

SUMMARY
5-Hydroxymethylcytosine (5hmC) binding, ES-cell-specific (HMCES) crosslinks to apurinic or apyrimidinic (AP, abasic) sites in single-strand DNA (ssDNA). To determine whether HMCES responds to the ssDNA abasic site in cells, we exploited the activity of apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3A (APOBEC3A). APOBEC3A preferentially deaminates cytosines to uracils in ssDNA, which are then converted to abasic sites by uracil DNA glycosylase. We find that HMCES-deficient cells are hypersensitive to nuclear APOBEC3A localization. HMCES relocalizes to chromatin in response to nuclear APOBEC3A and protects abasic sites from processing into double-strand breaks (DSBs). Abasic sites induced by APOBEC3A slow both leading and lagging strand synthesis, and HMCES prevents further slowing of the replication fork by translesion synthesis (TLS) polymerases zeta (Polζ) and kappa (Polκ). Thus, our study provides direct evidence that HMCES responds to ssDNA abasic sites in cells to prevent DNA cleavage and balance the engagement of TLS polymerases.
In Brief
Mehta et al. use APOBEC3A to demonstrate that HMCES responds to ssDNA abasic sites in cells and prevents replication fork collapse. APOBEC3A-induced abasic sites slow both leading and lagging strand polymerization, and HMCES engagement prevents further fork slowing because of the action of TLS polymerases zeta (Polζ) and kappa (Polκ).
Graphical Abstract
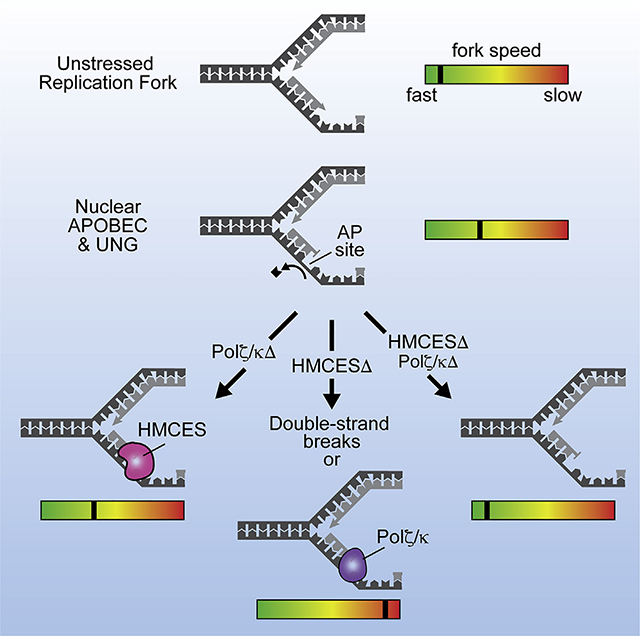
INTRODUCTION
Abasic sites, also known as apurinic or apyrimidinic sites (AP sites), occur between 10,000 and 50,000 times per cell per day. Base loss generating an abasic site can occur spontaneously or in response to DNA damage induced by endogenous and exogenous sources, including both oxidative and alkylation base damage. Abasic site repair in double-strand DNA (dsDNA) is performed primarily by base-excision repair (BER) using AP endonucleases (Boiteux and Guillet, 2004; Thompson and Cortez, 2020). During DNA replication, abasic sites that escape BER will stall the replicative polymerases, generating a single-strand DNA (ssDNA) abasic site. Replication-associated ssDNA abasic lesions can also form directly on the lagging-strand template because of cytosine deamination and removal by uracil N-glycosylase (UNG). These lesions have substantial mutagenic potential because they are often bypassed by translesion synthesis (TLS) polymerases. In addition, they are prone to undergoing β-elimination reactions or cleavage by endonucleases that result in DNA breaks (Talpaert-Borlé, 1987; Thompson and Cortez, 2020). Thus, the action of BER enzymes, such as AP endonuclease-1 and −2 (APEX1 and APEX2), would be deleterious in the context of ssDNA.
5-Hydroxymethylcytosine (5hmC) binding, ES-cell-specific (HMCES), travels with replication forks (Mohni et al., 2019), binds proliferating cell nuclear antigen (PCNA) and ssDNA (Mohni et al., 2019), and uses an N-terminal cysteine to form a DNA-protein crosslink (DPC) via a thiazolidine linkage with ssDNA abasic sites (Halabelian et al., 2019; Thompson et al., 2019; Wang et al., 2019). This linkage can be made at an abasic site positioned at a junction of ssDNA and dsDNA with a free 3′ terminus, similar to what would be formed when a polymerase stalls at an abasic site in template DNA (Thompson et al., 2019). HMCES-deficient cells are hypersensitive to ultraviolet radiation (UV), ionizing radiation (IR), the alkylating agent methyl methanesulfonate (MMS), and the oxidizing agent potassium bromate (Mohni et al., 2019). These DNA-damaging agents result in multiple types of lesions, but all can generate abasic sites. Furthermore, a HMCES DPC forms in cells treated with these agents. Thus, we hypothesized that HMCES is a shield for ssDNA abasic sites to prevent DNA cleavage and perhaps shunt ssDNA abasic site processing through a less mutagenic pathway (Mohni et al., 2019). Other studies suggest that HMCES is an epigenetic reader of 5-hydroxymethylcytosine, a cysteine protease, and an alternative non-homologous end-joining repair factor (Aravind et al., 2013; Kweon et al., 2017; Shukla et al., 2020; Spruijt et al., 2013). How these functions relate to an activity in abasic site processing is unclear.
