

Abstract
BX1 from Zea mays (zmBX1) is an enzyme of plant secondary metabolism that generates indole for the synthesis of plant defensins. It is a homolog of the tryptophan synthase α-subunit, TrpA. Whereas TrpA itself is a monomer in solution, zmBX1 is dimeric, confirmed in our work by native MS. Using cross-linking and mutagenesis, we identified the physiological dimerization interface of zmBX1. We found that homodimerization has only minor effects on catalysis and stability. A comparison of the zmBX1–zmBX1 homodimer and zmTrpA–zmTrpB heterodimer interfaces suggest that homodimerization in zmBX1 might, at an early point in evolution, have served as a mechanism to exclude the interaction with the tryptophan synthase β-subunit (zmTrpB), marking its transition from primary to secondary metabolism.
Introduction
Tryptophan synthase catalyzes the last two steps in the tryptophan biosynthesis pathway. It is a heterotetramer, where two α-subunits (TrpA) interact with the β-subunit (TrpB) dimer to form an αββα complex [1]. TrpA catalyzes the aldolytic cleavage of indole-3-glycerol phosphate (IGP) to glyceraldehyde-3-phosphate (GAP) and indole. The latter is channeled to the active site of TrpB, where it is converted with L-serine to L-tryptophan in a pyridoxal 5′-phosphate (PLP) dependent reaction [2]. Bi-directional allosteric communication between TrpA and TrpB ensures that the generation of indole is coupled with its subsequent conversion to L-tryptophan [3, 4]. TrpA by itself is almost inactive, but the interaction with TrpB results in an increase of its catalytic efficiency by several orders of magnitude [2, 5, 6]. Importantly, the TrpA-TrpB subunit orientation allows for allosteric communication between subunits as well as the interconnection of the two active sites. Whereas most bacteria contain just a single trpA gene, many plants have several trpA paralogs [7]. The corresponding proteins differ in their catalytic activity, ability to interact with TrpB, and physiological function. The TrpA homolog BX1 has been shown to catalyze the TrpA reaction with high efficiency in the absence of an interaction partner, thereby providing indole for the synthesis of benzoxazinoids, a class of plant defense molecules [8–10]. Two crystal structures of BX1 from Zea mays (zmBX1) have been solved and more recently, it has been shown that zmBX1 is a dimer in solution [8, 11]. Due to the ambiguity in identifying physiological protein-protein interactions from crystal structures solely based on computational analyses [12–15], we used cross-linking mass spectrometry and mutagenesis as complementary structural biology tools to determine the homodimerization interface of zmBX1.
Materials and Methods
Cloning, mutagenesis, expression and purification of zmBX1 and its mutants
Gene cloning, protein expression, and protein purification were performed as previously described [11]. Briefly, bx1 was amplified from cDNA of Zea mays W22 leaves and the amplicon was cloned into a pUR28a vector for protein expression with an N-terminal His6-tag. zmBX1 mutants were generated using the QuickChange™ site-directed mutagenesis kit. Oligonucleotides used for mutagenesis are listed in Table S1. Escherichia coli BL21 (DE3) Gold cells were transformed with the corresponding plasmid, grown in LB medium, induced by the addition of IPTG and grown overnight at 20 °C. The cells were then harvested by centrifugation and disrupted by sonication. The His6-tagged proteins in the soluble fraction were purified by immobilized metal ion chromatography (GE Healthcare, HisTrap FF Crude) followed by size exclusion chromatography (GE Healthcare, HiLoad 26/600 Superdex 75 PG). Amino acid sequences for all proteins are shown in Table S2.
Cross-linking and protein digestion
zmBX1 was either buffer-exchanged into 100 mM potassium phosphate buffer (pH 7.5) with 300 mM KCl or diluted in 100 mM potassium phosphate buffer (pH 7.5) with 300 mM KCl and 1 mM dithiothreitol (DTT). A 100-fold molar excess of MS-cleavable cross-linker disuccinimidyl sulfoxide (DSSO) (Cayman Chemical) [16] was mixed with zmBX1 (10 μM monomer concentration). The Dimethyl sulfoxide required to dissolve DSSO was diluted to less than 3% in the final reaction mixture. The cross-linking reaction was incubated at room temperature for 60 minutes and subsequently quenched by addition of Tris-HCl (pH 7.5) for a final concentration of 20 mM. Samples were analyzed by SDS-PAGE and / or subjected to in-solution digestion. For in-solution digestion, cross-linked zmBX1 was denatured with 8 M urea in 100 mM Tris-HCl (pH 7.7), reduced with dithiothreitol (final concentration: 20 mM) at 37 °C for 30 minutes, alkylated with iodoacetamide (final concentration: 40 mM) at 37 °C for 20 minutes, and finally diluted with 100 mM ammonium bicarbonate to reduce the urea concentration to less than 1 M. MS grade trypsin or Glu-C (Thermo Fisher Scientific), respectively, was added for a final protease to protein ratio of 1:50 (w/w) and proteins were digested at 37 °C overnight in separate digestion experiments. Peptides were desalted using SPEC Pt C18 cartridges (Agilent Technologies). Briefly, the C18 cartridges were equilibrated with 0.1% trifluoroacetic acid (TFA) (3 x 200 μL), the peptide mixture was loaded onto the C18 cartridge, the bound peptides were washed with 0.1% TFA (3 x 200 μL), eluted with 0.1% TFA, 50% acetonitrile (2 x 200 μL) and 0.1% TFA, 80% acetonitrile (1 x 200 μL), and eluted peptides were dried in a SpeedVac and resuspended in 0.1% TFA.
