, Luo R, Zhang H, Wang Z, Li Y, and Xu R (2010) DNA polymerase η protein expression predicts treatment response and survival of metastatic gastric adenocarcinoma patients treated with oxaliplatin-based chemotherapy. J. Transl Med 8, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Coggins GE, Maddukuri L, Penthala NR, Hartman JH, Eddy S, Ketkar A, Crooks PA, and Eoff RL (2013) N-Aroyl indole thiobarbituric acids as inhibitors of DNA repair and replication stress response polymerases. ACS Chem. Biol 8, 1722–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Penthala NR, Ketkar A, Sekhar KR, Freeman ML, Eoff RL, Balusu R, and Crooks PA (2015) 1-Benzyl-2-methyl-3-indolylmethylene barbituric acid derivatives: Anti-cancer agents that target nucleophosmin 1 (NPM1). Bioorg. Med. Chem 23, 7226–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sonar VN, Thirupathi Reddy Y, Sekhar KR, Sasi S, Freeman ML, and Crooks PA (2007) Novel substituted (Z)-2-(N-benzylindol-3-ylmethylene)quinuclidin-3-one and (Z)-(±)-2-(N-benzylindol-3-ylmethylene)quinuclidin-3-ol derivatives as potent thermal sensitizing agents. Bioorg. Med. Chem. Lett 17, 6821–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Eddy S, Ketkar A, Zafar MK, Maddukuri L, Choi J-Y, and Eoff R L. (2014) Human Rev1 polymerase disrupts G-quadruplex DNA. Nucleic Acids Res. 42, 3272–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zang H, Goodenough AK, Choi J-Y, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, and Guengerich FP (2005) DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem 280, 29750–29764. [DOI] [PubMed] [Google Scholar]
- (22).Choi J-Y, Eoff RL, Pence MG, Wang J, Martin MV, Kim E-J, Folkmann LM, and Guengerich FP (2011) Roles of the four DNA polymerases of the crenarchaeon Sulfolobus solfataricus and accessory proteins in DNA replication. J. Biol. Chem 286, 31180–31193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hogg M, Seki M, Wood RD, Doublie S, and Wallace SS (2011) Lesion bypass activity of DNA polymerase θ (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J. Mol. Biol 405, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Arrigo CJ, Singh K, and Modak MJ (2002) DNA polymerase I of Mycobacterium tuberculosis: functional role of a conserved aspartate in the hinge joining the M and N helices. J. Biol. Chem 277, 1653–1661. [DOI] [PubMed] [Google Scholar]
- (25).Ketkar A, Zafar MK, Maddukuri L, Yamanaka K, Banerjee S , Egli M, Choi J-Y, Lloyd RS, and Eoff RL (2013) Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity. Chem. Res. Toxicol 26, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dorjsuren D, Wilson DM, Beard WA, McDonald JP, Austin CP, Woodgate R, Wilson SH, and Simeonov A (2009) A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 37, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yamanaka K, Dorjsuren D, Eoff RL, Egli M, Maloney DJ, Jadhav A, Simeonov A, and Lloyd RS (2012) A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase κ. PLoS One 7, e45032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q Glover TW, and Canman CE (2010) Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol. Cell. Biol 30, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shinbrot E, Henninger EE, Weinhold N, Covington KR, Göksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, Sander C, Pursell ZF, and Wheeler DA (2014) Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 24, 1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pursell ZF, Isoz I, Lundstrom E-B, Johansson E, and Kunkel TA (2007) Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317, 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chou TC, and Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul 22, 27–55. [DOI] [PubMed] [Google Scholar]
- (32).Lin X, Okuda T, Trang J, and Howell SB (2006) Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol 69, 1748–1754. [DOI] [PubMed] [Google Scholar]
- (33).Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, 0rntoft T, Lukas J, and Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. [DOI] [PubMed] [Google Scholar]
- (34).Bartkova J, Hamerlik P, Stockhausen M-T, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, Lukas J, and Bartek J (2010) Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 29, 5095–5102. [DOI] [PubMed] [Google Scholar]
- (35).Hills SA, and Diffley JFX (2014) DNA replication and oncogene-induced replicative stress. Curr. Biol 24, R435–444. [DOI] [PubMed] [Google Scholar]
- (36).Zeman MK, and Cimprich KA (2014) Causes and consequences of replication stress. Nat. Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sale JE (2013) Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harbor Perspect. Biol 5, a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jackson SP, and Helleday T (2016) Drugging DNA repair. Science 352, 1178–1179. [DOI] [PubMed] [Google Scholar]
- (39).Strittmatter T, Brockmann A, Pott M, Hantusch A, Brunner T , and Marx A (2014) Expanding the scope of human DNA polymerase λ and β inhibitors. ACS Chem. Biol 9, 282–290. [DOI] [PubMed] [Google Scholar]
- (40).Strittmatter T, Bareth B, Immel TA, Huhn T, Mayer TU , and Marx A (2011) Small Molecule Inhibitors of Human DNA Polymerase λ. ACS Chem. Biol 6, 314–319. [DOI] [PubMed] [Google Scholar]
- (41).Inoue A, Kikuchi S, Hishiki A, Shao Y, Heath R, Evison BJ, Actis M, Canman CE, Hashimoto H, and Fujii N (2014) A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA crosslink, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem 289, 7109–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.