Because IR, UV, MMS, and potassium bromate only indirectly induce abasic sites and primarily generate other types of lesions that cause mutations and cell lethality, we sought a more-specific approach to testing the hypothesis that HMCES initiates an ssDNA abasic-site-repair mechanism. In this study, we used the cytidine deaminase APOBEC3A to test that idea. APOBEC proteins preferentially deaminate cytosines in ssDNA (Harris and Liddament, 2004). Thus, when aberrantly expressed in cancers, they target the lagging DNA template strand during replication and generate mutational signatures caused by misincorporation across from uracils and abasic sites (Burns et al., 2013; Haradhvala et al., 2016; Hoopes et al., 2016; Rebhandl et al., 2015; Seplyarskiy et al., 2016).
We find that HMCES-deficient cells are hypersensitive to nuclear APOBEC3A localization. HMCES prevents the processing of APOBEC3A-induced lesions into DSBs, supporting the idea that HMCES shields ssDNA abasic sites from aberrant processing to maintain genome integrity. In addition, we find that abasic sites induced by APOBEC3A slow replication elongation. The APOBEC3A-induced slowing is exacerbated by HMCES deficiency and abrogated when both TLS polymerization and HMCES are in-activated. Thus, HMCES facilitates fork progression that otherwise is slowed by TLS polymerase engagement at abasic sites.
RESULTS
HMCES-Deficient Cells Are Hypersensitive to ssDNA Abasic Site Induction
To test the hypothesis that HMCES responds directly to ssDNA abasic sites, we generated cell lines (hTERT-RPE-1 and HCT116) stably expressing GFP-tagged APOBEC3A fused to the mutant estrogen receptor (GFP-APOBEC3A-ERT2; Figure S1A). This system allows for rapid translocation of APOBEC3A into the nucleus upon treatment with 4-hydroxytamoxifen (4-OHT) (Figure S1B). In contrast to previous studies that used 24–48 h of transcriptional induction to achieve APOBEC3A and APOBEC3B overexpression (Burns et al., 2013; Landry et al., 2011), the localization system allows us to examine the consequences of nuclear APOBEC3A activity in minutes to hours. Once in the nucleus, APOBEC3A catalyzes the deamination of cytosine to uracil in ssDNA. The deaminated base is then removed by UNG, generating an abasic site (Stenglein et al., 2010).
In both hTERT-RPE-1 epithelial and HCT116 colorectal cancer cell lines, 4-OHT-induced nuclear localization of APOBEC3A led to a modest decrease in viability that was exacerbated by inactivating HMCES (Figures 1A–1C, S1C, and S1D). HMCES depletion, even in the absence of 4-OHT, also reduced the viability of the hTERT-RPE-1-APOBEC3A cells. This may indicate a greater dependency on HMCES in those cells or reflect some leakiness of the 4-OHT-regulated APOBEC3A fusion protein. Thus, HMCES promotes cell survival in response to nuclear APOBEC3A.
Figure 1. HMCES-Deficient Cells Are Hypersensitive to Nuclear APOBEC3A Expression, Which Promotes HMCES Localization to the Chromatin.

(A) Representative images of a colony-viability assay in hTERT-RPE-1 GFP-APOBEC3A-ERT2 cells transfected with non-targeting control (NT) or HMCES (H-1 and H-2) siRNAs and treated with 4-OHT for 24 h.
(B and C) Quantitation of clonogenic survival assays for RPE A3A (B) and HCT116 A3A (C). Means and SD of 3 experiments. p values were derived from an ANOVA with Holm-Sidak post-test.
(D and E) Quantitation of the insoluble HMCES PLA signal after 4-OHT treatment in EdU-positive (D) and EdU-negative (E) hTERT-RPE-1 cells, with and without stable expression of GFP-APOBEC3A-ERT2. (−1°, no primary HMCES antibody). Each data point represents the HMCES-PLA-integrated nuclear intensity in one cell. Bars represent the median, and p values were derived from an ANOVA with Dunn’s multiple comparisons post-test. See also Figures S1 and S2.
APOBEC3A Induces Re-localization of HMCES to Chromatin
HMCES-deficient cells are hypersensitive to DNA-damaging agents that promote HMCES localization to chromatin and DPC formation (Mohni et al., 2019). Because HMCES-deficient cells are hypersensitive to nuclear APOBEC3A, we wanted to determine whether APOBEC3A also produced a change in HMCES localization. Standard immunofluorescence imaging does not yield strong HMCES staining in cells extracted with detergent to remove soluble HMCES, whether or not they are treated with DNA-damaging agents. Therefore, we applied a high-content microscopy methodology based on the proximity ligation assay (PLA) to detect insoluble, chromatin-bound HMCES and first validated this method with UV radiation. Cells were irradiated, allowed to recover for 4 h, and soluble HMCES protein was removed by detergent pre-extraction before fixation. The amount of insoluble HMCES remaining was then quantified by PLA (Figure S2A). Consistent with our previous chromatin fractionation and rapid approach to DNA adduct recovery (RADAR) data (Mohni et al., 2019), this assay measures elevated levels of insoluble HMCES after exposure to UV radiation specifically in S-phase (EdU-positive) cells (Figures S2B and S2C). The HMCES PLA signal can be further increased upon incubation with the proteasome inhibitor MG132 (Figures S2B and S2D), which was shown to increase HMCES-DPC levels after UV radiation (Mohni et al., 2019). As expected, the HMCES PLA signal is not observed in HMCES knockout cells (HMCESD) or cells processed without a primary antibody (Figures S2B and S2D).