Liquid chromatography-tandem mass spectrometry (LC-MSn)
Peptides were analyzed by LC-MSn with an UltiMate™ 3000 RSLCnano coupled to an Orbitrap Fusion™ Tribrid™ Mass Spectrometer equipped with an EASY-Spray™ source. Roughly 2 μg of peptide were injected onto an EASY-Spray™ column (75 μm x 250 mm, 2 μm, C18, Thermo Fisher Scientific #ES802). The peptides were separated at a flow rate of 0.3 μL min−1 using the following gradient: 0-5 min = 5% B, 5-50 min = to 28% B, 50-60 min = to 40% B, 60-65 min = to 95% B, 65-70 min = 95% B, where solvent A = H2O with 0.1% formic acid and solvent B = acetonitrile with 0.1% formic acid. MS and MSn settings were used as described previously [17]. Data-dependent CID-MS2 scans (R = 30,000) with a normalized collision energy (NCE) of 25% and a dynamic exclusion duration of 30 seconds were performed on the precursor ions in the full MS1 scan (375 - 1600 m/z, R = 120,000). CID-MS3 scans (NCE 35%) were triggered for the CID-MS2 ions with a mass difference of 31.9721 Da and acquired in the ion trap. CID-MS2 scans were followed by EThcD-MS2 scans (R = 50,000) of the same MS1 precursor ions unless CID-MS2 resulted in the triggering of CID-MS3. Calibrated charge-dependent ETD parameters were used. Each MS experiment cycle was set to 5 seconds.
Data analysis of cross-linked peptides
Data were analyzed using the XLinkX v2.2 node in Proteome Discoverer v2.2 (Thermo Fisher Scientific) [17, 18]. Data were filtered for possible cross-links based on the MS2-MS3 acquisition strategy. The DSSO modification (158.004 Da) was set to be considered for the amino acids K, S, T, and Y. The protein database contained 72 proteins ranging from proteins commonly run on the analytical column to common contaminants that included zmBX1, BSA, ubiquitin, proteases, and human keratins. A complete list can be found in the supplementary text file. Depending on the protease used, either trypsin or Glu-C cleavage was selected for the searches. In the peptide search (Sequest HT) and XLinkX search, the maximum missed cleavages were 4, minimum peptide length was 4, precursor mass tolerance was 10 ppm, FTMS fragment mass tolerance was 20 ppm, and ITMS fragment mass tolerance was 0.5 Da. Carbamidomethylation of Cys was included as a static modification. Oxidation of Met, dead-end cross-link modifications quenched by water (176.014 Da) or Tris (279.078 Da), and MS-cleaved thiol (85.983 Da) and alkene (54.011 Da) cross-link modifications were all included as dynamic modifications. For peptide group modifications, a site probability threshold of 75 was used. For the cross-linked peptides, an FDR of 1% and initially a minimum XLinkX score cut-off of 20 was used. To increase the stringency of the cross-linking results, an XLinkX score cut-off of 50 was used in the reporting of these data. Additionally, the annotated spectra of the cross-linked peptides were manually checked for fragment ions that could unambiguously localize the cross-link modified residues.
Analytical size-exclusion chromatography (SEC).
Analytical size-exclusion chromatography was performed at 22 °C with an ÄKTAmicro liquid chromatography system (GE Healthcare) coupled to an autosampler (Spark Holland, Alias GE Bio Cool) with a sample cooling compartment (8 °C). 30 μM protein (monomer concentration) was injected onto a Superdex S75, 10 / 300 GL column and eluted with 50 mM potassium phosphate buffer (pH 7.5), 300 mM KCl at a flow rate of 0.5 mL min-1. The elution was monitored at 280 nm with an absorbance detector.
Size exclusion chromatography coupled to native mass spectrometry
For size exclusion chromatography, proteins were first buffer-exchanged into 300 mM ammonium acetate, pH 6.8 with Micro Bio-Spin™ 6 columns (Bio-Rad). 100 pmol protein was injected onto a bioZen™ 1.8 μm SEC-2 column (150 x 4.6 mm) equipped with a security guard column (Phenomenex), and eluted with 300 mM ammonium acetate at a flow rate of 0.2 mL min−1 using an UltiMate™ 3000 RSLC (Thermo Fisher Scientific) coupled to an Exactive™ Plus EMR Orbitrap™ mass spectrometer (Thermo Fisher Scientific) equipped with an ESI source and modified to incorporate a quadrupole mass filter and allow for surface-induced dissociation [19]. Mass spectra were recorded from 1000 - 10000 m/z at 8750 resolution as defined at 200 m/z. The ion injection time was set to 200 ms. Voltages applied to the transfer optics were optimized to allow for optimal ion transmission. Total ion chromatograms were smoothed (Boxcar; 15 points) and overlaid. Mass spectra were deconvoluted and plotted with UniDec version 4.0.0 beta [20] using the preset high-resolution native parameters adjusted to sample mass every 1 Da.
Steady-state enzyme kinetics
The activity of zmBX1 mutants was measured at 30 °C by monitoring the cleavage of IGP to GAP and indole with a coupled enzymatic assay [21]. The steady-state kinetics were performed as previously described [11]. The experimental conditions were as follows: final protein monomer concentration of 1 μM, 100 mM EPPS/KOH (pH 7.5), 180 mM KCl, 40 mM PLP, 6 mM NAD+, 20 mM NaAsO4, varying concentrations of IGP, and 5.5 μM GAP dehydrogenase from Thermotoga maritima. The data were fitted to a hyperbolic function using SigmaPlot 14.0 to determine values for vmax and KMIGP. The kcat values were determined by dividing vmax by the final monomer concentrations [E0]. The measurements for each zmBX1 mutant were performed in duplicate and analyzed separately.
Thermal denaturation followed by Differential Scanning Fluorimetry (nanoDSF)
Thermal denaturation was followed with a Prometheus NT.48 instrument (Nano-Temper Technologies, access provided by 2bind GmbH). Proteins (10 μM monomer concentration) were heated in 100 mM potassium phosphate buffer (pH 7.5), 300 mM KCl from 20 to 95 °C at a ramp rate of 1 °C min−1. The excitation power at 280 nm was 50%, and emission spectra were recorded at 330 and 350 nm. The change in the ratio of the fluorescence signal from 350 to 330 nm with increasing temperature was followed. Fluorescence transitions were fitted with the program supplied by the manufacturer. The apparent midpoint temperatures (TMapp) of the irreversible unfolding transition, defined by the inflection points of the curves, were determined. The experiments were performed in duplicate.
Results and Discussion
Identification of possible dimer interface
zmBX1 has been crystallized in two different conditions and the data have been deposited in the RCSB Protein Data Bank in 2004 as pdb id 1RD5 (space symmetry group: F 2 2 2, resolution: 2.02 Å, ligand: malonic acid) and pdb id 1TJR (space symmetry group: H 3, resolution: 2.30 Å, ligand: SO42−). The authors assigned the biological assemblies as monomers consistent with different protein-protein interactions being found in the crystal structures’ dimeric asymmetric units (Figure S1) [8]. However, recent data indicate that zmBX1 is a homodimer in solution [11]. This led us to revisit the structures to determine the physiologically relevant protein-protein interaction that might exist apart from the protein contacts that have been formed during the crystallization process [22]. We used PDBePISA to examine the interfaces in both crystal structures [23]. For 1RD5, chains A and B in the asymmetric unit were identified to most likely be the physiologically relevant dimer subunits having the largest interface area and the greatest Complex Formation Significance Score (CSS) of the identified protein interfaces. For 1TJR, chain A of the asymmetric unit and chain B found in the crystal packing by symmetry operation −y+1,x−y,z were identified to most likely be the physiologically relevant dimer subunits having the largest interface area and the only identified interface with a CSS. The two dimer structures were aligned in PyMOL (v2.3.2) [24]. A root mean square deviation of 1.975 Å for all alpha carbons shows that the subunit-subunit orientation within those two dimers is nearly identical in both crystal structures (Figure 1).
Figure 1.
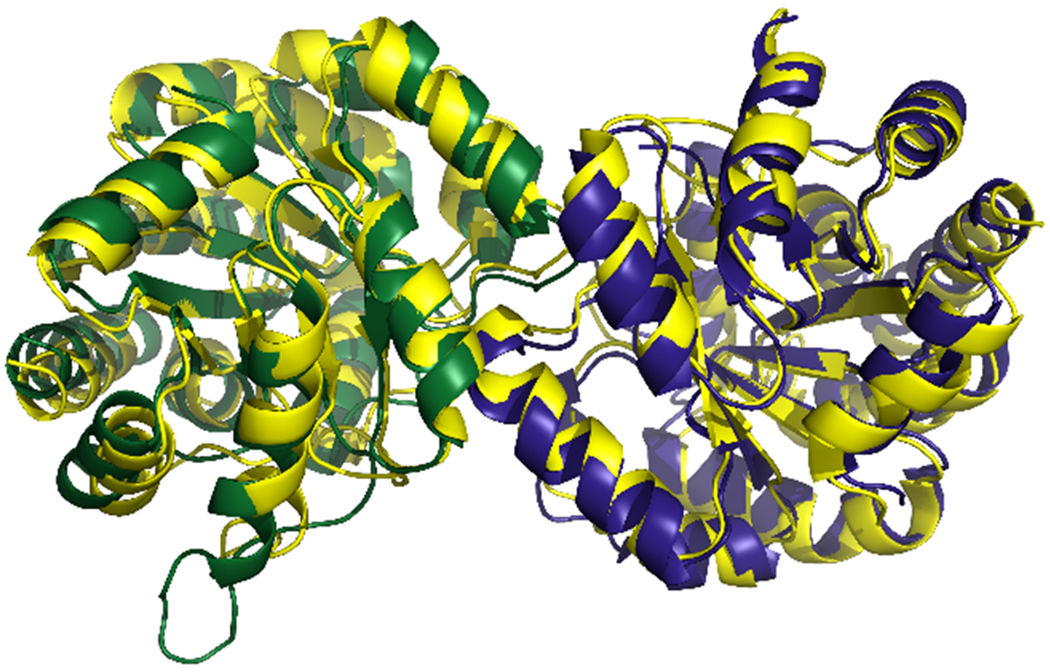
Superposition of 1RD5 (chain A of the asymmetric unit in green, chain B in blue) with 1TJR (chain A of the asymmetric unit and chain B with the symmetry operation -y+1,x-y,z (sy01) in yellow). Structures are shown in cartoon representation.
This suggests that the putative physiological protein-protein interaction is retained in both zmBX1 crystal structures, aside from the four additional crystal contacts in 1RD5 and the five additional crystal contacts in 1TJR.