We applied this assay to hTERT-RPE-1 cells with and without stable expression of APOBEC3A. The HMCES PLA signal is slightly elevated in S-phase cells expressing APOBEC3A compared with the parental cell line before the addition of 4-OHT and is further increased after 4-OHT treatment to induce nuclear APOBEC3A accumulation (Figures 1D and S2E). The elevation of signal before the addition of 4-OHT may again indicate some leakiness of the APOBEC3A fusion protein causing a low level of nuclear APOBEC3A localization. Unlike the S-phase-specific HMCES PLA signal observed after UV radiation, APOBEC3A nuclear localization also modestly increased the HMCES PLA signal in EdU-negative cells (Figure 1E). APOBEC3A can deaminate cytosines in ssDNA formed at transcription bubbles, possibly providing a source for the HMCES PLA signal in non-replicating cells (Hoopes et al., 2016). These results suggest that HMCES is recruited to chromatin in response to APOBEC3A nuclear localization and reveal that HMCES can function in contexts other than DNA replication.
HMCES Protects Abasic Sites from Generating DSBs
One rationale for the HMCES pathway is that it may shield ssDNA abasic sites from inappropriate nucleolytic processing that would generate DSBs. Previous biochemical experiments demonstrated that the HMCES DPC protects ssDNA abasic sites from APE1-dependent cleavage (Mohni et al., 2019; Thompson et al., 2019). To test whether this is also the case in cells, we measured DSB formation using neutral comet assays. 1 h of 4-OHT treatment to induce APOBEC3A nuclear localization generated a modest increase in DSB formation in control cells, which was significantly elevated when HMCES was inactivated (Figure 2A). The 4-OHT-dependent breaks could be further increased by exogenously treating the nuclei in the agarose plugs with AP endonuclease 1 (APE1) before electrophoresis (Figure 2B). The increase in DSBs after APE1 treatment is much larger in HMCES-deficient cells, suggesting that HMCES can prevent APE1 processing and DSB formation at abasic lesions.
Figure 2. HMCES Protects Cells from APOBEC3A-Induced DSBs.

(A)Cells were transfected with HMCESsiRNAs (H-1, H-2) or a non-targetingsiRNA (NT) and treated with 4-OHT for 1 h as indicated prior to neutral comet assay.
(B) Cells were transfected with siRNA and treated with 4-OHT. The agarose plugs in the neutral comet assay were treated with purified APE-I for the indicated samples. All bars represent the median, and p values were derived from an ANOVA with Dunn’s multiple comparisons post-test. See also Figure S3.
If HMCES protects abasic sites from BER-dependent processing, we reasoned that inactivating AP endonucleases in human cells lacking HMCES may reduce the frequency of DSBs. To test that hypothesis, we silenced the two major AP-endonucleases, APEX1 and APEX2, in HMCESD U2OS cells that were previously reported to contain elevated levels of DSBs compared with parental U2OS (Mohni et al., 2019). Inactivating APEX1 had little effect, whereas inactivating APEX2 caused a modest increase in breaks. Contrary to reducing DSB formation, inactivating both endonucleases simultaneously produced a significant increase in DSBs in HMCES-deficient cells (Figure S3). One likely explanation is that inactivating AP endonucleases greatly increases the frequency of abasic sites in dsDNA (Dumitrache et al., 2018; Mohni et al., 2019; Simeonov et al., 2009). These persistent dsDNA abasic sites would be converted to ssDNA abasic sites during replication or transcription and could undergo b-elimination or processing by endonucleases other than APEX1 and APEX2 when HMCES is not present to protect them.
APOBEC3A-Induced ssDNA Abasic Sites Slow Replication Elongation
Because APOBEC enzymes deaminate cytosines on the lagging strand ssDNA template (Hoopes et al., 2016; Seplyarskiy et al., 2016) and HMCES localizes to DNA in response to APOBEC3A nuclear localization, we examined whether nuclear APOBEC3A expression alters replication kinetics. Surprisingly, APOBEC3A nuclear localization is sufficient to significantly slow replication elongation (Figure 3A). This slowing was observed in three independent clones of HCT116 cells expressing APOBEC3A and is not an off-target effect of 4-OHT treatment since replication kinetics were unchanged in parental HCT116 cells (Figure 3A). Treatment with 4-OHT for as little as 30 min was sufficient to slow elongation (Figure 3B). Because DNA combing monitors both lagging and leading strand synthesis, both must be slowed by nuclear APOBEC3A expression.
Figure 3. APOBEC3A-Induced Abasic Sites Slow Replication Elongation.

(A) Three clones (c2, c9, and c11) of HCT116 GFP-APOBEC3A-ERT2 cells, or parental HCT116 cells, were incubated with 4-OHT, CldU, and IdU, as indicated, and processed for DNA combing.
(B) The uracil-DNA glycosylase inhibitor (UGI) or an empty vector (EV) was introduced by lentiviral infection before DNA combing.
(C) hTERT-RPE-1 cells were harvested and lysed 16 h after UGI was introduced by lentiviral infection to assess UNG inhibition.