Confirmation of proposed dimerization interface by cross-linking MS
In order to confirm that this protein-protein interaction is physiologically relevant and present in solution, we used cross-linking mass spectrometry. We selected disuccinimidyl sulfoxide (DSSO) as the cross-linking reagent due to the relatively high number of N-hydroxysuccinimide (NHS) ester reactive residues in zmBX1 (14 K, 19 S, 13 T, and 7 Y per subunit) [25], and due to the characteristic fragmentation of such an MS-cleavable cross-linker [16], as this simplifies data analysis [17, 26] without having a significant decrease in cross-linking efficiency when compared to non-MS-cleavable cross-linkers with similar spacer length like DSS and BS3 [27, 28]. In a first step, we performed the cross-linking reaction in 100 mM potassium phosphate buffer (pH 7.5) with 300 mM KCl. Analysis by SDS-PAGE after cross-linking showed two bands corresponding to monomer and dimer, indicating the successful formation of intermolecular cross-links. We performed in-solution digestion with either trypsin or Glu-C, respectively, in two separate digestion reactions, followed by LC-MS analysis using an MS3 method on the Orbitrap Fusion with subsequent data analysis using the XLinkX node on Proteome Discoverer. Cross-linking followed by Glu-C digestion resulted in the identification of more cross-links than obtained by trypsin digestion (Figure S2). A combination of two reasons for this could be that this protein has glutamate cleavage sites for Glu-C digestion that result in cross-linked peptides of an appropriate size for MS analysis, and the cross-linking reactions at lysine residues removed too many potential trypsin cleavage sites needed to generate cross-linked peptides. When these cross-links were mapped onto the putative zmBX1 dimer structure (asymmetric unit of 1RD5), we found that 25% of the cross-links violated a maximum Cα-Cα distance restraint of 30 Å [29]. This suggested i) the existence of a different zmBX1 quaternary structure or ii) the presence of multiple zmBX1 dimer structures in solution under the existing conditions. The latter seemed plausible given that zmBX1 contains three cysteine residues on the surface, making it to some extent susceptible to artificial subunit-subunit association in the absence of a reducing reagent. Consequently, we also performed the cross-linking reaction in the presence of dithiothreitol (DTT) with subsequent Glu-C digestion followed by LC-MS analysis. A comparison of cross-links identified after reaction in the absence and presence of DTT is shown in Figure S3. Artificial oligomerization due to cysteine oxidation might be of general concern as it can result in misinterpretation of cross-linking results. Our data suggest that this is rather easily preventable by performing the cross-linking reaction under reducing conditions. The cross-links identified from the reaction in the presence of DTT as mapped on the putative zmBX1 dimer structure are depicted in Figure 2a. The zmBX1 construct sequence numbering was adjusted to match the numbering of the 1RD5 crystal structure and will be referred to as such throughout (Figure S4). Manual inspection of the fragment ions in the cross-linked spectra showed that, in all but one case, the cross-link sites could be localized to a single residue within the peptide. For the cross-link T163-T200, the peptide containing T163 did not have an annotated fragment ion that could be any further localized than to either T163 or K164. However, these neighboring amino acids both fall under the maximum allowable cross-linking distance (either of them with T200 (<30 Å)).
Figure 2.
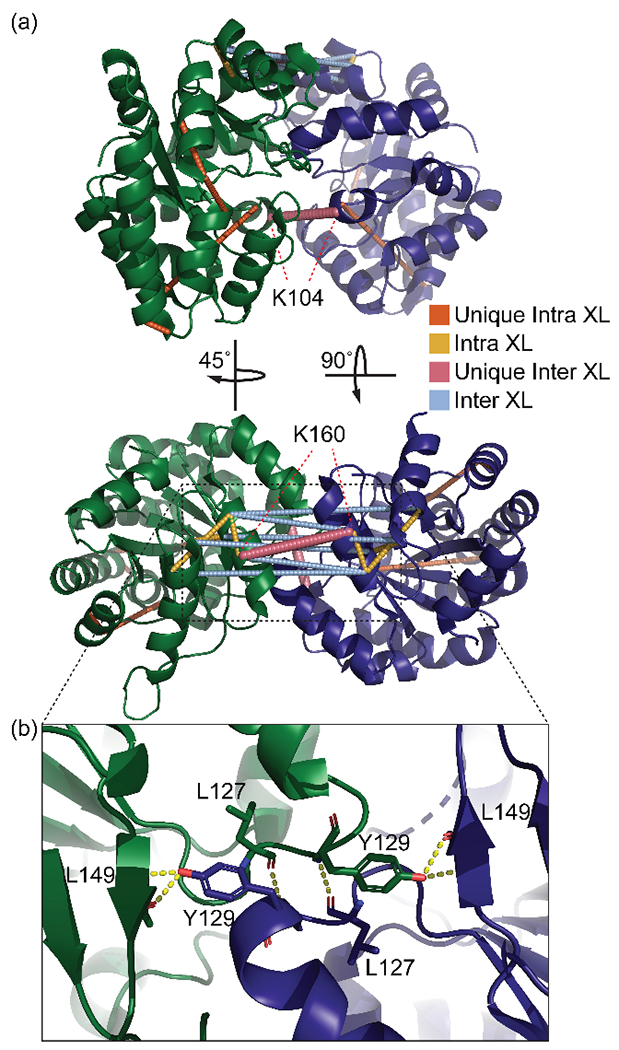
(a) Cross-links mapped onto the putative zmBX1 dimer (asymmetric unit of 1RD5). All distances are classified as unique intra- or intermolecular or as both when they fit Cα-Cα distances < 30 Å. The two unique intermolecular cross-links between K160 and K104 of each chain are labeled. A unique cross-link is defined as having only one possible distance that is cross-linkable within a structure. (b) zoom-in onto tyrosine 129 (Y129) from each subunit forming hydrogen bonding networks with backbone leucine residues of the opposite subunit. XL= cross-linkage.
Mapping the distances on the two additional possible dimers with the second and third largest interface areas within the crystal structure of 1RD5 and 1TJR revealed that only the original proposed dimer structure satisfied all cross-links (Table S3 and S4). Importantly, this is also true for cross-links K104-K104 and K160-K160, which are unique and can only be formed between the two subunits. Annotated spectra for these two critical cross-links are shown in Figure S5 and S6.
Additional confirmation of proposed dimerization interface by mutagenesis
In the proposed zmBX1 dimer structure, Y129 of each subunit fits inside the pocket of the opposite subunit, where it has hydrogen bonding interactions with the backbones of leucine 127 and 149 (Figure 2b). This tyrosine is conserved in BX1 proteins from different plants as indicated by the multiple sequence alignment shown in Figure 3.