(D) hTERT-RPE-1 GFP-APOBEC3A-ERT2 cells were infected with EV or UGI before DNA combing. All bars represent the median, and p values were derived from an ANOVA with Dunn’s multiple comparisons post-test.
APOBEC3A relies on UNG to convert the deaminated cytosines (uracils) to abasic sites. To confirm that the replication fork slowing is due to abasic sites and not simply uracil formation, we used a uracil-DNA glycosylase inhibitor (UGI) (Bennett et al., 1993). The UGI was expressed in APOBEC3A cells by lentiviral infection, and in vitro assays with cell extracts confirmed complete UNG inactivation within 16 h of infection (Figure 3C). UNG inhibition fully rescued the fork-elongation defect associated with nuclear APOBEC3A in both HCT116 and hTERT-RPE-1 cells (Figures 3B and 3D). Thus, abasic sites, not uracils, are responsible for slowing replication elongation.
HMCES and TLS Control Replication Elongation in Response to Abasic Sites
To determine how HMCES influences APOBEC3A-induced replication-elongation slowing, we repeated the DNA combing experiments in cells lacking HMCES. HMCES inactivation alone has minor effects on replication elongation in HCT116 or hTERTRPE-1 cells (Figure 4A). However, HMCES deficiency in combination with nuclear APOBEC3A localization significantly slowed replication elongation in both HCT116 and hTERT-RPE-1 cells relative to HMCES-proficient cells treated with 4-OHT (Figure 4A). This replication-elongation defect was rescued by UNG inhibition, indicating abasic sites are again the relevant DNA lesion that slows fork elongation (Figure 4B). Thus, the HMCES pathway helps to maintain elongation rates in response to increased levels of ssDNA abasic sites.
Figure 4. HMCES and TLS Slow Replication Elongation in Response to Nuclear APOBEC3A.

(A) Cells were transfected with HMCES (H-1 and H-2), POLK, or a non-targeting siRNA (NT) and were incubated with 4-OHT before DNA combing.
(B) UGI or empty vector (EV) was introduced by lentiviral infection.
(C) Cells were transfected with HMCES or non-targeting siRNA, treated with DMSO, 4-OHT, and 10 μM JH-RE-06 for 30 min as indicated, and processed for DNA combing.
(D) Cells were transfected with HMCES and POLK siRNA, treated with 4-OHT, and processed for DNA combing. Bars represent the median, and p values were derived from an ANOVA with Dunn’s multiple comparisons post-test.
iPOND (isolation of proteins on nascent DNA) stable isotope labeling with amino acids in cell culture (SILAC)-mass spectrometry analysis, comparing HMCES-proficient and HMCESΔ cells, indicated that the TLS proteins REV1 (a TLS scaffold) and REV3 (the catalytic subunit of Polζ) are enriched at replication forks when HMCES is inactivated (Mohni et al., 2019). Therefore, we considered whether the elongation kinetics in HMCES-deficient cells expressing nuclear APOBEC3A could be influenced by use of Polζ.
JH-RE-06 is a selective inhibitor of the REV1 and REV7 interaction, thereby inhibiting Polζ (Wojtaszek et al., 2019). Polζ inhibition by JH-RE-06 does not change elongation rates by itself and does not exacerbate or rescue the slow elongation kinetics caused by APOBEC3A-mediated abasic sites in HMCES-proficient cells (Figure 4C). However, JH-RE-06 treatment largely restores normal elongation rates when HMCES is inactivated (Figure 4C).
Polζ can insert nucleotides across from an abasic site, but its most characterized function is to extend 3′ termini after other TLS polymerases insert a base across from the lesion (Gan et al., 2008; Stone et al., 2011). In vitro studies indicate TLS polymerase kappa (Polκ) can insert a base across from a template abasic site in some sequence contexts, although other studies indicate this incorporation is the least efficient of all TLS polymerases (Choi et al., 2010; Zhang et al., 2000). Furthermore, Polκ was shown to cause fork slowing in other contexts (Jones et al., 2012) Therefore, we tested whether Polκ could be involved in slowing the elongation in HMCES-deficient cells. Indeed, small interfering RNA (siRNA) depletion of Polκ suppresses the decreased elongation rate caused by HMCES inactivation in nuclear APOBEC3A-expressing cells, although it does not fully restore the elongation kinetics to normal (Figure 4D). These results suggest that, in the absence of HMCES, TLS polymerases Polζ and Polκ slow elongation by engagement of the abasic-site-containing DNA template.
DISCUSSION
HMCES-deficient cells are hypersensitive to multiple DNA-damaging agents that can generate a common lesion, an abasic site (Mohni et al., 2019). However, whether HMCES actually responds to abasic sites in cells was unclear because these agents generate many types of DNA lesions, and other studies reported various functions for HMCES, unrelated to abasic sites (Aravind et al., 2013; Kweon et al., 2017; Shukla et al., 2020; Spruijt et al., 2013). We now find that HMCES does indeed respond to abasic sites and shields them from being processed into DSBs. HMCES helps to maintain replication fork speed by preventing TLS-induced replication fork slowing in response to ssDNA abasic site damage. Inactivating both HMCES and TLS returns elongation kinetics to normal. However, this improvement in fork speeds comes at the expense of increased DSBs and decreased cell viability since inactivating HMCES and TLS polymerases generates synthetic lethality (Mohni et al., 2019).