Figure 3.
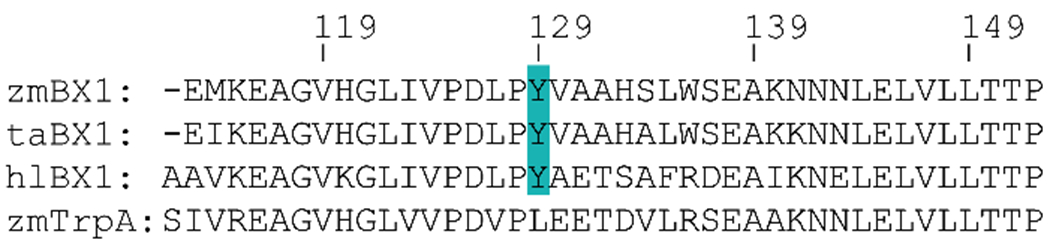
Multiple sequence alignment (MSA) of BX1 from Zea mays (zmBX1; UniProtID: P42390), Triticum aestivum (taBX1; UniProtID: Q7XAK6), and Hordeum lechleri (hlBX1; UniProtID: Q5QITQ0) as well as TrpA from Zea mays (zmTrpA; UniProtID: A0A3L6E3U7). Numbering is adjusted for zmBX1 and the conserved Y is highlighted. The MSA was calculated with Clustal Omega [30].
This suggests a similar quaternary structure for BX1 proteins from other plants as well, and the importance of Y129 for zmBX1 homodimerization. We generated two BX1 mutants, Y129A and Y129R, targeted to disrupt the dimer interface and cause monomerization. The alanine substitution was selected to cause the pocket and hydrogen bonding to not be filled, and the arginine substitution was selected to cause steric clash within the pocket. The amino acid substitutions were individually introduced by site-directed mutagenesis and the mutants were then expressed in E. coli and purified. Following purification, the mutants along with the wild type were analyzed by size-exclusion chromatography (SEC) as well as by SDS-PAGE after cross-linking in the presence of 1 mM DTT (Figure 4).
Figure 4.
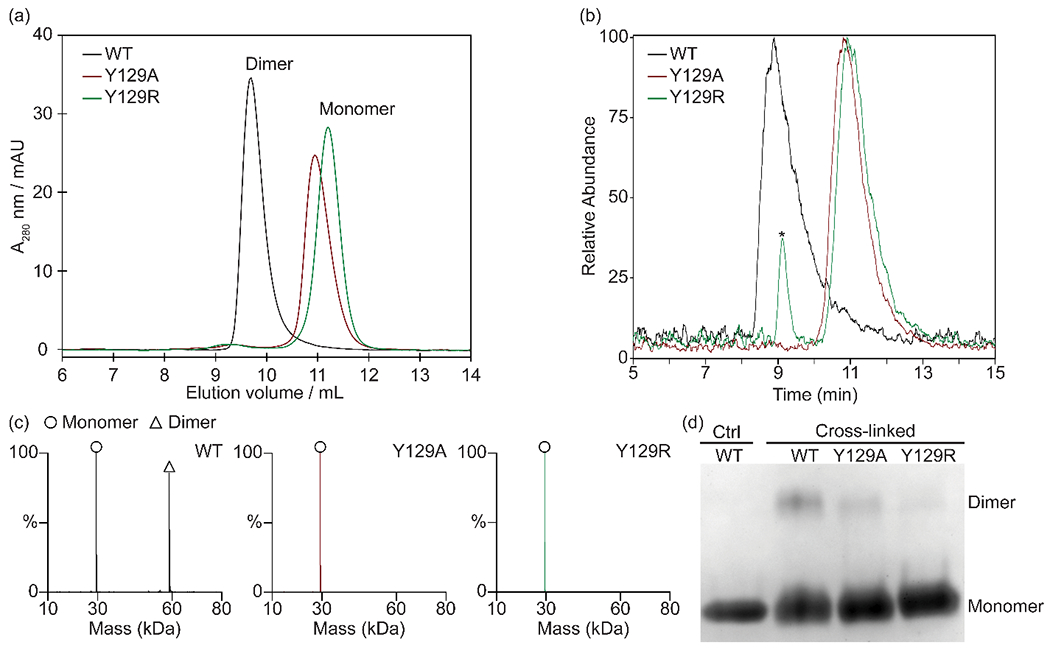
(a) SEC-UV chromatograms of zmBX1 wild type and mutants Y129A and Y129R. Oligomeric states as judged by elution time are indicated. (b) SEC-MS total ion chromatograms of zmBX1 wild type and mutants Y129A and Y129R. (c) Zero-charge mass spectra of SEC-MS peaks labeled with the oligomeric states. SEC-MS peak labeled with an asterisk was identified as a contaminant. Annotated mass spectra are shown in Figure S7. (c) SDS-PAGE of non-cross-linked (Ctrl) zmBX1 wild type and cross-linked zmBX1 wild type, Y129A and Y129R. Monomer and cross-linked dimer band positions on the gel are indicated.