Because APOBEC3A preferentially targets the lagging template strand and the DNA-combing methodology monitors synthesis on both strands, our fork-slowing data suggest that lagging-strand abasic sites may not only impair lagging-strand synthesis but also slow leading strand polymerization. This observation is unexpected because lagging strand lesions are generally thought to be bypassed easily by re-priming because of the discontinuous nature of lagging-strand synthesis (Cortez, 2019; Higuchi et al., 2003; Yeeles et al., 2013). There may be coupling of lagging and leading strand synthesis such that lagging-strand blocks also slow leading strand polymerization. Alternatively, nuclear APOBEC3A expression could induce abasic sites in both the leading and lagging strand templates. HMCES is recruited to chromatin in response to APOBEC3A expression even in cells not incorporating EdU, reflecting the ability of APOBECs to act in contexts other than replication. If these lesions escape excision repair, they could be present in the leading strand template to slow leading strand synthesis.
HMCES inactivation exacerbates the fork-elongation defect caused by APOBEC3A-induced abasic sites, but that slowing can be rescued by inactivating Polζ or Polκ. These results suggest that HMCES helps to maintain fork movement by preventing engagement of these TLS polymerases at forks stalled by abasic sites. These results are consistent with the observation that HMCES-deficient cells have elevated levels of Polζ at replication forks and increased Polζ-dependent mutation frequencies (Mohni et al., 2019). Polκ has been reported to inefficiently bypass abasic sites, and its activity slows replication (Choi et al., 2010; Jones et al., 2012; Zhang et al., 2000). The inefficiency of Polκ abasic site bypass may directly contribute to slower replication elongation. Whether this is also true for other TLS polymerases will be important to test because many polymerases can bypass abasic sites (Thompson and Cortez, 2020). The slowing caused by Polζ, which largely acts in the extension step of TLS after the action of another polymerase, suggests a more general effect of TLS pathways on fork movement and also indicates that TLS polymerases likely engage the replication fork directly and not only at post-replicative gaps. One possibility is that TLS engagement prevents faster pathways like re-priming from acting.
Removal of the uridine after cytidine deamination to generate an abasic site can reduce mutagenesis because uracil always leads to a C to T mutation, whereas conversion to an abasic site preserves the possibility that error-free repair mechanisms can operate (Hoopes et al., 2017). However, abasic sites increase the likelihood of strand cleavage. Thus, HMCES binding the abasic site may preserve genome stability by preventing DSB formation and providing time for error-free repair. The mechanism that generates the DSBs in the absence of HMCES is not clear. Although biochemical studies indicate that HMCES blocks the action of AP endonucleases and we find that AP endonucleases can cleave the abasic sites when HMCES is not present, the two major AP endonucleases (APEX1 or APEX2) are not required to generate the DSBs in cells. In fact, inactivating those enzymes actually generates more abasic sites and more DSBs. Many other enzymes are capable of strand cleavage at abasic sites or can process stalled forks into DSBs. Nucleophiles also promote β-elimination reactions at abasic sites yielding strand cleavage. Thus, there may be multiple sources of the DSBs in HMCES-deficient cells.
In addition to DSBs, abasic sites can generate other forms of DNA damage including interstrand and DNA protein crosslinks. Given the frequency of abasic sites and their potential to generate genome instability, organisms have many alternative mechanisms to deal with this lesion in different DNA contexts. In addition to BER, HMCES, and TLS, the Shu complex in yeast can shield ssDNA abasic sites from digestion by AP endonucleases (Rosenbaum et al., 2019). Further studies will be needed to understand how pathway choice is regulated and the mechanism by which the HMCES-bound abasic site is ultimately resolved.
In conclusion, HMCES responds to APOBEC3A-induced abasic sites, maintains genome stability, and facilitates replication elongation that would otherwise be slowed by the engagement of TLS polymerases. Because deregulated APOBEC activity is a source of mutagenesis in multiple cancer types, future studies should examine the balance among HMCES, TLS, and endonuclease processing to understand their importance in this process of tumorigenesis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu).
Materials Availability
Plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HCT116 GFP-APOBEC3A-ERT2 cells were cultured in 7.5% McCoy’s 5A supplemented with 7.5% Charcoal-Stripped FBS. hTERT-RPE-1 GFP-APOBEC3A-ERT2 were cultured in DMEM:F12 supplemented with Charcoal-Stripped FBS. U2OS were cultured in DMEM supplemented with 7.5% FBS. Cells were culture at 37C and 5% CO2 with humidity. All cell lines were regularly tested for mycoplasma and verified using short tandem repeat profiling. U2OS and hTERT-RPE-1 are female. HCT116 are male.
METHOD DETAILS
Generation of APOBEC3A stable cell lines
HCT116 cells were transfected with pACGFP-APOBEC3A-ERT2, selected with 500 mg/ml G418, and plated for individual clones using GFP fluorescent activated cell sorting (FACS). HCT116 and hTERT-RPE-1 were transfected with PiggyBac (PB510 Puro or PB533 Neo)-GFP-APOBEC3A-ERT2 along with the Super PiggyBac Transposase expression vector (PB210PA), selected with 2 μg/ml puromycin or 1 mg/ml G418, and populations of GFP-positive cells were subsequently sorted using FACS. GFP-APOBEC3A-ERT2 expression was verified by immunoblot.