Mutation of Y129 to alanine and to arginine led to an increase in elution time for SEC, consistent with the disruption of the zmBX1 dimer interface (Figure 4a). By using SEC coupled to native MS, we further confirmed zmBX1 Y129A (expected: 29,339 Da, determined: 29,338 Da) and Y129R (expected: 29,424 Da, determined: 29,423 Da) to be monomeric, and zmBX1 wild type (expected: 58,861 Da, determined: 58,861 Da) to be predominantly dimeric (Figure 4b and c). The SEC-UV and SEC-MS chromatograms demonstrate the consistency in the protein oligomeric states even when in an MS-compatible solution like ammonium acetate. The SEC results also agree with the SDS-PAGE data (Figure 4d) having a clear loss of intermolecular cross-links upon mutation of Y129 to alanine and to arginine. The presence of some cross-linked dimer in the mutant samples might be due to remaining weak interactions between the monomers that are still sufficient enough to drive the equilibrium towards dimer when DSSO is added due to the irreversible nature of the cross-linking reaction. Whereas the probability of intermolecular cross-linking is clearly higher for the wild type dimer compared to the monomerized variants, the overall cross-linking efficiency is still rather low as indicated by a large amount of monomer being detected by SDS-PAGE after denaturation. This is consistent with a relatively small number of intermolecular residues within DSSO cross-linking distances.
Physiological role of dimerization
We compared zmBX1 wild type with Y129A and Y129R to determine whether dimerization has a significant effect on enzymatic activity and thermal stability. We found that the catalytic efficiency of zmBX1 Y129A and Y129R is only one order of magnitude lower than that of zmBX1 wild type (Figure S8 and Table S5), suggesting that dimerization has only a moderate impact on enzyme catalysis. Thermal denaturation of zmBX1 wild type and the two mutants were carried out using differential scanning fluorimetry. Compared to wild type, the melting temperature (54 °C) decreased by roughly 4 °C for the mutants (Figure S9 and Table S5). Monomerizing zmBX1 only resulted in small changes to the biophysical properties of zmBX1 which suggests that reasons besides activity and stability may play a role in the observed difference in oligomeric state between zmBX1 and TrpA [11]. Taking into account that enzymes from secondary metabolism have most likely evolved from those of primary metabolism through gene duplication and subsequent specialization [31, 32], it is likely that an early BX1 predecessor might have been monomeric like TrpA and capable of interacting with the TrpA partner, the tryptophan synthase β-subunit, TrpB. In this context, BX1 homodimerization might have provided a way to prevent its interference with tryptophan biosynthesis for which the conserved orientation of the TrpA–TrpB interfaces is critical for the successful conversion of indole to L-tryptophan. In order to investigate this further, we examined the identified zmBX1 dimer structure and compared the zmBX1–zmBX1 and zmTrpA–zmTrpB interactions. As no structure of the tryptophan synthase from Zea mays (zmTS) was available, we generated a homology model of zmTS by SWISS-MODEL [33] using the closest related TS with known structure (last bacterial common ancestor TS; pdb id 5EY5) as the template. The zmTS model that we obtained had a Global Model Quality Estimate (GMQE) of 0.76 and QMEAN of −0.07 with 1 and 0, respectively, representing the highest reliability and agreement between the model and experimental structure. This model was used for a comparison of the zmBX1 and zmTrpA homo- and heterooligomerization interfaces, respectively (Figure 5).
Figure 5.
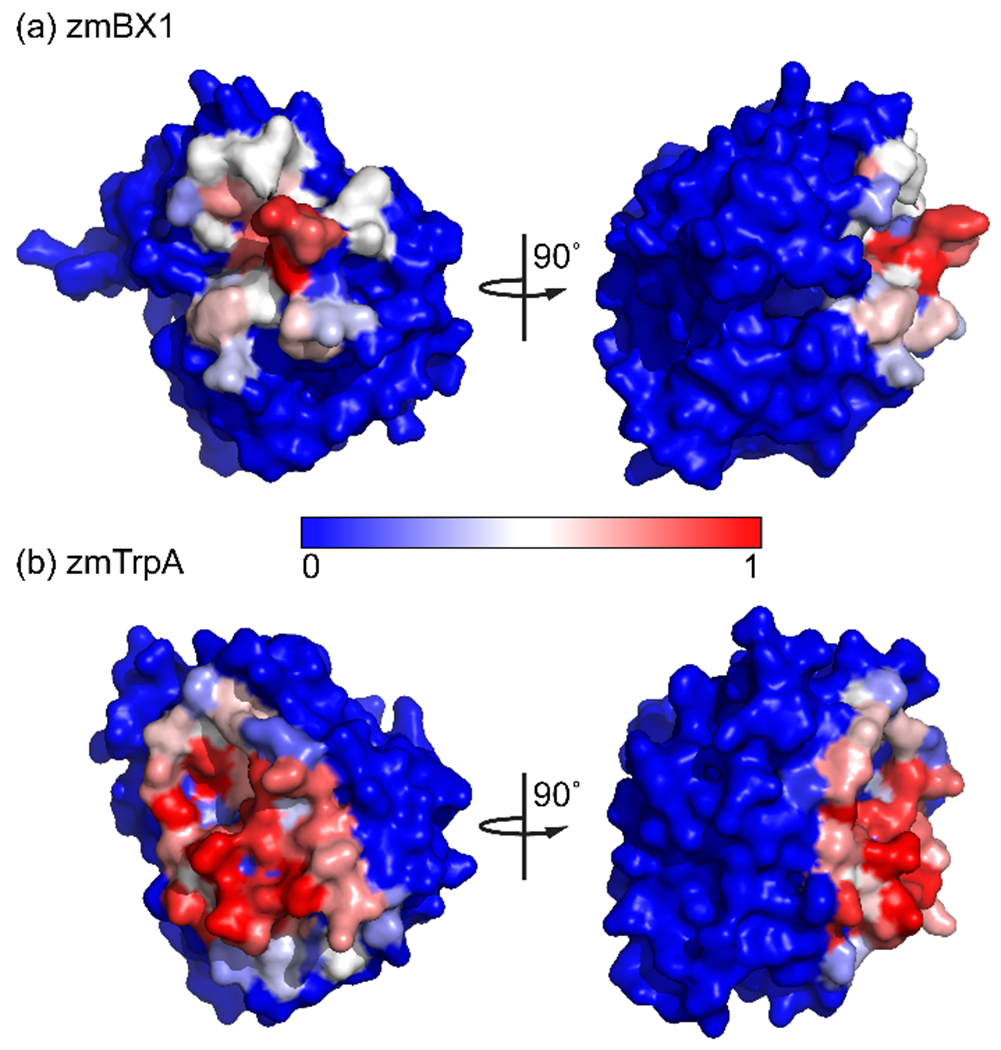
Comparison of (a) the homooligomerization interface of zmBX1 and (b) the heterooligomerization interface of zmTrpA. Surface representations of the proteins are shown for the same protein orientations after superposition in PyMOL. Coloring is according to the fraction of buried surface area as determined by PDBePISA, with fully exposed residues in blue and fully buried residues in red.