Cell Transfections
Plasmid transfections were performed with Fugene 6 (pAcGFP-APOBEC3A-ERT2) or PureFection Transfection Reagent (System Biosciences, PiggyBac-GFP-APOBEC3A-ERT2, PB510B-1). siRNA transfections were performed with Dharmafect-1 (Dharmacon).
Plasmids
The APOBEC3A cDNA was obtained from the Thermo Scientific Open Biosystems Human ORFeome collection (Cat# OHS5894-9916829). The pACGFP-ERT2 vector was provided by Dr. Neils Mailand (Haahr et al., 2016). PiggyBac transposon vectors were purchased from Systems Biosciences. pUGI-NLS UDG Inhibitor (UGI) was purchased from Addgene (Cat#101091).
siRNAs and Antibodies
siRNAs were obtained from QIAGEN or Dharmacon as specified. AllStars Negative Control siRNA QIAGEN cat#1027281. siHMCES-1 Cat#D-020333–01, siHMCES-2 Cat#J-020333–19. siPolymerase Kappa Cat#D-021038–01, Cat#D-021038–02, Cat#D-021038–03, Cat#D-021038–04. siAPEX1 Cat#D-010237–01, Cat#D-010237–02, Cat#D-010237–03, Cat#D-010237–05. siAPEX2 Cat#D-013730–01, Cat#D-013730–02, Cat#D-013730–03, Cat#D-013730–04. Antibodies: APOBEC3A (Sigma, Cat#SAB4500753), HMCES (Sigma, Cat#HPA044968), GFP (Abcam, Cat#ab290), BrdU (Abcam, Cat#ab6326), BrdU (BD Cat#347580), Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 (Life Technologies, Cat#A32723), Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (Life Technologies, Cat#A11007).
Live Cell Fluorescence Assays
Live cells were plated in a 96-well glass bottom dish. Following treatment with 10 μM 4-hydroxytamoxifen (4-OHT), cells were incubated with 1 μg/ml Hoechst 33342 in PBS for 10 min, rinsed with PBS and imaged using an ImageXpress (Molecular Devices).
Viability Assays
Cells were plated for the long-term clonogenic survival assays 48 h after siRNA transfection, and were subsequently treated with 10 μM 4-OHT for 24 h (Tocris, Cat#3412/10). 4-OHT was washed out and colonies were allowed to grow for approximately two weeks prior to scoring by methylene blue staining (48% methanol, 2% methylene blue, 50% water). All clonogenic survival assays were completed in triplicate with three experimental replicates per biological replicate.
Western Blot
Whole-cell lysates were extracted using Igepal lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Igepal CA-630, 1 mM EDTA, pH 8.5) enriched with sodium fluoride (1 mM), sodium vanadate (1 mM), protease inhibitor cocktail (Roche), phenylmethylsulfonyl fluoride (PMSF, 1 mM), 25 units of Pierce Universal Nuclease, and 1 mM MgCl2. Proteins were analyzed by SDS-PAGE and immunoblotting.
Proximity ligation assay
Cells were plated in a 96-well glass bottom dish. Following the indicated treatments, cells were pre-extracted in ice-cold pre-extraction buffer (20 mM HEPES, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X-100), and subsequently fixed with 3% paraformaldehyde, 2% sucrose. Cells were then permeabilized (20 mM HEPES, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X-100), and blocked (1 mg/ml BSA, 5% Goat Serum, 0.1% Triton X-100, 1mM EDTA). EdU incorporation was detected by a click reaction using Alexa Fluor 488 azide (Invitrogen). The proximity ligation assay was then performed as per manufacturer’s instructions (Sigma) using a HMCES polyclonal antibody (Sigma HPA044968). Images were acquired using the 20X objective on an ImageXpress (Molecular Devices) and analyzed with MetaXpress software.
UNG cellular activity assay
The UDG inhibitor (UGI; Addgene, Cat#101091) was cloned into the lentiviral plasmid pLX301 and co-transfected with pPAX2 and pCMV-VSV-G into 293(F)T packaging cells. Supernatants were collected 46 h after transfection, filtered, diluted 1:5 in growth media, and supplemented with 4 μg/ml polybrene. Target cells were infected once, harvested 16 h post-infection, and lysed in 50 mM Tris-Cl pH 7.4, 1% Igepal, 0.1% sodium deoxycholate, 10% glycerol, 150 mM NaCl, 1 mM DTT, 20 μg/ml RNaseA with an EDTA-free protease inhibitor cocktail tablet (Roche). To test for inhibition, 16 μl each lysate (or buffer) was incubated with 100 nM of a 42 nucleotide ssDNA oligonucleotide with a single deoxyuridine and a 5′ fluorescein tag (5′ (6-FAM)-CTA TGA TGA CTC TTC TGG TCU GGA TGG TAG TTA AGT GTT GAG 3′) at 37°C and 8 μl was removed at 15 min and 30 min. As a control, 5 units of purified UDG from E. coli (New England Biolabs) was added to one reaction. The DNA was then heated at 95°C for 10 min in the presence of 0.2 M NaOH to cleave abasic sites. Samples were mixed with an equal volume of formamide/EDTA loading buffer, resolved on a 10% TBE-Urea gel, and scanned using a Typhoon imager (GE Healthcare).
DNA molecular combing
Cell were labeled with 20 μM CldU (Sigma, C6891) followed by 100 μM IdU (Sigma, l7125), for the time indicated, with or without 10 μM 4-OHT (Tocris, 3412/10), JH-RE-06 (Vanderbilt Chemical Synthesis Core), or following 16 h infection with UGI. Approximately 400,000 cells were embedded in agarose plugs and the assay was performed as per Genomic Vision’s manufacturer instructions. The DNA was stained with antibodies that recognize IdU and CldU (Abcam Cat#ab6326, BD Cat#347580) for 1 h, washed in PBS, and probed with secondary antibodies for 45 min. Images were obtained using a 40X oil objective (Nikon Eclipse Ti). Analysis of fiber lengths performed using Nikon Elements software.
Neutral Comet Assay
Trevigen CometAssay ESII system was utilized to detect DNA double-strand breaks, and the assay was performed as per manufacturer’s instructions (Trevigen). Neutral comet assay with exogenous APE-I digestion was performed as per manufacturer’s instructions, however, agarose plugs were treated with 10 units per ml APE-I (NEB, M0282S) at 37°C prior to electrophoresis. Tail moments were scored using the open source Fiji and OpenComet software (Gyori et al., 2014; Schindelin et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were completed using Prism 8 (GraphPad). An ANOVA test was used when comparing more than two groups followed by a Holm-Sidak or Dunn’s post-test. No statistical methods or criteria were used to estimate sample size or to include/exclude samples. Multiple siRNAs, multiple clones, multiple approaches, and multiple cell lines were analyzed to confirm results were not caused by off-target effects or clonal variations. Unless otherwise stated, all experiments were performed at least twice and representative experiments are shown.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti HMCES | Sigma | Cat#HPA044968; RRID:AB_2679160 |
| Rabbit polyclonal anti APOBEC3A | Sigma | Cat# SAB4500753; RRID:AB_10742606 |
| Rabbit polyclonal anti GFP | Abcam | Cat#ab290; RRID:AB_303395 |
| Mouse monoclonal anti BrdU | BD | Cat#347580; RRID:AB_10015219 |
| Rat monoclonal anti BrdU | Abcam | Cat#ab6326; RRID:AB_305426 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Life Technologies | Cat# A32723; RRID:AB_2633275 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies | Cat# A11007; RRID:AB_10561522 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| JHRE06 | VICB synthesis core | N/A |
| CldU | Sigma Aldrich | Cat#C6891 |
| IdU | Sigma Aldrich | Cat#l7125 |
| EdU | VICB synthesis core | N/A |
| 4-Hydroxytamoxifen | Tocris | Cat#3412/10 |
| MG-132 | Sigma Aldrich | Cat#M7449 |
| Hoescht 33342 | ThermoFisher | Cat#62249 |
| Alexa Fluor 488 Azide | ThermoFisher | Cat#A10266 |
| APE-I | NEB | Cat#M0282S |
| Uracil-DNA Glycosylase (UDG) | NEB | Cat#M0280S |
| Critical Commercial Assays | ||
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat#DU092002 |
| Duolink In Situ PLA Probe Anti-Rabbit MINUS | Sigma-Aldrich | Cat#DU092004 |
| Duolink In Situ Detection Reagents, Red | Sigma-Aldrich | Cat#DU092008 |
| Comet Assay Kit | Trevigen | Cat#4250–050-K |
| FiberPrep Kit | Genomic Vision | Cat#EXTR-001 |
| Combi Coverslips | Genomic Vision | Cat#COV-002-RUO |
| U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| hTERT-RPE-1 | ATCC | Cat#CRL-4000; RRID:CVCL_4388 |
| HCT-116 | ATCC | Cat#CCL-247; RRID:CVCL_0291 |
| Oligonucleotides | ||
| HMCES siRNA | Dharmacon | Cat#D-020333–01 |
| HMCES siRNA | Dharmacon | Cat#J-020333–19 |
| PolK siRNA | Dharmacon | Cat#D-021038–01 |
| PolK siRNA | Dharmacon | Cat#D-021038–02 |
| PolK siRNA | Dharmacon | Cat#D-021038–03 |
| PolK siRNA | Dharmacon | Cat#D-021038–04 |
| APEX1 siRNA | Dharmacon | Cat#D-010237–01 |
| APEX1 siRNA | Dharmacon | Cat#D-010237–02 |
| APEX1 siRNA | Dharmacon | Cat#D-010237–03 |
| APEX1 siRNA | Dharmacon | Cat#D-010237–05 |
| APEX2 siRNA | Dharmacon | Cat#D-013730–01 |
| APEX2 siRNA | Dharmacon | Cat#D-013730–02 |
| APEX2 siRNA | Dharmacon | Cat#D-013730–03 |
| APEX2 siRNA | Dharmacon | Cat#D-013730–05 |
| AllStars Negative Control siRNA | QIAGEN | Cat#1027281 |
| Recombinant DNA | ||
| pAcGFP-ERT2 | Haahr et al., 2016 | Mailand Lab |
| APOBEC3A cDNA | Dharmacon | Cat# OHS5894–9916829 |
| pAcGFP-APOBEC3A-ERT2 | This paper | N/A |
| PiggyBac Transposon Vectors | Systems Biosciences | Cat# PB510B-1 |
| PiggyBac-GFP-APOBEC3A-ERT2 | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_000306 |
| Fiji | Schindelin et al., 2012 | http://fiji.sc RRID:SCR_002285 |
| OpenComet | Gyori et al., 2014 | N/A |
| Other |
Highlights.