This analysis revealed an overlap of the interfaces of zmBX1 and zmTrpA. By considering the overlap in the TrpA–TrpB and BX1–BX1 interfaces, zmBX1 homodimerization suggests a mechanism of excluding interactions with TrpB to allow for the transition of this enzyme into a secondary metabolism pathway [11]. This makes it plausible that the dimerization of zmBX1 might have – at least at an early point in protein evolution – served to prevent the interaction with TrpB and thus eliminate cross-talk between primary and secondary metabolism.
Conclusions
During protein crystallization, initial protein-protein interactions can rearrange and additional interactions (crystal contacts) are formed. This makes it challenging to derive the physiologically relevant quaternary structure from protein crystal structures alone. We revisited the two zmBX1 crystal structures that are publicly available after it became apparent that zmBX1 is a dimer in solution. We found a single zmBX1-zmBX1 interface common in both protein crystals and confirmed the existence of the dimer by native MS and the physiological dimer structure using cross-linking MS as well as mutagenesis of a hotspot residue within this protein-protein interaction. The combination of crystal structure analysis with complementary structural biology techniques is, in our view, of general use to re-evaluate deposited crystal structures to reduce the number of proteins with mis-assigned quaternary states. The interface of zmBX1 makes it plausible that homodimerization might have been driven by the requirement to exclude the interaction with the tryptophan synthase β-subunit, TrpB, at an early time in evolution. This mechanism might have helped to prevent enzyme cross-talk and separated primary and secondary metabolism at an early stage after trpA duplication.
Supplementary Material
Figure S1: Alignment of the asymmetric units of 1RD5 and 1TJR
Figure S2: Venn diagram comparing trypsin versus Glu-C digestion
Figure S3: Cross-links identified with and without DTT mapped onto 1RD5 and compared in a Venn diagram
Figure S4: ZmBX1 construct with residue numbering to fit 1RD5
Figure S5: Annotated spectra for cross-link K104-K104
Figure S6: Annotated spectra for cross-link K160-K160
Figure S7: SEC-MS of zmBX1 wild type and mutants
Figure S8: Steady-state kinetics plots for zmBX1 wild type and mutants
Figure S9: Thermal denaturation plots of zmBX1 wild type and mutants
Table S1: Primers used for site-directed mutagenesis
Table S2: Amino acid sequences for the zmBX1 wild type and mutant constructs.
Table S3: Identified cross-links and their measured distances for the top three interfaces of 1RD5
Table S4: Identified cross-links and their measured distances for the top three interfaces of 1TJR
Table S5: Kinetic parameters and apparent midpoint temperatures determined for zmBX1 wild type and mutants
Acknowledgements
This work was supported by a grant from the National Institutes of Health to V.H.W. (NIH P41GM128577). The Fusion Orbitrap instrument was supported by NIH Award Number Grant S10 OD018056. The cDNA of Z. mays W22 was a generous donation from Dr. Kevin Begcy (University of Regensburg). We thank Liwen Zhang, Sonja Fuchs, Christiane Endres, Jeannette Ueckert for expert technical assistance, Federica Accornero for access to the ChemiDoc Touch Imaging System (Bio-Rad Laboratories), and 2bind GmbH for access to the Prometheus NT.48 instrument (NanoTemper Technologies).
References
- 1.Hyde C, Ahmed SA, Padlan EA, Miles EW, Davies DR: Three-dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263, 17857–17871 (1988) [PubMed] [Google Scholar]
- 2.Dunn MF: Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys. 519, 154–166 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miles EW: The tryptophan synthase α2β2 complex: a model for substrate channeling, allosteric communication, and pyridoxal phosphate catalysis. J. Biol. Chem. 288, 10084–10091 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyde CC, Miles EW: The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Biotechnology. 8, 27–32 (1990) [DOI] [PubMed] [Google Scholar]
- 5.Hettwer S, Sterner R: A novel tryptophan synthase β-subunit from the hyperthermophile Thermotoga maritima: quaternary structure, steady-state kinetics, and putative physiological role. J. Biol. Chem. 277, 8194–8201 (2002) [DOI] [PubMed] [Google Scholar]
- 6.Ngo H, Kimmich N, Harris R, Niks D, Blumenstein L, Kulik V, Barends TR, Schlichting I, Dunn MF: Allosteric regulation of substrate channeling in tryptophan synthase: modulation of the L-serine reaction in stage I of the β-reaction by α-site ligands. Biochemistry. 46, 7740–7753 (2007) [DOI] [PubMed] [Google Scholar]
- 7.Kriechbaumer V, Weigang L, Fießelmann A, Letzel T, Frey M, Gierl A, Glawischnig E: Characterisation of the tryptophan synthase alpha subunit in maize. BMC Plant Biol. 8 (44), 1–11 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulik V, Hartmann E, Weyand M, Frey M, Gierl A, Niks D, Dunn MF, Schlichting I: On the structural basis of the catalytic mechanism and the regulation of the alpha subunit of tryptophan synthase from Salmonella typhimurium and BX1 from Maize, two Evolutionarily Related Enzymes. J. Mol. Biol. 352, 608–620 (2005) [DOI] [PubMed] [Google Scholar]
- 9.Frey M, Chomet P, Glawischnig E, Stettner C, Grün S, Winklmair A, Eisenreich W, Bacher A, Meeley RB, Briggs SP, Simcox K, Gierl A: Analysis of a chemical plant defense mechanism in grasses. Science. 277, 696–699 (1997) [DOI] [PubMed] [Google Scholar]
- 10.Frey M, Schullehner K, Dick R, Fiesselmann A, Gierl A: Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry. 70, 1645–1651 (2009) [DOI] [PubMed] [Google Scholar]
- 11.Schupfner M, Busch F, Wysocki V, Sterner R: Generation of a stand-alone tryptophan synthase α-subunit by mimicking an evolutionary blueprint. ChemBioChem. 20, 2742–2751(2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capitani G, Duarte JM, Baskaran K, Bliven S, Somody JC: Understanding the fabric of protein crystals: computational classification of biological interfaces and crystal contacts. Bioinformatics. 32, 481–489 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krissinel E: Macromolecular complexes in crystals and solutions. Acta Crystallogr. D. Biol. Crystallogr. 67, 376–385 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy ED: PiQSi: Protein Quaternary Structure Investigation. Structure. 15, 1364–1367 (2007) [DOI] [PubMed] [Google Scholar]
- 15.Peterhoff D, Zellner H, Guldan H, Merkl R, Sterner R, Babinger P: Dimerization determines substrate specificity of a bacterial prenyltransferase. ChemBioChem. 13, 1297–1303 (2012) [DOI] [PubMed] [Google Scholar]
- 16.Kao A, Chiu C, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L: Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics. 10, M110002212 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu F, Lössl P, Scheltema R, Viner R, Heck AJRR: Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 8:15473, 1–8 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, Rijkers DTS, Post H, Heck AJR: Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 12, 1179–1184 (2015) [DOI] [PubMed] [Google Scholar]
- 19.Vanaernum ZL, Gilbert JD, Belov ME, Makarov AA, Horning SR, Wysocki VH: Surface-induced dissociation of noncovalent protein complexes in an extended mass range orbitrap mass spectrometer. Anal. Chem. 91, 3611–3618 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV: Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creighton TE: A steady-state kinetic investigation of the reaction mechanism of the tryptophan synthetase of Escherichia coli. Eur. J. Biochem. 13, 1–10 (1970) [DOI] [PubMed] [Google Scholar]
- 22.Tsuchiya Y, Nakamura H, Kinoshita K: Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinforma. Chem. 1, 99–113 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krissinel E, Henrick K: Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007) [DOI] [PubMed] [Google Scholar]
- 24.Schrodinger LLC: The PyMOL Molecular Graphics System, Version 2.3.2, (2015)
- 25.Mädler S, Bich C, Touboul D, Zenobi R: Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J. Mass Spectrom. 44, 694–706 (2009) [DOI] [PubMed] [Google Scholar]
- 26.Sinz A: Divide and conquer: cleavable cross-linkers to study protein conformation and protein–protein interactions. Anal. Bioanal. Chem. 409, 33–44 (2017) [DOI] [PubMed] [Google Scholar]
- 27.Viner R, Bomgarden R, Singhal K, Snovida S, Gutierrex C, Huang L: Enrichment strategies for improvement of mass spec analysis of chemical cross-linked peptides. 290105, Thursday, Proceedings of the 65th ASMS Conference on Mass Spectrometry and Allied Topics, Indianapolis, Indiana, June 4–8 (2017) [Google Scholar]
- 28.Bomgarden R, Raja E, Etienne C, Liu F, Heck AJR, Mueller M, Viner R: Optimization of crosslinked peptide analysis on an orbitrap fusion lumos mass spectrometer. 280999, Tuesday, Proceedings of the 64th ASMS Conference on Mass Spectrometry and Allied Topics, San Antonio, Texas, June 5–9 (2016) [Google Scholar]
- 29.Klykov O, Steigenberger B, Pektaş S, Fasci D, Heck AJR, Scheltema RA: Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 13, 2964–2990 (2018) [DOI] [PubMed] [Google Scholar]
- 30.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG: Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7 (539), 1–6 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T: Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in arabidopsis. Plant Cell. 13, 681–693 (2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moghe GD, Last RL: Something old, something new: Conserved enzymes and the evolution of novelty in plant specialized metabolism. Plant Physiol. 169, 1512–1523 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T: SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Alignment of the asymmetric units of 1RD5 and 1TJR
Figure S2: Venn diagram comparing trypsin versus Glu-C digestion
Figure S3: Cross-links identified with and without DTT mapped onto 1RD5 and compared in a Venn diagram
Figure S4: ZmBX1 construct with residue numbering to fit 1RD5
Figure S5: Annotated spectra for cross-link K104-K104
Figure S6: Annotated spectra for cross-link K160-K160
Figure S7: SEC-MS of zmBX1 wild type and mutants
Figure S8: Steady-state kinetics plots for zmBX1 wild type and mutants
Figure S9: Thermal denaturation plots of zmBX1 wild type and mutants
Table S1: Primers used for site-directed mutagenesis
Table S2: Amino acid sequences for the zmBX1 wild type and mutant constructs.
Table S3: Identified cross-links and their measured distances for the top three interfaces of 1RD5
Table S4: Identified cross-links and their measured distances for the top three interfaces of 1TJR
Table S5: Kinetic parameters and apparent midpoint temperatures determined for zmBX1 wild type and mutants