HMCES-deficient cells are hypersensitive to nuclear APOBEC3A (A3A) localization
HMCES protects abasic sites from processing into double-strand breaks
A3A-induced abasic sites slow both leading and lagging strand replication
TLS polymerases zeta (Polζ) and kappa (Polκ) slow fork elongation in the absence of HMCES
ACKNOWLEDGMENTS
We thank Drs. Niels Mailand and John Maciejowski for providing reagents; Drs. James Dewar, Kareem Mohni, and Madison Adolph for helpful discussions; and Dr. Petria Thompson for critical reading of this manuscript. This research was supported by grants to D.C. from the National Institutes of Health (NIH) (R01ES030575 and R01GM116616). K.P.M.M. was supported by 5T32CA009582 and 1F32GM136096.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107705.
REFERENCES
- Aravind L, Anand S, and Iyer LM (2013). Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems. Biol. Direct 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SE, Schimerlik MI, and Mosbaugh DW (1993). Kinetics of the uracil-DNA glycosylase/inhibitor protein association. Ung interaction with Ugi, nucleic acids, and uracil compounds. J. Biol. Chem 268, 26879–26885. [PubMed] [Google Scholar]
- Boiteux S, and Guillet M (2004). Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst.) 3, 1–12. [DOI] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. (2013). APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Lim S, Kim EJ, Jo A, and Guengerich FP (2010). Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases α, δ, η, ι, κ, and REV1. J. Mol. Biol 404, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2019). Replication-coupled DNA repair. Mol. Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrache LC, Shimada M, Downing SM, Kwak YD, Li Y, Illuzzi JL, Russell HR, Wilson DM 3rd, and McKinnon PJ (2018). Apurinic endonuclease-1 preserves neural genome integrity to maintain homeostasis and thermoregulation and prevent brain tumors. Proc. Natl. Acad. Sci. USA 115, E12285–E12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan GN, Wittschieben JP, Wittschieben BO, and Wood RD (2008). DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 18, 174–183. [DOI] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis.Redox Biol. 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Räschle M, and Mailand N (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol 18, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L, and Arrowsmith CH (2019). Structural basis of HMCES interactions with abasic DNA and multivalent substrate recognition. Nat. Struct. Mol. Biol 26, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. (2016). Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell 164, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, and Liddament MT (2004). Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol 4, 868–877. [DOI] [PubMed] [Google Scholar]
- Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, and Maki H (2003). Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 8, 437–449. [DOI] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, and Roberts SA (2016). APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 14, 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JI, Hughes AL, Hobson LA, Cortez LM, Brown AJ, and Roberts SA (2017). Avoidance of APOBEC3B-induced mutation by error-free lesion bypass. Nucleic Acids Res. 45, 5243–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MJ, Colnaghi L, and Huang TT (2012). Dysregulation of DNA polymerase k recruitment to replication forks results in genomic instability. EMBO J. 31, 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon SM, Zhu B, Chen Y, Aravind L, Xu SY, and Feldman DE (2017). Erasure of Tet-oxidized 5-methylcytosine by a SRAP nuclease. Cell Rep. 21, 482–494. [DOI] [PubMed] [Google Scholar]
- Landry S, Narvaiza I, Linfesty DC, and Weitzman MD (2011). APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 12, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019). HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 176, 144–153.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebhandl S, Huemer M, Greil R, and Geisberger R (2015). AID/APOBEC deaminases and cancer. Oncoscience 2, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JC, Bonilla B, Hengel SR, Mertz TM, Herken BW, Kazemier HG, Pressimone CA, Ratterman TC, MacNary E, De Magis A, et al. (2019). The Rad51 paralogs facilitate a novel DNA strand specific damage tolerance pathway. Nat. Commun 10, 3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, and Nikolaev SI (2016). APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 26, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V, Halabelian L, Balagere S, Samaniego-Castruita D, Feldman DE, Arrowsmith CH, Rao A, and Aravind L (2020). HMCES functions in the alternative end-joining pathway of the DNA DSB repair during class switch recombination in B cells. Mol. Cell 77, 384–394.e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, and Wilson DM 3rd. (2009). Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS ONE 4, e5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Mu€nzel M, Wagner M, Mu€ller M, Khan F, et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159. [DOI] [PubMed] [Google Scholar]
- Stenglein MD, Burns MB, Li M, Lengyel J, and Harris RS (2010). APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol 17, 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Kumar D, Binz SK, Inase A, Iwai S, Chabes A, Burgers PM, and Kunkel TA (2011). Lesion bypass by S. cerevisiae Pol ζ alone. DNA Repair (Amst.) 10, 826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpaert-Borlé M (1987). Formation, detection and repair of AP sites. Mutat. Res 181, 45–56. [DOI] [PubMed] [Google Scholar]
- Thompson PS, and Cortez D (2020). New insights into abasic site repair and tolerance. DNA Repair (Amst.), Published online April 30, 2020 10.1016/j.dnarep.2020.102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PS, Amidon KM, Mohni KN, Cortez D, and Eichman BF (2019). Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nat. Struct. Mol. Biol 26, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Bao H, Chen L, Liu Y, Li Y, Wu B, and Huang H (2019). Molecular basis of abasic site sensing in single-stranded DNA by the SRAP domain of E. coli yedK. Nucleic Acids Res. 47, 10388–10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, et al. (2019). A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178, 152–159.e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Poli J, Marians KJ, and Pasero P (2013). Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, and Wang Z (2000). Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 28, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.